

Abstract
Objective
We examined the effect of glatiramer acetate (GA) on B-cell maturation, differentiation, and antigen presentation in MS and experimental autoimmune encephalomyelitis (EAE).
Methods
A cross-sectional study of blood samples from 20 GA-treated and 18 untreated patients with MS was performed by flow cytometry; 6 GA-treated patients with MS were analyzed longitudinally. GA-mediated effects on B-cell antigen-presenting function were investigated in EAE, or, alternatively, B cells were treated with GA in vitro using vehicle as a control.
Results
In MS, GA diminished transitional B-cell and plasmablast frequency, downregulated CD69, CD25, and CD95 expression, and decreased TNF-α production, whereas IL-10 secretion and MHC Class II expression were increased. In EAE, we observed an equivalent dampening of proinflammatory B-cell properties and an enhanced expression of MHC Class II. When used as antigen-presenting cells for activation of naive T cells, GA-treated B cells promoted development of regulatory T cells, whereas proinflammatory T-cell differentiation was diminished.
Conclusions
GA immune modulates B-cell function in EAE and MS and efficiently interferes with pathogenic B cell–T cell interaction.
Glatiramer acetate (GA), a synthetic random basic copolymer composed of glutamic acid, lysine, tyrosine, and alanine, is widely used in the treatment of MS.1 GA has been shown to reduce relapse rates and progression of neurologic disability.2 The precise mechanism of action by which GA mediates this benefit is still not fully understood. Studies showed a preferential differentiation of CD4+ T cells into T helper (Th)-2 cells,3,4 downregulation of Th17 cell differentiation,5 increased frequency and function of CD4+CD25+FoxP3+ regulatory T (Treg) cells,6,7 and modulation of CD8+ T cells.8 Moreover, GA was found to promote M2 monocyte differentiation7,9 and to reduce activation and proinflammatory cytokine secretion in monocytes9,10 and plasmacytoid dendritic cells.11
Several lines of evidence highlight essential roles of B cells in the pathogenesis of MS.12,13 This is broadly supported by the beneficial effect of B cell–depleting therapies, both in relapsing-remitting (RR)MS14,15 and primary progressive MS.16,17 Some studies have also shown immunomodulatory properties of GA on B cells, including reduction in the number of circulating B cells and a shift from a proinflammatory to an anti-inflammatory B-cell phenotype.18–20 The functional consequences of these phenotypical changes yet remained to be elucidated. Accordingly, we here focused on the question to what extent GA-mediated effects on B cells can change their ability to act as antigen-presenting cells (APCs) for the activation of T cells, an assumed key process in the development and propagation of MS.
Methods
Human
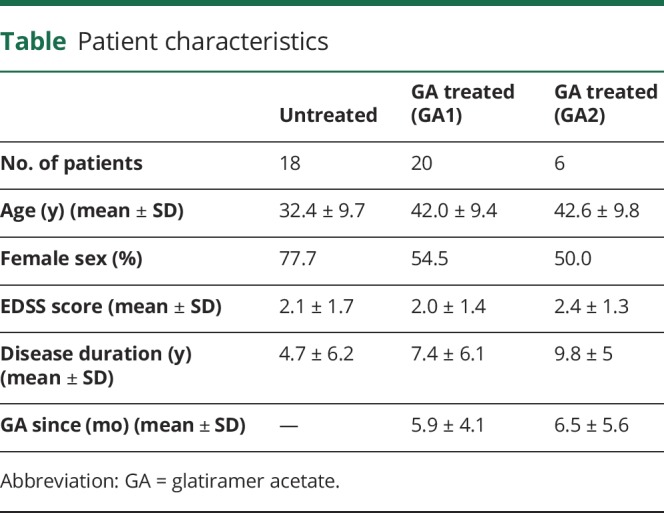
Blood samples were collected from patients with relapsing-remitting MS at the Clinical MS Center of the Universitätsmedizin Göttingen (UMG) in Germany between 2015 and 2018. The diagnosis of RRMS was based on the McDonald criteria. Twenty patients with MS naive to approved disease-modifying therapy were treated with GA for ≥1 month. Eighteen untreated patients with MS served as controls. Six GA-treated patients were analyzed longitudinally, having had blood samples taken at 2 different time points with an interval of at least 3 months. Demographic and disease-related information is summarized in the table.
Table.
Patient characteristics
Mice
Six- to ten-week-old female wild-type (WT) C57BL/6 mice were purchased from Charles River. MOG p35-55 TCR transgenic 2D2 mice were kindly provided by Dr. Kuchroo (Boston, MA).
Ethical approvals
Ethical approvals for all human samples used were given by the ethical review committee of the UMG (approval number 27/4/14). All animal experiments were performed in accordance with the guidelines of the Central Department for Animal Experiments, UMG, and approved by the Office for Consumer Protection and Food Safety of the State of Lower Saxony (protocol number 33.9-42502-04-17/2615).
EAE induction and scoring
Female WT mice were immunized with 50 μg MOG peptide35-55 MEVGWYRSPFSRVVHLYRNGK emulsified in complete Freund's adjuvant followed by intraperitoneal injections of 100 ng of Bordetella pertussis toxin at the day of immunization and 2 days thereafter. Experimental autoimmune encephalomyelitis (EAE) severity was assessed daily and scored on a scale from 0 to 5 scale as described.21
GA treatment
GA was provided by Teva Pharmaceutical Industries. Mice received daily SC injections of 150 μg GA suspended in 0.1 mL phosphate-buffered saline (PBS) or PBS alone.
Detection of anti-GA antibodies
Ninety-six-well plates were coated with 10 μg/mL GA in PBS overnight. Thereafter, diluted serum samples were incubated for 2 hours. After washing, plate-bound antibodies were detected with horseradish peroxidase–conjugated anti-mouse IgG, directed against the Fc part of the bound antibodies. Absorbance was measured at 450 nm with subtraction of a 540-nm reference wavelength on the iMark Microplate Reader.
Isolation of human and murine leukocytes
Human immune cell counts were determined in our hospital's routine laboratory. Human peripheral blood mononuclear cells (PBMCs) were isolated after Ficoll gradient centrifugation and cryopreserved at −80°. Splenic B cells were purified by negative magnetic activated cell sorting (MACS) separation using a mouse lineage panel. Splenic T cells were isolated by negative MACS separation using a mouse pan T-cell isolation kit II.
Flow cytometry
Pregating and gating strategy for human B-cell subsets and surface molecule expression was done as described in figure e-1 (links.lww.com/NXI/A218). Fc receptors were blocked using human TruStain FcX. Dead cells were stained with a fixable viability kit. Human B-cell differentiation was determined using CD19 (HIB19), CD20 (L27), CD24 (ML5), CD27 (O323), and CD38 (HIT2). B-cell activation was evaluated by CD25 (BC96), CD40 (5C3), CD69 (FN50), CD80 (L307.4), CD86 (FUN-1), CD95 (DX2), and major histocompatibility complex (MHC) II (Tü36) after 2 μg/mL CpG stimulation for 20 hours. T cells and monocytes were determined using CD4 (RPA-T4), CD8 (HIT-8a), and CD14 (M5E2), respectively. To investigate B-cell cytokine production, cell suspensions were stimulated with 1 μg/mL CpG, 500 ng/mL ionomycin and 20 ng/mL phorbol 12-myristate 13-acetate in the presence of 1 μL/mL brefeldin A for 22 hours followed by cell fixation/permeabilization and intracellular staining for IL-6 (MQ2-13A5), IL-10 (JES3-19F1), and TNF-α (MAb11). For murine experiments, Fc receptors were blocked using monoclonal antibody specific for CD16/CD32 (93). Dead cells were stained with a fixable viability kit. Splenic B-cell activation/differentiation was determined using CD19 (6D5), CD25 (PC61), CD69 (H1.2F3), CD40 (3/23), CD80 (16-10A1), CD86 (GL-1), and MHCII (AF6-120.1) after 1 μg/mL CpG stimulation for 20 hours. To investigate B-cell cytokine production, cell suspensions were stimulated with 1 μg/mL CpG in the presence of 1 μL/mL brefeldin A for 6 hours followed by cell fixation/permeabilization and intracellular staining for IL-10 (JES5-16E3) and IL-6 (MP5-20F3). Treg cell differentiation was evaluated by CD4 (GK1.5), CD25 (PC61), and by intracellular staining for FoxP3 (FJK-16s) after fixation and permeabilization using the fixation/permeabilization kit. To investigate Th1 and Th17 cell differentiation, cell suspensions were stimulated with 50 ng/mL phorbol 12-myristate 13-acetate and 0.5 μg/mL ionomycin in the presence of 1 μL/mL brefeldin A for 6 hours followed by a CD4 (GK1.5) staining. Cytokine production was analyzed by intracellular staining for IFN-γ (XMG1.2) and IL-17A (TC11-18H10). Samples were acquired on a BD LSRFortessa. All data evaluation was performed using FlowJo software.
ELISA
Production of cytokines was measured using ELISA MAX Standard Set kits. Absorbance was measured at 450 nm with subtraction of a 540-nm reference wavelength on the iMark Microplate Reader.
T-cell proliferation assay
MACS-purified splenic B cells were cocultured with MACS-purified MOG-specific CFSE-stained (CFSE Cell Division Tracker Kit) splenic T cells from 2D2 mice and were restimulated with MOG peptide35-55. After 72 hours, T-cell proliferation was evaluated by flow cytometry.
Statistical analysis
Statistical analysis was performed using the software GraphPad Prism 5.01 and 6.01. Human data sets were tested for Gauss distribution using the D'Agostino-Pearson omnibus normality test, Shapiro-Wilk normality test, and Kolmogorov-Smirnov normality test. For the comparison of 2 cross-sectional cohorts with Gauss distribution an unpaired and for longitudinal samples a paired t test was used, respectively. If the data were not Gauss distributed, a Mann-Whitney U test was applied in the cross-sectional analysis, and the Wilcoxon matched-pairs signed-rank test was used for the longitudinal data. Clinical scores and T-cell proliferation are depicted as mean ± SEM and were analyzed by the Mann-Whitney U test. GA antibody titers are shown as median and were analyzed using the Student t test. All other data are shown as median, and the statistical comparison was made using the Mann-Whitney U test. A value of p < 0.05 was considered significant and is shown by 1 asterisk. Two asterisks and 3 asterisks indicate significances of p < 0.01 and p < 0.001, respectively.
Data availability
The data that support the findings of this study are available from the corresponding author on reasonable request.
Results
In the present study, we compared 20 GA-treated patients with MS with 18 untreated MS controls (table). The mean GA therapy duration was 5.9 months and ranged from 1 to 14.5 months. Six GA-treated patients with MS were analyzed longitudinally at 2 different time points with an interval of >3 months. Patients were naive to any approved disease-modifying therapy and had not received steroids for at least 3 months before GA treatment.
GA alters PBMC composition
At first, we determined the impact of GA on PBMC composition. GA treatment resulted in a trend toward decreased absolute leukocyte and neutrophil numbers, whereas monocyte numbers became significantly elevated (figure 1, A and B). Frequency analysis of the cross-sectional and longitudinal study showed a tendency for decreased CD4+ T cells and significantly diminished CD19+ B cells, respectively (figure 1, C–F). This GA treatment effect did not correlate the respective treatment duration ranging between 1 and 14.5 months (figure e-2A, links.lww.com/NXI/A218).
Figure 1. GA treatment alters the composition of leukocytes in patients with MS.

Peripheral blood samples were taken from control (n = 18) and glatiramer acetate–treated (GA; n = 20) patients with MS. (A) Leukocyte counts and (B) neutrophil, lymphocyte, monocyte, eosinophil, and basophil counts were measured in routine clinical laboratory blood counts if available (*p < 0.05; unpaired t test). Next, peripheral blood mononuclear cells (PBMCs) were isolated from the samples. (C) Cell frequencies of CD4+ T cells (TC), CD8+ TC, CD14+ monocytes (Mo), and CD19+ B cells (BC) were determined using flow cytometry (ns; unpaired t test). (D) CD4+, CD8+ TC, CD14+ Mo and BC of patients with MS at 2 time points during GA medication; line connects an individual patient (n = 5; *p < 0.05; Wilcoxon matched-pairs signed-rank test). (E and F) Fold changes of the horizontal and longitudinal cell frequency changes. GA = glatiramer acetate.
GA decreases B-cell activation, differentiation, and proinflammatory cytokine production, whereas IL-10 secretion and MHC Class II expression are increased
To assess whether GA has an effect on B-cell activation, differentiation, and cytokine production, we analyzed peripheral blood B cells (figure 2, A–I, figure e-2, B and C, links.lww.com/NXI/A218). GA therapy was associated with a reduction in plasmablasts, both in the cross-sectional and longitudinal analyses of blood samples (figure 2, A and B). In addition, frequency of immature transitional B cells was decreased in the longitudinal study (figure 2B) and correlated with GA treatment duration (figure 2C). GA longitudinally downregulated CD25, CD69, and CD95 expression on B cells, whereas MHC Class II expression was upregulated as compared to untreated MS controls (figure 2, D–G). Other molecules involved in antigen presentation such as CD40, CD80, and CD86 showed no difference (figure 2, F and G). IL-6 production was not altered by GA treatment, both in the cross-sectional and longitudinal studies, whereas GA increased anti-inflammatory IL-10 and decreased proinflammatory TNF-α cytokine production in the longitudinal analysis; however, no correlation with longer GA treatment duration was found (figure 2, H and I, figure e-2B, links.lww.com/NXI/A218).
Figure 2. GA therapy changes the phenotype of human B cells in patients with MS.

Human peripheral blood mononuclear cells (PBMCs) were isolated from glatiramer acetate (GA; n = 20) or non-GA (control; n = 18) treated patients with MS. In addition, 6 patients were analyzed longitudinally on GA treatment. Red circles represent GA treatment, squares control treatment. (A) Mean frequency ± SEM of B-cell subpopulations defined as follows: transitional B cells (CD24high CD38high; transitional), mature B cells (CD24var CD38low; mature), antigen-activated B cells (CD27+; ag-activated), memory B cells (CD27var CD38−; memory), and plasmablasts (CD20− CD27+ CD38+; *p < 0.05; unpaired t test). (B) B-cell subset frequencies of patients with MS at 2 time points during GA therapy; line connects an individual patient (n = 6; *p < 0.05; Wilcoxon matched-pairs signed-rank test). (C) The individual patients' frequencies of BC subsets were correlated with the duration of GA treatment (*p < 0.05; linear regression). (D) MFI ± SEM of activation molecules expressed on B cells (ns; unpaired t test). (E) B-cell activation marker expression of patients with MS at 2 time points during GA therapy; line connects an individual patient (n = 6; *p < 0.05; Wilcoxon matched-pairs signed-rank test). (F) Mean MFI of molecules involved in antigen presentation expressed on B cells (*p < 0.05; unpaired t test). (G) Expression of molecules involved in antigen presentation of patients with MS at 2 time points during GA medication; line connects an individual patient (ns; Wilcoxon matched-pairs signed-rank test). (H) Shown is the frequency of positive cells regarding the respective cytokine (tumor necrosis factor [TNF], interleukin [IL]-6, and IL-10; mean ± SEM; ns; unpaired t test). (I) TNF, IL-6, and IL-10-positive B cells of patients with MS at 2 time points during GA medication; line connects an individual patient (*p < 0.05; Wilcoxon matched-pairs signed-rank test). GA = glatiramer acetate.
GA upregulates MHC Class II B-cell expression independent of EAE
To identify whether the observed reduction on B-cell activation, TNF-α production and the increase in IL-10 secretion and MHC Class II expression in patients with MS is a result of GA treatment or a concomitant disease-related effect, we initially administered daily subcutaneous GA to naive unimmunized wild-type mice (figure 3A). GA had no impact on B-cell activation or cytokine production; however, MHC Class II was significantly upregulated after ex vivo stimulation (figure 3, B–H).
Figure 3. GA upregulates MHC Class II expression on B cells.

(A) Naive mice received a daily SC injection of 150 μg GA. On day 10 post-treatment onset, splenic B cells were isolated and analyzed (B and C) for expression of activation markers, (D–F) costimulatory molecules, and (G) the antigen-presenting molecule MHC Class II as well as (H) for secretion of cytokines. Data are shown as median; n = 4; *p < 0.05; Mann-Whitney U test. GA = glatiramer acetate.
GA downregulates B-cell activation and ameliorates clinical severity of active EAE
To investigate the effect of GA on B-cell phenotype and function during pathologic conditions, mice received a daily subcutaneous GA injection, starting 7 days before immunization (figure 4A). GA ameliorated EAE (figure 4B), which was associated with a production of antibodies against GA (figure 4C), a decrease in expression of the early activation marker CD69 on B cells, and diminished secretion of IL-6, whereas the expression of costimulatory molecule CD86 and MHC Class II was upregulated (figure 4D).
Figure 4. GA prevents B-cell activation and ameliorates clinical severity of active EAE.

(A) GA therapy was performed by a daily SC injection of 150 μg, starting 7 days before MOG peptide35-55 immunization. Serum and splenic B cells were isolated on day 23 post-immunization. (B) Mean group EAE severity is given as mean ± SEM; disease incidence is indicated in brackets; n = 15; *p < 0.05; Mann-Whitney U test. (C) GA antibody titers were measured at 450 nm (data given as median; n = 3–4; ***p < 0.001; Student t test). (D) B-cell activation, expression of molecules involved in antigen presentation, and cytokine secretion were analyzed by FACS (data given as median; n = 5; *p < 0.05, **p < 0.01; Mann-Whitney U test). (E) B cells were cocultured with CFSE-labeled myelin-specific (2D2) naive T cells in the presence of 5, 25, or 100 μg/mL MOG peptide35-55. T-cell proliferation was evaluated by CFSE dilution and stratified by division frequency as follows: few divisions (1–2; black), intermediate divisions (3; medium gray), and many divisions (≥4; light gray). T-cell divisions are shown as mean ± SEM; n = 5; *p < 0.05; Mann-Whitney U test. Differentiation of myelin-specific naive T cells into (F) Treg cells (CD25+FoxP3+CD4+) or (G) Th1- (IFN-γ+CD4+) and Th17 cells (IL-17+CD4+) was analyzed by FACS (data given as median; n = 5). EAE = experimental autoimmune encephalomyelitis; GA = glatiramer acetate.
GA increases B-cell antigen-presenting capacity resulting in regulatory T-cell induction
To elucidate whether our findings on B-cell properties have mechanistic consequences on antigen-presenting function, B cells were isolated 30 days post-GA treatment onset and 23 days post-EAE induction and cocultured with MOG-specific (2D2) T cells in the presence of increasing MOG peptide35-55 concentrations (figure 4A). As shown in figure 4E, B cells purified from GA-treated mice triggered a significantly higher proliferation of myelin-specific T cells. Importantly, this related to an expansion of Treg cells (figure 4F), whereas Th1- and Th17 cell frequencies remained unaffected (figure 4G). Based on these findings, we next assessed the direct effect of GA exposure on B-cell APC function in vitro. Purified naive B cells were preincubated with GA following coculture with myelin-specific T cells in the presence of MOG peptide35-55 (figure 5A). GA pre-incubation resulted in a B-cell stimulatory effect (figure e-3, links.lww.com/NXI/A218), which was accompanied by enhanced capacity to generate Treg cells, paralleling our ex vivo findings on GA treatment (figure 5, B–E).
Figure 5. GA-treated B cells preferentially generate T regs, whereas development of proinflammatory T cells is diminished.

(A) Naive B cells purified from WT mice were incubated with 50 μg/mL GA or vehicle at 37°C for 3 hours. After washing, B cells were cocultured with CFSE-labeled myelin-specific (2D2) naive T cells in the presence of 5, 25, or 100 μg/mL MOG peptide35-55. (B) T-cell proliferation was evaluated by CFSE dilution and stratified by division frequency as follows: few divisions (1–2; black), intermediate divisions (3; medium gray), and many divisions (≥4; light gray). T-cell divisions are shown as mean ± SEM; n = 4; *p < 0.05; Mann-Whitney U test. Differentiation of myelin-specific naive T cells into (C) Treg cells (CD25+FoxP3+CD4+) or (D) Th1- (IFN-γ+CD4+) and (E) Th17 cells (IL-17+CD4+) was analyzed by FACS (data given as median; n = 4; *p < 0.05; Mann-Whitney U test). GA = glatiramer acetate.
Discussion
GA has been shown to reduce the relapse rate and progression of neurologic disability in MS.2 Past studies demonstrated anti-inflammatory properties of GA on T cells4,6,8 and myeloid cells.9,10,22 First lines of evidence indicate an immunomodulatory effect on B cells,18–20 although it remained unclear whether this may affect the ability of B cells to act as APCs. In this article, we investigated the phenotype and APC function of B cells in MS and its murine model on treatment with GA. We found decreased frequencies of immature (transitional) B cells and plasmablasts in GA-treated patients with MS. A reduction in circulating CD19+ B cells in GA-treated patients with RRMS has been also described previously,23 which could reflect diminished B-cell survival factors such as BAFF and APRIL after GA therapy, as it was observed in EAE.20 In this regard of interest may be that we found a correlation between high baseline B-cell frequencies, an active disease course, and a poor GA treatment response (figure e-2C, links.lww.com/NXI/A218), possibly suggesting that patients with MS with increased peripheral blood B-cell numbers might not properly respond to GA therapy.
By longitudinally analyzing the GA effect on B-cell phenotype, we observed a downregulation of the activation marker CD69, CD95, and CD25 and a decrease in TNF-α production and an increase in IL-10 secretion, which was supported by a recent study showing a shift toward anti-inflammatory cytokine production by B cells on GA therapy.19 Of interest, we found a modest but significant upregulation of MHC Class II expression on B cells in GA-treated patients with MS. B cells are thought to act as APCs for presentation of GA to T cells.24 Direct binding of GA to multiple murine and human MHC Class II epitopes25,26 has been shown, raising the question whether our observation might have consequences in terms of B-cell APC function. To address this pivotal issue, we first administered GA to naive WT mice to rule out a disease-related effect and indeed noticed an upregulation of MHC Class II expression on B cells, without any effect on other markers of activation. During pathologic conditions following EAE induction, GA treatment decreased clinical severity, B-cell activation, and proinflammatory cytokine production, whereas the costimulatory molecule CD86 and MHC Class II were again upregulated. To further elucidate the observed B-cell immune modulation with focus on B-cell antigen presentation, we used a coculture in which purified B cells from GA-treated mice or alternatively naive B cells following GA preincubation in vitro were used as APCs to activate naive myelin-specific T cells. GA-treated B cells triggered a significantly higher proliferation of naive myelin-specific T cells, composed of increased CD4+CD25+FoxP3+ Treg cells. As TGF-ß is associated with the development of Treg cells, we also measured TGF-ß production by B cells in our model, however at no detectable levels. This mechanistic observation, which is well supported by earlier reports on an expansion of Treg cells on GA treatment in MS,6 and indicates that GA centrally interferes with pathogenic B cell–T cell interaction in development and propagation of CNS demyelinating disease.
Our findings indicate common features to IFN-β, which also have been shown to exert immunomodulatory properties on B cells by abrogating proinflammatory and by fostering anti-inflammatory cytokine production.27 However, IFN-β is thought to primarily downregulate costimulatory molecules and MHC-Class II,27–29 our findings suggest the modulation of B-cell antigen presentation by GA as a key role for B cell–fostered Treg cell development.
Anti–CD20-mediated B-cell depletion has been shown to be a very efficient therapy in MS,14–17 however, treatment cessation may lead to a recovery of highly differentiated pathogenic B cells,30 and long-term treatment may lower immunoglobulin production, possibly raising the risk of infections over time.31 Our data support the concept that GA could act as a suitable maintenance therapy after cessation of anti-CD20 treatment by fostering regulatory properties in repopulating B cells. The first trial in humans provided inconclusive results.32 Although the beneficial effect by GA as maintenance therapy showed superior efficacy than GA therapy alone, this benefit seemed to wane within the study period. More trials are needed, as that study was limited due to a small number of patients and the lack of a control group receiving no maintenance therapy after rituximab cessation.
Moreover, GA could also have beneficial effects in other B cell–mediated diseases such as neuromyelitis optica (NMO). Although aquaporin-4 antibody (AQP4-IgG)-seropositive patients showed inefficient results,33,34 first lines of evidence indicate that patients with AQP4-IgG–seronegative NMO may respond to GA therapy.33,35,36
In conclusion, our data indicate that the pleotropic immunomodulatory effect of GA includes B cells and B-cell antigen presentation resulting in a normalization of MS-specific pathogenic B-cell differentiation and in an expansion of Treg cells. These novel findings may complement other established effects of GA in MS, may pioneer its preferential use after B-cell depletion, and may lastly be of clinical relevance in other B cell–driven CNS autoimmune diseases.
Acknowledgment
The authors thank Katja Grondey and Julian Koch for excellent technical support.
Glossary
- APC
antigen-presenting cell
- EAE
experimental autoimmune encephalomyelitis
- GA
glatiramer acetate
- NMO
neuromyelitis optica
- PBMC
peripheral blood mononuclear cell
- PBS
phosphate-buffered saline
- RRMS
relapsing-remitting MS
- WT
wild type
Appendix. Authors
Study funding
D. Häusler is supported by the Startprogramm of the UMG. J. W. Traub is supported by the VorSPrUNG program of the UMG. S. S. Zamvil is supported by research grants from the US NIH (1 RO1 NS092835-01, 1 R01 AI131624-01A1, 1 R21 NS108159-01, and 1 R21AI142186-01A1), the US National Multiple Sclerosis Society (1 RG1701-26628), the Weill Institute, and the Maisin Foundation. P.H. Lalive is supported by the Swiss National Science Foundation (SNSF_ 310030_176078). M.S. Weber receives research support from the National Multiple Sclerosis Society (NMSS; PP 1660), the Deutsche Forschungsgemeinschaft (DFG; WE 3547/5-1), from Novartis, Teva, Biogen Idec, Roche, Merck, and the ProFutura Programm of the UMG.
Disclosure
D. Häusler, Z. Hajiyeva, J.W. Traub, S.S. Zamvil, P.H. Lalive, and W. Brück report no disclosures. M.S. Weber is serving as an editor for PLoS One. Go to Neurology.org/NN for full disclosures.
References
- 1.Weber MS, Menge T, Lehmann-Horn K, et al. . Current treatment strategies for multiple sclerosis—efficacy versus neurological adverse effects. Curr Pharm Des 2012;18:209–219. [DOI] [PubMed] [Google Scholar]
- 2.Johnson KP, Brooks BR, Cohen JA, et al. . Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology 1995;45:1268–1276. [DOI] [PubMed] [Google Scholar]
- 3.Duda PW, Schmied MC, Cook SL, Krieger JI, Hafler DA. Glatiramer acetate (Copaxone) induces degenerate, Th2-polarized immune responses in patients with multiple sclerosis. J Clin Invest 2000;105:967–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neuhaus O, Farina C, Yassouridis A, et al. . Multiple sclerosis: comparison of copolymer-1- reactive T cell lines from treated and untreated subjects reveals cytokine shift from T helper 1 to T helper 2 cells. Proc Natl Acad Sci USA 2000;97:7452–7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aharoni R, Eilam R, Stock A, et al. . Glatiramer acetate reduces Th-17 inflammation and induces regulatory T-cells in the CNS of mice with relapsing-remitting or chronic EAE. J Neuroimmunol 2010;225:100–111. [DOI] [PubMed] [Google Scholar]
- 6.Hong J, Li N, Zhang X, Zheng B, Zhang JZ. Induction of CD4+CD25+ regulatory T cells by copolymer-I through activation of transcription factor Foxp3. Proc Natl Acad Sci USA 2005;102:6449–6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weber MS, Prod'homme T, Youssef S, et al. . Type II monocytes modulate T cell-mediated central nervous system autoimmune disease. Nat Med 2007;13:935–943. [DOI] [PubMed] [Google Scholar]
- 8.Karandikar NJ, Crawford MP, Yan X, et al. . Glatiramer acetate (Copaxone) therapy induces CD8(+) T cell responses in patients with multiple sclerosis. J Clin Invest 2002;109:641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weber MS, Starck M, Wagenpfeil S, Meinl E, Hohlfeld R, Farina C. Multiple sclerosis: glatiramer acetate inhibits monocyte reactivity in vitro and in vivo. Brain 2004;127:1370–1378. [DOI] [PubMed] [Google Scholar]
- 10.Kim HJ, Ifergan I, Antel JP, et al. . Type 2 monocyte and microglia differentiation mediated by glatiramer acetate therapy in patients with multiple sclerosis. J Immunol 2004;172:7144–7153. [DOI] [PubMed] [Google Scholar]
- 11.Stasiolek M, Bayas A, Kruse N, et al. . Impaired maturation and altered regulatory function of plasmacytoid dendritic cells in multiple sclerosis. Brain 2006;129:1293–1305. [DOI] [PubMed] [Google Scholar]
- 12.Weber MS, Hemmer B. Cooperation of B cells and T cells in the pathogenesis of multiple sclerosis. Results Probl Cell Differ 2010;51:115–126. [DOI] [PubMed] [Google Scholar]
- 13.Kinzel S, Weber MS. B cell-directed therapeutics in multiple sclerosis: rationale and clinical evidence. CNS Drugs 2016;30:1137–1148. [DOI] [PubMed] [Google Scholar]
- 14.Hauser SL, Waubant E, Arnold DL, et al. . B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med 2008;358:676–688. [DOI] [PubMed] [Google Scholar]
- 15.Kappos L, Li D, Calabresi PA, et al. . Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet 2011;378:1779–1787. [DOI] [PubMed] [Google Scholar]
- 16.Hawker K, O'Connor P, Freedman MS, et al. . Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol 2009;66:460–471. [DOI] [PubMed] [Google Scholar]
- 17.Montalban X, Belachew S, Wolinsky JS. Ocrelizumab in primary progressive and relapsing multiple sclerosis. N Engl J Med 2017;376:1694. [DOI] [PubMed] [Google Scholar]
- 18.Kala M, Rhodes SN, Piao WH, Shi FD, Campagnolo DI, Vollmer TL. B cells from glatiramer acetate-treated mice suppress experimental autoimmune encephalomyelitis. Exp Neurol 2010;221:136–145. [DOI] [PubMed] [Google Scholar]
- 19.Ireland SJ, Guzman AA, O'Brien DE, et al. . The effect of glatiramer acetate therapy on functional properties of B cells from patients with relapsing-remitting multiple sclerosis. JAMA Neurol 2014;71:1421–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Begum-Haque S, Sharma A, Christy M, et al. . Increased expression of B cell-associated regulatory cytokines by glatiramer acetate in mice with experimental autoimmune encephalomyelitis. J Neuroimmunol 2010;219:47–53. [DOI] [PubMed] [Google Scholar]
- 21.Häusler D, Torke S, Peelen E, et al. . High dose vitamin D exacerbates central nervous system autoimmunity by raising T-cell excitatory calcium. Brain 2019;142:2737–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vieira PL, Heystek HC, Wormmeester J, Wierenga EA, Kapsenberg ML. Glatiramer acetate (copolymer-1, copaxone) promotes Th2 cell development and increased IL-10 production through modulation of dendritic cells. J Immunol 2003;170:4483–4488. [DOI] [PubMed] [Google Scholar]
- 23.Carrieri PB, Carbone F, Perna F, et al. . Longitudinal assessment of immuno-metabolic parameters in multiple sclerosis patients during treatment with glatiramer acetate. Metabolism 2015;64:1112–1121. [DOI] [PubMed] [Google Scholar]
- 24.Jackson LJ, Selva S, Niedzielko T, Vollmer T. B cell receptor recognition of glatiramer acetate is required for efficacy through antigen presentation and cytokine production. J Clin Cell Immunol 2014;5:185. [Google Scholar]
- 25.Fridkis-Hareli M, Teitelbaum D, Gurevich E, et al. . Direct binding of myelin basic protein and synthetic copolymer 1 to class II major histocompatibility complex molecules on living antigen-presenting cells—specificity and promiscuity. Proc Natl Acad Sci USA 1994;91:4872–4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fridkis-Hareli M, Strominger JL. Promiscuous binding of synthetic copolymer 1 to purified HLA-DR molecules. J Immunol 1998;160:4386–4397. [PubMed] [Google Scholar]
- 27.Ramgolam VS, Sha Y, Marcus KL, et al. . B cells as a therapeutic target for IFN-beta in relapsing-remitting multiple sclerosis. J Immunol 2011;186:4518–4526. [DOI] [PubMed] [Google Scholar]
- 28.Niino M, Hirotani M, Miyazaki Y, Sasaki H. Memory and naive B-cell subsets in patients with multiple sclerosis. Neurosci Lett 2009;464:74–78. [DOI] [PubMed] [Google Scholar]
- 29.Jiang H, Milo R, Swoveland P, Johnson KP, Panitch H, Dhib-Jalbut S. Interferon beta-1b reduces interferon gamma-induced antigen-presenting capacity of human glial and B cells. J Neuroimmunol 1995;61:17–25. [DOI] [PubMed] [Google Scholar]
- 30.Häusler D, Häusser-Kinzel S, Feldmann L, et al. . Functional characterization of reappearing B cells after anti-CD20 treatment of CNS autoimmune disease. Proc Natl Acad Sci USA 2018;115:9773–9778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marcinno A, Marnetto F, Valentino P, et al. . Rituximab-induced hypogammaglobulinemia in patients with neuromyelitis optica spectrum disorders. Neurol Neuroimmunol Neuroinflamm 2018;5:e498 doi:10.1212/NXI.0000000000000498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Honce JM, Nair KV, Sillau S, et al. . Rituximab vs placebo induction prior to glatiramer acetate monotherapy in multiple sclerosis. Neurology 2019;92:e723–e732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ayzenberg I, Schollhammer J, Hoepner R, et al. . Efficacy of glatiramer acetate in neuromyelitis optica spectrum disorder: a multicenter retrospective study. J Neurol 2016;263:575–582. [DOI] [PubMed] [Google Scholar]
- 34.Stellmann JP, Krumbholz M, Friede T, et al. . Immunotherapies in neuromyelitis optica spectrum disorder: efficacy and predictors of response. J Neurol Neurosurg Psychiatry 2017;88:639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bergamaschi R. Glatiramer acetate treatment in Devic's neuromyelitis optica. Brain 2003;126(pt 6):1E; author reply 1E-a. [DOI] [PubMed] [Google Scholar]
- 36.Gartzen K, Limmroth V, Putzki N. Relapsing neuromyelitis optica responsive to glatiramer acetate treatment. Eur J Neurol 2007;14:e12–e13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author on reasonable request.