

Abstract
Objective
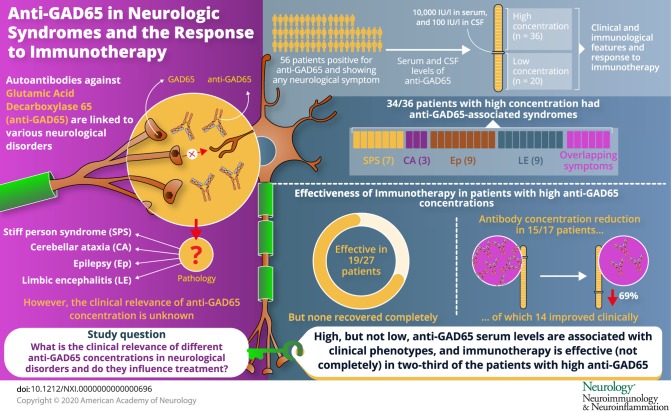
Antibodies against glutamic acid decarboxylase 65 (anti-GAD65) are associated with a number of neurologic syndromes. However, their pathogenic role is controversial. Our objective was to describe clinical and paraclinical characteristics of anti-GAD65 patients and analyze their response to immunotherapy.
Methods
Retrospectively, we studied patients (n = 56) with positive anti-GAD65 and any neurologic symptom. We tested serum and CSF with ELISA, immunohistochemistry, and cell-based assay. Accordingly, we set a cutoff value of 10,000 IU/mL in serum by ELISA to group patients into high-concentration (n = 36) and low-concentration (n = 20) groups. We compared clinical and immunologic features and analyzed response to immunotherapy.
Results
Classical anti–GAD65-associated syndromes were seen in 34/36 patients with high concentration (94%): stiff-person syndrome (7), cerebellar ataxia (3), chronic epilepsy (9), limbic encephalitis (9), or an overlap of 2 or more of the former (6). Patients with low concentrations had a broad, heterogeneous symptom spectrum. Immunotherapy was effective in 19/27 treated patients (70%), although none of them completely recovered. Antibody concentration reduction occurred in 15/17 patients with available pre- and post-treatment samples (median reduction 69%; range 27%–99%), of which 14 improved clinically. The 2 patients with unchanged concentrations showed no clinical improvement. No differences in treatment responses were observed between specific syndromes.
Conclusion
Most patients with high anti-GAD65 concentrations (>10,000 IU/mL) showed some improvement after immunotherapy, unfortunately without complete recovery. Serum antibody concentrations' course might be useful to monitor response. In patients with low anti-GAD65 concentrations, especially in those without typical clinical phenotypes, diagnostic alternatives are more likely.
Autoantibodies against glutamic acid decarboxylase (GAD) 65 have been linked to different types of syndromes. These antibodies are widely used as biomarkers for diabetes mellitus type 1 (DM1) diagnosis because they are present in 80% of patients at diagnosis.1,2 However, it is well known that anti-GAD65 can also be associated with specific neurologic disorders, including stiff-person syndrome (SPS), cerebellar ataxia (CA), epilepsy (Ep), and limbic encephalitis (LE).3–6
The pathophysiologic role of anti-GAD65 in neuroinflammation is still unclear. It is hard to understand whether there is a direct antibody-associated pathogenic effect because the target antigen is located intracellularly. Moreover, responses to immunotherapy seem to be poorer than in patients with neurologic disorders caused by most other antineuronal antibodies.7,8
In studies evaluating treatment effects in anti–GAD65-positive patients, methods used are variable, and patient cohorts are often restricted to one of the specific clinical phenotypes.9–11 In addition, some studies describing patients with neurologic symptoms and anti-GAD65 also include patients with low antibody concentrations. In these patients, clinical relevance of anti-GAD65 is questionable because low antibody concentrations are regularly found among patients with DM1 (without neurologic symptoms) and rarely in healthy individuals.1,2,12
The aim of this cohort study is to evaluate the clinical relevance of low and high anti-GAD65 concentrations in patients with neurologic symptoms, to establish clinically relevant cutoff values (in serum and CSF), and to evaluate clinical and serologic treatment responses.
Methods
Patients
We retrospectively included patients with neurologic symptoms and an increased anti-GAD65 concentration detected in serum and/or CSF, from January 2015 until June 2018. Anti-GAD65 was routinely detected at the Department of Immunology (Laboratory Medical Immunology) of the Erasmus University Medical Center by using ELISA and reported as negative or positive. Clinical information was obtained from medical files. Thirty of 56 patients (54%) were seen by one of the authors.
Standard protocol approvals, registrations, and patient consents
The institutional review board of the Erasmus University Medical Center approved the study protocol. Written informed consent was obtained from all patients.
Laboratory tests
Anti-GAD65 was determined in serum and CSF when available, using 3 assays. Paired serum and CSF samples were used if possible. Otherwise, serum samples drawn closest to the CSF tap were used, provided they were preimmunotherapy samples. First, automated quantitative ELISA was performed according to the manufacturer's instructions (Medizym anti-GAD; Medipan, Berlin, Germany). Calibration curves based on 5 calibrators (5, 18, 35, 120, and 250 IU/mL) were used to infer antibody concentrations. Samples were considered positive with anti-GAD65 concentrations above 5 IU/mL. When concentrations were over 250 IU/mL, we tested serial dilutions (1:10; 1:100; 1:1,000; 1:10,000) and chose the most reliable result (i.e., optical density value in the linear part of the calibration curve) to determine the IU/mL end concentration. Second, immunohistochemistry (IHC) was used as a screening method to determine immunoreactivity of patients' serum (diluted 1:200) or CSF (diluted 1:2) against rat hippocampal brain tissue. A detailed description can be found elsewhere.13 GAD65 antibody binding causes a characteristic staining pattern.13 Finally, cell-based assay (CBA) (Euroimmun, Lübeck, Germany; REF: FA 1022-1005-50) was performed according to the manufacturer's instructions, using human embryonal kidney cells (HEK293) expressing recombinant GAD65. Serum was diluted 1:10, and CSF was used undiluted.
ELISA provided quantitative results. IHC and CBA were used as confirmatory qualitative techniques to determine clinically relevant cutoff values for serum and CSF. Samples positive by ELISA and confirmed with positive IHC and CBA were considered high-concentration samples.14 Samples showing a positive staining pattern on IHC, but no typical GAD pattern, were tested more extensively with commercial and in-house CBAs using fixed or live cells (Euroimmun kits and in-house CBAs).
Defining clinical phenotypes and clinical relevance of anti-GAD65
Patients were allocated into 6 groups based on the clinical phenotypes described in the literature: (1) SPS, (2) CA, (3) Ep, (4) LE, (5) overlap, and (6) other. The overlap category consisted of patients who had developed more than 1 anti–GAD65-associated neurologic syndrome over the disease course. Patients with LE, presenting with seizures or developing seizures following LE, were not considered as overlap syndromes and were all classified as LE.
After determining cutoff values, patients were classified into a high-concentration or low-concentration group. Patients in the high-concentration group were studied more thoroughly to assess clinical and serologic response to immunotherapy. Immunotherapy responses were evaluated by one of the authors during follow-up or were retrospectively assessed from medical files. A seizure frequency reduction of at least 50% was considered as improvement in patients with Ep. For the other clinical phenotypes, the modified Rankin Scale (mRS) score and the Scale for the Assessment and Rating of Ataxia (SARA) score were used,15,16 when available. One-point improvement in the mRS score or 3 points at the SARA score was considered clinically relevant. In the absence of absolute scores, patients' and physicians' evaluations, as measured by the clinical global impression—improvement scale,17 were taken into account.
Serologic response to treatment was measured comparing pre- and post-immunotherapy ELISA antibody concentrations. A reduction of at least 25% following immunotherapy was considered a relevant concentration reduction.
Patients with isolated DM1
To explore the spectrum of concentrations of anti-GAD65 antibodies, we also assessed patients with DM1 without neurologic symptoms. At the Department of Immunology of the Erasmus University Medical Center, sera from 669 patients were tested for anti-GAD65 between January 2018 and December 2018. As the reason for testing was often unknown, filtering based on additional antibody testing for Islet Cell Cytoplasmic Autoantibodies (ICA) or tyrosine phosphatase antibodies (anti-IA2) was performed. This way, we selected samples from 198 patients that were sent specifically for a suspicion of insulin-dependent diabetes mellitus because neurologists would not request additional islet antigen antibodies. Seventy-three samples (37%) tested positive for anti-GAD65. Of these, 37 were analyzed more extensively with quantitative ELISA, IHC, and CBA. Patients with high concentrations and positive IHC and CBA (n = 3) were approached to identify associated neurologic disorders, of whom 2 could be traced.
Statistics
The Fisher exact test was used to compare categorical variables between the high-concentration and low-concentration groups. Age, CSF cell count, and protein count were compared with the Mann-Whitney U test. To compare antibody tests, the McNemar test was used. p Values less than 0.05 were considered significant. We used SPSS 24.0 and Graph Path 7.0 for analysis and data visualization.
Data availability
Any data not published within this article are available at the Erasmus University Medical Center. Patient-related data will be shared on request from any qualified investigator, maintaining anonymization of the individual patients.
Results
Laboratory tests
We identified 71 patients with neurologic symptoms and at least 1 positive serum or CSF anti-GAD65 test result, of whom 56 were included in our study. Eleven patients could not be reached to collect informed consent, and 4 patients refused to participate.
The median serum concentration measured with ELISA was 74,700 IU/mL (n = 54; interquartal range [IQR] 1,350–612,500 IU/mL; range 6–2,130,000 IU/mL), and the median CSF concentration was 2,430 IU/mL (n = 43; IQR 6–8,810 IU/mL; range 0–83,800 IU/mL). Serum and CSF samples were drawn a median of 25 months (IQR 6–69 months) and 19 months (IQR 1–69 months) after symptom onset, respectively.
IHC and CBA showed a high concordance (figure 1). A cutoff of 100 IU/mL showed a 100% concordance among tests in CSF. For serum, with a cutoff value of 10,000 IU/mL, 100% of low-concentration samples had a negative IHC, and 97% of high-concentration samples had a positive IHC. Similarly, 100% of high-concentration samples were positive with CBA, and 96% of low-concentration samples were negative. All CBAs and IHCs corresponded, except 3 diabetes samples with positive CBA and negative IHC. In those 3 samples, ELISA concentrations were between 6,220 and 15,400 IU/mL. Samples with an unspecific neuropil staining on IHC were excluded for calculations. Other antibodies were found in 1 patient in the high anti-GAD65 concentration (anti-gamma-aminobutyric acid B receptor [GABAbR]) and 2 patients with low anti-GAD65 concentration (1 anti-GABAbR and 1 anti-GlyR).
Figure 1. Comparison of laboratory techniques.

ELISA concentrations of serum (A and B) and CSF (A and D) of patients with neurologic disorders in comparison to IHC and CBA. Patients identified with dark gray and yellow squares in A showed a neuropil staining on IHC, instead of the typical GAD pattern. Samples with a neuropil staining and high ELISA concentration had a positive GAD65-CBA result, whereas CBA was negative for low-concentration samples (A). In B and D, dark gray and yellow dots are used in both IHC and CBA columns to identify these samples. In the dark gray dotted patients, a different antibody was found. Serum or CSF of the yellow dotted patients showed a neuropil staining, but no known antibody was found. Serum results from patients with antibody testing for diabetes (DM-1) are shown (C). Logarithmic transformation was used for charts. Concordance rates for ELISA, IHC, and CBA are provided for serum and CSF (E). CBA = cell-based assay; GAD = glutamic acid decarboxylase; IHC = immunohistochemistry.
In the DM1 group without overt neurologic symptoms, the median serum concentration of 68 patients with DM1 (35% of 192 samples) was 86 IU/mL (IQR 51–3,670 IU/mL; range 15–145,400 IU/mL). Three samples (1.6%) showed an anti-GAD65 concentration >10,000 IU/mL and also tested positive by IHC and CBA. The 2 patients traceable had DM1.One also had multiple other autoimmunities (vitiligo, idiopathic thrombocytopenic purpura, and thyroiditis), but in both patients, no neurologic or psychiatric symptoms were present.
Patients
Thirty-six patients were allocated into the high-concentration group (serum concentration >10,000 IU/mL or CSF concentration >100 IU/mL) and 20 patients into the low-concentration group. Clinical characteristics are shown in table 1.
Table 1.
Clinical and paraclinical characteristics of the high-concentration and low-concentration groups

In the high-concentration group, 34/36 (94%) patients had a typical anti–GAD-associated neurologic syndrome: SPS (n = 7), CA (n = 3), Ep (n = 9), LE (n = 9), or an overlap syndrome (n = 6). In the overlap group, 4 patients initially had drug-resistant focal Ep and developed CA, SPS, or a combination of both between 2 and 7 years after seizure onset (figure 2). One patient initially had CA and developed SPS 5 years later, and 1 SPS patient developed prominent cerebellar symptoms 7 years after onset. One patient with LE, no seizures and associated extralimbic involvement in MRI, had concomitant GABAbR antibodies, without a tumor. Of the remaining 2 patients, both with DM type 2, one had a pseudo-orthostatic tremor, and the other one had optic neuropathy.
Figure 2. Patients treated with immunotherapy.

Disease courses and treatment regimens of the 27 patients that were treated with immunotherapy. In patients with overlapping syndromes, the specific syndromes are indicated below the gy bar with the corresponding color at the relative time when they were diagnosed. Symbols inside the color bars show specific treatments or the moment they were initiated. A line with the same color is used for chronic (continuous line) and periodic (discontinuous line) treatments to show their duration. Symbols on the left side correspond to those in Figure 5.
In the low-concentration group, 12/20 patients had an alternative diagnosis, including 1 LE with anti-GABAbR and a pancreatic adenocarcinoma and 1 progressive encephalomyelitis with rigidity and myoclonus with anti-GlyR without an associated tumor, different variants of subacute or chronic polyradiculoneuropathies, and other non–immune-mediated diseases (glioblastoma, hemispastic syndrome after brain surgery, multiple system atrophy, or functional disorder). The remaining 8 patients had chronic Ep, otherwise seronegative LE or nonspecific ataxia and gait disorder (see table e-1, links.lww.com/NXI/A209).
Figure 3 shows patients distributed according to their clinical phenotype and antibody concentration in serum and CSF. The median concentrations of the different clinical phenotypes (in the high-concentration group) were comparable in serum (p = 0.210) and CSF (p = 0.067). No association was found between concentration levels and clinical severity between patients (data not shown).
Figure 3. Antibody concentrations and clinical syndromes.

Patients were grouped according to their clinical phenotype into 1 of the 4 classical anti–GAD65-associated syndrome categories, in the overlap or other category. Serum (A) and CSF (B) ELISA concentrations are shown in a logarithmic scale. Small black dots represent patients with low antibody concentrations. The specific symptoms of these patients are listed in the supplementary material. Orange dots in the SPS column were patients with progressive encephalomyelitis with rigidity and myoclonus, which is considered an extended form of SPS. (C) Serum/CSF ratio of anti-GAD65 ELISA values within the different syndromes in the high-concentration group. The blue ribbon represents the ratio-frame where intrathecal antibody synthesis would be expected to start. CA = cerebellar ataxia; Ep = epilepsy; GAD = glutamic acid decarboxylase; LE = limbic encephalitis; SPS = stiff-person syndrome.
Within the high-concentration group, the median serum/CSF anti-GAD65 ratio was 53 (IQR 171; range 7–1,761, n = 28). After excluding overlaps, there were no significant ratio differences between the clinical syndromes (p = 0.29), although the median ratio of SPS was more than 2-fold that of the other phenotypes (figure 3C).
Twenty-seven patients from the high-concentration group were treated with immunotherapy, of them 26 (96%) received IV immunoglobulins (IVIg) in different treatment regimes. Most used regime was 0.4 g/kg for 5 days, repeated monthly for at least 2 more times, to assess response. Eight patients (30%) were treated with IV methylprednisolone, and 3 patients received plasma exchange. Four patients were additionally treated with second-line immunotherapy, including rituximab (n = 3) and cyclophosphamide (n = 1). Chronic immunosuppression was given in 11 patients, consisting of combinations of azathioprine (n = 9), oral steroids (n = 8), mycophenolate mofetil (n = 5), and cyclosporine (n = 1). Figure 2 shows individual timelines of the high-concentration group patients treated with immunotherapy. Nineteen patients (70%) improved. Eight of 10 patients with SPS (including patients with overlap syndromes) improved according to themselves and their physicians, accompanied by the mRS score decrease in 7 of them (table 2). All patients with CA (including patients with overlap syndromes) except one felt better and obtained a reduction in the mRS score. SARA scores improved ≥3 points in the only 2 patients in whom scores were available. In patients with chronic Ep, >50% seizure frequency reduction was obtained in 4 of 7 patients. Immunotherapy was efficacious in 5 of 7 patients with LE, and 2 of them had relapses despite chronic treatment. Those 2 responded to more intensive therapies. Four typical patient examples are provided in figure 4.
Table 2.
Effects of immunotherapy in patients with high antiglutamic acid decarboxylase 65 concentrations



Figure 4. Disease course and treatment response of 4 patients in the high-concentration group.

Horizontal bars containing treatment abbreviations represent the time periods of the corresponding treatments. Blue vertical lines represent seizure frequency over time. Green discontinuous line connected by green circles shows evolution of serum anti-GAD65 concentration. Thick dark-blue line connected by rhomboids represents functional status over time, according to the visual score at the right most part of the charts. AED = antiepileptic drug; AZA = azathioprine; GAD = glutamic acid decarboxylase; IVIg = IV immunoglobulin; IVMP = IV methylprednisolone; MMF = mycophenolate mofetil; PDN = prednisone; RTX rituximab.
In 17/36 high concentration patients, pre- and post-treatment samples were available for comparison. Antibody concentrations showed a median concentration reduction of 69% (range 27%–99%), and decrease was over 25% in 15 of the 17 serum samples after immunotherapy (figure 5). A clear clinical response was observed in 14 of these 15 treated patients. The 2 patients without consistent decrease in concentration showed no clinical improvement. Both had focal Ep for years.
Figure 5. Antibody concentration response to immunotherapy.

Concentration responses to treatment of the 17 patients of whom pretreatment and posttreatment samples were available. Red lines represent the patients lacking clinical improvement. The blue dashed line in the right graph represent CSF samples obtained pretreatment and at clinical relapse. A reduction of at least 25% following immunotherapy was considered a relevant concentration reduction. GAD = glutamic acid decarboxylase.
In none of the patients, complete recovery was reached. Similarly, in none of the patients, antibodies disappeared in serum. In 6/17 patients with pre- and post-treatment samples, serum concentrations became lower than 10.000 IU/mL. Despite periodic or chronic immunotherapy, a stagnation, both clinically and serologically, was observed after initial improvement. However, after withdrawal or tapering of immunotherapy symptoms deteriorated, syndromes relapsed, or concentrations increased in 6 patients (figure 2 and figure 4). Four patients also had relapses or clinical worsening despite stable chronic or periodic immunotherapy.
Discussion
This study aimed to determine the clinical relevance of GAD65 antibodies in patients with neurologic syndromes and evaluate their responses to immunotherapy. We showed that patients with high anti-GAD65 concentrations presented with a limited number of specific neurologic syndromes, whereas the neurologic symptoms observed in patients with low concentrations were less specific, necessitating additional investigations. The laboratory tests used showed a high concordance and a clear cutoff value for the high and low antibody concentrations. Around two thirds of the patients responded to immunotherapy, although all incompletely. The clinical improvement was reflected in the reduction of anti-GAD65 concentrations in both serum and CSF.
Patients with high antibody concentrations had well-defined clinical phenotypes that have been classically associated with GAD65 antibodies, such as stiff-person syndrome, CA, Ep, or LE. In addition, 1 of 5 patients had overlap syndromes. Remarkably, in these patients, the relapse tended to present with different phenotypes. Another interesting feature was a different, not previously described phenotype, including pseudo-orthostatic tremor and optic neuropathy in 2 patients in the high-concentration group. Unfortunately, these 2 patients were not seen by one of the authors, so diagnosis and absence of more typical symptoms should be taken with caution.
From a different perspective, patients with low antibody concentrations presented with a diversity of neurologic syndromes. Some of them had a phenotype that is similar to the classically anti–GAD65-associated syndromes, which was probably the reason why anti-GAD65 was requested in these patients. However, frequently other diagnoses were identified in the workup or during the follow-up, like neurodegenerative diseases or tumors. In these cases, positive ELISA was not confirmed by IHC or CBA, and concentrations were low and comparable to those seen in samples tested for type 1 diabetes mellitus. In these patients, extensive workup to identify other diseases is warranted, including requesting other antibodies. Anti-GABAbR antibodies were identified in a few patients with high and low concentrations of GAD65 antibodies, in line with previous studies.18 Around 40% of the patients in both high- and low-concentration groups had been diagnosed with type 1 DM before the onset of neurologic symptoms, which is also in line with previous studies.19–22
To define which antibody concentration should be considered high and relevant, we compared 3 different laboratory techniques and found highly concordant results. ELISA concentrations above 10,000 IU/mL for serum and 100 IU/mL for CSF can be detected by IHC and CBA, with comparable results. CBA, as expected, is more specific, especially if a different antibody is concomitantly present in the tested sample (leading to an unspecific IHC staining pattern). Another cutoff value (of 2,000) has been suggested before,13,23 but these studies used a radioimmunoassay with different test calibration, currently not widely used anymore.
The role of GAD65 antibodies in neuroinflammation is debatable and still unknown. An obvious theory would be that antibodies block GAD65, interfering with GABA synthesis and impairing the inhibitory GABAergic circuits. This would result in a hyperexcitability state of the CNS. There are studies supporting this theory, demonstrating a reduced GABA concentration in the CSF and in the cerebral cortex of patients with SPS.24,25 However, GAD65 is located intracellularly and as such not accessible for extracellular antibodies. As an explanation, it has been hypothesized that GAD65 might transiently appear on the cell surface in the synaptic cleft during the process of neurotransmission and exocytosis.26 In rats, in vivo injection of GAD65 antibodies from patients with SPS induced electrophysiologic changes in myelinated neurons, whereas GAD65 antibodies from patients with DM1 did not.27 On the other hand, injection of GAD65-specific T cells in mice caused death even in mice without B cells.28 This suggests that the mechanism might be comparable to paraneoplastic syndromes, in which T cell–mediated responses primarily lead to symptoms.29 Similarly, despite several efforts, there are no convincing successful animal models to support the pathogenicity of GAD65 antibodies.
Moreover, studies report poor responses to immunotherapy targeting different GAD syndromes,6,11,30 as compared to other neurologic disorders caused by antibodies targeted to extracellular neuronal structures, again questioning the direct pathogenic role of anti-GAD65. Although the mechanisms remain questionable, our results revealed that many patients show improvement after immunotherapy (mostly IVIg), coupled to a simultaneous concentration decrease. Many patients improve not immediately after initiating treatments but might need some months of treatment before improvement. This might explain some lack of treatment response from the literature. Repeated doses or combinations of different treatments might be considered before classifying a patient as a nonresponder. Nevertheless, some of the patients do not respond at all to immunotherapy. In addition, it is humbling that even in responders, improvement is generally incomplete, and often patients have stagnation in both the clinical and the serologic response after a few months with chronic immunotherapy. Unfortunately, no patient completely recovered. A possible explanation might be a combination of functional and structural neuronal damage, as different research groups have demonstrated in CA.9,30,31 Immunotherapy helps to restore cell function, but cell death is nonreversible.
Despite clinical stabilization with immunotherapy, treatment intensity reductions or withdrawals frequently result in clinical progression (in CA) or relapses (in SPS, LE, and Ep). A few patients have relapses despite stable chronic immunotherapy. Fortunately, these patients usually respond to more intense treatments.
The limitations of this study are mainly linked to its retrospective design. In addition, not all patients were seen by one of the authors. Accordingly, it was sometimes difficult to assess outcome after treatment. Objective parameters such as mRS score, SARA score, and seizure frequency were used when applicable. As treatments were open label, a placebo effect or regression to the mean could explain part of the results. However, most patients had stable (or slowly progressive) disease for a longer period and showed a serious deviation from the time course before. Similarly, the association with concentrations also suggests some real effects. For future studies, it would be of additional value to study treatment outcome prospectively by using standardized questionnaires, examinations, serologic follow-up, and neuropsychological assessments.
To summarize, in patients with classical syndromes (stiff-person, CA, and encephalitis with seizures), detection of high concentrations of anti-GAD65 is practically diagnostic of an anti–GAD65-related syndrome. However, depending on the syndrome (e.g., LE), physicians should consider the possibility of concurrent antibodies, such as GABAbR. These classical syndromes should be treated with prolonged immunotherapy (as the current study suggests). On the other hand, the presence of atypical syndromes (or nonspecific encephalitic syndromes with seizures) or a typical syndrome but with low-concentration anti-GAD65 should raise concern for another underlying disease (anti-GAD65 as bystanders). In these patients, more extensive investigations for alternative diagnoses should be considered.
Acknowledgment
The authors thank all patients for their participation. They thank all referring physicians. A. Muñoz-Lopetegi received a Research Experience Fellowship grant from the European Academy of Neurology (EAN) for this research project. Authors R.F. Neuteboom, J.M. de Vries, M.W.J. Schreurs, P.A.E. Sillevis Smitt, and M.J. Titulaer of this publication are members of the European Reference Network for Rare Immunodeficiency, Autoinflammatory and Autoimmune Diseases–Project ID No 739543.
Glossary
- CA
cerebellar ataxia
- CBA
cell-based assay
- Ep
epilepsy
- GAD
glutamic acid decarboxylase
- IHC
immunohistochemistry
- IVIg
IV immunoglobulin
- LE
limbic encephalitis
- mRS
modified Rankin Scale
- SARA
Scale for the Assessment and Rating of Ataxia
- SPS
stiff-person syndrome
Appendix. Authors

Study funding
A. Muñoz-Lopetegi was supported by the EAN Experience Fellowship from the European Academy of Neurology. M.J. Titulaer was supported by an Erasmus MC fellowship and has received funding from the Netherlands Organization for Scientific Research (NWO, Veni incentive), from the Dutch Epilepsy Foundation (NEF, project 14-19), and from ZonMw (Memorabel program).
Disclosure
A. Muñoz-Lopetegi, M.A.A.M. de Bruijn, S. Boukhrissi, A.E.M. Bastiaansen, M.M.P. Nagtzaam, E.S.P. Hulsenboom, A.J.W. Boon, R.F. Neuteboom, J.M. de Vries, and M.W.J. Schreurs report no disclosures relevant to the manuscript. P.A.E. Sillevis Smitt holds a patent for the detection of anti-DNER and received research support from Euroimmun. M.J. Titulaer received research funds for serving on a scientific advisory board of MedImmune LLC and a travel grant for lecturing in India from Sun Pharma, India. M.J. Titulaer has filed a patent for methods for typing neurologic disorders and cancer, and devices for use therein, and has received research funds for serving on a scientific advisory board of MedImmune LLC, for consultation at Guidepoint Global LLC, and an unrestricted research grant from Euroimmun AG. Go to Neurology.org/NN for full disclosures.
References
- 1.Solimena M, de Camilli P, Blake R, et al. Autoimmunity to glutamic acid decarboxylase (GAD) in Stiff-Man syndrome and insulin-dependent diabetes mellitus. Trends Neurosci 1991;14:452–457. [DOI] [PubMed] [Google Scholar]
- 2.Sigurdsson E, Baekkeskov S. The 64-kDa beta cell membrane autoantigen and other target molecules of humoral autoimmunity in insulin-dependent diabetes mellitus. Curr Top Microbiol Immunol 1990;164:143–168. [DOI] [PubMed] [Google Scholar]
- 3.Giometto B, Miotto D, Faresin F, Argentiero V, Scaravilli T, Tavolato B. Anti-GABAergic neuron autoantibodies in a patient with stiff-man syndrome and ataxia. J Neurol Sci 1996;143:57–59. [DOI] [PubMed] [Google Scholar]
- 4.Peltola J, Kulmala P, Isojärvi J, et al. Autoantibodies to glutamic acid decarboxylase in patients with therapy-resistant epilepsy. Neurology 2000;55:46–50. [DOI] [PubMed] [Google Scholar]
- 5.Honnorat J, Saiz A, Giometto B, et al. Cerebellar ataxia with anti–glutamic acid decarboxylase antibodies. Arch Neurol 2001;58:225. [DOI] [PubMed] [Google Scholar]
- 6.Malter MP, Helmstaedter C, Urbach H, Vincent A, Bien CG. Antibodies to glutamic acid decarboxylase define a form of limbic encephalitis. Ann Neurol 2010;67:470–478. [DOI] [PubMed] [Google Scholar]
- 7.Mckeon A, Robinson MT, Mcevoy KM, et al. Stiff-man syndrome and variants clinical course, treatments, and outcomes. Arch Neurol 2012;69:230–238. [DOI] [PubMed] [Google Scholar]
- 8.Martinez-Hernandez E, Ariño H, McKeon A, et al. Clinical and immunologic investigations in patients with stiff-person spectrum disorder. JAMA Neurol 2016;73:714–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ariño H, Gresa-Arribas N, Blanco Y, et al. Cerebellar ataxia and glutamic acid decarboxylase antibodies: immunologic profile and long-term effect of immunotherapy. JAMA Neurol 2014;71:1009–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dalakas MC, Rakocevic G, Dambrosia JM, Alexopoulos H, McElroy B. A double-blind, placebo-controlled study of rituximab in patients with stiff person syndrome. Ann Neurol 2017;82:217–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malter MP, Frisch C, Zeitler H, et al. Treatment of immune-mediated temporal lobe epilepsy with GAD antibodies. Seizure 2015;30:57–63. [DOI] [PubMed] [Google Scholar]
- 12.Baekkeskov S, Aanstoot H, Christgau S, et al. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature 1900;347:151–156. [DOI] [PubMed] [Google Scholar]
- 13.Saiz A, Arpa J, Sagasta A, et al. Autoantibodies to glutamic acid decarboxylase in three patients with cerebellar ataxia, late-onset insulin-dependent diabetes mellitus, and polyendocrine autoimmunity. Neurology 1997;49:1026–1030. [DOI] [PubMed] [Google Scholar]
- 14.Saiz A, Blanco Y, Sabater L, et al. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: diagnostic clues for this association. Brain 2008;131:2553–2563. [DOI] [PubMed] [Google Scholar]
- 15.Quinn TJ, Dawson J, Walters MR, Lees KR. Functional outcome measures in contemporary stroke trials. Int J Stroke 2009;4:200–205. [DOI] [PubMed] [Google Scholar]
- 16.Weyer A, Abele M, Schmitz-Hübsch T, et al. Reliability and validity of the scale for the assessment and rating of ataxia: a study in 64 ataxia patients. Mov Disord 2007;22:1633–1637. [DOI] [PubMed] [Google Scholar]
- 17.Guy W. ECDEU Assessment Manual for Psychopharmacology. Rockville: US Department of Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, National Institute of Mental Health, Psychopharmacology Research Branch, Division of Extramural Research Programs; 1976. [Google Scholar]
- 18.Höftberger R, Titulaer MJ, Sabater L, et al. Encephalitis and GABA B receptor antibodies. Novel findings in a new case series of 20 patients. Neurology 2013;81:1500–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Solimena M, Folli F, Aparisi R, Pozza G, De Camilli P. Autoantibodies to GABA-ergic neurons and pancreatic beta cells in Stiff-Man syndrome. N Engl J Med 1990;322:1555–1560. [DOI] [PubMed] [Google Scholar]
- 20.Dalakas MC, Fujii M, Li M, McElroy B. The clinical spectrum of anti-GAD antibody-positive patients with stiff-person syndrome. Neurology 2000;55:1531–1535. [DOI] [PubMed] [Google Scholar]
- 21.Meinck HM, Thompson PD. Stiff man syndrome and related conditions. Mov Disord 2002;17:853–866. [DOI] [PubMed] [Google Scholar]
- 22.Rakocevic G, Raju R, Dalakas MC. Anti–glutamic acid decarboxylase antibodies in the serum and cerebrospinal fluid of patients with stiff-person syndrome. Arch Neurol 2004;61:902. [DOI] [PubMed] [Google Scholar]
- 23.Pittock SJ, Yoshikawa H, Ahlskog JE, et al. Glutamic acid decarboxylase autoimmunity with brainstem, extrapyramidal, and spinal cord dysfunction. Mayo Clin Proc 2006;81:1207–1214. [DOI] [PubMed] [Google Scholar]
- 24.Dalakas MC, Li M, Fujii M, Jacobowitz DM. Stiff person syndrome: quantification, specificity, and intrathecal synthesis of GAD65 antibodies. Neurology 2001;57:780–784. [DOI] [PubMed] [Google Scholar]
- 25.Levy LM, Levy-Reis I, Fujii M, Dalakas MC. Brain γ-aminobutyric acid changes in stiff-person syndrome. Arch Neurol 2005;62:970–974. [DOI] [PubMed] [Google Scholar]
- 26.Dalakas MC. Progress and stiff challenges in understanding the role of GAD-antibodies in stiff-person syndrome. Exp Neurol 2013;247:303–307. [DOI] [PubMed] [Google Scholar]
- 27.Manto M-U, Laute M-A, Aguera M, Rogemond V, Pandolfo M, Honnorat J. Effects of anti-glutamic acid decarboxylase antibodies associated with neurological diseases. Ann Neurol 2007;61:544–551. [DOI] [PubMed] [Google Scholar]
- 28.Burton AR, Baquet Z, Eisenbarth GS, et al. Central nervous system destruction mediated by glutamic acid decarboxylase-specific CD4+ T cells. J Immunol 2010;184:4863–4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bien CG, Vincent A, Barnett MH, et al. Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain 2012;135:1622–1638. [DOI] [PubMed] [Google Scholar]
- 30.Mitoma H, Hadjivassiliou M, Honnorat J. Guidelines for treatment of immune-mediated cerebellar ataxias. Cerebellum Ataxias 2015;2:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mitoma H, Manto M, Hampe CS. Immune-mediated cerebellar ataxias: from bench to bedside. Cerebellum Ataxias 2017;4:2–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Any data not published within this article are available at the Erasmus University Medical Center. Patient-related data will be shared on request from any qualified investigator, maintaining anonymization of the individual patients.