

Abstract
Vascular endothelial growth factor (VEGF) contributes to blood-retinal barrier (BRB) dysfunction in several blinding eye diseases, including diabetic retinopathy. Signaling via the secreted protein norrin through the frizzled class receptor 4 (FZD4)/LDL receptor–related protein 5–6 (LRP5–6)/tetraspanin 12 (TSPAN12) receptor complex is required for developmental vascularization and BRB formation. Here, we tested the hypothesis that norrin restores BRB properties after VEGF-induced vascular permeability in diabetic rats or in animals intravitreally injected with cytokines. Intravitreal co-injection of norrin with VEGF completely ablated VEGF-induced BRB permeability to Evans Blue-albumin. Likewise, 5-month diabetic rats exhibited increased permeability of FITC-albumin, and a single norrin injection restored BRB properties. These results were corroborated in vitro, where co-stimulation of norrin with VEGF or stimulation of norrin after VEGF exposure restored barrier properties, indicated by electrical resistance or 70-kDa RITC-dextran permeability in primary endothelial cell culture. Interestingly, VEGF promoted norrin signaling by increasing the FZD4 co-receptor TSPAN12 at cell membranes in an MAPK/ERK kinase (MEK)/ERK-dependent manner. Norrin signaling through β-catenin was required for BRB restoration, but glycogen synthase kinase 3 α/β (GSK-3α/β) inhibition did not restore BRB properties. Moreover, levels of the tight junction protein claudin-5 were increased with norrin and VEGF or with VEGF alone, but both norrin and VEGF were required for enriched claudin-5 localization at the tight junction. These results suggest that VEGF simultaneously induces vascular permeability and promotes responsiveness to norrin. Norrin, in turn, restores tight junction complex organization and BRB properties in a β-catenin–dependent manner.
Keywords: vascular endothelial growth factor (VEGF), tight junction, permeability, retina, endothelium, cell signaling, tetraspanin, Wnt signaling, blood-retinal barrier, diabetic retinopathy, norrin, tetraspanin 12 (TSPAN12)
Introduction
Müller cells (1–3) and endothelial cells (4) of the developing retina express norrin that contributes to proper angiogenesis and the formation of the blood-retinal barrier (BRB)2 (5). Norrin is a secreted 131-amino acid protein from the cysteine-knot growth factor superfamily that includes transforming growth factor β (6) and utilizes the wingless/integrated (Wnt) signaling pathway. Norrin forms a dimer that binds to the N-terminal extracellular cysteine-rich domain of the frizzled class receptor 4 (FZD4) and the β-propeller domains of the low-density lipoprotein receptor–related protein 5/6 (LRP5/6) co-receptor (7), activating the β-catenin canonical signaling pathway. In addition, the co-activator tetraspanin 12 (TSPAN12) binds and stabilizes FZD4 receptor at the cell membrane and enhances norrin-induced, but not Wnt-induced, β-catenin signaling (8–10). The canonical pathway of FZD4 signaling involves β-catenin–mediated transcriptional regulation. The APC destruction complex, formed by adenomatosis polyposis coli (APC), axin, protein phosphatase 2a, casein kinase 1α, and glycogen synthase kinase 3α/β (GSK-3α/β), phosphorylates and targets β-catenin for ubiquitination and proteosomal degradation. Norrin-binding FZD4 receptor complex inactivates the APC degradation complex and inhibits GSK-3α/β kinase, stabilizing β-catenin, which migrates to the nucleus and promotes gene transcription (reviewed in Ref. 11).
Mutations in norrin and its receptors may cause a spectrum of inherited exudative retinopathies. Mutations in the norrin gene (NDP) cause an X-linked retinal dysplasia on the severe end of the spectrum that presents with congenital or early childhood blindness, called Norrie disease (12–15). The retinal hypovascularization disorders, referred to as familial exudative vitreopathy (FEVR), are caused by mutations in the genes encoding for norrin receptor FZD4 (16–18), co-receptors LRP5 (19–21) and TSPAN12 (16, 17, 23, 24), the signaling molecule β-catenin (25, 26), and some norrin mutations (27). The hypovascular phenotype observed in both Norrie and FEVR diseases has been recapitulated in knockout mice models of norrin or the FZD4 receptor complex (1, 5, 8, 28–34), in which retinal vascular growth, mural cell recruitment, endothelial differentiation, and barrier properties are affected as a consequence of a low Sox7, Sox17, and Sox18 gene expression (35) or due to an increased expression of the Wnt signaling inhibitor APCDD1 (APC down-regulated 1 protein) (36). Importantly, these retinas show high retinal vascular permeability that correlates with reduced border immunostaining of the tight junction protein claudin-5 and increased expression of the transcytosis marker PLVAP, a phenotype that can be reversed by the expression of stabilized β-catenin (5). Together, these data indicate a requirement of norrin signaling through β-catenin in deep capillary angiogenesis, BRB formation and maintenance.
Loss of norrin or TSPAN12 in mouse retina reveals a phenotype that resembles some of the pathological features of diabetic retinopathy. Retinas from mice with norrin gene deletion showed formation of cystoid edema, neovascularization, and inflammation (37), whereas endothelium-specific loss of TSPAN12 induces cystoid edema formation and basement membrane collagen IV deposition (29). Other studies suggest that norrin (38) and β-catenin (39) increase in diabetes as determined in retinal sections of post-mortem human eyes, whereas in Akita mice and streptozotocin (STZ)-induced diabetic rats, both animal models of diabetes, β-catenin, and LRP5/6 were elevated (39). Nevertheless, the role of norrin signaling during diabetic retinopathy is not completely understood.
The release of cytokines, including vascular endothelial growth factor (VEGF), contributes to the pathophysiology of diabetic retinopathy through the disruption of the BRB (40). At a molecular level, it is known that VEGF signaling through protein kinase Cβ promotes occludin phosphorylation, which results in disorganization of the retinal endothelial tight junction complex and vascular dysfunction (41–43). However, limited studies have investigated restoration of the retinal endothelial barrier after VEGF-targeted disruption. The role of norrin signaling in BRB formation suggests that it may have the potential to restore BRB properties after induced retinal vascular permeability. Here, we demonstrate that VEGF and norrin interact in regulating barrier properties. VEGF induces permeability but simultaneously primes norrin signaling by promoting TSPAN12 co-receptor localization at the cell membrane. Subsequent norrin signaling promotes claudin-5 organization at the cell border and BRB restoration. This response is demonstrated in two in vivo models of BRB loss and in a cell culture model, thus supporting the potential of norrin as a therapeutic option to restore the BRB in diseases such as diabetic retinopathy.
Results
Norrin restores BRB properties in vivo
To determine the ability of norrin to counteract VEGF-induced BRB dysfunction, retinal permeability to Evans Blue dye was determined in rat retinas, 24 h after their intravitreal (IVT) injection with 40 ng of norrin or 50 ng of VEGF or with the co-injection of the two cytokines, VEGF/norrin. Control rats (vehicle-injected) showed an average of 2.5 μl/g/h of accumulated dye, and all conditions were normalized to this value. We have found that permeability in response to norrin injection was not different from control values, whereas, as expected, retinas injected with VEGF showed a significant increase in the accumulation of Evans Blue dye. Strikingly, the simultaneous injection of VEGF/norrin completely ablated the VEGF effect (Fig. 1A), thus indicating that norrin can inhibit VEGF-induced leakiness in the retina.
Figure 1.
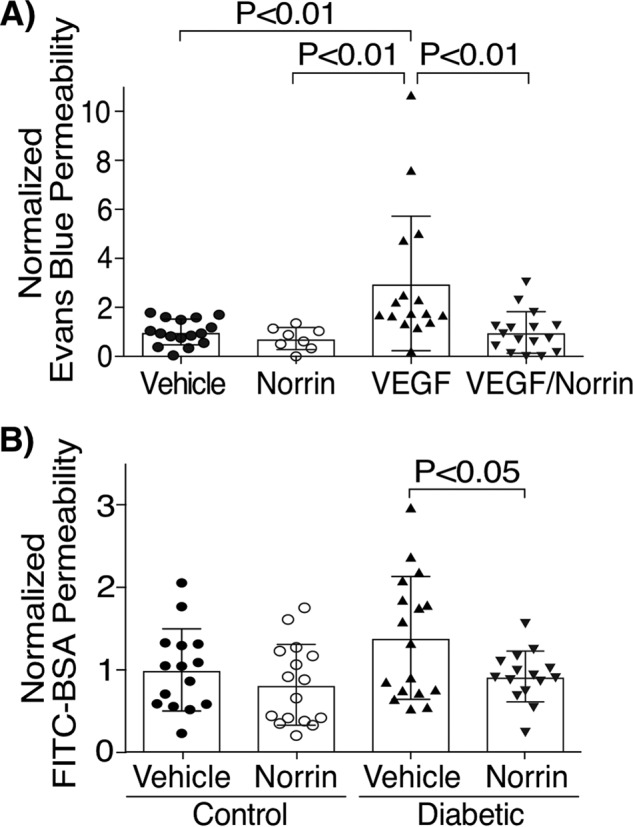
Norrin restores blood-retinal barrier permeability in vivo. A, Evans Blue accumulation in rat retinas with an IVT injection of vehicle, 40 ng of norrin, 50 ng of VEGF, or both (VEGF/norrin) for 24 h; n = 8–16. B, FITC-BSA accumulation in rat retinas of control (nondiabetic) or STZ-induced diabetic rats that were treated after 5 months with an IVT injection of vehicle or 40 ng of norrin for 24 h; n = 15–17. p values were calculated by one-way ANOVA, followed by Tukey's post hoc test (A) or t test analysis (B). Error bars, S.D.
To determine whether norrin is able to restore BRB properties, we also performed permeability assays in diabetic rats. After 5 months of STZ-induced diabetes, rats were treated with an IVT injection of vehicle in right eyes (OD) or 40 ng of norrin in left eyes (OS), and permeability was measured 24 h later by determining the retinal accumulation of intravenously injected FITC-BSA. Nondiabetic rats (control) injected with vehicle showed a 15.5-μl/g/h average of dye accumulation, and this was used for data normalization. Similar permeability to FITC-BSA was found in control rat eyes injected with norrin (Fig. 1B). Notably, diabetic rats had higher permeability of FITC-BSA, and this was completely restored to the control levels by norrin injection. Together, these results demonstrate the ability of norrin to restore BRB properties in two rat models of vascular dysfunction.
Norrin rescues VEGF-induced permeability in bovine retinal endothelial cells (BREC)
We further corroborated norrin effect in a primary culture of BREC. Cells were grown until confluence for 24 h and BRB formation was induced for the next 48 h with medium supplemented with 1% serum and 100 mm hydrocortisone (Fig. 2A). Subsequently, BREC monolayers were stimulated with vehicle, 40 ng/ml norrin (N), 50 ng/ml VEGF (V), or norrin together with VEGF (VEGF/norrin), and real-time measurements of trans-endothelial electrical resistance (TEER) (Fig. 2B) or diffusive permeability of 70-kDa RITC-dextran (Fig. 2C) were determined. All monolayers reached TEER values above 3,500 ohms (Fig. 2A) before stimulation, and data were normalized to time 0 baseline for each sample. In dextran flux assays, control monolayers had an average diffusive permeability (Po) of 1.5 × 10−6, and all conditions were normalized to this control value. Our results show that VEGF induced a reduction of TEER of ∼40% of the control value from 24 to 72 h and a 2-fold increase in the permeability of 70-kDa RITC-dextran by 72 h, whereas basal levels were not affected by norrin stimulation. The addition of both VEGF/norrin simultaneously led to an initial loss of TEER over the first 24 h, followed by complete barrier restoration by 72 h and complete restoration of dextran flux to control values at 72 h. These results indicate that norrin restores BRB properties after 24 h of VEGF stimulation.
Figure 2.
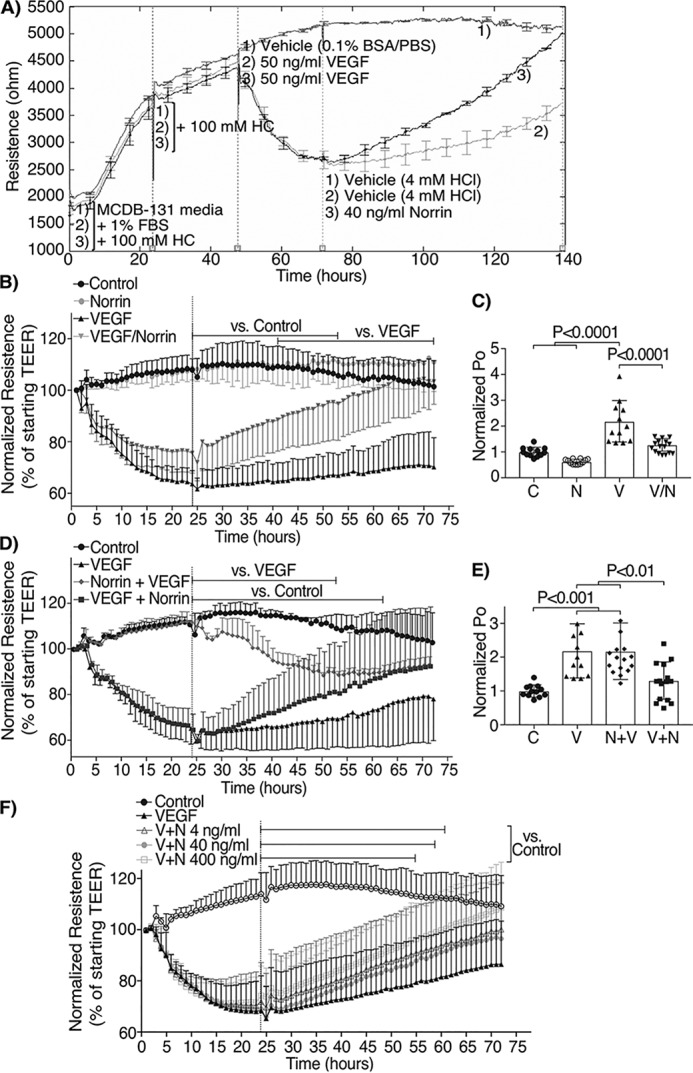
Norrin reverses and restores barrier properties after VEGF-induced permeability of BREC monolayers. A, B, D, and F, real-time TEER measurements; n = 4–7. A, sample experiment showing TEER values in ohms. First vertical line, start of experiment in MCDB-131 with 1% FBS and hydrocortisone (HC); second line, the addition of hydrocortisone; third line, the addition of VEGF or vehicle; fourth line, the addition of norrin or vehicle. B, D, and F, average of multiple TEER experiments normalized to t = 0. C and E, flux to 70-kDa RITC-dextran (Po), measured 72 h after last stimuli and over 4 h; n = 12–16. BREC were stimulated with vehicle (control, C), 40 ng/ml norrin (N), 50 ng/ml VEGF (V), or their combination. In B and C, both VEGF and norrin (V/N) were added at t = 0; in D and E, norrin stimulation was at t = 0 and VEGF at 24 h (N+V), or VEGF at t = 0 and norrin at 24 h (V+N). F, dose-response of norrin added 24 h after VEGF with TEER measurements. p values were calculated using two-way (B, D, and F) or one-way (C and E) ANOVA, followed by Tukey's post hoc test. The top lines in B, D, and F indicate the time points where there was a significant difference from 24 to 72 h, comparing the indicated monolayers with control or VEGF conditions. Monolayers stimulated only with VEGF were different from control at all time points. Error bars, S.D.
To better understand the requirement of VEGF activation in norrin-induced BRB restoration, BREC monolayers were prestimulated with norrin and then with VEGF 24 h later (N + V) or prestimulated with VEGF and then norrin 24 h later (V + N). As shown in Fig. 2 (D and E), prestimulation with norrin failed to prevent the VEGF-induced permeability to dextran, although the effect on TEER appeared attenuated by 48–72 h with a reduction of only 15% of control. However, when norrin was added 24 h after the addition of VEGF, norrin promoted barrier restoration measured both by TEER and dextran permeability. This effect of norrin restoration after VEGF addition was demonstrated to be dose-responsive (Fig. 2F). Because 40 ng/ml was enough to recover BRB properties in vivo and in vitro, we choose 40 ng/ml norrin as a working concentration for all other experiments. Together, these results indicate that norrin is able to restore the endothelial barrier properties after VEGF-induced permeability.
VEGF signaling facilitates TSPAN12 membrane localization
TEER results and dextran flux experiments indicate that norrin increases barrier properties only after VEGF stimulation, suggesting a requirement for VEGF signaling for norrin action in the endothelial cells. To evaluate this possibility, we determined whether VEGF regulates the expression of norrin receptor FZD4 or the LRP5 and TSPAN12 co-receptors. BREC monolayers were stimulated as indicated and qRT-PCR was performed to assess mRNA content. As shown in Fig. 3A, VEGF alone or norrin stimulation 24 h after VEGF (V + N), significantly increased TSPAN12 mRNA at 48 and 72 h, whereas FZD4 or LRP5 mRNA content was not significantly affected (Fig. S1, A and B). However, this increase occurred later than the observed VEGF/norrin recovery of TEER (Fig. 2B). Therefore, we next examined TSPAN12 protein content and localization. BREC monolayers were separated into fractions based on centrifugation using a compartmental protein extraction kit. Cell fractionation was demonstrated to selectively isolate markers: GAPDH in the cytoplasmic fraction, histone H3 in the nuclear fraction, and EGFR in the membrane fraction (Fig. 3B). Whole-cell lysates demonstrate the detection of TSPAN12 at the predicted molecular mass (35 kDa) and an additional band around 37 kDa (Fig. 3C). The total content of both bands was not affected by the addition of norrin and/or VEGF (Fig. 3D). However, in cell fractionation studies, VEGF and VEGF/norrin promoted TSPAN12 accumulation at the membrane fractions, particularly for the 37 kDa band. TSPAN12 also demonstrated a slight but significant increase in the cytosol with the addition of VEGF or VEGF/norrin together.
Figure 3.

VEGF facilitates norrin signaling by the induction of TSPAN12 expression and localization at the membrane. A, qRT-PCR of TSPAN12 after the addition of vehicle (C), norrin (N), VEGF (V) or both (V/N) for 12, 24, 48, and 72 h or VEGF for the first 24 h and norrin for the next 24 or 48 h (V+N; 48 and 72 h total). All samples were normalized to control (C) for 12 h; n = 4–6. B, representative image of lysates of BREC monolayers stimulated as indicated and then separated in cytosolic, nuclear, and membrane cell fractions, followed by blotting with antibodies against GAPDH, histone H3, and EGFR. C, BREC stimulated for 24 h were separated into cytoplasmic or membrane fractions and processed for Western blotting and the detection of TSPAN12. D, quantification of TSPAN12 content in cell fractions; n = 7–8. E, Western blotting of TSPAN12 on cell fractions of BREC monolayers preincubated with the protein synthesis inhibitor CHX (6 μg/ml) for 2 h before the addition of VEGF, the lysosome inhibitor hydroxychloroquine sulfate (Chl; 25 μm), or the proteasome inhibitor lactacystin (Lac; 5 μm) for 24 h. E, quantification of independent experiments; n = 8. p values were calculated by one-way ANOVA, followed by Tukey's post hoc test. Error bars, S.D.
Experiments suggest that VEGF increases TSPAN12 membrane content through translational control. Inhibition of protein synthesis with the addition of cycloheximide (CHX) was sufficient to prevent the VEGF-induced accumulation of TSPAN12 at the membrane (Fig. 3, E and F), whereas the presence of lysosome or proteasome inhibitors (hydroxychloroquine sulfate (Chl) or lactacystin (Lac), respectively) had no additional effect. Because TSPAN12 mRNA was not altered by VEGF at this time point (Fig. 3A) and the increase in protein was inhibited by the protein synthesis inhibitor, the results suggest that VEGF regulates TSPAN12 translation. Moreover, VEGF effect was specific for TSPAN12 because the content of FZD4 or LRP5/6 receptors in cytosol or membrane fractions was not significantly affected (Fig. S1, C and D).
The VEGF effect on TSPAN12 membrane content depended on MEK/ERK signaling. The specific MEK kinase inhibitor U0126 was added 30 min before VEGF addition, and cell fractions were isolated. As expected, U0126 completely blocked ERK phosphorylation in response to VEGF. Further, the MEK inhibitor prevented VEGF-induced TSPAN12 accumulation at the membrane (Fig. 4, A and B). These results were further corroborated by immunostaining the extracellular portion of TSPAN12 in nonpermeabilized and permeabilized BREC monolayers (Fig. 4C). Again, VEGF increased the amount of TSPAN12 exposed to the cell surface that was detected with a specific antibody against the extracellular regions of TSPAN12, and this effect was prevented by U0126 (Fig. 4D).
Figure 4.
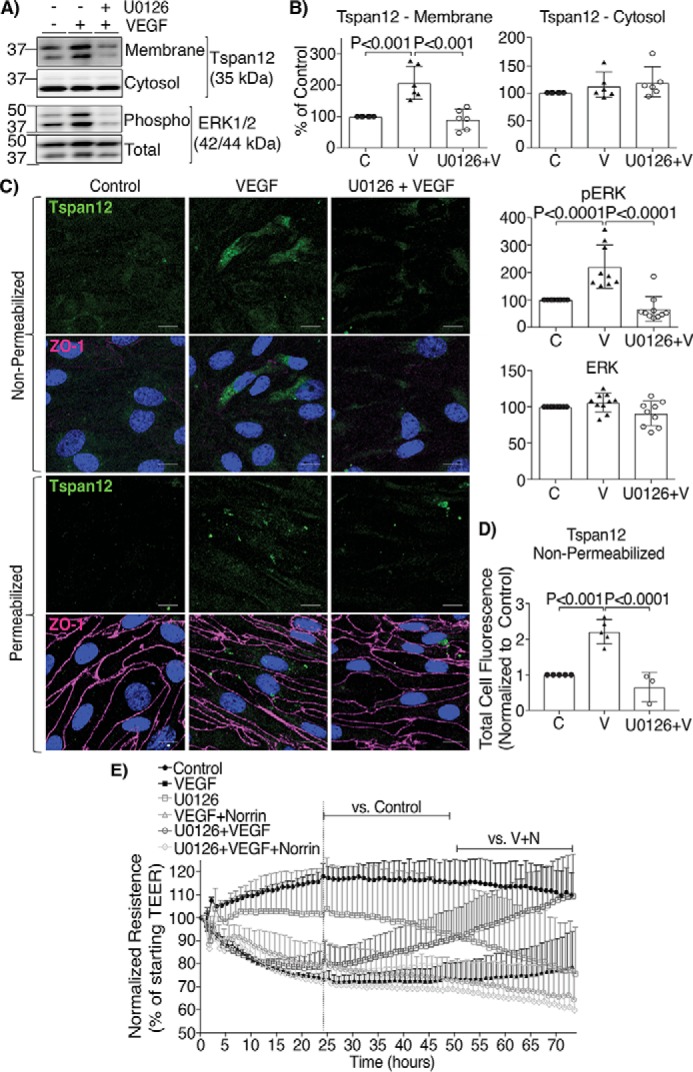
VEGF effect on norrin signaling depended on MEK/ERK1/2 signaling. A–D, BREC monolayers were preincubated for 30 min with a specific inhibitor of MEK/ERK1/2 (U0126, 10 μm) followed by the addition of VEGF for 24 h (U0126+V). A and B, TSPAN12 (cell fractions), phospho and total ERK (whole cell lysates) were quantified by Western blot; n=6. C, BREC monolayers were processed for the immunostaining of TSPAN12 extracellular region (green), ZO-1 (magenta), and Hoechst (blue). Some monolayers were not permeabilized to detect TSPAN12 localization only at the membrane, and some monolayers were permeabilized as controls; scale bar = 10 μm. D, quantification of total green fluorescence staining from nonpermeabilized monolayers. Two images per coverslip were taken from at least three independent experiments with 2–3 coverslips/condition. E, TEER of BREC stimulated with vehicle (Control), VEGF, U0126, VEGF for the first 24 h and then norrin (VEGF+Norrin; V+N), U0126 for 30 min and then VEGF at t = 0 (U0126+VEGF), or U0126 + VEGF followed by the addition of norrin at 24 h (U0126+VEGF+Norrin); n = 4–6. p values were calculated by one-way (B and D) or two-way (E) ANOVA, followed by Tukey's post-hoc test. The top lines in E indicate the time points where there was a significant difference from 24 to 72 h, comparing the condition indicated at the left with control or V + N. All VEGF-stimulated monolayers were significantly different from control. Error bars, S.D.
Inhibiting TSPAN12 membrane localization prevented norrin barrier restoration after VEGF. TEER measurements revealed that prestimulation of BREC with U0126 does not prevent VEGF-induced permeability, and, in fact, U0126 alone decreased barrier properties at about 50 h (Fig. 4E). However, MEK/ERK signaling inhibition, which prevented VEGF-induced increase in TSPAN12 membrane content, completely ablated the ability of norrin to restore barrier properties after VEGF (Fig. 4E). Together, these results suggest that VEGF primes norrin signaling by inducing TSPAN12 accumulation at the membrane, in a mechanism dependent on MEK/ERK signaling, and that over time, VEGF and norrin together induce TSPAN12 expression.
Norrin signaling through β-catenin is required to restore BRB properties
We also evaluated whether norrin activation of the Wnt/β-catenin signaling pathway is required for barrier restoration in BREC. First, we demonstrated that stimulation of endothelial cells with norrin did not alter VEGF signaling (Fig. S2). Next, we examined the Wnt signaling pathway by measuring β-catenin stabilization and nuclear translocation after norrin signaling by blotting for total β-catenin in whole-cell lysates and cell fractions. As shown in Fig. 5 (A and B), VEGF/norrin together promoted β-catenin stabilization and increased total β-catenin content. This was reflected in significantly increased β-catenin in both the cytosol and nuclear fractions after VEGF/norrin co-stimulation (Fig. 5, C and D). Axin2 mRNA was quantified as a target of β-catenin signaling (Fig. 5E). Norrin alone was sufficient to increase axin2 mRNA at 24 h, which was maintained for 72 h, revealing intact norrin signaling in the BREC. Interestingly, co-stimulation with VEGF and norrin demonstrated an inhibitory effect on norrin-induced axin2 mRNA expression despite the observation that accumulation of β-catenin in the nuclear fraction was strongest under these conditions, suggesting that VEGF may affect axin2 mRNA content in a mechanism separate from β-catenin stabilization. Collectively, these results indicate that norrin activates canonical Wnt signaling in BREC and that VEGF and norrin together promote β-catenin stabilization and nuclear translocation, consistent with VEGF-induced TSPAN12 plasma membrane localization promoting norrin signaling.
Figure 5.

Norrin and VEGF promote β-catenin stabilization. Lysates of BREC stimulated with vehicle (C), norrin (N), VEGF (V), or both (V/N) for 72 h were blotted for phospho-β-catenin Ser-33/Ser-47/Thr-41 and total protein (n = 5) (A and B) or separated in cytosolic, nuclear, and membrane cell fractions (C and D), followed by blotting with antibody against total β-catenin (n = 7). E, qRT-PCR of axin2 from BREC stimulated as indicated; n = 5–11. All samples were normalized to control (C), 12 h. p values were calculated by one-way ANOVA, followed by Tukey's post hoc test. Error bars, S.D.
To assess whether the canonical Wnt/β-catenin signaling pathway is required to restore BRB properties in BREC, we used XAV-939, a potent tankyrase inhibitor that promotes axin stabilization and subsequently β-catenin degradation. Axin2 mRNA content was used as a measure of β-catenin signaling activity, and XAV-939 stimulation reduced axin2 mRNA as expected (Fig. S3A). TEER measurements of BREC monolayers stimulated with VEGF, followed by the addition of XAV-939 at 24 h (VEGF+XAV-939) (Fig. 6A), showed that loss of β-catenin signaling further reduced barrier properties of VEGF-stimulated monolayers. Moreover, in BREC stimulated with VEGF for 24 h, followed by the simultaneous addition of XAV-939 and norrin (VEGF+XAV-939/norrin), the ability of norrin to restore barrier properties was reduced.
Figure 6.
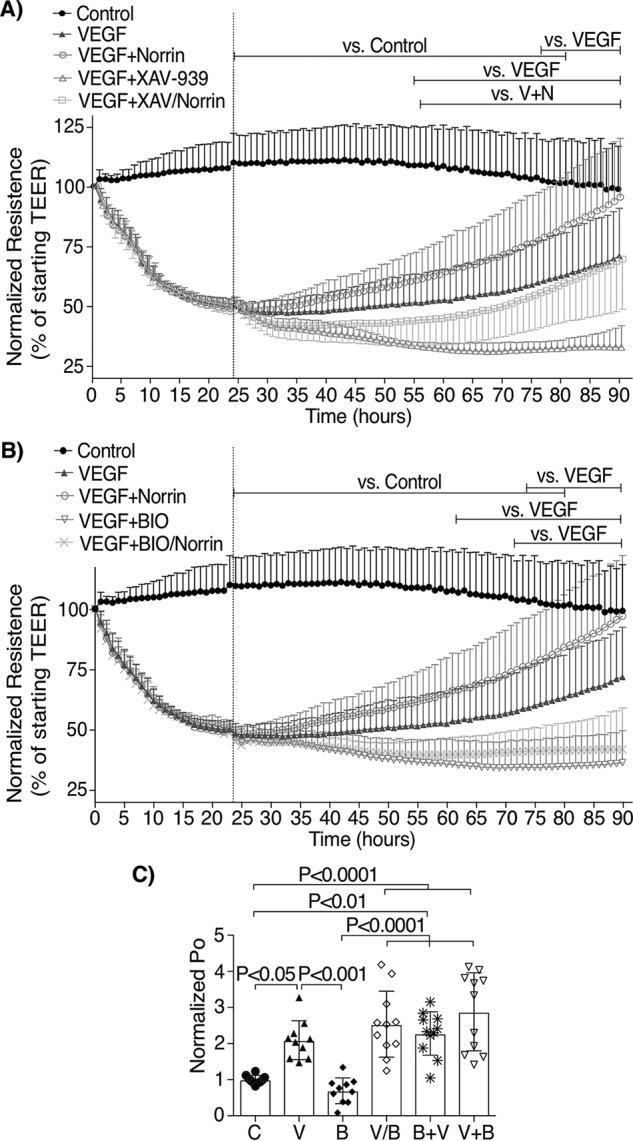
β-Catenin inhibition prevents norrin-induced restoration of TEER, but GSK-3α/β inhibition is not sufficient to reverse VEGF-induced permeability in BREC monolayers. A, TEER of BREC stimulated with vehicle (Control), VEGF (V), VEGF + norrin at 24 h (V+N), VEGF + XAV-939 (1 μm) at 24 h (VEGF+XAV), or VEGF followed by XAV-939 and norrin together at 24 h (VEGF+XAV/Norrin); n = 4–5. B, TEER of BREC monolayers stimulated with vehicle (Control), VEGF, VEGF + norrin at 24 h, VEGF + BIO (0.2 μm) at 24 h (V+B), or VEGF followed by BIO and norrin together at 24 h (VEGF+BIO/Norrin); n = 4–5. C, permeability of 70-kDa RITC-dextran (Po) of BREC monolayers stimulated with vehicle (C), VEGF (V), BIO (B), or BIO and VEGF together (V/B) for 72 h or stimulated with BIO (72 h total) and then VEGF for the last 48 h (B+V) or stimulated with VEGF (72 h total) before BIO for the last 48 h (V+B); n = 10–11. p values were calculated using two-way (A and B) or one-way (C) ANOVA, followed by Tukey's post hoc test. The top lines in A and B indicate the time points where there was a significant difference from 24 to 72 h, comparing the condition indicated at the left with control, VEGF, or V + N. All VEGF-stimulated monolayers were significantly different from control. Error bars, S.D.
To determine whether β-catenin activation is sufficient to restore barrier properties after VEGF-induced permeability, the GSK-3α/β inhibitor BIO was tested. Fig. S3B reveals that BIO addition increased β-catenin signaling, as observed by axin2 mRNA expression; however, inhibition of GSK-3α/β with BIO not only failed to restore VEGF-induced permeability (Fig. 6B), BIO also disrupted the barrier more than VEGF alone and completely blocked norrin's restorative effect. Further, dextran flux assays showed that BIO-stimulated cells had similar permeability to control monolayers (Fig. 6C), and consistent with TEER results, the addition of BIO before, after, or at the same time as VEGF was unable to rescue the VEGF-induced permeability. When BIO and XAV-939 were added alone, both had little effect on TEER until ∼48 h, when they began to decrease the barrier properties modestly (Fig. S3C). These data reveal the requirement of β-catenin signaling in norrin-induced BRB restoration; nevertheless, β-catenin stabilization by GSK-3α/β inhibition in BREC monolayers is insufficient and, in fact, detrimental for barrier restoration.
The restoration of BRB properties after VEGF is an effect specific to norrin but not Wnt signaling
TSPAN12 is a co-receptor that enhances norrin-induced but not Wnt-induced β-catenin signaling (8–10). To test the specificity of norrin signaling on the restoration of VEGF-induced permeability, FZD4 receptor was activated in BREC monolayers with the addition of Wnt3a and Wnt5a ligands. Measurements of axin2 mRNA (Fig. 7A and Fig. S4A) or phosphorylation of JNK (Fig. 7 (B and C) and Fig. S4 (B and C)) in BREC revealed that Wnt3a is able to activate both canonical/β-catenin and noncanonical/JNK signaling, whereas Wnt5a only promoted JNK phosphorylation. We tested the ability of Wnt ligands to restore TEER, and we found that Wnt3a or Wnt5a alone at 100 ng/ml did not have any effect on TEER as compared with control monolayers (Fig. 7D). When Wnt3a and Wnt5a were added together with VEGF, separately (VEGF/Wnt3a or VEGF/Wnt5a) or in combination (VEGF/Wnt3a/Wnt5a), there was little effect on VEGF-induced permeability, unlike norrin, which restored barrier properties. Interestingly, higher concentrations (200 ng/ml) of Wnt3a or Wnt5a alone decreased basal TEER (Fig. S4D) and, together with VEGF (Fig. S4E), had a greater effect on permeability. Together, these results suggest that restoration of BRB properties after VEGF is an effect specific for norrin signaling through FZD4 and again is consistent with the requirement of TSPAN12 membrane localization after VEGF.
Figure 7.

Wnt3a and Wnt5a do not restore barrier properties after VEGF. Activation of canonical β-catenin signaling (A) was corroborated by qRT-PCR of axin2 (n = 7–10), and the activation of the noncanonical JNK pathway by Wnt (B and C) was assessed by the phosphorylation of JNK (n = 6), whereas changes in barrier properties were evaluated by TEER measurements (D) (n = 6–14). BREC were stimulated with vehicle (Control), VEGF, norrin Wnt3a (100 ng/ml), Wnt5a (100 ng/ml), or their combination. p values were calculated by one-way (A–C) or two-way (D) ANOVA, followed by Tukey's post hoc test. The top lines in D indicate the time points where there was a significant difference, comparing the condition indicated at the left with VEGF or VEGF/norrin. All VEGF-stimulated monolayers were significantly different from control, Wnt3a, or Wnt5a. Error bars, S.D.
VEGF up-regulates claudin-5 expression, but only norrin promotes its localization at cell contacts
Claudin-5 is largely restricted to the vascular endothelium and contributes to blood-brain barrier properties (44). Previous development studies reveal that norrin signaling promotes increased claudin-5 in retinal endothelial cells (5). Because tight junction protein expression and organization at the cell border may both be regulated, the effect of VEGF and norrin on the expression and organization of claudin-5 was examined. We found that the addition of norrin in combination with or after VEGF increased the amount of claudin-5 mRNA at 72 h (Fig. 8A). Surprisingly, VEGF alone or in combination with norrin was able to increase claudin-5 protein (Fig. 8, B and C). However, because VEGF increases the permeability of BREC monolayers, we then examined the localization of claudin-5 by immunostaining. Fig. 8 (D and E) demonstrates that the addition of VEGF for 72 h yielded reduced border staining of claudin-5 as determined by masked scoring of confocal images. Importantly, the discontinuous borders between endothelial cells (arrows) were reduced after the addition of VEGF/norrin. Together, these data suggest that VEGF promotes claudin-5 expression, but norrin induces the BRB properties by the organization of claudin-5 specifically at the cell borders.
Figure 8.

Norrin and VEGF both increase the expression of claudin-5, but only norrin promotes its localization at cell contacts. BREC stimulated with vehicle (control, C), norrin (N), VEGF (V), both (V/N), or norrin after VEGF (V+N) for 12, 24, 48, and 72 h were processed for qRT-PCR with specific primers for claudin-5; n = 5–6 (A). All samples were normalized to control (C) for 12 h. B and C, Western blotting of claudin-5 (Cl-5); n = 5–6. D, immunostaining of claudin-5 in BREC stimulated at 72 h; n = 3. Arrows point to typical sites of tight junction disruption; scale bar, 10 μm. E, quantification of four images per experiment by masked scoring combined from three individuals. Data show the frequency of claudin-5 loss at the cell contacts ranked in five categories, where 100% corresponds to completely lost. p values were calculated by one-way ANOVA, followed by Tukey's post hoc test. Error bars, S.D.
Discussion
In retinal vascular development, angiogenesis concludes with maturation of the BRB. But in retinal pathologies, the factors that control BRB maintenance become unbalanced with increased VEGF expression (45). The role of norrin signaling has been studied in some models of retinopathies (reviewed in Ref. 46); however, little was known about the interaction between VEGF and norrin signals. Following the paradigm of vascular development, we tested the hypothesis that norrin may act to restore BRB after VEGF. Indeed, our in vivo experiments demonstrate that norrin can completely prevent and restore BRB function in two models of vascular dysfunction. These studies suggest potential novel treatment paradigms for patients with diabetic retinopathy or other retinal vascular diseases driven by VEGF-induced permeability, which focus on restoration of proper vascular function rather than prevention of further damage through binding VEGF.
The importance of the TSPAN12 co-activator in norrin signaling has recently emerged (8, 29). Gene deletion studies of TSPAN12 demonstrate reduced retinal capillary development and loss of barrier properties in adult mice with a phenotype similar to aspects of humans with diabetic retinopathy, including increased solute accumulation and cystoid edema (29). Consistent with the role of TSPAN12 as a co-activator for norrin signaling through FZD4, the studies described herein suggest a novel mechanism in which VEGF primes endothelial cells to respond to norrin by regulating the specific expression of TSPAN12 through translational control and by inducing its membrane accumulation in a MEK/ERK-dependent manner. This in turn promotes FZD4 response to norrin and subsequent β-catenin activation. VEGF also promotes an increase in claudin-5 content that organizes at tight junctions in response to norrin signaling. Together, these studies emphasize the importance of norrin signaling through FZD4 receptor and TSPAN12 co-receptor. Because there is ongoing research examining the use of antibodies against TSPAN12 in angiogenesis (47, 48), it is important to understand the specific role of norrin receptor molecules in retinal barrier biology.
Previous research has demonstrated that the expression of constitutively active β-catenin largely reverses the retinal developmental defect of norrin gene deletion (5, 49). The current study supports a requirement for β-catenin for barrier restoration because reduction of β-catenin activity with tankyrase inhibitor impeded the ability of norrin to promote barrier properties. Further, activation of β-catenin signaling induced by norrin and VEGF co-stimulation revealed increased localization of β-catenin at the nuclear fractions. However, despite the ability of norrin to promote β-catenin signaling, norrin alone failed to improve barrier properties compared with control monolayers. In addition, GSK-3α/β inhibition leading to activation of the β-catenin pathway, as measured by axin2 mRNA, failed to reverse VEGF-induced permeability. Wnt3a also promoted a robust axin2 expression without altering barrier properties, and its combination with Wnt5a only slightly improved TEER when it was added with VEGF but failed to completely restore VEGF-induced permeability like norrin. Collectively, the current research reveals a required role for β-catenin in norrin-induced barrier restoration after VEGF and suggests that norrin provides a specific signal beyond the canonical β-catenin pathway that is required for BRB restoration. Further, while in the brain, Wnt3a (50), Wnt7a/Wnt7b (51–58), and norrin (49) ligands regulate blood-brain barrier integrity with redundancy; in the retina, these Wnt ligands only have a small contribution (10), suggesting that, distinct from the brain, the retinal vascular barrier properties are largely regulated by norrin/FZD4/TSPAN12 signaling.
Interestingly, the current studies demonstrate that VEGF, while promoting endothelial permeability, simultaneously primes the endothelial cells for response to norrin for barrier restoration. In addition to increasing TSPAN12 at the cell membrane, VEGF promoted β-catenin stabilization and claudin-5 accumulation. This is consistent with a previous report demonstrating that VEGF can promote S-nitrosylation of β-catenin by endothelial nitric oxide synthase, resulting in a decreased interaction with VE-cadherin, thus promoting β-catenin accumulation at the cytoplasmic and nuclear fractions in bovine aortic endothelial cells (59). Moreover, previous studies demonstrated that β-catenin activation increases claudin-3 (50) and claudin-5 (60) expression in the brain and retina, respectively. Here, VEGF alone increased claudin-5 protein content, but the greatest amount of claudin-5 mRNA and protein was obtained in the presence of VEGF and norrin together. Studies exploring claudin-5 regulation describe a mechanism in which VE-cadherin promotes AKT (also known as protein kinase B or PKB) phosphorylation on Thr-308 that induces FoxO1 (forkhead box protein O1) activation, β-catenin accumulation, and claudin-5 expression. A similar mechanism involving β-catenin accumulation may explain the VEGF or combined norrin and VEGF induction of claudin-5 expression.
Because VEGF promoted an increase in both TSPAN12 membrane protein content and total claudin-5 protein without altering mRNA, the data suggest that VEGF has translational control of these proteins, potentially through MEK/ERK signaling. Although other mechanisms might be involved as well, the activation of MAPK through growth factor stimulation has been demonstrated to control translation (61, 62). Here, we have found that MEK/ERK signaling was required for the promotion of TSPAN12 membrane accumulation and for norrin-induced BRB properties, thus suggesting a novel potential mechanism that can be the focus of future research.
Norrin promotes BRB formation by the induction of tight junction organization. In retinal endothelial cells of norrin, FZD4, LRP5, or TSPAN12 knockout mice, the fenestration marker PLVAP was increased, whereas claudin-5 staining at the cell border was decreased (5, 10, 29, 31, 49, 63, 64), thus indicating that in the absence of norrin signaling, both paracellular and transcellular routes are compromised. In addition, recent studies using siRNA suggest that PLVAP contributes to VEGF-induced permeability (65). However, preliminary studies in BREC revealed no observable changes in PLVAP by fluorescent immunostaining after VEGF addition, and no further studies on this protein were carried out. Conversely, here we have found that VEGF increased claudin-5 protein expression, but the border organization was decreased after VEGF compared with control. However, when both VEGF and norrin signaling were activated, claudin-5 was observed localized at the cell borders. The mechanisms by which VEGF/norrin together promote claudin-5 translocation are currently unknown and will be the focus of future research.
Recent publications also suggest a role of norrin in hypoxia response. Norrin ablation decreases expression of proangiogenic transcription factors, such as Sox7, Sox17, and Sox18 (35), or VEGF-dependent expression of hypoxia-inducible factor (66), resulting in early specific loss of retinal ganglion cells (RGC) (67). Interestingly, in an oxygen-induced retinopathy model, Wnt3a, Wnt7a, and Wnt10a, but not norrin, are elevated (33), and norrin injection promotes revascularization of the hypoxic area in a mechanism dependent on insulin-like growth factor 1 expression (67–70), resulting in RGC survival (71). Together, these studies reveal the relationship of VEGF and angiogenic factors with norrin during hypoxia-driven angiogenesis.
Norrin may also provide nonvascular benefit. In a model of RGC damage, norrin was able to increase the numbers of optic nerve axons and perikarya of surviving RGC (72). Additionally, norrin signaling through β-catenin and endothelin-2 can protect photoreceptors from light damage (73). In the brain, norrin protects the blood-brain barrier integrity in a rat model of subarachnoid hemorrhage (74), and by activating FZD4 signaling, it can reduce the preneoplasic lesions of medulloblastoma (75).
Overall, our results demonstrate that VEGF both induces retinal vascular permeability and simultaneously primes endothelial cells to respond to norrin, which can subsequently promote BRB properties. These studies support the potential of norrin to be used as a therapeutic option for the treatment of VEGF-related pathologies, including diabetic retinopathy.
Experimental procedures
In vivo studies
All animals were treated in accordance with the Association for Research in Vision and Ophthalmology (ARVO) Statement on the Use of Animals in Ophthalmic and Visual Research and the guidelines established by the University of Michigan Institutional Animal Care and Use Committee. Male Long–Evans rats (Charles River Laboratories, Wilmington, MA) weighting 200–250 g were anesthetized with ketamine/xylazine (66.7 and 6.7 mg/kg of body weight, respectively), to receive an IVT injection of vehicle (0.1% BSA/PBS + 4 mm HCl), 50 ng of rat recombinant VEGF (diluted in 0.1% BSA/PBS) (R&D Systems (Minneapolis, MN), 564-RV), and/or 40 ng of recombinant human norrin (diluted in 4 mm HCl) (R&D Systems, 3014-NR) per eye. Evans Blue retinal permeability was assessed 24 h after injection, as reported previously (76). Briefly, 45 mg/kg body weight of Evans Blue dye (Sigma–Aldrich, E-2129) was injected via femoral vein, and after 2 h, 0.3 ml of blood was drawn from the vena cava to obtain plasma. Rats were then perfused with warm saline at 66 ml/min for 2 min via the left ventricle; retinas were harvested and dried overnight in a SpeedVac vacuum concentrator (Savant, Thermo Fisher Scientific); and the dye was extracted with formamide (Sigma-Aldrich, 47671) at 70 °C overnight. Dye concentrations in retina extracts and plasma samples were determined by absorbance at 620 and 740 nm using the FLUOstar Omega microplate reader (BMG Labtech Inc., Cary, NC).
Diabetes was induced in male Long–Evans rats by the intraperitoneal injection of STZ (65 mg/kg, 10 mm sodium citrate buffer, pH 4.5; Sigma-Aldrich, S0130). 3 days later, blood glucose levels were measured, and rats with >250 mg/dl were considered diabetic. After 5 months, rats received an IVT injection of vehicle (4 mm HCl) in right eyes or 40 ng of norrin in left eyes, and 24 h before the retinal permeability assay with 100 mg/kg body weight FITC-BSA (Sigma-Aldrich, A9771), exactly as described (77). Briefly, FITC-BSA accumulation in the retina was measured following the Evans Blue procedure, except the dye was extracted with 1% Triton-PBS by overnight shaking in an incubator (New Brunswick Scientific, Edison, NJ), and its accumulation was measured using the FLUOstar Omega fluorescence spectrophotometer (485-nm excitation and 520-nm emission).
The retinal permeability data were expressed as microliters of plasma equivalent/g dry retina weight/h of circulation (μl/g/h). The amounts of dye (Evans Blue or FITC-BSA) extracted from the retina were normalized with the plasma levels of dye from each animal and the dry weight of each retina and then divided by the circulation time.
Primary BREC culture
Primary BREC preparations used for in vitro studies were grown as described previously (78). Briefly, electrical cell-substrate impedance–sensing (ECIS) chambers, Transwell® inserts, 60-mm Petri dishes, or plastic coverslips were coated with 1 μg/cm2 of fibronectin bovine plasma (Sigma-Aldrich, F1141) for 1 h at room temperature. BREC from passage 2–8 were grown until confluence at 37 °C in MCDB-131 complete medium (Sigma–Aldrich, M8537) with 10% fetal bovine serum (FBS) (HycloneTM, Thermo Fisher Scientific, Sh30071.03) and supplemented with 10 ng/ml EGF (Sigma–Aldrich, E9644), 0.1 mg/ml EndoGro (Vec Technologies Inc. (Rensselaer, NY), EG-5), 0.045 mg/ml heparin (Thermo Fisher Scientific, H19), 8 μg/ml Tylosin (Sigma-Aldrich, T3397), and 0.01 ml/ml antibiotic/antimycotic (Gibco, Thermo Fisher Scientific, 15240062). Then medium was changed to MCDB-131 with 1% FBS and 100 mm hydrocortisone (Sigma-Aldrich, H0888) for an additional 2 days. At this point, cells were stimulated as indicated under “Results” with the recombinant human proteins: VEGF165 (293-VE), norrin (3014-NR), Wnt3a (5036-WN), or Wnt5a (645-WN) (R&D Systems) diluted in 0.1% BSA/PBS, except for norrin, which was diluted in 4 mm HCl and/or with BIO (3194), XAV-939 (3748), U0126 (1144) (Tocris Bioscience, Bristol, UK), cycloheximide (2112S) (Cell Signaling, Danvers, MA), hydroxychloroquine sulfate (H1306) (Tokyo Chemical Industry Co., Ltd.), or lactacystin (70980) (Cayman Chemical, Ann Arbor, MI), which were diluted in DMSO. All control monolayers were stimulated with the volume equivalent to drug or protein diluents (vehicle) used in each experiment.
TEER and solute flux
TEER measurements were performed at 4,000 Hz once every hour, using the ECIS Z-θ system. 45,000 BREC were plated in 8-well chamber slides (Applied Bio Physics Inc. (Troy, NY), 8W10E+) equipped with two sets of 20 circular 250-μm diameter gold-plated active electrodes (79). These are located on interdigitated fingers among a substrate area of 0.8 cm2, and 400 μl of medium was used per well.
Measurements of solute flux were carried out 72 h after last stimuli and over 3.5 h, using 10 μm 70-kDa RITC-dextran (Sigma-Aldrich, R9379). 1 × 105 cells/well were grown on 12-mm diameter, 0.4-μm pore size, polyester membrane Corning®Costar® Transwell® cell culture inserts (Corning, Thermo Fisher Scientific, 3460), and Po (cm/s) was calculated as described previously (80) using the equation, Po = ((FL/Δt)VPL)/(FAA), where Po is in cm/s, FL is basolateral fluorescence, FA is apical fluorescence, Δt is the change in time, A is the surface area of the filter, and VL is the volume of the basolateral chamber.
Immunofluorescence staining
BREC monolayers were plated on ThermanoxTM plastic round coverslips with a 13-mm diameter (Thermo Fisher Scientific, 174950). After the indicated stimulation, monolayers were washed twice with Dulbecco's PBS/calcium/magnesium chloride (Gibco, Thermo Fisher Scientific, 14040-133), fixed for 10 min with 1% paraformaldehyde (Electron Microscopy Sciences (Hatfield, PA), 15710), permeabilized for 10 min with 0.2% Triton X-100, blocked for 1 h with 2% normal goat serum (Life Technologies, Inc., Thermo Fisher Scientific, 50062Z) and 0.1% Triton X-100, and incubated with the primary antibodies polyclonal rabbit α-claudin-5 (Invitrogen, Thermo Fisher Scientific, 34-1600), polyclonal rabbit α-TSPAN12-FITC–conjugated (Aviva Systems Biology Corp. (San Diego, CA), ARP46887_P050), and/or monoclonal rat α-ZO-1 clone R40.76 (Millipore, Sigma–Aldrich, MABT11) for 2 days at 4 °C, followed by its detection with secondary fluorescent antibodies goat α-rabbit Alexa Fluor 488 (A11034) or goat α-rat Alexa Fluor 647 (A21247) (Life Technologies, Thermo Fisher Scientific) and Hoechst (Invitrogen, Thermo Fisher Scientific; H3570) for nuclear staining overnight at 4 °C. Samples were imaged using confocal microscopy. Claudin-5 cell border staining was analyzed by a semiquantitative ranking score system on a scale of five categories that indicate the percentage of loss in cell border staining (0–100%). Three independent observers were asked to assign a ranking score to four images per condition in a masked fashion. Results of three independent experiments were summed, and the frequency of each ranking score was calculated to determine differences between conditions. For TSPAN12 staining analysis, total green fluorescence was quantified from not permeabilized monolayers using ImageJ software.
Cell fraction assays
Cytoplasmic, nuclear, and membrane fractions were isolated using a compartment protein extraction kit (Millipore, Sigma–Aldrich, 2145). BREC monolayers were plated on 60-mm Petri dishes (Corning, Thermo Fisher Scientific, 353002) and harvested exactly as described in the manufacturer's protocol following the instructions for adherent cells. Briefly, cells were scrapped from cell culture plates with extraction buffer containing sucrose (concentration not specified) and vortexed. After 20 min of rotation at 4 °C, cells were passed through a 27-gauge needle until the nucleus was released from cells, as observed by light microscopy. Homogenates were centrifuged (15,000 × g for 20 min at 4 °C), and the supernatant containing the cytoplasmic fraction was collected. Pellets were washed and resuspended by pipetting in nuclear extraction buffer (without sucrose). After 20 min of shaking (4 °C) and 15,000 × g centrifugation (20 min at 4 °C), the supernatant containing nuclear fraction was collected. A sodium deoxycholate and Nonidet P-40 buffer was added to the pellet, and after 20 min of shaking (4 °C) and 15,000 × g centrifugation (20 min at 4 °C), the supernatant containing the membrane fraction was collected, and the pellet was discarded. Equal amounts of protein were loaded in SDS-PAGE and processed for Western blot analysis.
Western blotting
Cells were harvested in a Triton X-100-deoxycolate-SDS–based lysis buffer. The insoluble material was pelleted by centrifugation for 13 min at 13,000 × g at 4 °C, and the supernatants were used to determine the protein concentration by the Bio-Rad DCTM protein colorimetric assay (Bio-Rad, 500-0113). Equal amounts of protein were prepared with NuPAGETM LDS sample buffer (Thermo Fisher Scientific, NP0007) and NuPageTM sample-reducing agent (Thermo Fisher Scientific, NP0009), and Western blotting was carried out in a NuPAGETM (Invitrogen, Thermo Fisher Scientific) system exactly as described (22). Primary antibodies (Table S1) were diluted 1:1,000 in 2% ECLTM prime blocking agent (Amersham Biosciences, GE Healthcare, RPN418) and TBS-Tween, incubated overnight, and detected with horseradish peroxidase–conjugated secondary antibodies diluted 1:10,000 (Table S1) and chemiluminescence. Results were analyzed using AlphaView software FluorChemTM systems, by obtaining the mean gray intensity of each band in a fixed area. The background value was subtracted from all bands and then reported as a percentage of the control of each experiment.
qRT-PCR
RNA was extracted and genomic DNA was removed using the RNeasy Plus mini kit (Qiagen Inc. (Hilden, Germany), 74134), and 1 μg of RNA per sample was processed with the Omniscript reverse transcription kit (Qiagen Inc., 205113) to obtain cDNA. Quantitative PCR was performed with TaqManTM Real-Time PCR master mix (Applied Biosystems, Thermo Fisher Scientific, 4304437) and using specific TaqManTM gene expression assays (Thermo Fisher Scientific) to detect FZD4 (Bt04293845_m1), LRP5 (Mm01227476_m1), TSPAN12 (Bt03240118_m1), axin2 (Bt04311243_g1), or claudin-5 (Mm00727012_s1). Results were normalized to β-actin (4351319) mRNA and expressed as relative change using the ΔΔCt method.
Statistical analysis
Data were analyzed using GraphPad Prism software in at least three independent experiments. Graphs represent the mean ± S.D. p values were calculated by t test and one-way or two-way ANOVA, as indicated for each figure, and p ≤ 0.05 was considered significant.
Author contributions
M. D.-C. and D. A. A. conceptualization; M. D.-C., C.-M. L., and S. L. data curation; M. D.-C., C.-M. L., S. L., and D. A. A. formal analysis; M. D.-C. and D. A. A. funding acquisition; M. D.-C. and D. A. A. validation; M. D.-C., C.-M. L., S. L., and D. A. A. investigation; M. D.-C. and D. A. A. visualization; M. D.-C., C.-M. L., S. L., and D. A. A. methodology; M. D.-C., C.-M. L., S. L., and D. A. A. writing-original draft; M. D.-C., C.-M. L., S. L., and D. A. A. writing-review and editing; D. A. A. resources; D. A. A. supervision; D. A. A. project administration.
Supplementary Material
Acknowledgments
We thank Dr. Xuwen Liu (University of Michigan) for providing exceptional primary retinal endothelial cells and Alyssa Dreffs (University of Michigan) for technical support.
This work was supported by National Institutes of Health Grant EY012021 (to D. A. A.) and Core Grants P30EY007003 and DK020572, the Jules and Doris Stein Professorship from Research to Prevent Blindness (to D. A. A.), American Diabetes Association Minority Postdoctoral Fellowship Award 4-16-PMF-003 (to M. D.-C.) and the Deutsche Forschungsgemeinschaft (DFG) research group FOR2325, “The Neurovascular Interface Grant LI 911/5–1; the Excellence Cluster Cardio-Pulmonary Institute (CPI); the European Union HORIZON 2020 Grant MSCA-ITN-2015 675619 “BtRAIN”; and the German Centre for Heart and Circulation Research (DZHK) Grant 81X2800146 (to S. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1 and Figs. S1–S4.
- BRB
- blood-retina barrier
- APC
- adenomatosis polyposis coli
- CHX
- cycloheximide
- BREC
- bovine retinal endothelial cell(s)
- EGF
- epidermal growth factor
- EGFR
- epidermal growth factor receptor
- ERK
- extracellular signal–regulated kinase
- FEVR
- familial exudative vitreopathy
- FZD4
- frizzled class receptor 4
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GSK-3α/β
- glycogen synthase kinase 3α/β
- IVT
- intravitreal
- LRP5/6
- low-density lipoprotein receptor–related protein 5/6
- MAPK
- mitogen-activated protein kinase
- MEK
- MAPK/ERK kinase
- PLVAP
- plasmalemma vesicle-associated protein
- RGC
- retinal ganglion cell(s)
- RITC
- rhodamine B isothiocyanate
- JNK
- c-Jun N-terminal kinase
- STZ
- streptozotocin
- TEER
- trans-endothelial electrical resistance
- TSPAN12
- tetraspanin 12
- VE-cadherin
- vascular endothelial cadherin
- VEGF
- vascular endothelial growth factor
- ECIS
- electrical cell-substrate impedance–sensing
- Po
- diffusive permeability
- N
- norrin
- V
- VEGF
- qRT-PCR
- quantitative RT-PCR
- FBS
- fetal bovine serum
- ANOVA
- analysis of variance.
References
- 1. Ye X., Wang Y., Cahill H., Yu M., Badea T. C., Smallwood P. M., Peachey N. S., and Nathans J. (2009) Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell 139, 285–298 10.1016/j.cell.2009.07.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ye X., Smallwood P., and Nathans J. (2011) Expression of the Norrie disease gene (Ndp) in developing and adult mouse eye, ear, and brain. Gene Expr. Patterns 11, 151–155 10.1016/j.gep.2010.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berger W., van de Pol D., Bächner D., Oerlemans F., Winkens H., Hameister H., Wieringa B., Hendriks W., and Ropers H. H. (1996) An animal model for Norrie disease (ND): gene targeting of the mouse ND gene. Hum. Mol. Genet. 5, 51–59 10.1093/hmg/5.1.51 [DOI] [PubMed] [Google Scholar]
- 4. Lee H., Jo D. H., Kim J. H., and Kim J. H. (2013) Norrin expression in endothelial cells in the developing mouse retina. Acta Histochem. 115, 447–451 10.1016/j.acthis.2012.10.008 [DOI] [PubMed] [Google Scholar]
- 5. Zhou Y., Wang Y., Tischfield M., Williams J., Smallwood P. M., Rattner A., Taketo M. M., and Nathans J. (2014) Canonical WNT signaling components in vascular development and barrier formation. J. Clin. Invest. 124, 3825–3846 10.1172/JCI76431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Meitinger T., Meindl A., Bork P., Rost B., Sander C., Haasemann M., and Murken J. (1993) Molecular modelling of the Norrie disease protein predicts a cystine knot growth factor tertiary structure. Nat. Genet. 5, 376–380 10.1038/ng1293-376 [DOI] [PubMed] [Google Scholar]
- 7. Ke J., Harikumar K. G., Erice C., Chen C., Gu X., Wang L., Parker N., Cheng Z., Xu W., Williams B. O., Melcher K., Miller L. J., and Xu H. E. (2013) Structure and function of Norrin in assembly and activation of a Frizzled 4-Lrp5/6 complex. Genes Dev. 27, 2305–2319 10.1101/gad.228544.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Junge H. J., Yang S., Burton J. B., Paes K., Shu X., French D. M., Costa M., Rice D. S., and Ye W. (2009) TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt-induced FZD4/β-catenin signaling. Cell 139, 299–311 10.1016/j.cell.2009.07.048 [DOI] [PubMed] [Google Scholar]
- 9. Lai M. B., Zhang C., Shi J., Johnson V., Khandan L., McVey J., Klymkowsky M. W., Chen Z., and Junge H. J. (2017) TSPAN12 is a Norrin co-receptor that amplifies Frizzled4 ligand selectivity and signaling. Cell Rep. 19, 2809–2822 10.1016/j.celrep.2017.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang Y., Cho C., Williams J., Smallwood P. M., Zhang C., Junge H. J., and Nathans J. (2018) Interplay of the Norrin and Wnt7a/Wnt7b signaling systems in blood-brain barrier and blood-retina barrier development and maintenance. Proc. Natl. Acad. Sci. U.S.A. 115, E11827–E11836 10.1073/pnas.1813217115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nusse R., and Clevers H. (2017) Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–999 10.1016/j.cell.2017.05.016 [DOI] [PubMed] [Google Scholar]
- 12. Berger W., van de Pol D., Warburg M., Gal A., Bleeker-Wagemakers L., de Silva H., Meindl A., Meitinger T., Cremers F., and Ropers H. H. (1992) Mutations in the candidate gene for Norrie disease. Hum. Mol. Genet. 1, 461–465 10.1093/hmg/1.7.461 [DOI] [PubMed] [Google Scholar]
- 13. Richter M., Gottanka J., May C. A., Welge-Lüssen U., Berger W., and Lütjen-Drecoll E. (1998) Retinal vasculature changes in Norrie disease mice. Invest. Ophthalmol. Vis. Sci. 39, 2450–2457 [PubMed] [Google Scholar]
- 14. Meindl A., Berger W., Meitinger T., van de Pol D., Achatz H., Dörner C., Haasemann M., Hellebrand H., Gal A., and Cremers F. (1992) Norrie disease is caused by mutations in an extracellular protein resembling C-terminal globular domain of mucins. Nat. Genet. 2, 139–143 10.1038/ng1092-139 [DOI] [PubMed] [Google Scholar]
- 15. Parzefall T., Lucas T., Ritter M., Ludwig M., Ramsebner R., Frohne A., Schöfer C., Hengstschläger M., and Frei K. (2014) A novel missense NDP mutation [p.(Cys93Arg)] with a manifesting carrier in an Austrian family with Norrie disease. Audiol. Neurootol. 19, 203–209 10.1159/000358866 [DOI] [PubMed] [Google Scholar]
- 16. Musada G. R., Syed H., Jalali S., Chakrabarti S., and Kaur I. (2016) Mutation spectrum of the FZD-4, TSPAN12 AND ZNF408 genes in Indian FEVR patients. BMC Ophthalmol. 16, 90 10.1186/s12886-016-0236-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schatz P., and Khan A. O. (2017) Variable familial exudative vitreoretinopathy in a family harbouring variants in both FZD4 and TSPAN12. Acta Ophthalmol. 95, 705–709 10.1111/aos.13411 [DOI] [PubMed] [Google Scholar]
- 18. Fei P., Zhu X., Jiang Z., Ma S., Li J., Zhang Q., Zhou Y., Xu Y., Tai Z., Zhang L., Huang L., Yang Z., Zhao P., and Zhu X. (2015) Identification and functional analysis of novel FZD4 mutations in Han Chinese with familial exudative vitreoretinopathy. Sci. Rep. 5, 16120 10.1038/srep16120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang L., Yang Y., Li S., Tai Z., Huang L., Liu Y., Zhu X., Di Y., Qu C., Jiang Z., Li Y., Zhang G., Kim R., Sundaresan P., Yang Z., and Zhu X. (2016) Whole exome sequencing analysis identifies mutations in LRP5 in Indian families with familial exudative vitreoretinopathy. Genet. Test Mol. Biomarkers 20, 346–351 10.1089/gtmb.2015.0322 [DOI] [PubMed] [Google Scholar]
- 20. Liu Y. Q., Zhu X., Li S. J., Yang Y. M., Yang M., Zhao P. Q., and Zhu X. J. (2017) Identification of LRP5 mutations in families with familial exudative vitreoretinopathy. Yi Chuan 39, 241–249 10.16288/j.yczz.16-339 [DOI] [PubMed] [Google Scholar]
- 21. Fei P., Zhang Q., Huang L., Xu Y., Zhu X., Tai Z., Gong B., Ma S., Yao Q., Li J., Zhao P., and Yang Z. (2014) Identification of two novel LRP5 mutations in families with familial exudative vitreoretinopathy. Mol. Vis. 20, 395–409 [PMC free article] [PubMed] [Google Scholar]
- 22. Aveleira C. A., Lin C. M., Abcouwer S. F., Ambrósio A. F., and Antonetti D. A. (2010) TNF-α signals through PKCζ/NF-κB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes 59, 2872–2882 10.2337/db09-1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu Y., Huang L., Li J., Zhang Q., Fei P., Zhu X., Tai Z., Ma S., Gong B., Li Y., Zang W., Zhu X., Zhao P., and Yang Z. (2014) Novel mutations in the TSPAN12 gene in Chinese patients with familial exudative vitreoretinopathy. Mol. Vis. 20, 1296–1306 [PMC free article] [PubMed] [Google Scholar]
- 24. Gal M., Levanon E. Y., Hujeirat Y., Khayat M., Pe'er J., and Shalev S. (2014) Novel mutation in TSPAN12 leads to autosomal recessive inheritance of congenital vitreoretinal disease with intra-familial phenotypic variability. Am. J. Med. Genet. A 164A, 2996–3002 10.1002/ajmg.a.36739 [DOI] [PubMed] [Google Scholar]
- 25. Panagiotou E. S., Sanjurjo Soriano C., Poulter J. A., Lord E. C., Dzulova D., Kondo H., Hiyoshi A., Chung B. H., Chu Y. W., Lai C. H. Y., Tafoya M. E., Karjosukarso D., Collin R. W. J., Topping J., Downey L. M., et al. (2017) Defects in the cell signaling mediator β-catenin cause the retinal vascular condition FEVR. Am. J. Hum. Genet. 100, 960–968 10.1016/j.ajhg.2017.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wu J. H., Liu J. H., Ko Y. C., Wang C. T., Chung Y. C., Chu K. C., Liu T. T., Chao H. M., Jiang Y. J., Chen S. J., and Chung M. Y. (2016) Haploinsufficiency of RCBTB1 is associated with Coats disease and familial exudative vitreoretinopathy. Hum. Mol. Genet. 25, 1637–1647 10.1093/hmg/ddw041 [DOI] [PubMed] [Google Scholar]
- 27. Musada G. R., Jalali S., Hussain A., Chururu A. R., Gaddam P. R., Chakrabarti S., and Kaur I. (2016) Mutation spectrum of the Norrie disease pseudoglioma (NDP) gene in Indian patients with FEVR. Mol. Vis. 22, 491–502 [PMC free article] [PubMed] [Google Scholar]
- 28. Ngo M. H., Borowska-Fielding J., Heathcote G., Nejat S., Kelly M. E., McMaster C. R., and Robitaille J. M. (2016) Fzd4 haploinsufficiency delays retinal revascularization in the mouse model of oxygen induced retinopathy. PLoS ONE 11, e0158320 10.1371/journal.pone.0158320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang C., Lai M. B., Pedler M. G., Johnson V., Adams R. H., Petrash J. M., Chen Z., and Junge H. J. (2018) Endothelial cell-specific inactivation of TSPAN12 (Tetraspanin 12) reveals pathological consequences of barrier defects in an otherwise intact vasculature. Arterioscler. Thromb. Vasc. Biol. 38, 2691–2705 10.1161/ATVBAHA.118.311689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu Q., Wang Y., Dabdoub A., Smallwood P. M., Williams J., Woods C., Kelley M. W., Jiang L., Tasman W., Zhang K., and Nathans J. (2004) Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell 116, 883–895 10.1016/S0092-8674(04)00216-8 [DOI] [PubMed] [Google Scholar]
- 31. Schäfer N. F., Luhmann U. F., Feil S., and Berger W. (2009) Differential gene expression in Ndph-knockout mice in retinal development. Invest. Ophthalmol. Vis. Sci. 50, 906–916 10.1167/iovs.08-1731 [DOI] [PubMed] [Google Scholar]
- 32. Xia C. H., Liu H., Cheung D., Wang M., Cheng C., Du X., Chang B., Beutler B., and Gong X. (2008) A model for familial exudative vitreoretinopathy caused by LPR5 mutations. Hum. Mol. Genet. 17, 1605–1612 10.1093/hmg/ddn047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen J., Stahl A., Krah N. M., Seaward M. R., Dennison R. J., Sapieha P., Hua J., Hatton C. J., Juan A. M., Aderman C. M., Willett K. L., Guerin K. I., Mammoto A., Campbell M., and Smith L. E. (2011) Wnt signaling mediates pathological vascular growth in proliferative retinopathy. Circulation 124, 1871–1881 10.1161/CIRCULATIONAHA.111.040337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zuercher J., Fritzsche M., Feil S., Mohn L., and Berger W. (2012) Norrin stimulates cell proliferation in the superficial retinal vascular plexus and is pivotal for the recruitment of mural cells. Hum. Mol. Genet. 21, 2619–2630 10.1093/hmg/dds087 [DOI] [PubMed] [Google Scholar]
- 35. Zhou Y., Williams J., Smallwood P. M., and Nathans J. (2015) Sox7, Sox17, and Sox18 cooperatively regulate vascular development in the mouse retina. PLoS ONE 10, e0143650 10.1371/journal.pone.0143650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mazzoni J., Smith J. R., Shahriar S., Cutforth T., Ceja B., and Agalliu D. (2017) The Wnt inhibitor Apcdd1 coordinates vascular remodeling and barrier maturation of retinal blood vessels. Neuron 96, 1055–1069.e6 10.1016/j.neuron.2017.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Beck S. C., Karlstetter M., Garcia Garrido M., Feng Y., Dannhausen K., Mühlfriedel R., Sothilingam V., Seebauer B., Berger W., Hammes H. P., Seeliger M. W., and Langmann T. (2018) Cystoid edema, neovascularization and inflammatory processes in the murine Norrin-deficient retina. Sci. Rep. 8, 5970 10.1038/s41598-018-24476-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Halfter W., Moes S., Asgeirsson D. O., Halfter K., Oertle P., Melo Herraiz E., Plodinec M., Jenoe P., and Henrich P. B. (2017) Diabetes-related changes in the protein composition and the biomechanical properties of human retinal vascular basement membranes. PLoS ONE 12, e0189857 10.1371/journal.pone.0189857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen Y., Hu Y., Zhou T., Zhou K. K., Mott R., Wu M., Boulton M., Lyons T. J., Gao G., and Ma J. X. (2009) Activation of the Wnt pathway plays a pathogenic role in diabetic retinopathy in humans and animal models. Am. J. Pathol. 175, 2676–2685 10.2353/ajpath.2009.080945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Frey T., and Antonetti D. A. (2011) Alterations to the blood-retinal barrier in diabetes: cytokines and reactive oxygen species. Antioxid. Redox. Signal. 15, 1271–1284 10.1089/ars.2011.3906 [DOI] [PubMed] [Google Scholar]
- 41. Murakami T., Felinski E. A., and Antonetti D. A. (2009) Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J. Biol. Chem. 284, 21036–21046 10.1074/jbc.M109.016766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Murakami T., Frey T., Lin C., and Antonetti D. A. (2012) Protein kinase Cβ phosphorylates occludin regulating tight junction trafficking in vascular endothelial growth factor-induced permeability in vivo. Diabetes 61, 1573–1583 10.2337/db11-1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Muthusamy A., Lin C. M., Shanmugam S., Lindner H. M., Abcouwer S. F., and Antonetti D. A. (2014) Ischemia-reperfusion injury induces occludin phosphorylation/ubiquitination and retinal vascular permeability in a VEGFR-2-dependent manner. J. Cereb. Blood Flow Metab. 34, 522–531 10.1038/jcbfm.2013.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nitta T., Hata M., Gotoh S., Seo Y., Sasaki H., Hashimoto N., Furuse M., and Tsukita S. (2003) Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 161, 653–660 10.1083/jcb.200302070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rubio R. G., and Adamis A. P. (2016) Ocular angiogenesis: vascular endothelial growth factor and other factors. Dev. Ophthalmol. 55, 28–37 10.1159/000431129 [DOI] [PubMed] [Google Scholar]
- 46. Chen Q., and Ma J. X. (2017) Canonical Wnt signaling in diabetic retinopathy. Vision Res. 139, 47–58 10.1016/j.visres.2017.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bucher F., Zhang D., Aguilar E., Sakimoto S., Diaz-Aguilar S., Rosenfeld M., Zha Z., Zhang H., Friedlander M., and Yea K. (2017) Antibody-mediated inhibition of Tspan12 ameliorates vasoproliferative retinopathy through suppression of β-catenin signaling. Circulation 136, 180–195 10.1161/CIRCULATIONAHA.116.025604 [DOI] [PubMed] [Google Scholar]
- 48. Paes K. T., Wang E., Henze K., Vogel P., Read R., Suwanichkul A., Kirkpatrick L. L., Potter D., Newhouse M. M., and Rice D. S. (2011) Frizzled 4 is required for retinal angiogenesis and maintenance of the blood-retina barrier. Invest. Ophthalmol. Vis. Sci. 52, 6452–6461 10.1167/iovs.10-7146 [DOI] [PubMed] [Google Scholar]
- 49. Wang Y., Rattner A., Zhou Y., Williams J., Smallwood P. M., and Nathans J. (2012) Norrin/Frizzled4 signaling in retinal vascular development and blood brain barrier plasticity. Cell 151, 1332–1344 10.1016/j.cell.2012.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liebner S., Corada M., Bangsow T., Babbage J., Taddei A., Czupalla C. J., Reis M., Felici A., Wolburg H., Fruttiger M., Taketo M. M., von Melchner H., Plate K. H., Gerhardt H., and Dejana E. (2008) Wnt/β-catenin signaling controls development of the blood-brain barrier. J. Cell Biol. 183, 409–417 10.1083/jcb.200806024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cho C., Smallwood P. M., and Nathans J. (2017) Reck and Gpr124 are essential receptor cofactors for Wnt7a/Wnt7b-specific signaling in mammalian CNS angiogenesis and blood-brain barrier regulation. Neuron 95, 1056–1073.e5 10.1016/j.neuron.2017.07.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ulrich F., Carretero-Ortega J., Menéndez J., Narvaez C., Sun B., Lancaster E., Pershad V., Trzaska S., Véliz E., Kamei M., Prendergast A., Kidd K. R., Shaw K. M., Castranova D. A., Pham V. N., et al. (2016) Reck enables cerebrovascular development by promoting canonical Wnt signaling. Development 143, 147–159 10.1242/dev.123059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vanhollebeke B., Stone O. A., Bostaille N., Cho C., Zhou Y., Maquet E., Gauquier A., Cabochette P., Fukuhara S., Mochizuki N., Nathans J., and Stainier D. Y. (2015) Tip cell-specific requirement for an atypical Gpr124- and Reck-dependent Wnt/β-catenin pathway during brain angiogenesis. Elife 4 10.7554/eLife.06489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chang J., Mancuso M. R., Maier C., Liang X., Yuki K., Yang L., Kwong J. W., Wang J., Rao V., Vallon M., Kosinski C., Zhang J. J., Mah A. T., Xu L., Li L., et al. (2017) Gpr124 is essential for blood-brain barrier integrity in central nervous system disease. Nat. Med. 23, 450–460 10.1038/nm.4309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kuhnert F., Mancuso M. R., Shamloo A., Wang H. T., Choksi V., Florek M., Su H., Fruttiger M., Young W. L., Heilshorn S. C., and Kuo C. J. (2010) Essential regulation of CNS angiogenesis by the orphan G protein-coupled receptor GPR124. Science 330, 985–989 10.1126/science.1196554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cullen M., Elzarrad M. K., Seaman S., Zudaire E., Stevens J., Yang M. Y., Li X., Chaudhary A., Xu L., Hilton M. B., Logsdon D., Hsiao E., Stein E. V., Cuttitta F., Haines D. C., et al. (2011) GPR124, an orphan G protein-coupled receptor, is required for CNS-specific vascularization and establishment of the blood-brain barrier. Proc. Natl. Acad. Sci. U.S.A. 108, 5759–5764 10.1073/pnas.1017192108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhou Y., and Nathans J. (2014) Gpr124 controls CNS angiogenesis and blood-brain barrier integrity by promoting ligand-specific canonical wnt signaling. Dev. Cell 31, 248–256 10.1016/j.devcel.2014.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Posokhova E., Shukla A., Seaman S., Volate S., Hilton M. B., Wu B., Morris H., Swing D. A., Zhou M., Zudaire E., Rubin J. S., and St Croix B. (2015) GPR124 functions as a WNT7-specific coactivator of canonical β-catenin signaling. Cell Rep. 10, 123–130 10.1016/j.celrep.2014.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Thibeault S., Rautureau Y., Oubaha M., Faubert D., Wilkes B. C., Delisle C., and Gratton J. P. (2010) S-Nitrosylation of β-catenin by eNOS-derived NO promotes VEGF-induced endothelial cell permeability. Mol. Cell 39, 468–476 10.1016/j.molcel.2010.07.013 [DOI] [PubMed] [Google Scholar]
- 60. Taddei A., Giampietro C., Conti A., Orsenigo F., Breviario F., Pirazzoli V., Potente M., Daly C., Dimmeler S., and Dejana E. (2008) Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat. Cell Biol. 10, 923–934 10.1038/ncb1752 [DOI] [PubMed] [Google Scholar]
- 61. Kelleher R. J. 3rd, Govindarajan A., Jung H. Y., Kang H., and Tonegawa S. (2004) Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell 116, 467–479 10.1016/S0092-8674(04)00115-1 [DOI] [PubMed] [Google Scholar]
- 62. Roux P. P., and Topisirovic I. (2018) Signaling pathways involved in the regulation of mRNA translation. Mol. Cell Biol. 38, e00070–18 10.1128/MCB.00070-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Xia C. H., Lu E., Zeng J., and Gong X. (2013) Deletion of LRP5 in VLDLR knockout mice inhibits retinal neovascularization. PLoS ONE 8, e75186 10.1371/journal.pone.0075186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chen J., Stahl A., Krah N. M., Seaward M. R., Joyal J. S., Juan A. M., Hatton C. J., Aderman C. M., Dennison R. J., Willett K. L., Sapieha P., and Smith L. E. (2012) Retinal expression of Wnt-pathway mediated genes in low-density lipoprotein receptor-related protein 5 (Lrp5) knockout mice. PLoS ONE 7, e30203 10.1371/journal.pone.0030203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wisniewska-Kruk J., van der Wijk A. E., van Veen H. A., Gorgels T. G., Vogels I. M., Versteeg D., Van Noorden C. J., Schlingemann R. O., and Klaassen I. (2016) Plasmalemma vesicle-associated protein has a key role in blood-retinal barrier loss. Am. J. Pathol. 186, 1044–1054 10.1016/j.ajpath.2015.11.019 [DOI] [PubMed] [Google Scholar]
- 66. Rattner A., Wang Y., Zhou Y., Williams J., and Nathans J. (2014) The role of the hypoxia response in shaping retinal vascular development in the absence of Norrin/Frizzled4 signaling. Invest. Ophthalmol. Vis. Sci. 55, 8614–8625 10.1167/iovs.14-15693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ohlmann A., Scholz M., Goldwich A., Chauhan B. K., Hudl K., Ohlmann A. V., Zrenner E., Berger W., Cvekl A., Seeliger M. W., and Tamm E. R. (2005) Ectopic norrin induces growth of ocular capillaries and restores normal retinal angiogenesis in Norrie disease mutant mice. J. Neurosci. 25, 1701–1710 10.1523/JNEUROSCI.4756-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ohlmann A., Seitz R., Braunger B., Seitz D., Bösl M. R., and Tamm E. R. (2010) Norrin promotes vascular regrowth after oxygen-induced retinal vessel loss and suppresses retinopathy in mice. J. Neurosci. 30, 183–193 10.1523/JNEUROSCI.3210-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zeilbeck L. F., Müller B. B., Leopold S. A., Senturk B., Langmann T., Tamm E. R., and Ohlmann A. (2016) Norrin mediates angiogenic properties via the induction of insulin-like growth factor-1. Exp. Eye Res. 145, 317–326 10.1016/j.exer.2015.12.001 [DOI] [PubMed] [Google Scholar]
- 70. Zeilbeck L. F., Müller B., Knobloch V., Tamm E. R., and Ohlmann A. (2014) Differential angiogenic properties of lithium chloride in vitro and in vivo. PLoS ONE 9, e95546 10.1371/journal.pone.0095546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dailey W. A., Drenser K. A., Wong S. C., Cheng M., Vercellone J., Roumayah K. K., Feeney E. V., Deshpande M., Guzman A. E., Trese M., and Mitton K. P. (2017) Norrin treatment improves ganglion cell survival in an oxygen-induced retinopathy model of retinal ischemia. Exp. Eye Res. 164, 129–138 10.1016/j.exer.2017.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Seitz R., Hackl S., Seibuchner T., Tamm E. R., and Ohlmann A. (2010) Norrin mediates neuroprotective effects on retinal ganglion cells via activation of the Wnt/β-catenin signaling pathway and the induction of neuroprotective growth factors in Muller cells. J. Neurosci. 30, 5998–6010 10.1523/JNEUROSCI.0730-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Braunger B. M., Ohlmann A., Koch M., Tanimoto N., Volz C., Yang Y., Bösl M. R., Cvekl A., Jägle H., Seeliger M. W., and Tamm E. R. (2013) Constitutive overexpression of Norrin activates Wnt/β-catenin and endothelin-2 signaling to protect photoreceptors from light damage. Neurobiol. Dis. 50, 1–12 10.1016/j.nbd.2012.09.008 [DOI] [PubMed] [Google Scholar]
- 74. Chen Y., Zhang Y., Tang J., Liu F., Hu Q., Luo C., Tang J., Feng H., and Zhang J. H. (2015) Norrin protected blood-brain barrier via frizzled-4/β-catenin pathway after subarachnoid hemorrhage in rats. Stroke 46, 529–536 10.1161/STROKEAHA.114.007265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bassett E. A., Tokarew N., Allemano E. A., Mazerolle C., Morin K., Mears A. J., McNeill B., Ringuette R., Campbell C., Smiley S., Pokrajac N. T., Dubuc A. M., Ramaswamy V., Northcott P. A., Remke M., et al. (2016) Norrin/Frizzled4 signalling in the preneoplastic niche blocks medulloblastoma initiation. Elife 5, e16764 10.7554/eLife.16764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Xu Q., Qaum T., and Adamis A. P. (2001) Sensitive blood-retinal barrier breakdown quantitation using Evans Blue. Invest. Ophthalmol. Vis. Sci. 42, 789–794 [PubMed] [Google Scholar]
- 77. Lin C. M., Titchenell P. M., Keil J. M., Garcia-Ocaña A., Bolinger M. T., Abcouwer S. F., and Antonetti D. A. (2018) Inhibition of atypical protein kinase C reduces inflammation-induced retinal vascular permeability. Am. J. Pathol. 188, 2392–2405 10.1016/j.ajpath.2018.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Antonetti D. A., and Wolpert E. B. (2003) Isolation and characterization of retinal endothelial cells. Methods Mol. Med. 89, 365–374 10.1385/1-59259-419-0:365 [DOI] [PubMed] [Google Scholar]
- 79. Tiruppathi C., Malik A. B., Del Vecchio P. J., Keese C. R., and Giaever I. (1992) Electrical method for detection of endothelial cell shape change in real time: assessment of endothelial barrier function. Proc. Natl. Acad. Sci. U.S.A. 89, 7919–7923 10.1073/pnas.89.17.7919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Harhaj N. S., Barber A. J., and Antonetti D. A. (2002) Platelet-derived growth factor mediates tight junction redistribution and increases permeability in MDCK cells. J. Cell Physiol. 193, 349–364 10.1002/jcp.10183 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.