

Abstract
During homeostasis, interactions between tolerogenic dendritic cells (DCs), self-reactive T cells and T regulatory cells (Tregs) contribute to maintaining mammalian immune tolerance. In response to infection, immunogenic DCs promote the generation of pro-inflammatory effector T cell subsets. However, Autoimmune diseases can develop when complex homeostatic mechanisms that maintain the balance between regulatory and effector functions become impaired. Here, we discuss the newest advances on the mechanisms of physiopathologic homeostasis and strategies that may be employed to restore a dysregulated immune equilibrium. We compare approaches that may lead to toxic side effects or which lack cost effectiveness, and focus on the promising strategy of engineered targeted nanoparticles -- envisioned to achieve long-term remission in patients with…, and potentially, to preventautoimmunity in susceptible individuals. (120 words)
Keywords: Autoimmune Diseases, Immune Regulation, Immunotherapy, Nanoparticles
An emerging paradigm for the treatment of autoimmune diseases in humans.
In the maintenance of mammalian host defense, the immune system must not only combat infections, but also prevent the emergence of pathologic self-reactive immune cells that have not been eliminated during ontogeny. This emergence can result in various autoimmune diseases [1, 2]. At present, the prevention and cure of many autoimmune diseases represent major unmet needs. Current treatments can halt autoimmune disease progression but are rarely capable of achieving long -term remission. Here, we elaborate on the challenges of those therapies and review new strategies that have the potential to realize unmet objectives.
The human immune system is constantly active in nature. In the steady-state, billions of its half trillion T cells divide every day and are replaced by an equal number of cells that die by apoptosis [3]. Homeostatic mechanisms, where cytokines such as TGF-β play an essential role, maintain the T cell pool at a relatively constant number [4]. Among T cell subsets, the most rapidly dividing population are memory and CD4+ T regulatory cells (Tregs) [3], capable of suppressing pathogenic, self-reactive cells escaping deletional mechanisms of thymic selection [3]. Similarly, antigen-presenting dendritic cells (DCs) that support the development, function and homeostasis of Tregs, also divide at a high rate [5]. These actions in the steady state greatly contribute to the active maintenance of immune tolerance.
In response to a microbial infection, tissue injury, or vaccination, the immune balance of the steady state shifts from a tolerogenic state to an immunogenic/inflammatory one. After the immunogen has been cleared, homeostatic regulatory mechanisms bring the system back to its initial tolerogenic state. However, if these mechanisms fail, where prevalent immunogenicity is maintained, associated proinflammatory effector mechanisms can contribute to the breach of self-tolerance and subsequent trigger of autoimmune diseases. Due to impaired immunoregulatory circuits, potentially pathologic self-reactive T cells can become activated and overcome control by protective immune cell subsets.
The development of therapeutic strategies that could reset a dysregulated immune system back to a normal tolerogenic state has long been pursued and has met many challenges. Fortunately, in recent times, it has become possible to prevent and reverse established autoimmune disease for certain pathologies,. Here, we overview the main cellular immune components and interactions that contribute to the maintenance of immune homeostasis. and briefly discuss co-stimulatory receptors and signaling pathways that determine whether naïve T cells differentiate into effector cells or regulatory cells. We highlight new advances in therapeutic strategies that can rebalance a dysregulated immune system, and focus on an emerging approach that employs biodegradable nanoparticles to target both T cells and DCs in vivo, safely restoring, long-term, Treg suppression of pathologic cells. This type of approach could be fine-tuned, and if successful, might serve as a platform to investigate its feasibility in therapeutically targeting impaired immune homeostasis in a variety of human autoimmune disorders.
Immune homeostasis: Maintaining a balance between immune tolerance and immunogenicity
Positive and negative immune regulation
The immune system includes cells that canact positively (e.g., to eliminate pathogens), or negatively (e.g., to protect the host from inadvertent or excessive activation) (Fig.1). Although over-simplified, and depending on context, Innate immune cells with positive functions might include immunogenic so-called M1 inflammatory macrophages and antigen-presenting DCs, as well as cytotoxic NK cells that produce IFN-γ. Their counterparts might include so-called M2 anti-inflammatory macrophages [6], tolerogenic dendritic cells (DCs) [7] and NK cells that produce TGF-β and IL-10 [8]. Effector cells might include pro-inflammatory CD4+ helper T cell Th1, Th2 and Th17 subsets, CD8+ cytotoxic killer T cells, NK-T cells and antibody-secreting B cells. corresponding inhibitory populations might include CD4+ or CD8+ regulatory T cell (Treg) subsets, regulatory NK-T cells [9], and albeit controversial, regulatory B cells [10].
Figure 1.
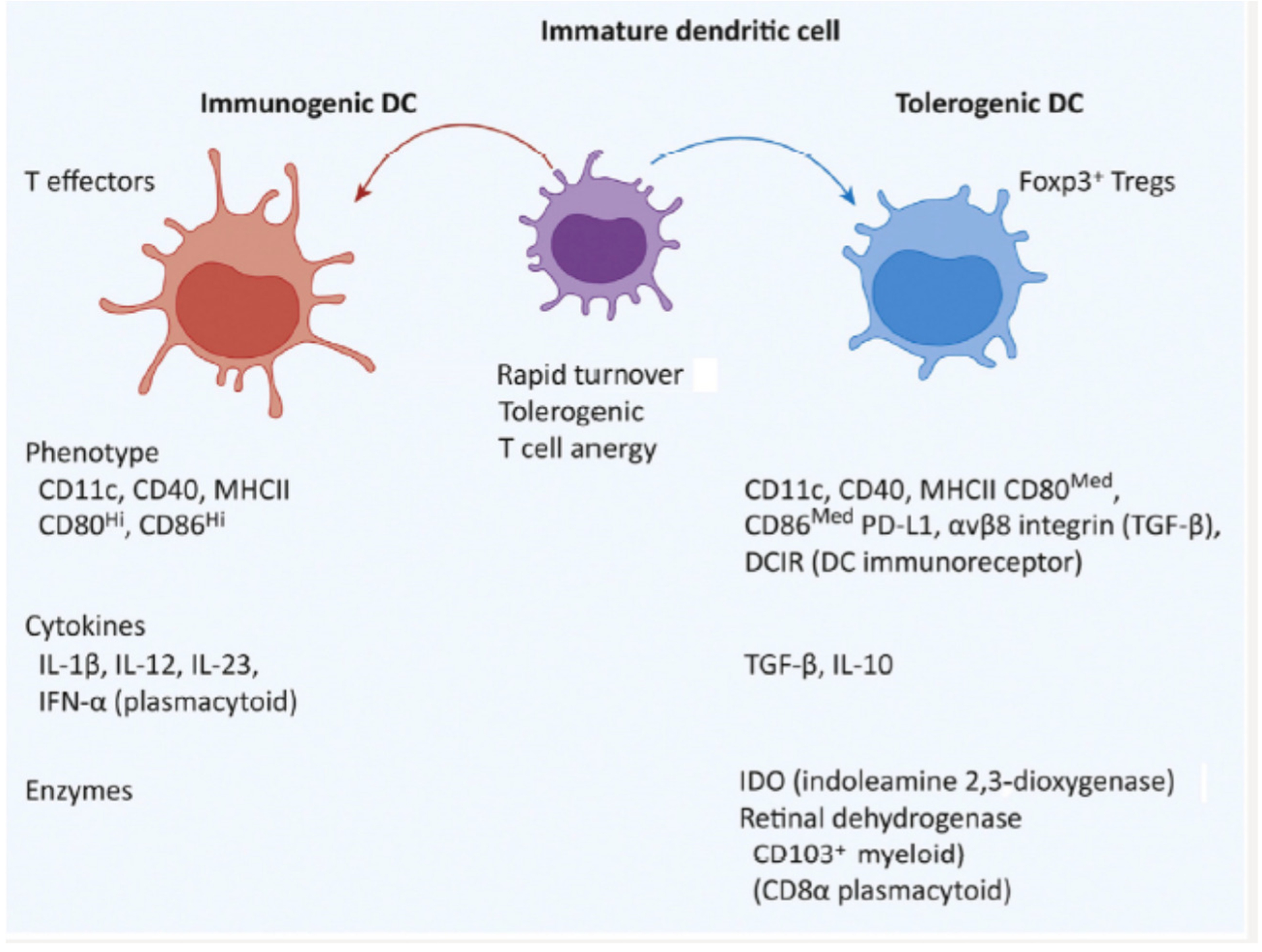
The Precursors of Migratory cDCs Differentiate into Tolerogenic or Immunogenic Antigen-Presenting Cells in Lymphoid Tissues in Response to Environmental Cues and Cytokines. The properties of each are shown.
Effector and regulatory cell types express characteristic receptors and cytokines related to their functional activity. Many cell types can share the expression a given receptor or cytokine. For example, in addition to T cells that readily express the negative co-stimulatory CTLA-4, PD-1 and TIM-1 receptors following TCR activation, NK cells that express these same receptors have also been described [11, 12]. Although this Yin/Yang concept describes effectors and regulators, it is important to emphasize that these are not mutually exclusive cellular phenotypes, and effector functions frequently overlap.
Thus, both pro-inflammatory and counteracting anti-inflammatory cells can be activated simultaneously when the immune system is stimulated. The balance between positive and negative activation is context-dependent and determines the ensuing magnitude of an immune response. For example, the cellular expression of negative co-stimulatory molecules such as CTLA-4 and PD-1 is critical since the genetic deletion of these molecules can result in overstimulation and generalized autoimmunity [13, 14]. At present pharmaceutical agents used to treat certain autoimmune conditions might only halt progression, and pathology might generally flare when treatment is stopped. In simple terms, this might be in part because while targeting proinflammatory cells, drugs can also counteract regulatory cells and pathways needed to sustain remission.
Tolerogenic and immunogenic immune responses
In both the thymus and in the periphery, antigen presentation, such as by DCs, can lead to clonal deletion of potentially pathogenic self-reactive T cells, or the generation of self-peptide specific Tregs. Self-reactive T cells expressing low affinity for self-peptides, however, can escape thymic deletion and become part of the peripheral T cell repertoire.
Besides their role in antibody production, B cells in the periphery have the capacity to present low-affinity or cryptic peptides to T cells, rendering them tolerant. Even activated B cells can acquire important tolerogenic effects mediated by cytokines such as TGF-β, and can induce Tregs [15, 16]. Similar to Tregs, activated B cells can also express the TGF-β binding protein GARP [17] which enables cells to convert latent TGF-β to its active form, in turn, promoting local immunosuppression. However, strongly activated B cells can also become accessory cells for T cell activation [18]. When self-reactive B cell tolerance is broken in a variety of autoimmune diseases, B cells can acquire immunogenic properties. Indeed, Their presentation of low-affinity peptides to T cells along with certain cytokines/chemokines and co-stimulatory molecules/ligands that stimulate the relevant T helper cells can become a driving force in these diseases. The antigen-presenting capacity of B cells might contribute to explaining why B cell depletion by anti-CD20 monoclonal antibodies can successfully treat or control a subset T cell-dependent autoimmune diseases [19].
Dendritic Cells
Mouse and human DCs have been classified functionally according to their maturation state. Although immature/semi-mature DCs were considered tolerogenic cells and fully mature DCs were considered immunogenic [24], increasing evidence has shown that immature DCs can acquire with maturation either a tolerogenic or an immunogenic phenotype, each characterized by specific cytokine production and cell surface receptors. Although both tolerogenic and immunogenic DCs express MHC class II and the co-stimulatory molecules CD40, CD80 and CD86, immunogenic DCs express higher amounts of these receptors. This allows them to provide strong co-stimulatory signals required for immunogenic responses. Additionally, to counteract inhibitory receptors, immunogenic DCs also express CD70 that binds CD27 on T cells [25].
Interestingly, although both tolerogenic and immunogenic DCs express ICOS ligand (ICOS-L), ICOS signaling by DCs is predominantly tolerogenic. On the other hand, tolerogenic DCs typically express the negative co-stimulatory molecules PD-1 ligand (PD-L1) and CD70 [26]. Tolerogenic DCs also express the integrin αvβ8 that enables activation of latent TGF-β with subsequent production of the immunoregulatory enzyme indoleamine 2,3-dioxygenase (IDO) which promotes immune tolerance in autoimmunity and allergic inflammation by suppressing T cell responses, mostly through induction of Tregs (Box 1, Fig. I).
Box 1.
Immunogenic versus Tolerogenic DCs. Mouse DC subsets have been well characterized, but human DCs have been characterized to a much lesser extent [125,126]. In steady-state, mouse DCs can be classified into two main categories: plasmacytoid DCs (pDCs) and conventional or classical DCs (cDCs). pDCs secrete type I interferons (IFNs) and help to control viral infection [127]. Similarly to the subset of monocyte-derived DCs, pDCs are fully differentiated in the bone marrow (BM), with a half-life of 8–9 days in the periphery [128]. cDCs are instead phagocytic and short-lived (turnover of about 2 days) [5]. cDCs can be subdivided into lymphoid organ-resident DCs and migratory DCs [129]. Because immature cDCs lack strong costimulatory receptors and promote anergy (non-responsiveness) of self-reactive T cells upon contact, they are generally considered to be tolerogenic (Figure I). Lymphoid organ-resident DCs are phenotypically immature because they express low amounts of costimulatory receptors. Instead, migratory DCs display a mature phenotype (high expression of costimulatory molecules) following recruitment by inflammatory stimuli, and carry processed antigen. They can present MHC II/peptide complexes to CD4+ T cells and cross-present MHC I/peptides to CD8+ T cells; these migratory DCs can become mature tolerogenic DCs that can induce immune tolerance more efficiently than resident DCs in mice [130].
T regulatory cells (Tregs)
In order to control innate and adaptive immune responses in an efficient and context-dependent manner, the immune system has evolved multiple mechanisms, including using immunosuppresive regulatory T (Treg) cells,(Box 2). Tregs comprise heterogeneous cell populations, which broadly include CD4+, CD8+, DN-T cells and NK-T cells [9, 28–31]. Regulatory B cells and NK cells have also been reported, although these remain poorly understood [10, 32]
Box 2.
Characteristics of CD4+ CD25+ Foxp3+ Tregs. These CD4+ T cells in mice and humans are crucial mediators of peripheral self-tolerance and immune homeostasis, as well as being key modulators of immune responses directed towards a spectrum of non-self-antigens derived from allergens, tumors, and microbes [41,131]. Induction and sustained expression of Foxp3 is essential for the development of Tregs, and establishes the core transcriptional program that ensures their phenotype, suppressive function, and key homeostatic processes, including cytokine signaling pathways [41,131]. There are three major subsets of Foxp3+ Tregs: (i) to as us Tregs developing in the thymus are referred as thymus-derived tTregs (formerly nTregs) [132], and are selected by high-affinity for self-peptides in thymic development; (ii) Tregs induced by exogenous antigens in the periphery from conventional T cells, especially in the intestinal tract, are called peripheral Tregs (pTregs); (iii) Tregs induced ex vivo/in vitro are called inducible Tregs (iTregs) [132]. In the mouse, TCR stimulation in conjunction with Foxp3-inducing signals (e.g., IL-2, TGF-b, retinoic acid) promotes iTreg or pTreg cell development [133]. In humans, inducers such as retinoic acid or rapamycin must be added to IL-2 and TGF-b to induce functional iTregs [67,107]. In humans, Foxp3+ Tregs can be divided according to antigenic memory status, namely CD45RA+CD25bright (naive precursor cells) and CD45RA-CD25mod (rapidly dividing cells) [3]. The phenotypic and functional similarities or differences between tTregs, pTregs, and iTregs have previously been reviewed [134]. Endogenous Tregs in circulation, lymphoid or non-lymphoid tissues represent a pool of cells originating from tTregs and pTregs. Although Foxp3 is frequently used as a Tregmarker, in humans (unlikemice) Foxp3 is also an activation marker; thus, not all Foxp3+ T cells are Tregs in humans [135].
Thymic development and sustained function of Tregs in peripheral tissues requires stable and high Foxp3 expression, and waning Foxp3 expression in Tregs in various inflammatory contexts can compromise their functional stability and/or redirect their differentiation towards a pathogenic-like T cell phenotype to favor their functional adaptation in situ [36]. Treg-specific Foxp3 expression is under tight epigenetic control and involves an evolutionary conserved DNA element in the mouse and human Foxp3/FOXP3 locus, known as the Treg-specific demethylated region (TSDR), which is demethylated in Tregs, but heavily methylated in effector T (Teff) cells [136]. The alarmins IL-1 and −33 differentially regulate the functional specialization of Foxp3+ Tregs during mucosal inflammation [137]. Thus, local inflammatory signals can modulate this epigenetic landscape, alter Foxp3 expression, and redirect the functional fate of Tregs. Environmental cues may selectively affect transcriptional or mRNA translational mechanisms, and alter ensuing protein expression and activity in effector or Treg cell subsets [138].
Tregs use multiple modes of suppression, including contact-dependent, cytokine-independent mechanisms, and soluble cytokines such as IL-10, TGF-β and IL-35. For instance,They can modulate the activity of DCs by contact-dependent inhibitory receptors such as CTLA-4, PD-1 and lymphocyte activation gene 3 (LAG3) [44]. Through expression of the ectoenzymes CD39 and CD73 they can also break down ATP to adenosine, which has marked anti-inflammatory effects [45]. In addition, they can suppress NK, B cells and even non-lymphoid cells in adipose tissue [46, 47]. (Box 2)
Key role for IL-2 and TGF-β in Treg-mediated immune homeostasis
IL-2 and TGF-β are the two most important cytokines involved in the generation, function and expansion of CD4+ Tregs, especially pTregs and iTregs [60] (Box 2). Before Foxp3 was identified, our group reported that these two cytokines could generate CD4+ and CD8 Tregs [61, 62]. Although IL-2 was initially described as a T cell growth factor, this cytokine is critical for strong Foxp3 expression by Tregs and requires CD25 for this purpose. The CD25 α chain, through its association with the CD122 β chain and CD132 γ chain mediates high affinity IL-2 signaling and IL-2 positively regulates CD25 expression. Thus, with inadequate IL-2, CD25 expression will decrease, leading to decreased expression of Foxp3 and reduced Treg suppressor activity. Therefore, decreased IL-2 production in certain autoimmune diseases can result in impaired Treg function [65]. The crucial role of IL-2 and TGF-β on the properties of CD4+ Tregs has been extensively reviewed [63, 64]. Of note, In addition to maintaining Foxp3 expression, IL-2 also upregulates PD-1 expressed by Tregs [66].
It is well established that TGF-β is a critical regulator of thymic T cell development as well as a crucial player in peripheral T cell homeostasis, tolerance to self-antigens, and T cell differentiation during immune responses. TGF-ß promotes T cell self-tolerance through direct regulation of both effector T cells and nTreg cells [67]. Tregs are a major source of TGF-β [68], but IL-2 must be provided by other T cells [36] [37]. Furthermore, in addition to IL-2 and TGF-β, CD4+ Tregs require continuous antigen stimulation for their survival and expansion [60, 69].
DC/T cell interactions are required for the maintenance of immune homeostasis
In the steady state, motile T cells make frequent contacts with DCs [70] and these interactions have crucial effects on immune regulation. T cells recirculate through lymphoid tissues and in one hour, 500–5,000 T cells can interact with a single DC [71]. The tonic signaling resulting from these interactions significantly enhances CD4+ and CD8+ T cell responsiveness to foreign antigenic stimuli, which is lost following depletion of DCs [72, 73]. Similarly, continuous tonic Treg/DC interactions are required for functional tolerogenic DCs, and DCs become immunogenic when Tregs are depleted [72, 73]. Those tolerogenic effects critically depend on the negative co-stimulatory molecules PD-1 and CTLA-4 [71].
Differences in the affinities of MHC/self-peptide with T cell receptors (TCR) can lead to differences in T cell/DC interactions in peripheral lymphoid tissues. As in the thymus, high-affinity MHC/self-peptide recognition leads to T cell deletion. In the periphery, high-affinity MHC/self-peptide recognition can lead to T cell anergy or Treg induction. Low/intermediate affinity MHC/self-peptide recognition maintains tonic T cell responsiveness and the priming of Teff cells [71].
Using high-resolution imaging techniques to study Treg/DC interactions in the steady state for the maintenance of tolerogenic DCs, workers have identified lymphoid tissue clusters comprising DCs, Tregs and self-reactive, IL-2-producing T cells. These tolerogenic DCs expresshigh amounts of CD86 and activated self-reactive T cells to produce IL-2 in amounts that can sustain and expand Tregs. Disruption of TCR signaling in Tregs, or local depletion on local of IL-2, broke tolerance, indicating that in the steady state self-reactive T cells are continuously activated, and that when there is disruption of Tregs that control them autoimmunity will result [44, 74]. Similarly, constitutive ablation of DCs broke self-tolerance of CD4+ T cells and resulted in fatal autoimmunity [75].
In the steady state, feedback circuits can support the continuous differentiation of immature DCs towards a mature tolerogenic phenotype (Fig.2). TGF-β and IL-10 produced by Tregs and other cells induce immature DCs to become tolerogenic [22]. TGF-β induces DCs to express PD-L1, which is critical for the generation of pTregs [76]. TGF-β and IL-10 are also produced by tolerogenic DCs. The inhibitory effects of TGF-β weaken strong TCR stimulation during generation of Tregs (see below), also facilitating the expression of CTLA-4 and PD-1. CTLA-4 binds CD80/86 for the induction of inhibitory effects which include additional TGF-β production. PD-1 binding to PD-L1, and weaker CD80 signals help DCs to generate more CD4+ Tregs. IL-10 produced by tolerogenic DCs and Tregs also has positive feedback effects and induces DCs to induce the regulatory T cell subset of Tr1 cells that in turn produce large quantities of IL-10 [77].
Figure 2.
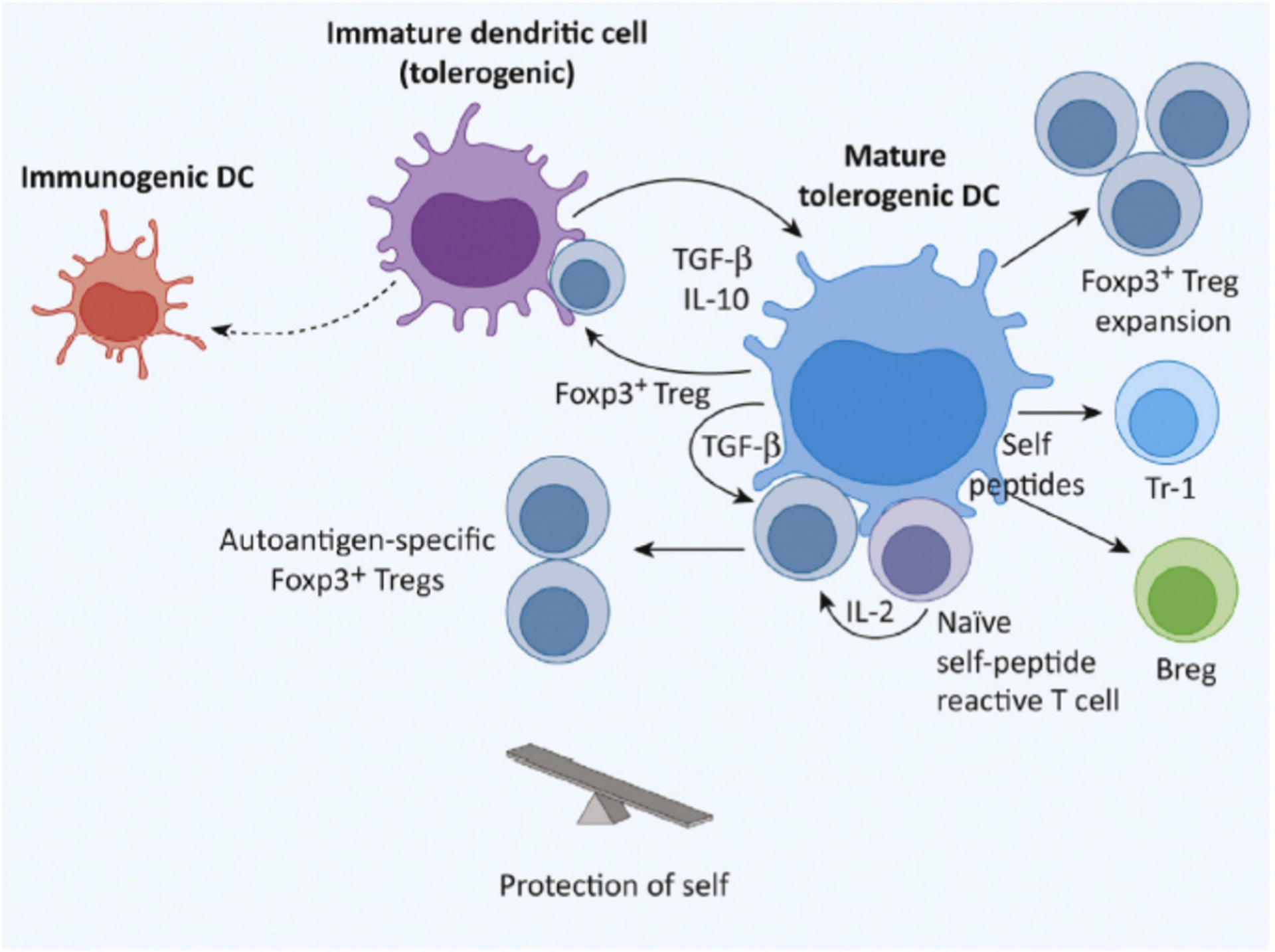
Tolerogenic Dendritic Cells (DCs) in Steady-State. Regulatory T cells (Tregs) producing IL-10 and TGF-b interact with immature DCs and induce them to become tolerogenic. These DCs express medium amounts of costimulatory CD80 and CD86 that are sufficient to expand Tregs. In clusters of DCs, Tregs, and self-reactive T cells in lymphoid tissues, self-reactive T cells produce the IL-2 required for Treg expansion. IL-2 and TGF-b produced by the DCs can induce naı¨ve CD4+ T cells to become self-antigen-specific Tregs. Tolerogenic DCs produce the IL-10 necessary to induce Tr-1 Tregs and regulatory B cells (Bregs).
In a mouse model of systemic lupus erythematosus (SLE), direct Treg help enabled DC populations to become tolerogenic. iTregs induced ex vivo with IL-2 and TGF-β prevented a lupus syndrome in mice via a TGF-β – dependent mechanism. Transfer of these DCs to secondary mice increased Foxp3+ cells and protected these mice from lupus also via a TGF-β mechanism [78]. Thus, Treg/DC interactions can have important tolerogenic effects as a consequence of positive feedback circuits that can involve rapidly dividing immature DCs and Tregs [3, 5].
Disruption of immune homeostasis can trigger autoimmune disease in susceptible hosts
In the steady state the immune system may be poised toward a tolerogenic state; however, in response to an infectious agent, or after antigen/adjuvant immunization, activation innate immunity can result in immature cDC populations shifting from tolerogenic to immunogenic phenotypes (Fig. 3). Immunogenic DCs produce pro-inflammatory cytokines that are needed for the clonal expansion of T and B cells and clearance of the pathogen. Once the pathogen has been eliminated, homeostatic mechanisms can restore the number of cells to baseline - which now include a subset of memory cells [3]. For example, activated T cells that express negative co-stimulatory molecules such as CTLA-4 and/or PD-1 can undergo activation-induced apoptosis,s and are phagocytosed by immature DCs;These apoptotic cells induce DCs to produce IL-10 and TGF-β and become tolerogenic [77, 79] (Fig. 4). The result is ideally the return to the homeostatic balance that was initially present under steady state conditions. If the microbial agent cannot be eliminated, the infection can become chronic.
Figure 3.
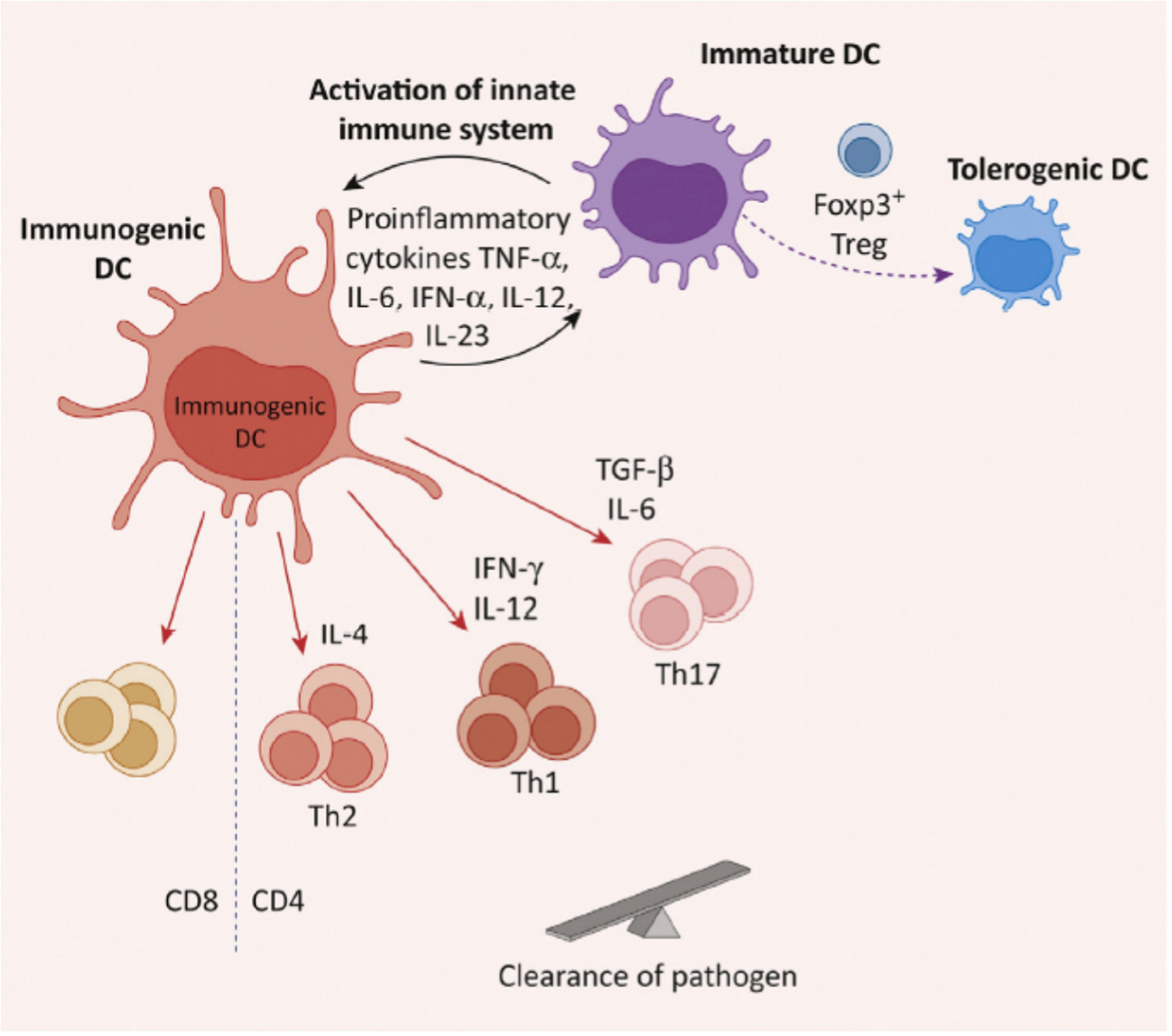
Immunogenic Dendritic Cells (DCs) in Infections. Immature DCs become immunogenic in response to microbial infections, tissue injury, or proinflammatory cytokines. This strong stimulation activates components of the innate immune system which then trigger Toll-like receptors on immature DCs. The result is their differentiation to a mature immunogenic state. Cytokines produced by these DCs facilitate the induction of T effector cell populations that clear the pathogen. Abbreviation: Th1/2/17, type 1/2/17 T helper cells.
Figure 4.
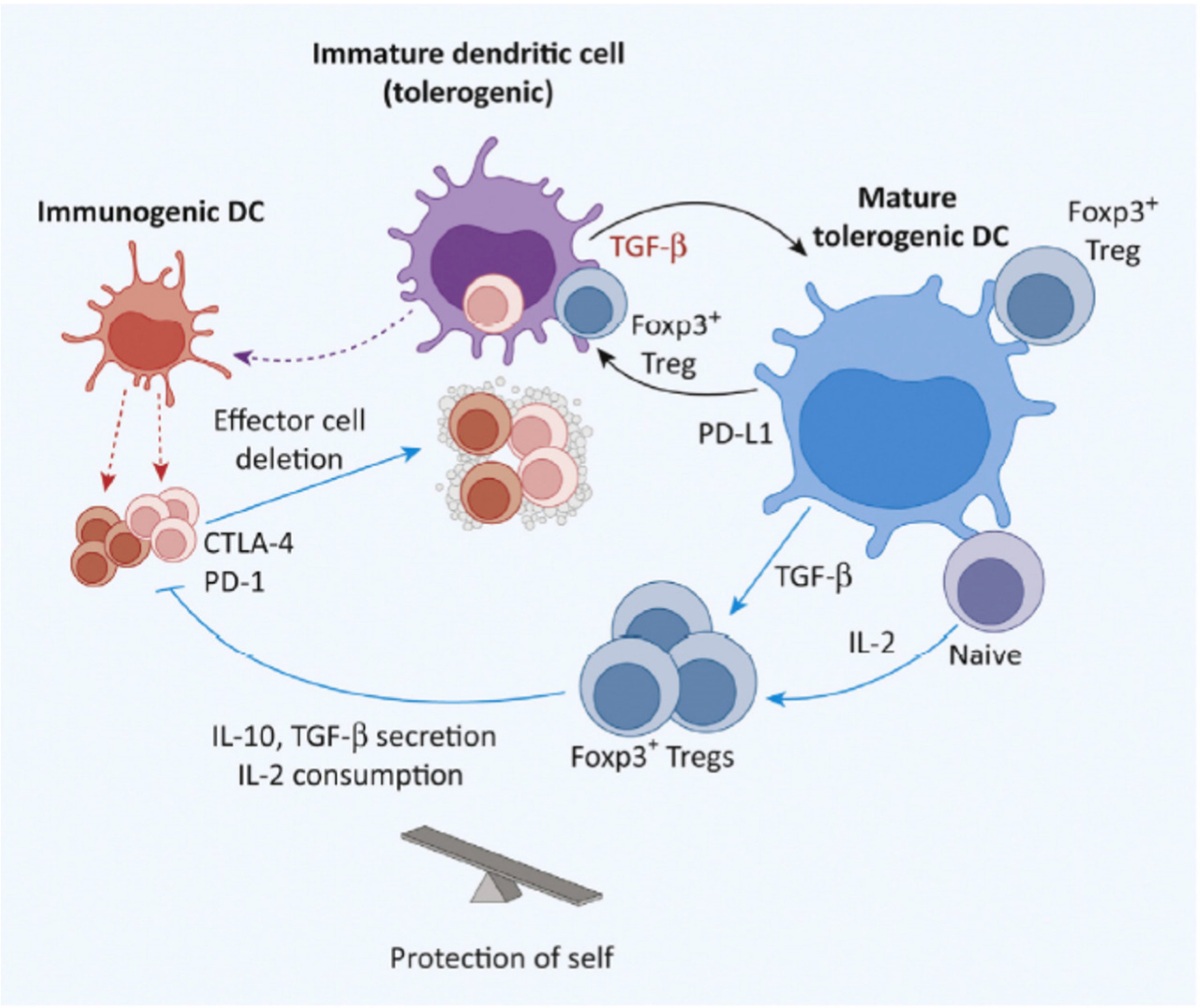
Tolerance Returning Post-Infection. With resolution, most clonally expanded T cells undergo apoptosis to restore the total number of cells to normal baseline. Phagocytosis of these dendritic cells (DCs) by macrophages and immature DCs generates TGF-b – that is important in switching DCs from immunogenic to mature tolerogenic cells. The steady-state is then ideally restored. Abbreviation: Treg, regulatory T cell.
However, other multifactorial reasons may prevent the immune system from returning to the steady state, including genetic, epigenetic, and environmental elements. The strong stimulation of an acute infection can not only result in the expansion of antigen-specific T cells but also provide bystander activation that recruits immune cells with additional antigenic specificities. These may include some cells with self-reactive potential, as suggested by the presence of autoantibodies during an acute infection.
The persistent antigen stimulation from immunogenic DCs sustains the activity of T effector cells, and the pro-inflammatory cytokines these cells produce can stimulate immunogenic DCs to establish a continuous feedback circuit. If latent pathologic self-reactive cells are a component of the bystander cells, and do not undergo apoptosis, these cells can persist, remain activated, and trigger an autoimmune response in susceptible individuals (Fig. 5).
Figure 5.
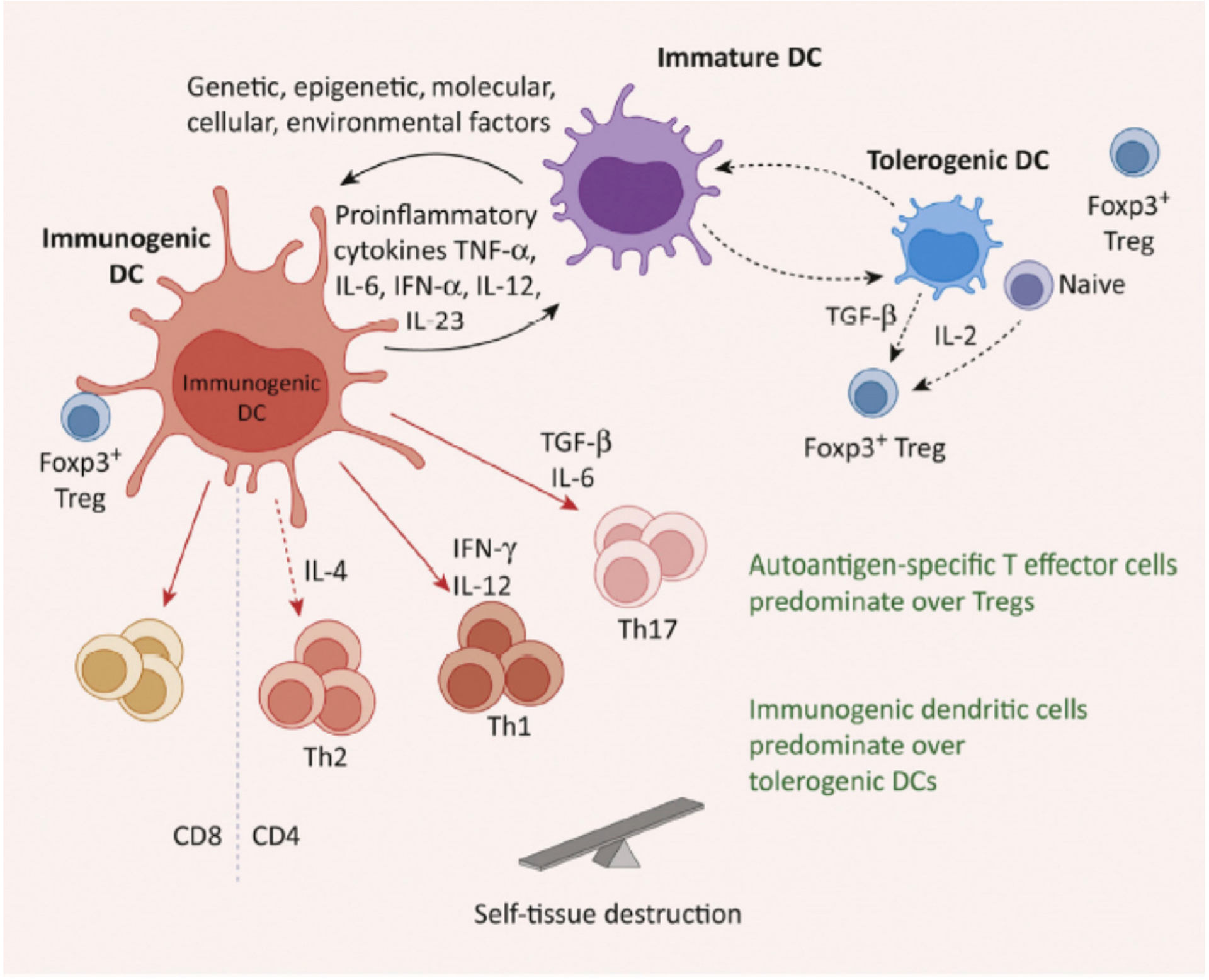
Failure To Return to Steady-State Can Result in Autoimmunity. For multifactorial reasons, the failure of a homeostatic compensatory response (following e.g., infection) can result in the persistence of potentially pathogenic self-reactive cells triggered by infection, and might initiate autoimmune disease in susceptible individuals. Abbreviations: DE, dendritic cell; Th1/2/17, type 1/2/17 T helper cells; Treg, regulatory T cell.
While this dysregulation of the immune system is a feature of adaptive immunity, the innate immune system strongly contributes to the generation of autoantigens responsible for a prodromal or priming phase of autoimmune diseases. Infections and/or inflammation stimulate myeloid cells to release proteases that cleave proteins into peptides. In addition, microbial proteases and posttranslational modifications of proteins such as citrullination and glycosylation also create altered peptides [80]. In individuals bearing a specific MHC polymorphism (e.g…..), these peptides can be recognized as autoantigens and initiate autoimmunity. In SLE, rheumatoid arthritis and type 1 diabetes (T1D), autoantibodies can be detected many years before the onset of clinical disease [80, 81].
In certain cases, Acute infections have been associated with the subsequent development of autoimmune disease, as in the case of…. [81]. While molecular mimicry between pathogens and self has been proposed to explain the role of infection in autoimmunity, it is likely as well that infectious agents that are not totally eliminated, such as the Epstein Barr (EB) virus, can promote the disruption of immune homeostasis and trigger pathologic immunity.
In such cases, the disruption of immunological homeostasis induced by an infection could convert the prodromal state of an autoimmune disease into a full developed active disease [80]. Here, the original latent naïve pathologic self-reactive cells may become memory cells with a much lower activation threshold than naïve cells. This result adds considerable additional difficulty to a successful treatment.
Molecular Basis of Therapeutic Immune Regulation
Cytokine-based therapies, targeting signaling pathways, and other
For many years corticosteroids and immunosuppressive drugs have been the predominant drugs used to treat a variety of autoimmune diseases. However, because of their broad non-specific effects and toxic side effects, alternative approaches have emerged. One used with certain success has been to block proinflammatory cytokines. Examples include anti-TNF-α for RA), psoriatic arthritis and inflammatory bowel disease, as well as IL-1 antagonists for adult Still’s disease. It is well established that for many subjects with these diseases, this approach has been successful. Although IL-10 and TGF-β have strong anti-inflammatory effects, they are too toxic for clinical use, especially TGF-β. A major problem with cytokines is that their effect is context-dependent. For example, although IL-2 and TGF-β induce Tregs, IL-17 and TGF-β induce pro-inflammatory Th17 cells. Even anti-TNF-α has opposite effects; In addition to its strong pro-inflammatory actions when present in conjunction with IL-1 and IL-6, TNF-α can also serve as a Treg activator [93].
Since IL-2 production or bioavailability is impaired in autoimmune diseases such as SLE and T1D, and this cytokine is critical for Treg function, low-dose IL-2 has been used to correct this defect [94, 95], and five clinical trials are currently in progress to treat…. (see https://clinicaltrials.gov for a complete list).
When further considering Cytokine signaling, the protein tyrosine kinase JAK/STAT inhibitor tofacitinib has been approved by the US FDA for the treatment of RA. It markedly weakens strong cytokine signaling and thereby reduces the production of many pro-inflammatory cytokines, while favoring production of anti-inflammatory IL-10, although tolerogenic effects of tofacitinib have not been described [96]. Unlike cytokine antagonists, JAK inhibitors are given orally.
Anti-CD3 antibodies have also been used to treat subjects with T1D. This therapy induces a population of non-cytotoxic CD8+CD25+Foxp3+TNFR2+ Tregs in vivo [97]. A phenotypically similar human CD8+ Treg population has been induced ex vivo with anti-CD3/28 beads [30]. Anti-CD2 can also has tolerogenic properties, especially in combination with anti-CD3 [98], by inducing suppressive concentrations of TGF-β [99]. Alefacept, (fusion protein, CD2 ligand), has been used with certain success inpatients T1D [100]. Finally, intravenous immunoglobulin (IVIG) administration has been used to treat several immune-mediated diseases and can expand CD4+ Tregs, and IVIG can contain peptides that stimulate Tregs, and sialylated IgG that induces tolerogenic DCs [27].
Strategies aiming to rebalance the immune system against autoimmunity
High dose immunosuppression and autologous hematopoietic stem cell transplantation
The first demonstration that a dysregulated immune system could be reset originated when depletion of T and B cells followed by autologous hematopoietic stem cell transplantation (HSCT) resulted in complete remission of rapidly progressive scleroderma [101] and long-term remission of autoimmune diseases including multiple sclerosis and SLE [102]. followed by(e.g. high dose cyclophosphamide).. As the immune system reconstitutes with new thymic-derived, naïve T cells, it encompasses a vastly diversified T cell repertoire. Pathogenic self-reactive T cells and pre-existing autoantibodies are eradicated and are replaced by a renewed T cell compartment [102]. In this scenario, one might speculate that immunogenic DCs are shifted to a tolerogenic phenotype and the resulting Tregs predominate over pathogenic T cells, Persistence of this effect appears to be responsible for the long-term therapeutic benefits.
Studies of peripheral blood mononuclear cells (PBMCs) from subjects with SLE harvested before and after HSCT provide strong support for the concept of immune resetting., In one study, increased numbers of both functional CD4+ and CD8+ Tregs were observed following HSCT. The newly generated CD8+ T cells in SLE contained suppressor cells with a TGF-β-dependent suppressive activity. Although anti-nucleosomal antibodies in SLE were no longer detectable after HSCT, when CD8+ cells were depleted, these antibodies reappeared, suggesting that CD8+ Tregs are likely to play an important role in controlling SLE autoimmunity [104]. Further studies should further elucidate this point.
While HSCT benefits suggests that a dysregulated immune system can be reset – to a certain extent -- the toxicity associated with this procedure has been considerable in patients with autoimmune diseases. In scleroderma patients, 3–10% mortality was documented, generally due to cardiac complications [101]. Mortality was also high in SLE patients. These outcomes addto the concerns reagrding the potential side effects of high-dose cyclophosphamide (e.g. infertility and decreased host defense against infection).
Tolerogenic peptides
Coupling peptides to chemically-treated splenocytes has been shown to result in antigen-specific tolerance [106] and elaboration of this approach has been used to prevent and treat animal models of autoimmune disease [107]. In mouse models of SLE, immunization of mice with high dose histone nucleosomal peptides with adjuvant was reported to be pathogenic, but low doses of the same peptides were tolerogenic [108]. Thus, Low dose peptides can induce CD4+ and CD8+ Tregs in lupus-prone mice, and in humans, can induce Tregs that suppress autoantibody production [108, 109]. Others have also identified peptides derived from anti-DNA antibodies in SLE that are tolerogenic [110].
Moreover, Antigen-specific Tregs have been induced using methods that recapitulate the homoeostatic compensatory response of immunogenic DCs to tolerogenic APCs in animal models of multiple sclerosis and autoimmune diabetes. Specifically, Apoptotic cells taken up by macrophages and immature DCs induce these cells to produce TGF-β; This results in the generation of tolerogenic APCs. The uptake of apoptotic cells by APCs is tolerogenic [105] Accordingly, massive apoptosis of immune cells can be elicited by irradiation or depletion of B cells and CD8+ T cells in mice. This can lead to the generation of antigen-specific CD4+ Tregs, decreased production of IL-17 and IFN-γ, and the suppression of autoimmune disease. Similarly to other studies [78] neutralization of TGF-β can abolishthese therapeutic effects [111].
Treg-based therapies
An approach to induce Treg predominance over Teff cells is by adoptive transfer of large numbers of Tregs expanded ex vivo. Ideally, the suppressive population should be antigen-specific, stable in inflammatory environments, and not cause global immunosuppression. Unfortunately, antigen-specific Tregs are rare and technically difficult to isolate and expand. The hurdles that must be overcome for Treg-based therapy are numerous and have been reviewed elsewhere [112] (Box 4).The emphasis to date has been to isolate and expand the small numbers of CD4+CD25+Foxp3+ cells circulating in human blood. For therapeutic indications, using naïve CD4+ T cells to induce and expand polyclonal iTregs would be in principle, technically less difficult than expanding the much smaller number of CD4 Tregs in human blood, and which require isolation by cell sorting.
Box 4.
Molecular Control of T Cell Tolerance. The phosphoinositide 3-kinase (PI3K) pathway is a principal path utilized in Teff and Tregs, and TCR, CD28, and IL-2 signals can all activate this pathway [141,142]. Signaling through PI3K and downstream Akt and mTOR is a predominant pathway determining the fate of T cell differentiation [143,144]. mTORC-1 is required for Treg functional competency (its deficiency in mice can lead to increased sensitivity to TGF-b and enhanced Foxp3 expression by Tregs) [145]. mTORC2 deficiency in mice can lead to Treg instability [145]. Rapamycin attenuates mTOR signaling (mainly through mTORC-1).
Strong, continuous stimulation of PI3K induces Teff formation, whereas weaker stimulation results in Treg differentiation [146–148] (see Figures 2 and 3 in main text). CD28 signals are required for both Teff and Treg generation [149]. High amounts of CD28 ligands CD80 and CD86 on immunogenic DCs provide the strong signaling needed for Teff formation, whereas lower amounts on tolerogenic DCs support Treg generation [149]. By attenuating signaling through mTOR, rapamycin can promote Treg differentiation [146]. In steady-state, rapid turnover of Tregs is stimulated by self-peptides, IL-2, and IL-7 [150]. These Tregs have built-in mechanisms to control PI3k/Akt/mTOR signaling [151]. The primary negative regulator is phosphatase and tensin homolog (PTEN) [152,153]. Deletion of PTEN releases mTORC-2 inhibition; this results in the activation of B cells and T follicular helper (Tfh) cells in germinal centers, as well as lupus-like disease in Pten and Foxp3 genetically deficient mice [153,154].
The negative costimulatory molecules CTLA-4 and PD-1 attenuate strong TCR stimulation by DCs, leading to weaker signals that favor Treg generation and upregulate various ubiquitin ligases [155]. Ligation of CTLA-4 can increase inhibitory TGF-b, and ligation of PD-1 enhances TGF-b activity. In parallel, APC ligation of CTLA-4 induces IDO – which degrades tryptophan (an essential amino acid needed for T cells to respond to other DCs) [156]. Ligation of PD-1 in human Tregs upregulates PTEN activity, which attenuates Akt/mTOR signaling [157].
Upon stimulation of innate immunity by a pathogen, Toll-like receptor ligands in DCs can induce the adaptor molecules MyD88 (myeloid differentiation primary response 88), p21Ras/Raf, and PI3K/Akt/mTOR. The subsequent strong TCR and CD28 costimulation in T cells induces NF-kB, whose c-Rel component partly inhibits IL-2/TGF-b-induced iTregs, resulting in the promotion of immunogenic DCs that induce Teff cells [148] (see Figure 3 in main text). Activated Teff cells also have high energy requirements and upregulate glucose transporters such as Glut1 for glycolytic metabolism; strong CD28 and PI3K signaling through mTOR generates ATP by glycolysis to meet energy demands [158]. Tregs, by contrast, with their lower energy requirements, use lipid oxidation as their principal form of metabolism [159]. This increases AMP-activated protein kinase (AMPK) activation, which inhibits mTOR signaling and promotes Treg differentiation [151].
Currently, many clinical trials are ongoing to test Treg-based therapies in subjects with T1D, SLE, inflammatory bowel disease, autoimmune hepatitis, and amyotrophic lateral sclerosis. Other indications include acute and chronic GVHD, and liver and kidney transplantation. (see https://clinicaltrials.gov for a complete list). A clinical trial with polyclonal, autologous CD4 Tregs in T1D has been completed and showed long-term survival of about 25% of expanded Tregs, with no serious adverse side effects [113]. Recent attempts have been made to test chimeric antigen receptor (CAR) T cells to target Tregs to specific antigens or broader alloantigens [114]. The recent use of allosteric modifiers of Foxp3-binding proteins such as TIP60, a histone actetyltransferase, has proven to be very efficient augmenting Treg function and correcting the functional defect in Treg cells from IPEX patients. Similar “Treg adjuvants” might be envisaged in future Treg cell-based therapies [41] (Box 4).
Immunotherapeutic nanoparticles
Nanoparticles are currently being tested for the treatment of autoimmune disease because they can be engineered for three distinct uses: they can be anti-inflammatory, tolerogenic, and function as carriers of biologic agents. Nanoparticles targeted to macrophages can polarize these cells to become anti-inflammatory if encapsulated with cytokines such as IL-10, statins, angiotensin receptor antagonists or peroxisome proliferator-activated receptor-γ (PPARγ) agonists [115]. Nanoparticles loaded with biological agents such as tumor necrosis factor antagonists ameliorate inflammatory arthritis [116].
The materials used for the preparation of nanoparticles can include metals, liposomes and synthetic and natural polymers [117, 118]. Metal oxide nanoparticles can be conjugated with antigens, targeting ligands and immunomodulators on the cell surface. However, these nanoparticles cannot encapsulate agents and are not biodegradable. Thus, long-term use could possibly cause safety issues. Liposomes that carry antigen, NF-kB inhibitors, or immunosuppressive drugs have been used to suppress arthritis and lupus [119, 120]. The most widely used materials to date, however, are natural and synthetic polymers, such as polylactic acid (PLA) or poly (lactic-co-glycolic acid) (PLGA). Unlike liposomes, these are solid particles which can encapsulate hydrophobic molecules that include rapamycin, vitamin D3 and dexamethasone [121].
Tolerogenic nanoparticles can be made antigen-specific or non-specific. There are three broad approaches to achieve antigen-specific tolerance that have been reviewed in detail [118]. Most of these approaches are directed to induce tolerogenic APCs. First, the tolerogenic peptides relevant to specific autoimmune diseases are attached to, or encapsulated into nanoparticles. These serve as artificial APCs that present antigen without strong co-stimulatory signals, mimicking the effect of apoptotic cells on APCs. Some concentrate in the liver which has a tolerogenic environment. One group used peptides presented on MHC molecules complexed to iron-oxide nanoparticles to induce antigen-specific CD4+ Tr1-like cells [122]. However, this approach must overcome the obstacle of MHC polymorphism in human autoimmune diseases.
Second, antigen-containing nanoparticles can be targeted to pro-tolerogenic receptors such as anti-inflammatory cytokines or inhibitory CD22 expressed by B cells. Peptides delivered orally can induce oral tolerance. Although this approach has been successful in treating animal models of autoimmune disease, human translation has thus far been disappointing [118].
Third, nanoparticles can be loaded with immunomodulators that drive immature APCs to become tolerogenic. These agents include NFκB inhibitors, protein kinase C (PKC) inhibitors and mTOR inhibitors [24, 123]. PLGA nanoparticles loaded with rapamycin and specific peptides complexed to their surface have been promising in reversing autoimmunity in animal disease models [118]. PLGA nanoparticles with rapamycin and a pegylated uricase fixed to their surface are currently being investigated in a phase 2 clinical trial for the normalization of serum uric acid concentrations in patients with severe symptomatic gout. These individuals required uricase to lower serum uric acid levels. Unfortunately, uricase is highly immunogenic and induces neutralizing antibodies that interfere with treatment. uricase loaded nanoparticles have been shown to be tolerogenic and inhibitanti-drug antibodies [116].
Fourth, in addition to targeting APCs, PLGA nanoparticles have been designed to induce and expand CD4+ Tregs by coating with anti-CD4 to target CD4+ cells and loading with IL-2 and TGF-β for the expansion of functional CD4 Tregs. With this approach in vivo, the concentrations of IL-2 and TGF-β required can be 100 to 1000-fold less than the concentrations of soluble cytokines needed to expand Tregs. These lower doses are anticipated to minimize the possibility of side effects observed when these cytokines are given by injection, although this remains to be tested. Tregs induced with this approach appear to be stable in an inflammatory environment [124]. Because Autoimmune diseases such as SLE and T1D are characterized by decreased IL-2 production [94, 95], judicious use of IL-2 loaded targeted nanoparticles might potentially provide the concentrations needed for maintenance and expansion of Tregs generated by antigen-specific tolerogenic DCs in vivo, although this warrants robust testing. Recently, anti-CD2 and anti-CD4 coated nanoparticles loaded with IL-2 and TGF-β were used to induce both CD4 and CD8 Foxp3+ Tregs in an animal model of lupus that prevented disease [125].We propose thatt is possible to induce multiple protective Treg subsets in vivo simultaneously. While these studies did not include direct investigations on the effects of the nanoparticles on the memory immune cell compartment,, nonetheless, a long-lasting protection was noted in the treated mice, as evidenced by a significant improvement in clinical disease manifestations, laboratory results and histopathology findings relative to controls [106].
While promising, these findings need to consider that even hypothetically perfect nanodelivery vehicles are likely to face future problems. The question of nanoparticle toxicology is a critical one requiring an exhaustive range of assays and metrics towards fully addressing the high or low probability of nanoparticle-mediated toxic effects in vivo and at what range of doses and by what route of administration. It will also be critical to understand possible nanoparticle treatement synergies or antagonisms with other drugs while in combination with ongoing therapies. Current investigations are aiming to yield controlled, finely-tuned targeted delivery of new-generation nanoparticles that might be used at low doses to treat certain autoimmune pathologies.
Taken together, these different avenues and emerging technologies may hold promise to potentially advance the clinical management of autoimmune patients.
Concluding Remarks
The long-standing interest in halting or reversing established autoimmune disease to induce long-term remission, practically and safely, has recently received new promise. Peptides that induce tolerogenic responses in SLE and T1D can be encapsulated in biodegradable nanoparticles with various Treg-promoting drugs.
Box 3.
DCs, Tregs, Self-Reactive T Cells, and Feedback Circuits. By using high-resolution imaging techniques to study Treg/DC interactions in steady-state for the maintenance of tolerogenic DCs, researchers have identified lymphoid tissue clusters comprising DCs, Tregs, and self-reactive IL-2-producing T cells. These tolerogenic DCs express high amounts of CD86 and activate self-reactive T cells to produce IL-2 in amounts that can sustain and expand Tregs. This IL-2-dependent effect was demonstrated via an IL-2-related transcriptional activator in the CD4+ T cells that are present in these clusters [45]. Disruption of TCR signaling in Tregs by blocking costimulation or blocking IL-2 activity can break tolerance, indicating that, in steady-state, self-reactive T cells are continuously activated; when Tregs that control these self-reactive T cells are dysregulated, autoimmunity can ensue [45,50,61]. Similarly, constitutive ablation of DCs in mice was shown to break self-tolerance of CD4+ T cells and resulted in fatal autoimmunity [139]. In steady-state, feedback circuits can support the continuous differentiation of immature DCs towards a mature tolerogenic phenotype which induces Tregs (see Figure 2 in main text). TGF-b and IL-10 produced by Tregs and other cells can then induce immature DCs to become tolerogenic [125]. TGF-b induces mouse DCs to express PD-L1, which is crucial for the generation of pTregs [140]. IL-10 produced by tolerogenic DCs and Tregs also exerts positive feedback effects, and can induce naıive mouse and human T cells to become Tr1 cells, that in turn produce large quantities of IL-10. Tregs can also induce DCs to become tolerogenic. In a mouse model of SLE, direct Treg help enabled DC populations to become tolerogenic. Ex vivo IL-2- plus TGF-b-induced iTregs prevented a lupus syndrome inmice via a TGFb (but not IL-10) -dependent mechanism. Transfer of these DCs to secondary mice increased Foxp3+ cells and protected thesemice from lupus [5,105]. Inhibiting both TGF-b and IL-10 significantly blocked the effect of these tolerogenic DCs. Thus, Treg/DC interactions can have important tolerogenic effects dependent on TGF-b and IL-10 as a consequence of positive feedback circuits on rapidly dividing immature DCs and Tregs [3,5].
Box 5.
Clinical Trials Testing Low-Dose IL-2 or DCs in Autoimmunity. Tolerogenic DCs have been prepared for clinical trials by treating human monocytes with granulocyte/macrophage colony-stimulating factor (GM-CSF) plus IL-4 to make immature DCs, in conjunction with a tolerogenic agent such as dexamethasone or vitamin D3 [160]. Currently, five Phase I studies listed are listed by ClinicalTrials.gov where patients are being recruited. These are open-label studies to assess safety: NCT03337165i (RA), NCT00445913ii (T1D), NCT02618902iii (MS), NCT02903537iv (MS), and NCT02283671v (MS and neuromyelitis optica). There have only been limited attempts to develop a commercial tolerogenic DC product. Because IL-2 production or bioavailability is impaired in autoimmune diseases such as SLE and T1D, and this cytokine is crucial for Treg function, low-dose IL-2 has been tested to correct this defect [161,162], and various clinical trials are currently in progress. Some include low-dose IL-2: NCT02084238vi is a completed open-label interventional study to test the efficacy and safety of low-dose recombinant human IL-2 in SLE. Forty participants received 1 million units every other day for 14 days and were followed for 10 weeks. A decreased Safety of Estrogens in Lupus National Assessment (SELANA)-SLE Disease Activity Index (SLEDAI) clinical score was reported (reduced from 11.3 to 3.8 after treatment), with no serious adverse side effects. NCT03312335vii is an ongoing Phase II open-label study to treat 16 subjects with SLE once daily by subcutaneous injection of 1.5million international units of aldesleukin in 5 day courses every 3 weeks for four cycles. The primary outcome will measure increases in CD4+Foxp3+ cells. NCT01988506viii is an ongoing open-label Phase II study measuring the safety and efficacy of low-dose IL-2 in 14 different autoimmune diseases. 132 subjects will receive 1 million IU of recombinant human (rh)IL-2 per day for 5 consecutive days, and then receive 1 million IU every 15 days for 6 months. Lupus patients will receive the maintenance dose every 7 days. The primary outcome will measure the percentage of Tregs. NCT02955615ix is a Phase II randomized double-blind trial involving 100 participants to study moderate to severely active SLE with a longer-acting IL-2 (measuring the SLE responder index, SRI). NCT03451422x is a randomized triple-blind Phase I/II SLE trial involving 132 participants to study the effects of a genetically altered IL-2 that binds selectively to Tregs (one of the primary outcome measurements is SRI). NCT03782636xi is a randomized triple-blind Phase II trial testing whether IL-2 (aldesleukin) can preserve insulin production in children and young adults with T1D. Participants (n = 45) will receive subcutaneously 0.2 3 106 IU/m2 twice weekly for 6 months. NCT02411253xii is a randomized quadruple-blind Phase II trial involving 138 T1D participants to block ongoing autoimmune destruction in T1D (primary outcome: serum C-peptide responses).
Box 6.
A Therapeutic Potential for CD4+ Treg Subsets? The adoptive transfer of large numbers of expanded Tregs (either ex vivo expansion of pre-existing Tregs, or de novo generation and expansion of iTregs) is an approach to treat autoimmune diseases. However, the potential therapeutic role of transferred Tregs cells has been challenged by both technical and theoretical difficulties even considering that both populations have successfully prevented or treated animal models of lupus, inflammatory bowel disease, myasthenia gravis, and MS [134,163]. Expanded Tregs are polyclonal, not antigen-specific. Their expansion is time-consuming and expensive. For Tregs to be a successful therapeutic, they must be functional in an inflammatory environment. As stated above, Tregs exhibit remarkable plasticity, and some Tregs can become Th1 or Th17 cells in an inflammatory environment [42]. Comparisons of tTregs and iTregs in murine and humanized mouse models have revealed a superiority of iTregs in resisting conversion in some instances. Mouse iTregs were more resistant to Th17 conversion than natural Tregs [167]. In a mouse model of collagen-arthritis, only iTregs reversed established arthritis [164]. In a mouse model of chronic gut inflammation, only iTreg transfer decreased IL-6 and IL-17 expression in the colons of iTreg- versus nTreg-treated mice [165]. In an asthma murine model induced by an environmental antigen, iTregs were superior to nTregs in decreasing airway resistance [166]. Treatment of human nTregs, but not iTregs, with proinflammatory cytokines abolished their protective activity on a human anti-mouse graft versus host disease in immunodeficient mice [107]. explanation for this may be that the combination of IL-2 and TGF-b decreases IL-6 receptor expression in CD4+ T cells, thus blocking IL-17 production and conferring resistance to iTregs [167]. Other investigators, however, do not believe that iTregs are a suitable therapeutic because the methylated TSDR region of the Foxp3/FOX3P locus renders them unstable [168]. However, this methylation does not always correlate with in vivo suppressive activity [169], and the continuous stimulation of iTregs in vivo might increase their stability [170]. One important difference between mouse and human iTregs is that the former can be induced with IL-2 and TGF-b only, whereas human iTregs require an additional agent to stabilize Foxp3 (such as retinoic acid, rapamycin, or IL-2/anti-IL-2 complexes) and to develop full functional activity [171,172]. Tregs induced with retinoic acid have been deemed stable in the presence of proinflammatory IL-1 and IL-6 [171]. To generate functional iTregs one must control the strength of TCR stimulation. Tregs need suboptimal or limited TCR stimulation to become Tregs rather than Teff cells [146]. Even with the appropriate conditioning agents, iTreg functional activity will be poor if the TCR stimulation to induce them is too strong [169]. Because the methodology to induce large numbers of iTregs ex vivo is technically less challenging than expanding the small numbers of tTregs present in blood, with recent improvements [173] this approach might be viable for the putative treatment of some autoimmune diseases.
Box 7.
Therapeutic Nanoparticles. Tolerogenic NPs can be made antigen- or non-specific. There are three broad approaches to achieve antigen-specific tolerance (reviewed in detail in [8]). Most of these approaches aim to induce tolerogenic APCs: (i) Tolerogenic peptides relevant to specific autoimmune diseases are attached to, or are encapsulated in, NPs. These can serve as artificial APCs that present antigen without strong costimulatory signals, mimicking the effect of apoptotic cells on APCs [174]. Some concentrate in the liver, which has a tolerogenic environment. One group used peptides presented on MHC molecules complexed to iron oxide NPs to induce antigen-specific CD4+ Tr1-like cells [175]. However, this approach must overcome the obstacle of MHC polymorphism in human autoimmune disease. (ii) Antigen-containing NPs can be targeted to pro-tolerogenic receptors such as anti-inflammatory cytokines or inhibitory CD22 expressed by B cells. Peptides delivered orally can induce oral tolerance. Although this approach has been successful in treating animalmodels of autoimmune disease, human translation has too date been disappointing [8]. (iii) NPs can be loaded with immunomodulators that drive immature APCs to become tolerogenic, for example NF-kB inhibitors, protein kinase C (PKC) inhibitors, and mTOR inhibitors [176,177]. PLGA NPs loaded with rapamycin and specific peptides complexed to their surface have been promising in reversing autoimmunity in animal disease models [8]. PLGA NPs with rapamycin and a polyethylene glycol-modified (PEGylated) uricase fixed to their surface are being investigated in a completed Phase II randomized, quadruple-blind clinical trial (NCT00325195xiii) testing the normalization of plasma uric acid (PLA) in 225 patients with severe symptomatic gout (primary outcome: PLA concentrations). These NPs might block the neutralizing Abs that interfere with treatment [178]. (iv) In addition to targeting APCs, PLGA NPs coated with anti-CD4 to target CD4+ T cells and loaded with IL-2 and TGF-b have been designed to induce and expand functional CD4+ Tregs. These Tregs appear to be stable in an inflammatory environment [9]. Because autoimmune diseases such as SLE and T1D are characterized by decreased IL-2 production [161,162], judicious use of IL-2-loaded targeted NPs might potentially provide the concentrations needed for the maintenance and expansion of Tregs generated by antigen-specific tolerogenic DCs in vivo, although this warrants robust testing. Recently, anti-CD2 and -CD4-coated NPs loaded with IL-2 and TGF-b were used to induce both CD4+ and CD8+ Foxp3+ Tregs in a mouse model of lupus, and were reported to suppress disease symptoms [10]. Thus, NPs might be ideally used to induce either tolerogenic DCs or Treg populations in vivo.
Acknowledgments
T.M.F. is supported by the National Institutes of Health (NIH; grant HD097531), and C.P. is supported by the Canadian Institutes of Health Research (CIHR; PJT-148821), a Canada Research Chair, and the Anna Maria Solinas Laroche Career Award in Immunology. A.L.C. is supported by the NIH grant HD097531.
Footnotes
Resources
Clinicaltrials.gov (insert exact url link)
References
- 1.Rees F et al. (2017) The worldwide incidence and prevalence of systemic lupus erythematosus: a systematic review of epidemiological studies. Rheumatology (Oxford) 56 (11), 1945–1961. [DOI] [PubMed] [Google Scholar]
- 2.Mayer-Davis EJ et al. (2017) Incidence Trends of Type 1 and Type 2 Diabetes among Youths, 2002–2012. N Engl J Med 377 (3), 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akbar AN et al. (2007) The dynamic co-evolution of memory and regulatory CD4+ T cells in the periphery. Nat Rev Immunol 7 (3), 231–7. [DOI] [PubMed] [Google Scholar]
- 4.Surh CD and Sprent J (2012) TGF-beta puts the brakes on homeostatic proliferation. Nat Immunol 13 (7), 628–30. [DOI] [PubMed] [Google Scholar]
- 5.Breton G et al. (2015) Circulating precursors of human CD1c+ and CD141+ dendritic cells. J Exp Med 212 (3), 401–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eguchi K and Nagai R (2017) Islet inflammation in type 2 diabetes and physiology. J Clin Invest 127 (1), 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iberg CA et al. (2017) Dendritic Cells As Inducers of Peripheral Tolerance. Trends Immunol 38 (11), 793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deniz G et al. (2008) Regulatory NK cells suppress antigen-specific T cell responses. J Immunol 180 (2), 850–7. [DOI] [PubMed] [Google Scholar]
- 9.Huijts CM et al. (2015) mTOR Inhibition Per Se Induces Nuclear Localization of FOXP3 and Conversion of Invariant NKT (iNKT) Cells into Immunosuppressive Regulatory iNKT Cells. J Immunol 195 (5), 2038–45. [DOI] [PubMed] [Google Scholar]
- 10.Mauri C and Menon M (2017) Human regulatory B cells in health and disease: therapeutic potential. J Clin Invest 127 (3), 772–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tallerico R et al. (2017) IL-15, TIM-3 and NK cells subsets predict responsiveness to anti-CTLA-4 treatment in melanoma patients. Oncoimmunology 6 (2), e1261242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y et al. (2017) Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene 36 (44), 6143–6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waterhouse P et al. (1995) Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 270 (5238), 985–8. [DOI] [PubMed] [Google Scholar]
- 14.Nishimura H et al. (1999) Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 11 (2), 141–51. [DOI] [PubMed] [Google Scholar]
- 15.Morlacchi S et al. (2011) Self-antigen presentation by mouse B cells results in regulatory T-cell induction rather than anergy or clonal deletion. Blood 118 (4), 984–91. [DOI] [PubMed] [Google Scholar]
- 16.Weissler KA and Caton AJ (2014) The role of T-cell receptor recognition of peptide:MHC complexes in the formation and activity of Foxp3(+) regulatory T cells. Immunol Rev 259 (1), 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wallace CH et al. (2018) B lymphocytes confer immune tolerance via cell surface GARP-TGF-beta complex. JCI Insight 3 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirokawa M et al. (1992) Human resting B lymphocytes can serve as accessory cells for anti-CD2-induced T cell activation. J Immunol 149 (6), 1859–66. [PubMed] [Google Scholar]
- 19.Franks SE et al. (2016) Targeting B cells in treatment of autoimmunity. Curr Opin Immunol 43, 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen M et al. (2007) Regulation of the lifespan in dendritic cell subsets. Mol Immunol 44 (10), 2558–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Idoyaga J et al. (2013) Specialized role of migratory dendritic cells in peripheral tolerance induction. J Clin Invest 123 (2), 844–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qian C and Cao X (2018) Dendritic cells in the regulation of immunity and inflammation. Semin Immunol 35, 3–11. [DOI] [PubMed] [Google Scholar]
- 23.Joffre OP et al. (2012) Cross-presentation by dendritic cells. Nat Rev Immunol 12 (8), 557–69. [DOI] [PubMed] [Google Scholar]
- 24.Hasegawa H and Matsumoto T (2018) Mechanisms of Tolerance Induction by Dendritic Cells In Vivo. Front Immunol 9, 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van de Ven K and Borst J (2015) Targeting the T-cell co-stimulatory CD27/CD70 pathway in cancer immunotherapy: rationale and potential. Immunotherapy 7 (6), 655–67. [DOI] [PubMed] [Google Scholar]
- 26.Hubo M et al. (2013) Costimulatory molecules on immunogenic versus tolerogenic human dendritic cells. Front Immunol 4, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Massoud AH et al. (2014) Dendritic cell immunoreceptor: a novel receptor for intravenous immunoglobulin mediates induction of regulatory T cells. J Allergy Clin Immunol 133 (3), 853–63 e5. [DOI] [PubMed] [Google Scholar]
- 28.Sakaguchi S et al. (2013) The plasticity and stability of regulatory T cells. Nat Rev Immunol 13 (6), 461–7. [DOI] [PubMed] [Google Scholar]
- 29.Ablamunits V and Herold KC (2008) Generation and function of human regulatory CD8+ T cells induced by a humanized OKT3 monoclonal antibody hOKT3gamma1 (Ala-Ala). Hum Immunol 69 (11), 732–6. [DOI] [PubMed] [Google Scholar]
- 30.Horwitz DA et al. (2013) Therapeutic polyclonal human CD8+ CD25+ Fox3+ TNFR2+ PD-L1+ regulatory cells induced ex-vivo. Clin Immunol 149 (3PB), 450–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suzuki M et al. (2008) Inhibitory CD8+ T cells in autoimmune disease. Hum Immunol 69 (11), 781–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoshida O et al. (2010) Regulatory natural killer cells in murine liver and their immunosuppressive capacity. Liver Int 30 (6), 906–12. [DOI] [PubMed] [Google Scholar]
- 33.Ohkura N et al. (2013) Development and maintenance of regulatory T cells. Immunity 38 (3), 414–23. [DOI] [PubMed] [Google Scholar]
- 34.Levings MK et al. (2001) IFN-alpha and IL-10 induce the differentiation of human type 1 T regulatory cells. J Immunol 166 (9), 5530–9. [DOI] [PubMed] [Google Scholar]
- 35.Abbas AK et al. (2013) Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol 14 (4), 307–8. [DOI] [PubMed] [Google Scholar]
- 36.Lu L et al. (2010) Characterization of protective human CD4CD25 FOXP3 regulatory T cells generated with IL-2, TGF-beta and retinoic acid. PLoS One 5 (12), e15150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jhunjhunwala S et al. (2012) Controlled release formulations of IL-2, TGF-beta1 and rapamycin for the induction of regulatory T cells. J Control Release 159 (1), 78–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mohr A et al. (2018) Human FOXP3(+) T regulatory cell heterogeneity. Clin Transl Immunology 7 (1), e1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Horwitz DA et al. (2008) Natural and TGF-beta-induced Foxp3(+)CD4(+) CD25(+) regulatory T cells are not mirror images of each other. Trends Immunol 29 (9), 429–35. [DOI] [PubMed] [Google Scholar]
- 40.Miyara M et al. (2011) Human FoxP3+ regulatory T cells in systemic autoimmune diseases. Autoimmun Rev 10 (12), 744–55. [DOI] [PubMed] [Google Scholar]
- 41.Bin Dhuban K et al. (2017) Suppression by human FOXP3(+) regulatory T cells requires FOXP3-TIP60 interactions. Sci Immunol 2 (12). [DOI] [PubMed] [Google Scholar]
- 42.Rudra D et al. (2012) Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat Immunol 13 (10), 1010–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Piccirillo CA et al. (2014) Translational control of immune responses: from transcripts to translatomes. Nat Immunol 15 (6), 503–11. [DOI] [PubMed] [Google Scholar]
- 44.Liu Z et al. (2015) Immune homeostasis enforced by co-localized effector and regulatory T cells. Nature 528 (7581), 225–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deaglio S et al. (2007) Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 204 (6), 1257–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kasper IR et al. (2016) Empowering Regulatory T Cells in Autoimmunity. Trends Mol Med 22 (9), 784–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Becker M et al. (2017) Adipose-tissue regulatory T cells: Critical players in adipose-immune crosstalk. Eur J Immunol 47 (11), 1867–1874. [DOI] [PubMed] [Google Scholar]
- 48.Hill JA et al. (2007) Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity 27 (5), 786–800. [DOI] [PubMed] [Google Scholar]
- 49.Floess S et al. (2007) Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol 5 (2), e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yurchenko E et al. (2012) Inflammation-driven reprogramming of CD4+ Foxp3+ regulatory T cells into pathogenic Th1/Th17 T effectors is abrogated by mTOR inhibition in vivo. PLoS One 7 (4), e35572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bhela S et al. (2017) The Plasticity and Stability of Regulatory T Cells during Viral-Induced Inflammatory Lesions. J Immunol 199 (4), 1342–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Q et al. (2011) IL-2 controls the stability of Foxp3 expression in TGF-beta-induced Foxp3+ T cells in vivo. J Immunol 186 (11), 6329–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou X et al. (2010) Cutting edge: all-trans retinoic acid sustains the stability and function of natural regulatory T cells in an inflammatory milieu. J Immunol 185 (5), 2675–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kong N et al. (2012) Antigen-specific transforming growth factor beta-induced Treg cells, but not natural Treg cells, ameliorate autoimmune arthritis in mice by shifting the Th17/Treg cell balance from Th17 predominance to Treg cell predominance. Arthritis Rheum 64 (8), 2548–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karlsson F et al. (2011) Ex vivo generation of regulatory T cells: characterization and therapeutic evaluation in a model of chronic colitis. Methods Mol Biol 677, 47–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang H et al. (2013) Comparison of induced versus natural regulatory T cells of the same TCR specificity for induction of tolerance to an environmental antigen. J Immunol 191 (3), 1136–43. [DOI] [PubMed] [Google Scholar]
- 57.Lu L et al. (2014) Critical role of all-trans retinoic acid in stabilizing human natural regulatory T cells under inflammatory conditions. Proc Natl Acad Sci U S A 111 (33), E3432–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Selvaraj RK and Geiger TL (2007) A kinetic and dynamic analysis of Foxp3 induced in T cells by TGF-beta. J Immunol 179 (2), 11 p following 1390. [PubMed] [Google Scholar]
- 59.Selvaraj RK and Geiger TL (2008) Mitigation of Experimental Allergic Encephalomyelitis by TGF-{beta} Induced Foxp3+ Regulatory T Lymphocytes through the Induction of Anergy and Infectious Tolerance. J Immunol 180 (5), 2830–2838. [DOI] [PubMed] [Google Scholar]
- 60.Li MO and Rudensky AY (2016) T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat Rev Immunol 16 (4), 220–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yamagiwa S et al. (2001) A role for TGF-beta in the generation and expansion of CD4+CD25+ regulatory T cells from human peripheral blood. J Immunol 166 (12), 7282–9. [DOI] [PubMed] [Google Scholar]
- 62.Gray JD et al. (1994) The role of transforming growth factor beta in the generation of suppression: an interaction between CD8+ T and NK cells. J Exp Med 180 (5), 1937–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chinen T et al. (2016) An essential role for the IL-2 receptor in Treg cell function. Nat Immunol 17 (11), 1322–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li MO et al. (2007) T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity 26 (5), 579–91. [DOI] [PubMed] [Google Scholar]
- 65.Smigiel KS et al. (2014) Regulatory T-cell homeostasis: steady-state maintenance and modulation during inflammation. Immunol Rev 259 (1), 40–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Asano T et al. (2017) PD-1 modulates regulatory T-cell homeostasis during low-dose interleukin-2 therapy. Blood 129 (15), 2186–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li MO et al. (2006) Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 25 (3), 455–71. [DOI] [PubMed] [Google Scholar]
- 68.Zheng SG et al. (2002) Generation ex vivo of TGF-beta-producing regulatory T cells from CD4+CD25- precursors. J Immunol 169 (8), 4183–9. [DOI] [PubMed] [Google Scholar]
- 69.Levine AG et al. (2014) Continuous requirement for the TCR in regulatory T cell function. Nat Immunol 15 (11), 1070–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tang Q et al. (2006) Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol 7 (1), 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garbi N et al. (2010) Tonic T cell signalling and T cell tolerance as opposite effects of self-recognition on dendritic cells. Curr Opin Immunol 22 (5), 601–8. [DOI] [PubMed] [Google Scholar]
- 72.Probst HC et al. (2005) Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat Immunol 6 (3), 280–6. [DOI] [PubMed] [Google Scholar]
- 73.Schildknecht A et al. (2010) FoxP3+ regulatory T cells essentially contribute to peripheral CD8+ T-cell tolerance induced by steady-state dendritic cells. Proc Natl Acad Sci U S A 107 (1), 199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tang Q et al. (2008) Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity 28 (5), 687–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ohnmacht C et al. (2009) Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. J Exp Med 206 (3), 549–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Francisco LM et al. (2009) PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med 206 (13), 3015–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Levings MK et al. (2002) The role of IL-10 and TGF-beta in the differentiation and effector function of T regulatory cells. Int Arch Allergy Immunol 129 (4), 263–76. [DOI] [PubMed] [Google Scholar]
- 78.Lan Q et al. (2012) Polyclonal CD4+Foxp3+ Treg cells induce TGFbeta-dependent tolerogenic dendritic cells that suppress the murine lupus-like syndrome. J Mol Cell Biol 4 (6), 409–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kushwah R and Hu J (2010) Dendritic cell apoptosis: regulation of tolerance versus immunity. J Immunol 185 (2), 795–802. [DOI] [PubMed] [Google Scholar]
- 80.Opdenakker G et al. (2016) Microbiomic and Posttranslational Modifications as Preludes to Autoimmune Diseases. Trends Mol Med 22 (9), 746–757. [DOI] [PubMed] [Google Scholar]
- 81.Ercolini AM and Miller SD (2009) The role of infections in autoimmune disease. Clin Exp Immunol 155 (1), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Huynh A et al. (2015) Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol 16 (2), 188–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sakaguchi S and Sakaguchi N (2005) Regulatory T cells in immunologic self-tolerance and autoimmune disease. Int Rev Immunol 24 (3–4), 211–26. [DOI] [PubMed] [Google Scholar]
- 84.Delgoffe GM et al. (2009) The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 30 (6), 832–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Huang N and Perl A (2018) Metabolism as a Target for Modulation in Autoimmune Diseases. Trends Immunol 39 (7), 562–576. [DOI] [PubMed] [Google Scholar]
- 86.Zeng H and Chi H (2017) mTOR signaling in the differentiation and function of regulatory and effector T cells. Curr Opin Immunol 46, 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shrestha S et al. (2015) Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat Immunol 16 (2), 178–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Semple K et al. (2011) Strong CD28 costimulation suppresses induction of regulatory T cells from naive precursors through Lck signaling. Blood 117 (11), 3096–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Molinero LL et al. (2011) High TCR stimuli prevent induced regulatory T cell differentiation in a NF-kappaB-dependent manner. J Immunol 186 (8), 4609–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fujiwara M et al. (2017) Cbl-b Deficiency Mediates Resistance to Programmed Death-Ligand 1/Programmed Death-1 Regulation. Front Immunol 8, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Huynh A et al. (2014) Signals and pathways controlling regulatory T cells. Immunol Rev 258 (1), 117–31. [DOI] [PubMed] [Google Scholar]
- 92.MacIver NJ et al. (2013) Metabolic regulation of T lymphocytes. Annu Rev Immunol 31, 259–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen X and Oppenheim JJ (2010) TNF-alpha: an activator of CD4+FoxP3+TNFR2+ regulatory T cells. Curr Dir Autoimmun 11, 119–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Humrich JY and Riemekasten G (2017) Low-dose IL-2 therapy - a complex scenario that remains to be further explored. Nat Rev Rheumatol 13 (6), 386. [DOI] [PubMed] [Google Scholar]
- 95.Grinberg-Bleyer Y et al. (2010) IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med 207 (9), 1871–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ghoreschi K et al. (2011) Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J Immunol 186 (7), 4234–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ablamunits V et al. (2008) Human regulatory CD8 T cells. Ann N Y Acad Sci 1150, 234–8. [DOI] [PubMed] [Google Scholar]
- 98.Chavin KD et al. (1993) Combined anti-CD2 and anti-CD3 receptor monoclonal antibodies induce donor-specific tolerance in a cardiac transplant model. J Immunol 151 (12), 7249–59. [PubMed] [Google Scholar]
- 99.Ohtsuka K et al. (1999) Cytokine-mediated down-regulation of B cell activity in SLE: effects of interleukin-2 and transforming growth factor-beta. Lupus 8 (2), 95–102. [DOI] [PubMed] [Google Scholar]
- 100.Rigby MR et al. (2015) Alefacept provides sustained clinical and immunological effects in new-onset type 1 diabetes patients. J Clin Invest 125 (8), 3285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sullivan KM et al. (2018) Autologous Stem-Cell Transplantation for Severe Scleroderma. N Engl J Med 378 (11), 1066–1067. [DOI] [PubMed] [Google Scholar]
- 102.Alexander T et al. (2016) Resetting the immune system with immunoablation and autologous haematopoietic stem cell transplantation in autoimmune diseases. Clin Exp Rheumatol 34 (4 Suppl 98), 53–7. [PubMed] [Google Scholar]
- 103.Horwitz DA, P.S., Ou JN, Chen M, Gray JD, Zheng SQ (2013) Therapeutic Polyclonal of human CD8+ Fox3+ TNFR2+ PD-L1+ Regulatory Cells Induced ex-vivo Clin Immunol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang L et al. (2009) Regulatory T cell (Treg) subsets return in patients with refractory lupus following stem cell transplantation, and TGF-beta-producing CD8+ Treg cells are associated with immunological remission of lupus. J Immunol 183 (10), 6346–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Skoberne M et al. (2005) Apoptotic cells at the crossroads of tolerance and immunity. Curr Top Microbiol Immunol 289, 259–92. [DOI] [PubMed] [Google Scholar]
- 106.Jenkins MK and Schwartz RH (1987) Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J Exp Med 165 (2), 302–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Getts DR et al. (2013) Exploiting apoptosis for therapeutic tolerance induction. J Immunol 191 (11), 5341–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kang HK et al. (2005) Very low-dose tolerance with nucleosomal peptides controls lupus and induces potent regulatory T cell subsets. J Immunol 174 (6), 3247–55. [DOI] [PubMed] [Google Scholar]
- 109.Zhang L et al. (2013) Major pathogenic steps in human lupus can be effectively suppressed by nucleosomal histone peptide epitope-induced regulatory immunity. Clin Immunol 149 (3), 365–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hahn BH et al. (2005) Tolerogenic treatment of lupus mice with consensus peptide induces Foxp3-expressing, apoptosis-resistant, TGFbeta-secreting CD8+ T cell suppressors. J Immunol 175 (11), 7728–37. [DOI] [PubMed] [Google Scholar]
- 111.Ando K et al. (2015) Discrimination of p53 immunohistochemistry-positive tumors by its staining pattern in gastric cancer. Cancer Med 4 (1), 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Marek-Trzonkowska N et al. (2013) Clinical application of regulatory T cells in type 1 diabetes. Pediatr Diabetes 14 (5), 322–32. [DOI] [PubMed] [Google Scholar]
- 113.Bluestone JA et al. (2015) Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med 7 (315), 315ra189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Dawson NAJ and Levings MK (2017) Antigen-specific regulatory T cells: are police CARs the answer? Transl Res 187, 53–58. [DOI] [PubMed] [Google Scholar]
- 115.Di Mascolo D et al. (2013) Rosiglitazone-loaded nanospheres for modulating macrophage-specific inflammation in obesity. J Control Release 170 (3), 460–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kishimoto TK et al. (2016) Improving the efficacy and safety of biologic drugs with tolerogenic nanoparticles. Nat Nanotechnol 11 (10), 890–899. [DOI] [PubMed] [Google Scholar]
- 117.Metcalfe SM and Fahmy TM (2012) Targeted nanotherapy for induction of therapeutic immune responses. Trends Mol Med 18 (2), 72–80. [DOI] [PubMed] [Google Scholar]
- 118.Kishimoto TK and Maldonado RA (2018) Nanoparticles for the Induction of Antigen-Specific Immunological Tolerance. Front Immunol 9, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Capini C et al. (2009) Antigen-specific suppression of inflammatory arthritis using liposomes. J Immunol 182 (6), 3556–65. [DOI] [PubMed] [Google Scholar]
- 120.Look M et al. (2014) The nanomaterial-dependent modulation of dendritic cells and its potential influence on therapeutic immunosuppression in lupus. Biomaterials 35 (3), 1089–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ilyinskii PO et al. (2018) Synthetic vaccine particles for durable cytolytic T lymphocyte responses and anti-tumor immunotherapy. PLoS One 13 (6), e0197694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Clemente-Casares X et al. (2016) Expanding antigen-specific regulatory networks to treat autoimmunity. Nature 530 (7591), 434–40. [DOI] [PubMed] [Google Scholar]
- 123.Getts DR et al. (2015) Harnessing nanoparticles for immune modulation. Trends Immunol 36 (7), 419–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Johnson BM et al. (2015) Acute exposure to ZnO nanoparticles induces autophagic immune cell death. Nanotoxicology 9 (6), 737–48. [DOI] [PubMed] [Google Scholar]
- 125.Horwitz DA et al. (2018) Suppression of Murine Lupus by CD4(+) and CD8(+) T Regulatory Cells Induced by T-Cell Targeted Nanoparticles Loaded with IL-2 and TGF-beta. Arthritis Rheumatol. [DOI] [PMC free article] [PubMed] [Google Scholar]