

Abstract
The small GTPase cell division cycle 42 (CDC42) plays essential roles in neurogenesis and brain development. Previously, using murine embryonic P19 cells as a model system, we showed that CDC42 stimulates mTOR complex 1 (mTORC1) activity and thereby up-regulates transcription factors required for the formation of neural progenitor cells. However, paradoxically, although endogenous CDC42 is required for both the initial transition of undifferentiated P19 cells to neural progenitors and their ultimate terminal differentiation into neurons, ectopic CDC42 overexpression promotes only the first stage of neurogenesis (i.e. the formation of neuroprogenitors) and not the second phase (differentiation into neurons). Here, using both P19 cells and mouse embryonic stem cells, we resolve this paradox, demonstrating that two splice variants of CDC42, differing only in nine amino acid residues in their very C-terminal regions, play distinct roles in neurogenesis. We found that a CDC42 splice variant that has a ubiquitous tissue distribution, termed here as CDC42u, specifically drives the formation of neuroprogenitor cells, whereas a brain-specific CDC42 variant, CDC42b, is essential for promoting the transition of neuroprogenitor cells to neurons. We further show that the specific roles of CDC42u and CDC42b in neurogenesis are due to their opposing effects on mTORC1 activity. Specifically, CDC42u stimulated mTORC1 activity and thereby induced neuroprogenitor formation, whereas CDC42b worked together with activated CDC42-associated kinase (ACK) in down-regulating mTOR expression and promoting neuronal differentiation. These findings highlight the remarkable functional specificities of two highly similar CDC42 splice variants in regulating distinct stages of neurogenesis.
Keywords: small GTPase, CDC42, stem cells, mTOR complex (mTORC), differentiation, ACK, CDC42b, CDC42u, neurogenesis, splice variant
Introduction
The Rho-family small GTPase cell division cycle 42 (CDC42)3 has been conserved throughout eukaryotic evolution from yeast to humans, suggesting that it plays fundamental roles in biology (1–3). Indeed, the total knockout of the Cdc42 gene in mice results in early embryonic lethality (4), whereas tissue-specific conditional knockouts of the Cdc42 gene have revealed critical roles in organ development and, in particular, in the development of the central nervous system (5, 6). When CDC42 is knocked out in the apical progenitor cells of the mouse telencephalon, including neuroepithelial and radial glial cells, these cells show defects in their ability to maintain epithelial structures and cell polarity and ultimately fail to adopt their proper cellular fates (7–9).
Previously, we showed that CDC42 is involved in the determination of Nestin-positive neural progenitor cell fate by controlling the expression of tissue specific transcription factors, using murine embryonal carcinoma P19 cells as a model system (10). CDC42 is activated by FGF- and Delta/Notch-dependent signaling pathways in the cell lineage specification phase of retinoic acid (RA)-induced neural differentiation of P19 cells. In turn, it promotes the activation status of mTORC1 (mechanistic target of rapamycin complex 1) and the resultant up-regulation of tissue-specific transcription factors, including neuroectodermal PAX6, which plays important roles in determining embryonic apical neural stem/progenitor cell fate. When WT or constitutively active CDC42 is ectopically overexpressed in P19 cells, it causes spontaneous differentiation into Nestin-positive neural progenitor cells, even in the absence of stimulation by RA. However, these cells lose the ability to terminally differentiate into post-mitotic neurons. Although ectopic overexpression of CDC42 inhibits terminal neural differentiation, the expression and activation levels of endogenous CDC42 continue to increase during the time window of the terminal differentiation of neural progenitors into neurons (10). This leads to a fundamental question: If CDC42 expression is required for terminal differentiation into neurons, why does ectopic expression of CDC42 only induce the formation of neural progenitor cells and prevent them from undergoing terminal differentiation?
One possible explanation for this apparent contradiction is related to the fact that vertebrates express two splice variants of CDC42, which might trigger distinct sets of cellular signals. Mammalian CDC42 was first identified from a human brain cDNA library in 1990 and referred to as G25K (11). Concurrently, our group identified a different form of CDC42 from human placenta and platelets, designating it as CDC42Hs (12, 13). Subsequent studies proved that G25K is the brain-specific splice variant expressed only in vertebrates (14–16), whereas CDC42Hs is the form conserved throughout eukaryotic evolution and shows ubiquitous distribution in general mammalian tissues (17) (to avoid confusion, hereafter we designate the total CDC42 population as CDC42, ubiquitously expressed CDC42 as CDC42u (ubiquitous), and brain-specific variant as CDC42b (brain) in this study). These two CDC42 splice variants differ only in the C-terminal nine amino acid residues and share an entire GTPase domain. Most of studies on mammalian CDC42 focused on the evolutionarily conserved CDC42u but not CDC42b. Thus far, only a few studies have reported on the biological functions of CDC42b in brain development (18–22), and the biological differences of two CDC42 splice variants still remain undetermined. Although the C-terminal amino acids of small GTPases, which represent hypervariable regions, are essential for their subcellular localization, the fact that the key functional domains of CDC42u and CDC42b are virtually identical suggests that they might share binding partners. These points raise some important questions. Specifically, are these two highly similar CDC42 splice variants capable of distinct functions, and does their potential competition to interact with common binding partners affect their functional and cellular activities?
In this study, we show that, despite their sequence similarity, the two CDC42 isoforms play functionally distinct roles in the neural differentiation of P19 embryonal carcinoma cells, as well as E14 mouse embryonic stem cells (ES cells). Our results suggest that the two isoforms have opposing actions on mTOR-dependent cellular signals, through which they control the transition of Nestin-positive neural progenitor cells into post-mitotic neurons. We further show that the distinct effects of CDC42u versus CDC42b on mTOR and neuronal differentiation are mediated through a nonreceptor tyrosine kinase, ACK (activated CDC42-associated kinase). Although both CDC42 splice variants are able to interact with ACK, only CDC42b promotes the ubiquitylation and degradation of mTOR in an ACK-dependent manner. These findings show that the two CDC42 splice variants are not functionally redundant. They provide new insights into how small GTPases exhibit their functional diversity and specificity and shed light on the molecular details of how Nestin-positive neural progenitor cells undergo proliferative arrest and terminally differentiate into post-mitotic neurons.
Results
CDC42b is highly up-regulated in post-mitotic neurons
The two known splice variants of CDC42, one that is ubiquitously distributed and the other that is brain-specific, differ only in their C-terminal nine amino acid residues, one residue in the alternative splicing site and eight residues in the hypervariable tail (Fig. 1A). Commercially available anti-CDC42 antibodies recognized both Myc-tagged CDC42u and CDC42b when expressed in NIH3T3 cells (Fig. 1B, second row from the bottom), whereas a CDC42b-specific antibody detected only Myc-CDC42b and not Myc-CDC42u (Fig. 1B, bottom row) (18). Tissue lysates from adult mouse brains showed abundant expression of a post-mitotic neuronal marker βIII-tubulin, a late-stage neuronal marker MAP2, as well as CDC42b (Fig. 1B, left lanes).
Figure 1.

A, an alignment of amino acid sequences indicates the differences between the two splice variants of CDC42, derived from isoform-specific exons. The differences in amino acid sequences are highlighted in gray boxes. The numbers and bars above the sequences indicate the numbers and positions of amino acid sequences. B, immunoblotting images showing the specificity of anti-pan-CDC42 and anti-CDC42b antibodies, using Western blotting. Tissue lysates from adult mouse brain served as an expression control of CDC42b. Vinculin served as a loading control. Myc-tagged CDC42 splice variants were expressed in NIH3T3 cells, and their expression was detected with anti-Myc, anti-pan-CDC42 (total CDC42), and anti-CDC42b antibodies. The bars and numbers beside each panel indicate the positions and sizes (kDa) of molecular markers, respectively. C and D, immunoblotting images showing the expression level of CDC42b in undifferentiated and differentiated P19 (C) and E14 cells (D). P19 and E14 cells were subject to the neural or cardiomyogenic differentiation protocols, and their differentiation status was monitored with the indicated differentiation markers. E, immunoblotting images showing the expression level of CDC42b in neural differentiated P19 cells. The cells were subjected to the RA-induced neural differentiation protocol until the indicated day. In days 6–9, neuronal differentiation was inhibited with serum stimulation (+RA/+serum, day 9), to maintain neural progenitor status, or promoted by switching medium into serum-free neuronal medium (+RA, day 9). F, immunoblotting images showing the expression (left) and activation levels (right) of CDC42b in neurally differentiated E14 cells. E14 cells were subjected to each culture condition, and the expression levels of differentiation markers and CDC42 proteins were monitored using Western blotting. The protein lysate from each culture condition was subjected to GST or GST-PBD pulldown assay, and the relative amount of the GTP-bound active form of CDC42 proteins was monitored. G, an epifluorescent image showing the differentiation status of E14-derived neurons. E14 cells were subjected to the monolayer neural differentiation protocol until day 21. Cells were fixed and stained with Hoechst (blue) and anti-βIII-tubulin antibody (green). The black bar below the image indicates scale (50 μm).
To examine the biological functions of the CDC42 splice variants, we used two cell differentiation model systems, P19 embryonal carcinoma cells and E14 mouse embryonic stem cells. Prior to induction, both P19 and E14 cells expressed the pluripotent transcription factor OCT4 (Fig. 1 (C and D), second rows from the top). Upon stimulation with RA to induce neural differentiation, P19 cells lost OCT4 expression and up-regulated βIII-tubulin and MAP2 expression (Fig. 1C, third and fourth rows from the top), indicative of differentiation into post-mitotic neurons. Neural differentiation of E14 cells was initiated by withdrawal of LIF and chemical inhibitors. Following this induction, E14 cells differentiated into βIII-tubulin– and MAP2–positive post-mitotic neurons when cultured under monolayer neural induction conditions (Fig. 1D, third and fourth rows from the top). In contrast, both P19 and E14 cells up-regulated GATA4 and cardiac troponin T (cTnT) when using cardiomyocyte differentiation protocols (Fig. 1, C and D, third and fourth rows from the bottom). Although total CDC42 expression was up-regulated in both neural and cardiomyogenic cells in each differentiation model system, CDC42b was up-regulated only during neural differentiation (Fig. 1, C and D, bottom rows).
RA induction prompted P19 cells to first differentiate into Nestin-positive neural progenitor cells, and then upon serum stimulation, the cells remained in that status. Switching the medium from serum-containing α-minimum essential medium to serum-free, B27-supplemented, Neurobasal medium caused the cells to terminally differentiate into βIII-tubulin–positive post-mitotic neurons more efficiently (Fig. 1E, third row from the top). The protein expression level of CDC42b was highly up-regulated at day 9 in P19 cells undergoing RA-dependent neural differentiation, but not in Nestin-positive neural progenitor cells (Fig. 1E, bottom row). When exposed to the monolayer neural differentiation protocol, E14 cells also showed a transition into OCT4−/Nestin+ cell status. Upon stimulation with epidermal growth factor and bFGF, the Nestin-positive cell population exhibited colony-forming ability in a suspension culture condition (data not shown), indicative of differentiation into neural stem cells (NSCs). The NSCs were then individually separated and further cultured to differentiate into βIII-tubulin– and MAP2–positive post-mitotic neurons (Fig. 1, F (left, second and third rows from the top) and G). The protein expression level of CDC42b was highly up-regulated in neuron-enriched cell populations but not in Nestin-positive NSCs (Fig. 1F, left, bottom row). Using a p21-binding domain (PBD)-pulldown assay, which detects the relative levels of the activated GTP-bound forms of the CDC42 proteins, we observed that activated, GTP-bound CDC42b was exclusively present in neurons, whereas total CDC42 protein (which includes CDC42u) was also detected in ESCs and NSCs (Fig. 1F, right). These results suggest that CDC42b is highly up-regulated and activated in post-mitotic neurons, but not in ESCs or NSCs.
The two CDC42 splice variants have distinct functions in the different stages of neural differentiation
Previously, we showed the importance of total CDC42 for the RA-induced neural differentiation of P19 cells, using siRNAs that recognized both CDC42 isoforms (10). To assess the functional differences between CDC42u and CDC42b, therefore, we designed siRNAs that selectively target each of the two CDC42 isoforms. First, prior to RA induction, we treated P19 cells with siRNAs targeting each of the CDC42 isoforms. Following the siRNA treatments, P19 cells were subjected to the RA-induced neural differentiation protocol, and at day 4, total mRNAs and cell lysates were collected and analyzed. CDC42-common siRNAs #1 and #2 (i.e. targeting both CDC42 isoforms) depleted the mRNA of both isoforms (Fig. S1A, columns 1–3), whereas the isoform-specific siRNAs selectively knocked down the mRNA expression level of the specific CDC42 isoform being targeted (Fig. S1A, columns 4 and 5).
We then examined the effects of the CDC42 isoform-specific knockdowns on the entire process of RA-induced neural differentiation by extending the neural differentiation protocol until day 9, when neurogenesis begins. Cells treated with control siRNA showed normal βIII-tubulin–positive neurite extension in immunofluorescence microscopy experiments (Fig. 2A, panel 1), whereas cells treated with CDC42-common siRNA #2 did not show βIII-tubulin staining (panel 2). Unlike cells treated with CDC42-common siRNA #2, a small number of cells treated with splice variant–specific siRNAs still showed βIII-tubulin staining. Whereas the resultant βIII-tubulin–positive neurons showed normal neurite extension when knocking down CDC42u (Fig. 2A, panel 3), this was not the case when knocking down CDC42b (panel 4).
Figure 2.

A, epifluorescence images showing the neuronal cell differentiation and morphology of siRNA-treated P19 cells. Cells were treated with the indicated siRNAs and then subjected to the RA-induced neural differentiation protocol. At day 9, cells were fixed and stained with Hoechst (blue) and anti-βIII-tubulin antibody (green). The black bar to the right of the images indicates scale (20 μm). In B–D, P19 cells were subjected to the RA-dependent neural differentiation protocol until day 4 and then treated with siRNAs and/or lentivirus. After transfection and/or transduction, cells were subjected to the neural differentiation protocol until day 9. B, immunoblotting images showing the expression levels of βIII-tubulin and total CDC42 proteins. Cell lysates were collected at day 9 in the RA-dependent neural differentiation protocol. Histograms show the percentages of βIII-tubulin–positive neurons in each treatment (C and D). Error bars, S.E. (n = 4). Significance of differences is indicated by n.s. (not significant, p > 0.05) and an asterisk (p < 0.05), using a t test (C) or Tukey's test (D). E, immunoblotting images showing the expression levels of βIII-tubulin, total CDC42 proteins, and CDC42b in E14 cells. The cells were subjected to neural differentiation until day 11 and then treated with lentivirus for 2 days. After lentivirus treatment, cells were subjected to neural differentiation until day 28.
Because defects in the establishment of neural progenitor cells might have affected the subsequent neuronal differentiation processes in the experiments described above, we next examined the effects of knocking down the two CDC42 isoforms during the terminal differentiation phase, by performing RNAi after the multipotent cells differentiated into neural progenitors (i.e. at day 4 of the RA-induced neural differentiation procedure). Treatment with the CDC42-common siRNAs resulted in decreased βIII-tubulin protein expression as well as a reduction in the number of βIII-tubulin–positive neurons generated (Fig. 2, B (lanes 1–3) and C (columns 1 and 2)), and in neurite extension (Fig. S2, panels 1 and 2). However, treatment of cells with CDC42u-specific siRNAs did not affect βIII-tubulin expression levels or the number of βIII-tubulin–positive neurons (Fig. 2, B (lane 4) and C (column 3)). Moreover, the βIII-tubulin–positive neurons showed normal neurite extension (Fig. S2, panel 3). In contrast, treatment with the CDC42b-specific siRNAs markedly inhibited βIII-tubulin expression (Fig. 2B, lane 5), reduced the number of βIII-tubulin–positive neurons (Fig. 2C, column 4), and showed defects in neurite extension (Fig. S2, panel 4). This indicates that the functions of the CDC42 splice variants are not redundant, but rather that CDC42b and not CDC42u is essential in the late stages of neural differentiation in P19 cells.
To further establish the functional differences between the two CDC42 isoforms during neuronal differentiation, we treated P19 cell-derived neural progenitor cells with both CDC42u- and CDC42b-targeting siRNAs and simultaneously expressed a YFP-tagged siRNA-resistant CDC42u or CDC42b (YFP-CDC42u* or YFP-CDC42b*, respectively), using a lentivirus-based expression system (Fig. S1B). Knocking down both CDC42 splice variants simultaneously reduced the number of βIII-tubulin–positive neurons, in the presence of YFP expression, and this effect was restored by expressing YFP-CDC42b* (Fig. 2D, column 4). Expression of YFP-CDC42u* was less effective (Fig. 2D, column 3), suggesting that CDC42b but not CDC42u is sufficient to restore the defects caused by knocking down both CDC42 splice variants in the P19-derived neurons.
The observed defects in neurogenesis could be partially due to effects on neuronal cell survival, because cells treated with the CDC42-common siRNA #2 showed increased apoptosis, as determined using annexin V and propidium iodide staining (Fig. S2C, second black column from the left). Interestingly, cells treated with CDC42u-specific siRNAs did not show a high degree of apoptosis (Fig. S1C, third black and white columns from the left), whereas cells treated with the CDC42b-specific siRNAs showed increased apoptosis in serum-free conditions (Fig. S1C, first black column from the right), but not in serum-stimulated conditions (first white column from the right). This indicates that CDC42b is essential for the survival of neurons but not for the survival of neural progenitor cells, consistent with the observed highly elevated expression of CDC42b in neurons (Fig. 1E).
In E14 cells, a lentivirus-based shRNA expression system was used to achieve efficient knockdown of the CDC42 splice variants. Because knocking down total CDC42 or CDC42u, but not CDC42b, caused apoptosis during the transition from undifferentiated ESCs into NSCs (data not shown), we performed RNAi experiments during the transition from NSCs into neurons, only after the neural cell lineage was specified. Knocking down total CDC42 significantly affected βIII-tubulin expression (Fig. 2E, lanes 1–3). Cells treated with shRNAs selectively targeting CDC42u did not show a significant change in βIII-tubulin expression, whereas knocking down CDC42b caused a severe reduction (Fig. 2E, lanes 4 and 5). Collectively, these results show that the CDC42b isoform, but not the CDC42u, is essential for the terminal differentiation from NSCs into post-mitotic neurons in both P19 and E14 cells.
CDC42 isoforms have antagonistic effects on the transition of Nestin-positive neural progenitor cells into post-mitotic neurons
We previously reported that ectopic expression of WT CDC42u or a constitutively active CDC42u mutant inhibits the ability of Nestin-positive neural progenitor cells to terminally differentiate into βIII-tubulin–positive post-mitotic neurons (10). The experiments described above show that CDC42u and CDC42b have distinct biological functions in neural progenitor cells and post-mitotic neurons. This prompted us to ask whether establishing a balance between CDC42u and CDC42b activities is critical in determining whether Nestin-positive neural progenitor cells maintain their progenitor status or undergo differentiation into post-mitotic neurons. To address this question, we established P19 cell lines stably expressing the two Myc-tagged CDC42 isoforms (Fig. S3) and examined their abilities to induce neural differentiation.
Vector control cells differentiated into Nestin-positive neural progenitor cells or βIII-tubulin–positive neurons when exposed to the RA-induced neural differentiation protocol (Fig. 3A, top panels). Cells ectopically expressing Myc-tagged CDC42u exhibited strong Nestin staining but failed to differentiate into neurons (Fig. 3A, middle panels). In contrast, cells expressing Myc-CDC42b were capable of undergoing normal RA-dependent neuronal differentiation (Fig. 3A, bottom panels). These results demonstrate that ectopic expression of CDC42u halts RA-treated P19 cells at the Nestin-positive neural progenitor stage, whereas ectopic expression of CDC42b permits terminal neuronal differentiation.
Figure 3.

A, epifluorescence images showing the differentiation status of the P19 stable cell lines that expressed Myc-tagged CDC42u or CDC42b at day 13 in the RA-induced neural differentiation protocol. Cells were fixed and stained with Hoechst (blue), anti-βIII-tubulin (green), and anti-Nestin (red) antibodies. The black bar below the image indicates scale (50 μm). B, immunoblotting images showing the neural differentiation of P19 stable cell lines. Vector control and Myc-CDC42u(F28L)– or Myc-CDC42b(F28L)–expressing cells were subjected to RA-induced neural differentiation, and their differentiation status was monitored with the indicated differentiation markers. Vinculin served as a loading control. C, histograms showing the percentages of βIII-tubulin–positive neurons. P19 stable cell lines were subjected to the RA-induced neural (−serum) or neural progenitor cell (+serum) differentiation protocol until day 9. Cells were fixed at day 9, and the numbers of βIII-tubulin–positive neurons were counted. Error bars, S.E. (n = 4). Significance of differences is indicated by n.s. (not significant, p > 0.05) and an asterisk (p < 0.05), using Tukey's test.
The biological differences caused by the two CDC42 isoforms were more pronounced in P19 cell lines expressing constitutively active F28L mutants of the CDC42 proteins. Myc-CDC42u(F28L)–expressing cells failed to express βIII-tubulin and MAP2 when exposed to the RA-induced neural differentiation protocol (Fig. 3B). In contrast, Myc-CDC42b(F28L)–expressing cells showed higher expression levels of βIII-tubulin and MAP2 (Fig. 3B). Furthermore, even upon stimulation with 10% serum, which is sufficient to block terminal differentiation of vector control cells, Myc-CDC42b(F28L)–expressing cells differentiated into βIII-tubulin–positive neurons (Fig. 3C, first white column from the right). Thus, whereas CDC42u promotes the differentiation of P19 cells into Nestin-positive neural progenitor cells, CDC42b stimulates their transition to post-mitotic neurons. Moreover, it apparently is important to maintain a balance between the signaling activities of the two CDC42 isoforms to ensure that Nestin-positive neural progenitor cells are able to terminally differentiate into post-mitotic neurons.
CDC42b suppresses mTOR-dependent signaling during neural differentiation
We previously reported that CDC42 drives P19 cells to differentiate into Nestin-positive neural progenitor cells, and P19 cells maintain their progenitor status, partially through the ability of CDC42 to modulate mTORC1-dependent cell signals (10). Therefore, we examined the specific effects exerted by the two CDC42 isoforms on mTORC1 activity. First, we knocked down each CDC42 isoform in P19 cells, prior to RA induction, to assess effects on mTORC1-dependent cell signals during the early stage of neural differentiation. Consistent with our previous finding, treatment with the CDC42-common and CDC42u-specific siRNAs caused a reduction in the RA-induced phosphorylation of mTOR (Ser-2448), which is specific to mTORC1 activation (Fig. 4A (columns 1–4) and Fig. S1D (lanes 1–4)), as well as in the expression of a downstream target, neuroectodermal transcription factor, PAX6 (Fig. 4B, columns 1–4). In contrast, treatment with the CDC42b-specific siRNAs had minimal effect (Fig. 4A (column 5) and Fig. S1D (lane 5)). To assess whether this signaling pathway was conserved in E14 cells, we knocked down the CDC42 splice variants in E14-derived NSCs. E14 cells first were subject to the neural differentiation protocol, and knockdowns were then performed on the derived NSCs. Consistently, knocking down total CDC42 or the CDC42u isoform, but not the CDC42b isoform, caused a significant reduction in the mTOR (Ser-2448) phosphorylation status and PAX6 expression (Fig. 4C).
Figure 4.

A, histograms showing densitometry analyses of phospho-mTOR (pSer-2448) blots. P19 cells were treated with siRNAs for 18 h and then subjected to the RA-dependent neural differentiation protocol. Cell lysates were collected at day 4 in the RA-induced neural differentiation protocol. Signal intensities are represented relative to that of control siRNA-treated cells. B, histograms showing the relative mRNA expression levels of PAX6 in the RNAi background. The mRNA levels are represented relative to that of control siRNA-treated cells. C, immunoblotting images showing the expression levels of total CDC42 proteins and the expression and activation levels (pSer-2448) of mTOR in E14-derived NSCs in the RNAi background. E14-derived NSCs were treated with lentivirus, containing an shRNA expression system. Cell lysates were collected at day 14 in the monolayer neural differentiation protocol. Vinculin served as a loading control. The numbers below the immunoblotting images of mTOR indicate the -fold changes and S.E., based on densitometry (n = 3). D, immunoblotting images showing total and phosphorylation levels of mTOR in the P19 stable cell lines. Vector control and Myc-CDC42u– and Myc-CDC42b–expressing P19 stable cell lines were subjected to the RA-induced neural differentiation protocol. Cell lysates were collected at day 4 and subjected to Western blotting. E, histograms showing the relative mRNA expression levels of PAX6 in the P19 stable cell lines. Error bars, S.E. (n = 4 in A, and n = 3 in B and E). Significance of differences is indicated by n.s. (not significant, p > 0.05) and an asterisk (p < 0.05) using a t test (A, B, and E). F, immunoblotting images showing the expression and activation levels of mTOR in E14-derived NSCs. E14 cells were subjected to the neural differentiation protocol until day 11, and the derived NSCs were treated with lentivirus, containing protein expression cassettes, for 2 days. Cell lysates were collected at day 18.
Because CDC42b is only weakly expressed during the early stage of neural differentiation, it is possible that the predominant CDC42u could compensate for the loss of CDC42b. To eliminate this possibility, we examined the effects of ectopic expression of the CDC42 isoforms on mTOR-dependent cell signals during the early stage of neural differentiation. Upon RA treatment, Myc-CDC42u–expressing cell lines showed an increase in the phosphorylation level of mTOR (Fig. 4D, lanes 1 and 2). In contrast, Myc-CDC42b–expressing cells showed a marked decrease in both mTOR expression and mTORC1 activation status (Fig. 4D, lane 3). These antagonistic effects on mTOR-dependent cell signaling pathways were also apparent when examining the expression levels of the downstream target, PAX6. Myc-CDC42u–expressing cells showed an up-regulation of PAX6 compared with vector control cells, whereas Myc-CDC42b–expressing cell lines showed a sharp decrease in PAX6 expression (Fig. 4E). Complementary to the results obtained using P19 cells, the antagonistic effects of CDC42u and CDC42b on mTOR-dependent signals and PAX6 were also observed in E14-derived NSCs (Fig. 4F). Collectively, these results demonstrate that the two CDC42 isoforms have opposing effects on mTOR-dependent signaling.
CDC42b promotes mTOR degradation in an ACK-dependent manner in nonneuronal cells
We next set out to determine how CDC42b mediates distinct signaling events and cellular responses from those triggered by CDC42u. We first examined ACK as a potential signaling partner for CDC42b, because ACK has been reported to bind to the CDC42 proteins and is highly up-regulated in the brain (23). We did not find any differences in the ability of C-terminal V5-tagged ACK (ACK-V5) to co-immunoprecipitate with the two Myc-tagged CDC42 isoforms or any changes in the autophosphorylation status of ACK when bound to either of the two CDC42 splice variants in COS7 cells (Fig. 5A). However, we discovered that CDC42b works together with ACK in a unique way to regulate mTOR. In HeLa cells, either Myc-tagged WT CDC42b or ACK-V5 alone caused a modest reduction in mTOR levels, but when combined, they synergistically down-regulated mTOR (Fig. 5B, lanes 1–4). In contrast, a kinase-defective mutant of ACK, ACK(K158R), did not promote mTOR degradation, either alone or when co-expressed with Myc-CDC42b (Fig. 5B, lanes 5 and 6), suggesting that the kinase activity of ACK is necessary for the mTOR down-regulation. Unlike Myc-CDC42b, Myc-CDC42u showed a protective effect on the ACK-V5–dependent down-regulation of mTOR (Fig. 5B, lane 7), demonstrating that the two CDC42 splice variants exert distinct functional effects, as mediated through ACK.
Figure 5.

A, immunoblotting images showing the results of immunoprecipitation (IP) experiments between N-terminal Myc-tagged CDC42 splice variants and C-terminal V5-tagged ACK (ACK-V5) in COS7 cells. Tagged proteins were immunoprecipitated using only beads or beads with the indicated antibodies, and the co-precipitated proteins were detected by Western blotting. The Input panel indicates the relative expression levels of the indicated proteins in the cell lysates used for the immunoprecipitation experiments. The tyrosine phosphorylation status of the immunoprecipitated ACK-V5 was also detected using anti-phosphotyrosine antibody. B, immunoblotting images showing the effects of ectopically expressed Myc-CDC42b, ACK-V5, and a kinase-defective mutant of ACK, ACK(K158R)-V5, on mTOR expression in HeLa cells. β-Actin served as a loading control. C, immunoblotting images showing the effects of a proteasome inhibitor, MG132, on mTOR levels, due to the co-expression of Myc-CDC42b and ACK-V5 in HeLa cells. The numbers below the immunoblotting images of mTOR indicate the -fold changes, based on densitometry. D, immunoblotting images showing the ubiquitylation status of mTOR in HeLa cells. HeLa cells were transfected with control and ACK-V5– and Myc–tagged CDC42 splice variant–expressing vectors and then subjected to serum starvation, in the presence of MG132. Endogenous mTOR proteins were immunoprecipitated using an anti-mTOR antibody, and the ubiquitylation level of mTOR was detected with an anti-ubiquitin antibody (Ub). The numbers below the immunoblotting images indicate the -fold changes and S.E., based on densitometry (n = 3 in B and C, and n = 2 in D).
The reduction in mTOR levels induced by CDC42b and ACK was prevented upon treatment with a proteasomal inhibitor MG132 (Fig. 5C), suggesting that CDC42b and ACK promote the degradation of mTOR in a proteasome-dependent manner. Because protein ubiquitylation is essential for proteasomal protein degradation, we next examined the ubiquitylation status of mTOR, in the presence or absence of the two CDC42 isoforms and ACK. The immunoprecipitated mTOR showed higher ubiquitylation levels in cells co-expressing Myc-CDC42b and ACK-V5, compared with vector control cells or cells expressing ACK-V5 and/or Myc-CDC42u, in the presence of MG132 (Fig. 5D, right). These results suggest that CDC42b, working together with ACK, is able to promote the ubiquitylation and proteasomal degradation of mTOR in nonneuronal epithelial cells.
ACK is essential for CDC42b-dependent neuronal differentiation
ACK is highly up-regulated in the mouse brain (23), and we found that the expression of ACK increased during RA-induced neural differentiation of P19 cells on day 11 (Fig. 6A). Undifferentiated E14 cells showed higher ACK expression than undifferentiated P19 cells, and the expression of ACK was further increased during neural differentiation into NSCs and neurons (Fig. 6B). To test the importance of ACK in neurogenesis, we knocked down ACK by treating P19 cells with ACK-targeting siRNAs at day 4 during the RA-induced neural differentiation protocol. When knocking down ACK, the expression level of βIII-tubulin was significantly decreased (Fig. S4A), and fewer cells showed βIII-tubulin staining on day 9 of the RA-dependent neural differentiation protocol (Fig. 6C). In serum-stimulated conditions, loss of ACK did not affect the survival of neural progenitor cells, with no changes in apoptosis detected (Fig. S4B, white columns). However, under serum-starved conditions promoting neuronal differentiation, higher numbers of ACK siRNA-treated cells showed apoptosis (Fig. S4B, black columns), suggesting that ACK is essential for neuronal cell survival. Although defects in neurite extension were less severe than in CDC42b-specific siRNA-treated cells (Fig. S2, panels 5–7), generally, the phenotypes caused by knocking down ACK were similar to those caused by knocking down CDC42b.
Figure 6.

Immunoblotting images showing the expression levels of ACK upon the neural differentiation of P19 cells (A) and E14 cells (B). Vinculin served as a loading control. In C and D, P19 cells or the indicated P19 cell lines were subjected to the RA-dependent neural differentiation protocol until day 4 and then treated with control or ACK-targeting siRNAs. After transfection, cells were subjected to the neural or neural progenitor cell differentiation protocol until day 9. Histograms show the percentages of βIII-tubulin–positive neurons. Error bars, S.E. (n = 4 in C, and n = 6 in D). Significance of differences is indicated by n.s. (not significant, p > 0.05) and an asterisk (p < 0.05), using a t test (C) and Tukey's test (D).
To test the involvement of ACK in CDC42b-dependent neuronal differentiation, vector control and Myc-CDC42b(F28L)-expressing P19 cells were treated with ACK-targeting siRNAs at day 4 following RA induction. As shown above, control P19 cells did not show significant βIII-tubulin expression when serum-stimulated under conditions of neural progenitor cell differentiation, whereas expression of Myc-CDC42b(F28L) promoted their differentiation into βIII-tubulin-positive neurons (Fig. 6D (columns 1 and 5) and Fig. S4C (lanes 1 and 5)). Knocking down ACK expression attenuated the ability of Myc-CDC42b(F28L)–expressing cells to differentiate into βIII-tubulin–positive neurons (Fig. 6D (columns 6–8) and Fig. S4C (lanes 6–8)). ACK knockdowns did not affect cell survival in either control or Myc-CDC42b(F28L)–expressing P19 cells in serum-stimulated conditions (Fig. S4D), eliminating the possibility that knocking down ACK attenuates neuronal differentiation by causing apoptosis in a CDC42b(F28L)-independent manner. These results indicate that the activation of CDC42b promotes the differentiation of neural progenitor cells into post-mitotic neurons in an ACK-dependent manner in P19 cells.
CDC42b reduces mTOR levels in an ACK-dependent manner
To further analyze the functions of ACK in the CDC42b-dependent neural differentiation of P19 cells, we established P19 stable cell lines expressing ACK-targeting shRNAs or V5-tagged ACK (Fig. S4, E and F). The ACK knockdown P19 cell lines showed higher expression of mTOR at day 4 in the RA-dependent differentiation protocol compared with control shRNA-expressing cells in neural differentiation conditions (Fig. 7A). ACK-V5–expressing P19 cells showed decreased levels of mTOR at day 4 of the RA-dependent differentiation protocol, whereas ACK(K158R)-expressing cells showed increased mTOR expression (Fig. 7B), suggesting that the kinase activity of ACK is necessary for the down-regulation of mTOR during the neural differentiation process of P19 cells. To determine whether CDC42b promotes decreased mTOR expression in an ACK-dependent manner, as we observed in nonneuronal epithelial cells, we examined mTOR expression by combining ectopic CDC42b expression with knockdowns of ACK. We first treated Myc-CDC42b–expressing P19 cell lines with control or ACK-targeting siRNAs, prior to subjecting the cells to RA-dependent differentiation. The Myc-CDC42b–expressing cells showed diminished levels of mTOR expression on day 4 of the RA-dependent differentiation, but knocking down ACK expression was sufficient to restore mTOR levels (Fig. 7C).
Figure 7.

Immunoblotting images showing the expression levels of ACK and/or mTOR in P19 stable cell lines. A, control shRNA- or ACK-targeting shRNA-expressing cells; B, vector control and ACK-V5– or ACK(K158R-V5)–expressing cells. P19 cells were subjected to the RA-dependent neural differentiation protocol until day 4 and serum-starved for days 2–4. C, immunoblotting images showing the expression levels of ACK and mTOR in P19 stable cell lines. Vector control and Myc-CDC42b–expressing cells were treated with the indicated siRNAs and then subjected to the normal RA-dependent neural differentiation protocol until day 4 and serum-starved for days 2–4. D–F, immunoblotting images showing the expression levels of ACK and mTOR in E14-derived NSCs. E14 cells were subjected to neural differentiation until day 11 and then treated with the indicated lentivirus and/or protein-expressing DNA vectors. D, control shRNA- or ACK-targeting shRNA-expressing lentivirus; E, vector control, ACK-V5-, or ACK(K158R)-expressing vectors; F, control and HA-CDC42b(F28L)–, control shRNA–, and ACK–targeting shRNA-expressing lentivirus. On day 14, cell lysates were collected and subjected to Western blotting experiments. The numbers below the immunoblotting images of mTOR indicate the -fold changes and S.E., based on densitometry (n = 3 in A, B, D, and E). The results shown in C and F are representative of three experiments.
We then examined whether CDC42b similarly down-regulates mTOR in an ACK-dependent manner in E14-derived NSCs. E14 cells were first subjected to the monolayer neural differentiation protocol until day 11, followed by ACK knockdown or ectopic protein expression. As observed in P19 cells, knocking down ACK up-regulated mTOR levels in E14-derived NSCs (Fig. 7D). Ectopic expression of ACK-V5 down-regulated mTOR expression, whereas the kinase-dead ACK(K158R)-V5 slightly up-regulated its expression (Fig. 7E). Upon ectopic expression of HA-CDC42b(F28L), mTOR levels were sharply down-regulated, and simultaneous knockdowns of ACK partially restored this effect in E14-derived NSCs (Fig. 7F). Thus, CDC42b down-regulates mTOR expression in an ACK-dependent manner during the neural differentiation of E14 cells as well as P19 cells.
Discussion
The Rho-family small GTPase CDC42 has been suggested to play a critical role in the development of the central nervous system, using conditional knockout mouse models (7–9). Among the essential roles played by CDC42 are the establishment of proper cellular polarity for neuroepithelial and radial glial cells as well as ensuring the correct maintenance of apical neural progenitor cells (7–9). It therefore comes as no surprise that the loss of CDC42 has severe consequences for neurogenesis and brain development. In these studies, however, the manipulation of the Cdc42 gene affects the expression of both its splice variants, and the biological differences between these two seemingly identical forms of the protein had been underappreciated and remain unanswered.
In a previous study, we focused on the involvement of CDC42 in the establishment of neural progenitor cell fate, by taking advantage of the embryonal carcinoma P19 cell line (10). In response to RA, P19 cells lose their multipotent character and the expression of OCT4, thereby undergoing the transition to neural progenitor cells and ultimately terminal differentiation into neurons and glial cells. Thus, P19 cells provide a tractable model for examining the actions of CDC42 in different stages of neurogenesis (10). We showed that CDC42 plays an essential part in the establishment of neural progenitor cell fate, when activated downstream from FGF receptors and Delta/Notch, by up-regulating mTOR activity and driving the increased expression of key transcription factors, including PAX6 (10). The existence of a signaling link between CDC42 and mTOR has been suggested to occur in various cellular contexts (10, 24, 25), and both proteins have been shown to play important roles in the maintenance of neural stem and progenitor cells in the developing mammalian brain (7–9, 26, 27). However, the P19 cell system enabled us to establish a clear functional connection between CDC42 and mTOR in neurogenesis.
Nonetheless, our previous study also presented a puzzling paradox. Specifically, although the expression of endogenous CDC42 was absolutely required both for the generation of neural progenitors from multipotent cells and for their progression to neurons, we found that the ectopic expression of CDC42, while being able to drive the transition of multipotent P19 cells to neural progenitors, completely blocked their further differentiation. What made this a particularly vexing observation is that the expression of CDC42 is not halted at the neural progenitor stage, but continues to increase through terminal neural differentiation. We felt that by uncovering the explanation behind these puzzling findings, we would obtain insights into how cell fate decisions are made during neurogenesis and, in particular, how neural progenitors undergo the transition to differentiated neurons. In fact, this would turn out to be the case, as we show in the present study that the answer to the puzzle is that a second splice variant form of CDC42, uniquely expressed in the brain and only differing from the ubiquitously distributed isoform of CDC42 within the C-terminal nine amino acids, accounts for the “CDC42 requirement” during terminal neural differentiation. Unlike the ubiquitously distributed form of CDC42 (CDC42u), which is expressed throughout both major stages of neurogenesis, the expression of the brain-specific isoform of CDC42 (CDC42b) predominantly occurs at the outset of terminal neuronal differentiation. Moreover, whereas the ectopic overexpression of WT CDC42, and particularly a constitutively active CDC42u(F28L) mutant, blocks the differentiation of neural progenitor cells into neurons, the analogous expression of WT or constitutively active CDC42b drives terminal differentiation. Therefore, despite their striking similarity in primary amino acid sequence, the two known isoforms of CDC42 play specific and distinct roles during neurogenesis, with CDC42u being essential for the first major step involving the transition of multipotent cells into neural progenitors, whereas CDC42b is uniquely able to ensure completion of the process by stimulating the second stage (i.e. the terminal differentiation of neural progenitors into neurons).
How do these highly similar forms of CDC42 mediate such distinct functional and biological outcomes? The answer is not likely to be a simple one, as the two major stages of neurogenesis may well involve the complex integration of multiple signaling outputs from the different isoforms of CDC42. There appears to be a requirement for the ubiquitous form of CDC42 in both stages of neurogenesis (i.e. during the formation of neural progenitors and in the generation of neurons), although these roles are apparently under tight regulation, as evidenced by the excessive expression and/or activation of CDC42u giving rise to a block in neural differentiation. Although CDC42b appears to be exclusively involved in the differentiation of neural progenitors into neurons, it is reasonable to assume that CDC42b uses more than one type of signal to influence multiple facets of this stage of neurogenesis, including potential roles in axon guidance, axon-dendrite specifications, and synaptogenesis (19, 22, 28, 29). Nonetheless, what does appear to be a critical signaling node for the two forms of CDC42 in the major stages of neurogenesis is the differential regulation of mTOR. Whereas CDC42u helps to ensure maximal mTOR activity, leading to the up-regulation of PAX6 expression during the transition of multipotent cells to neural progenitors, CDC42b appears to negatively regulate mTOR activity and markedly repress PAX6 expression.
It is still not clear how CDC42u signals the up-regulation of mTOR during the formation of neural progenitor cells, nor in any other cellular context where it has been shown to activate mTOR (10, 24, 25). However, the negative regulatory actions of CDC42b toward mTOR that are necessary for neuronal differentiation appear to require the participation of ACK, a nonreceptor tyrosine kinase that was identified as a specific binding partner for activated, GTP-bound CDC42u (30). Of course, this begs the question of how CDC42b is able to uniquely cooperate with ACK, when both CDC42u and CDC42b are capable of binding to this tyrosine kinase through their common effector loop (i.e. also known as their Switch 1 domain). The answer may come from the differences in the last nine amino acids of these two GTPases, which will likely influence their cellular localization. The C-terminal portions of the two forms of CDC42 each contain a polybasic region and a CAAX motif. For CDC42u, its CAAX motif, CVLL, specifies the attachment of a geranyl-geranyl moiety, following endoproteolytic cleavage of the VLL tripeptide and C-terminal methylation. Meanwhile, CDC42b, which has a double cysteine motif, CCIF, shows multiple options regarding its lipid modification: single isoprenylation in the first cysteine residue by geranyl-geranyl or farnesyl-moiety with further modifications (i.e. tripeptide cleavage and methylation); the combination of isoprenylation and palmitoylation in its first and second cysteine residues, respectively (18, 20); and also dual palmitoylation in both cysteine residues (19). It has been reported that the palmitoylation of the second cysteine residue in the CCIF sequence of geranyl-geranyl–modified CDC42b inhibits its ability to productively interact with RhoGDIα (20). During the later stage of neurogenesis, within specific regions of neurons where palmitoylation may be occurring to a high degree, such as the cholesterol-enriched dendritic protrusions (18, 19, 22), CDC42b might be more efficiently palmitoylated by up-regulated DHHC proteins. Because single or dual palmitoylated CDC42b is apparently unable to interact with RhoGDIα (20), it may more stably associate with the protrusion membrane, thereby helping to promote processes important for dendritic spine formation. Additionally, the hypervariable regions of RAC and RAS small GTPases have been also shown to function as a binding platform for their signaling partners (31–34). Taken together, these findings imply that the differential signaling outputs of CDC42u and CDC42b might indeed be due to their distinct abilities to associate with specific proteins within distinct membrane regions, such that CDC42b is preferentially located within the proximity of mTOR and is thus able to recruit ACK to negatively regulate mitogenic signaling and mTOR function.
In conclusion, we now highlight strikingly distinct biological functions for the two CDC42 isoforms during neurogenesis, when Nestin-positive neural progenitor cells terminally differentiate into post-mitotic neurons (summarized in Fig. 8). We recognize that the required actions of CDC42b during terminal neuronal differentiation likely involve additional signaling functions aside from negatively regulating mTOR activity. However, these findings now provide an answer to what had been a puzzling observation regarding how CDC42 could be required for both major stages of neurogenesis, while only appearing to be capable of promoting the first stage (the conversion of multipotent cells into neural progenitor cells). We also make the intriguing discovery that mTOR is the critical regulatory switch that receives distinct signals from the two isoforms of CDC42. We suspect that the ability of CDC42b to negatively regulate mTOR in some way aids the transition of neuroprogenitor cells from a highly proliferative state to a growth state that is more appropriate for terminal differentiation. Because the deregulation of mTOR activity is often observed in neurological disorders and neurodegenerative diseases (26, 27), these regulatory capabilities exhibited by the two CDC42 isoforms might have critically important consequences for maintaining healthy neurons both in the embryonic and adult brain.
Figure 8.
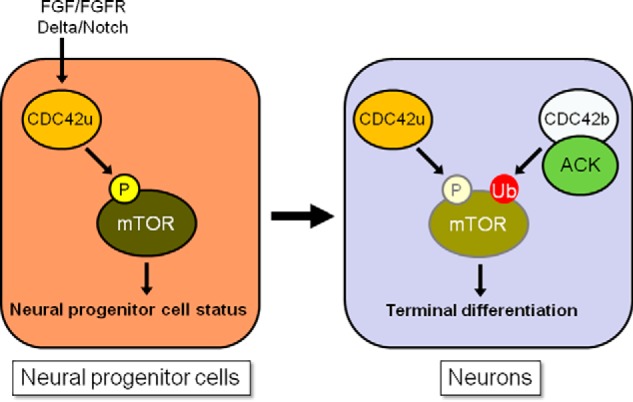
Schematic diagrams showing a model for how the two CDC42 splice variants control mTOR activity, thereby regulating the maintenance of neural progenitor cells and their transition into neurons. In neural progenitor cells, FGF-FGFR– and Delta-Notch–dependent cell signals activate CDC42u, which up-regulates mTOR activity (high phosphorylation level is indicated by the bright yellow–circled P) and promotes the maintenance of neural progenitor cell status. In neurons, the up-regulated CDC42b and ACK promote the ubiquitylation and degradation of mTOR (high ubiquitylation level is indicated by the red-circled Ub), counteracting the CDC42u-dependent mTOR activation. This tunes down mTOR-dependent signaling (lower activation and expression levels are indicated by the pale color–circled P and mTOR), thereby promoting the terminal differentiation of neural progenitor cells into post-mitotic neurons.
Experimental procedures
Antibodies and DNA constructs
Monoclonal anti-MAP2, anti-βIII-tubulin (TuJ1 or Tu20), and anti-phosphotyrosine (4G10) antibodies were from Millipore. Monoclonal and polyclonal anti-CDC42 antibodies were obtained from Millipore, Santa Cruz Biotechnology, Inc. (P1 and B8), Abcam, and BD Biosciences. Anti-CDC42b–specific polyclonal antibody was kindly provided by Dr. Rujun Kang in the laboratory of the late Dr. Alaa El-Husseini (University of British Columbia). Monoclonal anti-OCT4 (C10), anti-ACK (A-11), polyclonal anti-GFP (FL), and anti-GATA4 antibodies were from Santa Cruz Biotechnology. Monoclonal anti-cTnT antibody was from Thermo Scientific. Monoclonal anti-Vinculin antibody was from Sigma–Aldrich. Polyclonal antibodies that recognize total or phosphorylated mTOR (7C10 and D9C2) were from Cell Signaling. Monoclonal anti-Myc (9E10), polyclonal anti-V5, and anti-Nestin antibodies (NESTIN18) were from Covance. Polyclonal anti-ACK antibody was described previously (39).
The gene encoding CDC42b was obtained by PCR from the cDNA library that was made from RNA extracts of P19 cells subjected to the RA-induced neural differentiation protocol. Point mutants of CDC42b were generated using the QuikChange II site-directed mutagenesis kit (Agilent Technologies). Constructs expressing Myc-tagged CDC42 and CDC42b proteins were generated using the pCDNA3 vector by subcloning techniques. DNA constructs expressing CDC42u, cloned from human cDNA libraries, do not include the recognition site for the CDC42u-specific siRNA that targets the 3′-UTR region of the mouse Cdc42u mRNA. To establish CDC42b-expressing DNA constructs that were resistant to the CDC42b-specific RNAi, the codon usages of CDC42b were modified as follows: TGTTGCATCTTTTGA, in which the underlined letters indicate the modified residues.
The siRNAs that targeted both CDC42 splice variants were described previously (10). The CDC42 isoform-specific siRNAs are as follows: CAUUCCACUCCCUAGGUCUAGUUUA (CDC42u, sense), UAAACUAGACCUAGGGAGUGGAAUG (CDC42u, antisense), GAAGUGCUGUAUAUUCUAAACCGUU (CDC42b, sense), and AACGGUUUAGAAUAUACAGCACUUC (CDC42b antisense) (Invitrogen). The silencer select siRNAs targeting ACK were purchased from Invitrogen (Tnk2 MSS225066, MSS225067, and MSS284822).
The lentivirus-based mammalian expression system was established using the GATEWAY system (Invitrogen). pDONR-221 and -207 were used as entry vectors, and the acceptor vector was modified from pSIN-EF2-OCT4-Pur (Dr. James Thomson laboratory at the University of Wisconsin (Madison, WI), Addgene #16579) (35), whose OCT4 was replaced with YFP-tagged target proteins of interest. pLKO.1-puro vector was used for the lentivirus-based shRNA expression system targeting CDC42 and ACK, purchased from the Sigma–Aldrich MISSION shRNA Library: TRCN0000071684 (CDC42 common #1), TRCN0000071686 (CDC42 common #2), TRCN0000322228 (ACK #1), TRCN0000350731 (ACK #2), and TRCN0000322165 (ACK #3).
To establish DNA constructs including insulator sequences, preventing DNA silencing during differentiation, two tandem chicken β-globin HS4 insulator sequences (Dr. Ben Szaro laboratory at the State University of New York (Albany, NY), Addgene #74100) (36) were inserted upstream of the CMV promoter in pCDH-CMV-MCS-EF1-puro vector (System Biosciences), using subcloning techniques.
The shRNA sequences targeting CDC42 splice variants were inserted into the AgeI-EcoRI restriction enzyme sites of pLKO.1-puro vector, using subcloning techniques: CCGGAAGGCCTAAAGAATGTGTTTGCTCGAGCAAACACATTCTTTAGGCCTTTTTTTG (CDC42u-specific, sense), AATTCAAAAAAAGGCCTAAAGAATGTGTTTGCTCGAGCAAACACATTCTTTAGGCCTT (CDC42u-specific, antisense), CCGGAAGTGCTGTATATTCTAAACCCTCGAGGGTTTAGAATATACAGCACTTTTTTTG (CDC42b-specific, sense), AATTCAAAAAAAGTGCTGTATATTCTAAACCCTCGAGGGTTTAGAATATACAGCACTT (CDC42b-specific, antisense).
Cell culture, transfection, viral transduction, immunoblotting, and semiquantitative PCR
COS7 and HeLa cells were cultured in 10% fetal bovine serum–containing Dulbecco's modified Eagle's medium. In each experiment, serum was freshly thawed from frozen stocks to minimize compromised serum activities. Transfections were carried out using Lipofectamine with or without Plus reagent, as suggested by the manufacturer's instructions (Invitrogen). COS7 and HeLa cells were treated with 20 nm MG132 (Sigma–Aldrich), when preventing proteasomal protein degradation.
Cell culture and differentiation of P19 cells were performed as described previously (10). Transfections were carried out using Lipofectamine 2000, as suggested by the manufacturer's instructions (Invitrogen). To establish stable cell lines, cells were selected with 500 μg/ml G418 (Research Products International) and then maintained in growth medium supplemented with 250 μg/ml G418, with the drug being removed prior to experiments. Stable cell lines that expressed similar levels of the target proteins of interest were used for further experiments (Figs. S3 and S4F). P19 cells were transfected with Stealth siRNAs (Invitrogen) or control siRNA using Lipofectamine 2000 as described previously (10).
293T cells were transfected with transfer vectors, pCMV delta R8.2 packaging, and pMD2.G envelope plasmids, using polyethyleneimine. Cell debris was removed by centrifugation and filtration, and virus particles were concentrated using a Lenti-X concentrator (Takara Bio), following the manufacturer's instructions. P19 cells were treated with concentrated lentivirus, using spinoculation (1,600 × g for 1 h at room temperature). To establish stable cell lines, lentivirus-treated cells were selected with 500 ng/ml puromycin (Invitrogen) and then maintained in growth medium supplemented with 250 ng/ml puromycin, with the drug being removed prior to experiments. Because endogenous ACK protein expression levels were too low to measure the effects of ACK-targeting shRNAs in undifferentiated P19 cells, the effectiveness of ACK RNAi was assessed after RA induction.
Immunoblotting assays were performed as described previously (10). The immunoblotting images were scanned and quantified, using ImageJ. The densitometry measurement for the mTOR expression was normalized to a loading control, and those for assessing protein phosphorylation levels were normalized to the total target protein of interest.
RNA extraction, reverse transcriptase reaction, and semiquantitative real-time PCR were performed as described previously (10).
CDC42 activation assays were performed in pulldown experiments using GSH S-transferase (GST) fused to the PBD. Cell lysates were collected with lysis buffer (20 mm HEPES (pH 7.5), 150 mm NaCl, 1% Triton X-100, 1 mm sodium orthovanadate, 20 mm NaF, 20 mm β-glycerol phosphate, and 10 μg/ml leupeptin and aprotinin), containing 2 mm MgCl2. Lysate proteins (300 μg) were incubated with GSH-agarose beads bound to 50 μg of recombinant GST or GST-PBD at 4 °C for 90 min. The beads were washed three times with lysis buffer and then subjected to SDS-PAGE and immunoblotting.
Mouse embryonic stem cell culture, viral transduction, and differentiation
Mouse E14Tg2a embryonic stem cells were cultured in 0.1% gelatin-coated tissue culture dishes, with N2B27 medium (+LIF, +2i), including 49% (v/v) Neurobasal, 49% Dulbecco's modified Eagle's medium/F-12, 1% B27 (Invitrogen), 0.5% N2 (Invitrogen), 2 mm glutamine, 150 μm monothioglycerol (Sigma–Aldrich), 0.05% BSA, 103 units/ml LIF (Millipore), 1 μm PD035901 (Selleckchem), and 3 μm CHIR99021 (Selleckchem) (37). One-half of the medium was exchanged with new medium each day. Cells were dissociated with StemPro Accutase (Invitrogen) during cell passaging.
We performed neural differentiation in E14 cells, utilizing a modified version of a monolayer neural differentiation protocol, based on a previously reported study (38). In our monolayer neural differentiation protocol, E14 cells (2.0 × 103 cells/mm2) were placed in gelatin-coated tissue culture plates in N2B27 medium without LIF, PD035901, and CHIR99021 (−LIF, −2i). After 24 h, the cells were replated in gelatin-coated tissue culture plates in N2B27 medium (−LIF, −2i) with a lower cell concentration (0.4 × 103 cells/mm2). After 2 days in the second culture plate (day 3 from the start of differentiation), cells were dissociated with Accutase and further cultured in bacterial-grade dishes for another 4 days, in the presence of 5 ng/ml epidermal growth factor and bFGF (Invitrogen). The media and growth factors were exchanged with fresh media and factors on day 5. The floating cells formed cell aggregates, which were collected as an NSC-enriched cell population on day 7. For further terminal differentiation, collected NSCs were then dissociated with Accutase, and dissociated cells (0.4 × 103 cells/mm2) were placed in laminin-coated tissue culture plates in N2B27 medium (−LIF, −2i), in the presence of 5 ng/ml bFGF. The medium and bFGF were exchanged on day 9. On day 11, the cells were replated in poly-l-lysine–coated tissue culture plates or coverslips (1.0 × 103 cells/mm2) in N2B27 medium (−LIF, −2i), in the presence of 1% FBS. On day 13, the medium was replaced with B27 medium (Neurobasal medium with 1% B27 supplement and 2 mm glutamine), and then half of the medium was exchanged every other day.
Lentivirus transduction and DNA transfections were performed on day 11 in the monolayer neural differentiation protocol. Dissociated E14-derived NSCs were subject to spinoculation (1,600 × g for 1 h at room temperature) and then placed together with lentivirus in poly-l-lysine–coated dishes for 2 days in the presence of 1% FBS. For DNA transfections, dissociated E14-derived NSCs were reverse-transfected with protein expression vectors, using Lipofectamine 2000, in the presence of 1% FBS. After 8 h of Lipofectamine treatment, the medium was exchanged.
In the cardiac differentiation protocol, 400–1,000 cells were used to form a cell aggregate in 10 μl of N2B27 medium (−LIF, −2i), using the hanging-drop method. After 3 days of hanging-drop culture, cell aggregates were collected and then placed in a bacterial-grade dish, with Iscove's modified Dulbecco's medium, containing 20% CS. After another 4 days of culture, the medium was replaced with 20% CS-containing Iscove's modified Dulbecco's medium, and then the medium was exchanged every 3 days.
Mouse brain tissue collection
Mice used in this study were housed and handled in accordance with the Cornell University Institutional Animal Care and Use Committee regulations. For brain tissue collection, mice were euthanized by carbon dioxide inhalation. After euthanasia, the whole brain was harvested from mice, frozen in liquid nitrogen, and ground using a mortar and pestle. The tissue powder was then suspended in lysis buffer (50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate, 1 mm sodium vanadate, 10 μg/ml leupeptin and aprotinin), and after centrifugation, the supernatant was used as mouse brain lysates for immunoblotting experiments.
Immunofluorescence microscopy
Epifluorescence microscopy was performed as described previously (10). Briefly, P19 cells were subject to the RA-induced neural differentiation protocol and fixed with 3.7% formaldehyde or 2% paraformaldehyde at the indicated day. The fixed cells were then permeabilized with 0.1% Triton X-100 or 0.2% saponin (Sigma–Aldrich)-containing PBS. After blocking with 2% BSA, cells were stained with primary antibodies and, subsequently, the secondary antibodies that recognize mouse or rabbit IgG that are conjugated with fluorescence dyes (Molecular Probes). Epifluorescence images were captured with an Axioskop inverted microscope system (Carl Zeiss) equipped with a Sensicam qe charge-coupled device camera system (The COOKE Corp.), Plan-APOCHROMAT ×63 (Carl Zeiss), and LCPLFL ×40 (Olympus) objective lens. The captured images were processed with IPLab (SCANALYTICS Inc.).
In neural differentiation assays, cells were immunostained with anti-Nestin (1:2,000), βIII-tubulin (1:1,000), and MAP2 antibodies (1:3,000). In neuronal differentiation assays, ∼100 or more cells were counted for each condition, and the percentage of βIII-tubulin–positive cells was determined in each experiment.
Apoptosis assay
Apoptotic cells were detected using the annexin V: FITC apoptosis detection kit I (BD Biosciences), following the manufacturer's instructions. Annexin V-FITC+/propidium iodide+ apoptotic cells were counted using a fluorescence microscope system. Approximately 100 or more cells were counted for each condition, and the percentage of apoptotic cells was determined in each experiment.
Statistical analysis
Statistical differences between two samples were calculated using a paired Student's t test. Two-tailed p < 0.05 was considered to be significant. Statistical differences among more than two samples were calculated using Tukey's honestly significant difference test, after statistically significant differences among groups were calculated using a one-way analysis of variance (p < 0.01). In a Tukey test, p < 0.05 was considered to be significant.
Author contributions
M. E. and R. A. C. conceptualization; M. E. and J. E. D. formal analysis; M. E. validation; M. E., J. E. D., and R. A. C. investigation; M. E. methodology; M. E. writing-original draft; M. E. and R. A. C. writing-review and editing; R. A. C. supervision; R. A. C. funding acquisition; R. A. C. project administration.
Supplementary Material
Acknowledgments
We thank Cindy Westmiller for excellent secretarial assistance. We thank Dr. Carrie Stearns, Dr. Michael Lukey, and Dr. Kristin Wilson-Cerione for helpful discussion.
This work was supported by NIGMS, National Institutes of Health, Grant R35 GM122575 and NCI, National Institutes of Health, Grant R01 CA201402 (to R. A. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4.
- CDC42
- cell division cycle 42
- ACK
- activated Cdc42-associated kinase
- RA
- retinoic acid
- CDC42u
- CDC42 ubiquitous
- CDC42b
- CDC42 brain
- ESCs
- embryonic stem cells
- NSCs
- neural stem cells
- PBD
- p21-binding domain
- LIF
- leukemia-inhibitory factor
- FGF
- fibroblast growth factor
- bFGF
- basic FGF
- mTOR
- mechanistic target of rapamycin
- mTORC1
- mechanistic target of rapamycin complex 1
- FBS
- fetal bovine serum
- YFP
- yellow fluorescent protein.
References
- 1. Etienne-Manneville S., and Hall A. (2002) Rho GTPases in cell biology. Nature 420, 629–635 10.1038/nature01148 [DOI] [PubMed] [Google Scholar]
- 2. Jaffe A. B., and Hall A. (2005) Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247–269 10.1146/annurev.cellbio.21.020604.150721 [DOI] [PubMed] [Google Scholar]
- 3. Takai Y., Sasaki T., and Matozaki T. (2001) Small GTP-binding proteins. Physiol. Rev. 81, 153–208 10.1152/physrev.2001.81.1.153 [DOI] [PubMed] [Google Scholar]
- 4. Chen F., Ma L., Parrini M. C., Mao X., Lopez M., Wu C., Marks P. W., Davidson L., Kwiatkowski D. J., Kirchhausen T., Orkin S. H., Rosen F. S., Mayer B. J., Kirschner M. W., and Alt F. W. (2000) Cdc42 is required for PIP2-induced actin polymerization and early development but not for cell viability. Curr. Biol. 10, 758–765 10.1016/S0960-9822(00)00571-6 [DOI] [PubMed] [Google Scholar]
- 5. Heasman S. J., and Ridley A. J. (2008) Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 9, 690–701 10.1038/nrm2476 [DOI] [PubMed] [Google Scholar]
- 6. Melendez J., Grogg M., and Zheng Y. (2011) Signaling role of Cdc42 in regulating mammalian physiology. J. Biol. Chem. 286, 2375–2381 10.1074/jbc.R110.200329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cappello S., Attardo A., Wu X., Iwasato T., Itohara S., Wilsch-Bräuninger M., Eilken H. M., Rieger M. A., Schroeder T. T., Huttner W. B., Brakebusch C., and Götz M. (2006) The Rho-GTPase cdc42 regulates neural progenitor fate at the apical surface. Nat. Neurosci. 9, 1099–1107 10.1038/nn1744 [DOI] [PubMed] [Google Scholar]
- 8. Chen L., Liao G., Yang L., Campbell K., Nakafuku M., Kuan C. Y., and Zheng Y. (2006) Cdc42 deficiency causes Sonic hedgehog-independent holoprosencephaly. Proc. Natl. Acad. Sci. U.S.A. 103, 16520–16525 10.1073/pnas.0603533103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Peng X., Lin Q., Liu Y., Jin Y., Druso J. E., Antonyak M. A., Guan J. L., and Cerione R. A. (2013) Inactivation of Cdc42 in embryonic brain results in hydrocephalus with ependymal cell defects in mice. Protein Cell 4, 231–242 10.1007/s13238-012-2098-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Endo M., Antonyak M. A., and Cerione R. A. (2009) Cdc42-mTOR signaling pathway controls Hes5 and Pax6 expression in retinoic acid-dependent neural differentiation. J. Biol. Chem. 284, 5107–5118 10.1074/jbc.M807745200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Munemitsu S., Innis M. A., Clark R., McCormick F., Ullrich A., and Polakis P. (1990) Molecular cloning and expression of a G25K cDNA, the human homolog of the yeast cell cycle gene CDC42. Mol. Cell. Biol. 10, 5977–5982 10.1128/MCB.10.11.5977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hart M. J., Shinjo K., Hall A., Evans T., and Cerione R. A. (1991) Identification of the human platelet GTPase activating protein for the CDC42Hs protein. J. Biol. Chem. 266, 20840–20848 [PubMed] [Google Scholar]
- 13. Shinjo K., Koland J. G., Hart M. J., Narasimhan V., Johnson D. I., Evans T., and Cerione R. A. (1990) Molecular cloning of the gene for the human placental GTP-binding protein Gp (G25K): identification of this GTP-binding protein as the human homolog of the yeast cell-division-cycle protein CDC42. Proc. Natl. Acad. Sci. U.S.A. 87, 9853–9857 10.1073/pnas.87.24.9853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Olenik C., Barth H., Just I., Aktories K., and Meyer D. K. (1997) Gene expression of the small GTP-binding proteins RhoA, RhoB, Rac1, and Cdc42 in adult rat brain. Mol. Brain Res. 52, 263–269 10.1016/S0169-328X(97)00270-2 [DOI] [PubMed] [Google Scholar]
- 15. Olenik C., Aktories K., and Meyer D. K. (1999) Differential expression of the small GTP-binding proteins RhoA, RhoB, Cdc42u and Cdc42b in developing rat neocortex. Mol. Brain Res. 70, 9–17 10.1016/S0169-328X(99)00121-7 [DOI] [PubMed] [Google Scholar]
- 16. Makeyev E. V., Zhang J., Carrasco M. A., and Maniatis T. (2007) The microRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol. Cell 27, 435–448 10.1016/j.molcel.2007.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boureux A., Vignal E., Faure S., and Fort P. (2007) Evolution of the Rho family of ras-like GTPases in eukaryotes. Mol. Biol. Evol. 24, 203–216 10.1093/molbev/msl145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kang R., Wan J., Arstikaitis P., Takahashi H., Huang K., Bailey A. O., Thompson J. X., Roth A. F., Drisdel R. C., Mastro R., Green W. N., Yates J. R. 3rd, Davis N. G., and El-Husseini A. (2008) Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature 456, 904–909 10.1038/nature07605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wirth A., Chen-Wacker C., Wu Y. W., Gorinski N., Filippov M. A., Pandey G., and Ponimaskin E. (2013) Dual lipidation of the brain-specific Cdc42 isoform regulates its functional properties. Biochem. J. 456, 311–322 10.1042/BJ20130788 [DOI] [PubMed] [Google Scholar]
- 20. Nishimura A., and Linder M. E. (2013) Identification of a novel prenyl and palmitoyl modification at the CaaX motif of Cdc42 that regulates RhoGDI binding. Mol. Cell. Biol. 33, 1417–1429 10.1128/MCB.01398-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moutin E., Nikonenko I., Stefanelli T., Wirth A., Ponimaskin E., De Roo M., and Muller D. (2017) Palmitoylation of cdc42 promotes spine stabilization and rescues spine density deficit in a mouse model of 22q11.2 deletion syndrome. Cereb. Cortex 27, 3618–3629 10.1093/cercor/bhw183 [DOI] [PubMed] [Google Scholar]
- 22. Yap K., Xiao Y., Friedman B. A., Je H. S., and Makeyev E. V. (2016) Polarizing the neuron through sustained co-expression of alternatively spliced isoforms. Cell Rep. 15, 1316–1328 10.1016/j.celrep.2016.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ureña J. M., La Torre A., Martínez A., Lowenstein E., Franco N., Winsky-Sommerer R., Fontana X., Casaroli-Marano R., Ibáñez-Sabio M. A., Pascual M., Del Rio J. A., de Lecea L., and Soriano E. (2005) Expression, synaptic localization, and developmental regulation of Ack1/Pyk1, a cytoplasmic tyrosine kinase highly expressed in the developing and adult brain. J. Comp. Neurol. 490, 119–132 10.1002/cne.20656 [DOI] [PubMed] [Google Scholar]
- 24. Fang Y., Park I. H., Wu A. L., Du G., Huang P., Frohman M. A., Walker S. J., Brown H. A., and Chen J. (2003) PLD1 regulates mTOR signaling and mediates Cdc42 activation of S6K1. Curr. Biol. 13, 2037–2044 10.1016/j.cub.2003.11.021 [DOI] [PubMed] [Google Scholar]
- 25. Wang J. B., Sonn R., Tekletsadik Y. K., Samorodnitsky D., and Osman M. A. (2009) IQGAP1 regulates cell proliferation through a novel CDC42-mTOR pathway. J. Cell Sci. 122, 2024–2033 10.1242/jcs.044644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hoeffer C. A., and Klann E. (2010) mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 33, 67–75 10.1016/j.tins.2009.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Swiech L., Perycz M., Malik A., and Jaworski J. (2008) Role of mTOR in physiology and pathology of the nervous system. Biochim. Biophys. Acta 1784, 116–132 10.1016/j.bbapap.2007.08.015 [DOI] [PubMed] [Google Scholar]
- 28. Michaelson D., Ali W., Chiu V. K., Bergo M., Silletti J., Wright L., Young S. G., and Philips M. (2005) Postprenylation CAAX processing is required for proper localization of Ras but not Rho GTPases. Mol. Biol. Cell 16, 1606–1616 10.1091/mbc.e04-11-0960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Garvalov B. K., Flynn K. C., Neukirchen D., Meyn L., Teusch N., Wu X., Brakebusch C., Bamburg J. R., and Bradke F. (2007) Cdc42 regulates cofilin during the establishment of neuronal polarity. J. Neurosci. 27, 13117–13129 10.1523/JNEUROSCI.3322-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Manser E., Leung T., Salihuddin H., Tan L., and Lim L. (1993) A non-receptor tyrosine kinase that inhibits the GTPase activity of p21cdc42. Nature 363, 364–367 10.1038/363364a0 [DOI] [PubMed] [Google Scholar]
- 31. Dharmaiah S., Bindu L., Tran T. H., Gillette W. K., Frank P. H., Ghirlando R., Nissley D. V., Esposito D., McCormick F., Stephen A. G., and Simanshu D. K. (2016) Structural basis of recognition of farnesylated and methylated KRAS4b by PDEδ. Proc. Natl. Acad. Sci. U.S.A. 113, E6766–E6775 10.1073/pnas.1615316113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maxwell K. N., Zhou Y., and Hancock J. F. (2018) Rac1 nanoscale organization on the plasma membrane is driven by lipid binding specificity encoded in the membrane anchor. Mol. Cell. Biol. 38, e00186–18 10.1128/MCB.00186-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van Hennik P. B., ten Klooster J. P., Halstead J. R., Voermans C., Anthony E. C., Divecha N., and Hordijk P. L. (2003) The C-terminal domain of Rac1 contains two motifs that control targeting and signaling specificity. J. Biol. Chem. 278, 39166–39175 10.1074/jbc.M307001200 [DOI] [PubMed] [Google Scholar]
- 34. Lam B. D., and Hordijk P. L. (2013) The Rac1 hypervariable region in targeting and signaling—a tail of many stories. Small GTPases 4, 78–89 10.4161/sgtp.23310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu J., Vodyanik M. A., Smuga-Otto K., Antosiewicz-Bourget J., Frane J. L., Tian S., Nie J., Jonsdottir G. A., Ruotti V., Stewart R., Slukvin I. I., and Thomson J. A. (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920 10.1126/science.1151526 [DOI] [PubMed] [Google Scholar]
- 36. Wang C., and Szaro B. G. (2015) A method for using direct injection of plasmid DNA to study cis-regulatory element activity in F0 Xenopus embryos and tadpoles. Dev. Biol. 398, 11–23 10.1016/j.ydbio.2014.11.010 [DOI] [PubMed] [Google Scholar]
- 37. Ying Q. L., Wray J., Nichols J., Batlle-Morera L., Doble B., Woodgett J., Cohen P., and Smith A. (2008) The ground state of embryonic stem cell self-renewal. Nature 453, 519–523 10.1038/nature06968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ying Q. L., Stavridis M., Griffiths D., Li M., and Smith A. (2003) Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat. Biotechnol. 21, 183–186 10.1038/nbt780 [DOI] [PubMed] [Google Scholar]
- 39. Shen F., Lin Q., Gu Y., Childress C., and Yang W. (2007) Activated Cdc42-associated kinase 1 is a component of EGF receptor signaling complex and regulates EGF receptor degradation. Mol. Biol. Cell. 18, 732–742 10.1091/mbc.E06-02-0142 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.