

Abstract
Objective
To investigate the possible human leukocyte antigen (HLA) association of both myelin oligodendrocyte glycoprotein (MOG-IgG)-associated diseases (MOGAD) and aquaporin-4 antibody (AQP4-IgG)-positive neuromyelitis optica spectrum disorders (NMOSDs) in the Dutch population with European ancestry to clarify similarities or differences in the immunogenetic background of both diseases.
Methods
Blood samples from patients in the Dutch national MS/NMOSD expert clinic were tested for MOG-IgG and AQP4-IgG using a cell-based assay. HLA Class I and II genotyping was performed in 43 MOG-IgG–seropositive and 42 AQP4-IgG–seropositive Dutch patients with European ancestry and compared with those of 5,604 Dutch healthy blood donors.
Results
No significant HLA association was found in MOG-IgG–seropositive patients. The AQP4-IgG–seropositive patients had a significant higher frequency of HLA-A*01 (61.9% vs 33.7%, OR 3.16, 95% CI, 1.707–5.863, p after correction [pc] = 0.0045), HLA-B*08 (61.9% vs 25.6%, OR 4.66, 95% CI, 2.513–8.643, pc < 0.0001), and HLA-DRB1*03 (51.2% vs 27.6%, OR 2.75, 95% CI, 1.495–5.042, pc = 0.0199) compared with controls.
Conclusions
The present study demonstrates differences in the immunogenetic background of MOGAD and AQP4-IgG–positive NMOSD. The strong positive association with HLA-A*01, -B*08, and -DRB1*03 is suggestive of a role of this haplotype in the etiology of AQP4-IgG–positive NMOSD in patients with European ancestry, whereas in MOGAD no evidence was found for any HLA association in these disorders.
Myelin oligodendrocyte glycoprotein antibodies (MOG-IgG) are related to a spectrum of demyelinating syndromes of the CNS, both in adult and pediatric patients. These are currently known as MOG-IgG–associated diseases (MOGADs) and predominantly include acute disseminated encephalomyelitis (ADEM), optic neuritis (ON), and/or transverse myelitis (TM).1–4 A subset of these patients seropositive for MOG-IgG fulfill the criteria for neuromyelitis optica spectrum disorder (NMOSD),5 whereas the majority of NMOSDs are associated with aquaporin-4 antibodies (AQP4-IgG).6–9 This partial clinical overlap between patients seropositive for MOG-IgG and AQP4-IgG is interesting because the underlying disease mechanism is thought to be different, with autoantibodies targeting oligodendrocytes and astrocytes, respectively.10–12 The existence of these IgG autoantibodies, however, is indicative for a key role for B and CD4+ T cells in the disease process of both disorders and, consequently, for a potential role for human leukocyte antigen (HLA).
Possible HLA associations could further elucidate similarity or difference between MOG-IgG– and AQP4-IgG–related disorders based on their immunogenetic background. Previous studies have described different HLA associations in NMOSD including subgroups of AQP4-IgG–seropositive patients. Of interest, so far, a possible HLA association has never been studied in MOGAD. Furthermore, HLA associations vary between ethnic backgrounds, and the Dutch AQP4-IgG-seropositive patients with European ancestry have never been studied before. The aim of this study was to investigate the possible HLA association of both MOGAD and AQP4-IgG–positive NMOSD in the Dutch population with European ancestry to clarify the contribution of HLA to the pathophysiology in both of these diseases.
Methods
Study participants and definitions
This study was conducted at the Dutch national center for acquired demyelinating syndromes, which includes the NMOSD expert clinic (Erasmus MC) and Pediatric MS center (Erasmus MC-Sophia Children's Hospital) in Rotterdam, the Netherlands, and Sanquin Diagnostic Services in Amsterdam, the Netherlands. Serum samples from all patients who either presented primarily at our hospitals or were referred by other (non)academic hospitals were sent in for routine MOG-IgG and AQP4-IgG diagnostics as part of standard clinical care. Patients from all ages were included if they were (1) seropositive for either MOG-IgG or AQP4-IgG and (2) of European descent. Included patients presented with a demyelinating event within the acquired demyelinating syndromes spectrum, i.e., ON and/or (longitudinally extensive) transverse myelitis ([LE]TM), area postrema syndrome, or ADEM. Diagnosis at last follow-up was determined as NMOSD if fulfilling the Wingerchuk 2015 diagnostic criteria.5 Patients who had 1 or more ON following initial presentation with ADEM were diagnosed with ADEM-ON.13 Patients with a possible diagnosis of MS (clinically and radiologically) were excluded.
HLA allele frequencies of MOG-IgG– and AQP4-IgG–seropositive patients were compared with the HLA allele frequencies of 5,604 healthy Dutch blood donors from the Leiden area in the Netherlands.
Standard protocol approvals, registrations, and patient consents
The Medical Ethical Committee of Erasmus Medical Center in Rotterdam approved this study. All patients signed informed consent to store their blood samples and clinical characteristics in our Biobank.
Cell culture and cell-based assays
All serum samples were tested blindly, centrally, and in duplicate at Sanquin Diagnostics with the cell-based assay (CBA) as previously described.1,8 Briefly, patient serum was incubated with HEK293 cells transiently transfected with AQP4-M23 (final serum dilution 1:20) or LN18 cells stably transfected with full-length MOG (final serum dilution 1:200). After washing, cells were incubated with goat anti-human IgG allophycocyanin-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, Brunschwig Chemie B.V., Amsterdam, the Netherlands [specific for human IgG]) and analyzed using fluorescence-activated cell sorter. The cutoff was determined in every assay as average Delta MFI +10 SDs of 8 individual sera of negative controls. Our assay comprises an anti–IgG-specific detection antibody, and thus, no IgM anti-MOG or IgM anti-AQP4 is detected.
HLA analysis
HLA Class I and II genotyping was performed at the National Reference Center for Histocompatibility Testing at the Department of Immunohaematology and Blood Transfusion, Leiden University Medical Center, the Netherlands. DNA for HLA analysis was isolated from peripheral blood samples. HLA Class I typing (HLA-A*, -B*, -Cw*) was performed by the reverse sequence–specific oligonucleotide (SSO) method on a suspension array platform using microspheres as a solid support to immobilize oligonucleotide probes (Luminex bead technology; Immucor Transplant Diagnostics, Stamford, CT). HLA Class II (HLA-DRB1*, DQB1*, DRB3*/4*/5*) was genotyped using the PCR SSO probe technique,14 which results in medium-low resolution subtyping.
Statistical analysis
For patient characteristic group comparisons, the χ2 or Fisher exact test and the Mann-Whitney U test were used when appropriate. HLA allele frequencies were analyzed using the Fisher exact probability test. p Values were corrected with Sidak method for multiple testing. A corrected p value (pc) below 0.05 was considered significant. ORs were calculated using the Woolf method with Haldane modifications (Woolf-Haldane).
Data availability
The raw data used in this article are available at the Erasmus University Medical Center and could be shared by request from a qualified investigator, maintaining anonymization of the individual patients.
Results
In total, 85 Dutch patients with European ancestry were analyzed, including 43 MOG-IgG– and 42 AQP4-IgG–seropositive patients. Patient characteristics are shown in table 1.
Table 1.
Patient characteristics of MOG-IgG– and AQP4-IgG–seropositive patients

Patient characteristics of MOG-IgG–seropositive patients
MOG-IgG–seropositive patients were predominantly male (63%), and almost half of the MOG-IgG–seropositive patients (49%) had their onset of disease during childhood (<18 years) (table 1). Table 1 also gives a separate overview of included children and adults. Except for age at onset of disease, patient characteristics were not significantly different between children and adults.
As described above, the clinical phenotype of MOGAD consists of a spectrum of demyelinating disorders. In total, 12 MOG-IgG–seropositive patients (28%) presented with ON, of which 42% were bilateral, and 10 (23%) with TM, of which 60% showed an longitudinally extensive lesion on MRI. In addition, of the 7 patients (16%) presenting with a combination of ON and TM, 71% had a bilateral ON, and 67% an LETM on MRI.
Figure 1 shows an overview of diagnosis at last follow-up of the included MOG-IgG–seropositive patients, separated for children and adults. Within a median follow-up of 55 months (range: 1–463 months), in total, 28% of MOG-IgG–seropositive patients (including 4 children and 8 adults) were finally diagnosed with a combination of ON and MT. Of these, only 6 patients fulfilled the Wingerchuk 2015 criteria for NMOSD diagnosis (50%). Only pediatric patients were diagnosed with ADEM (62% of children vs 0% of adults, p < 0.001). Adults were more often diagnosed with a TM compared with pediatric patients (5% of children vs 32% of adults, p = 0.046).
Figure 1. Diagnosis at last follow-up of included MOG-IgG–seropositive children and adults.
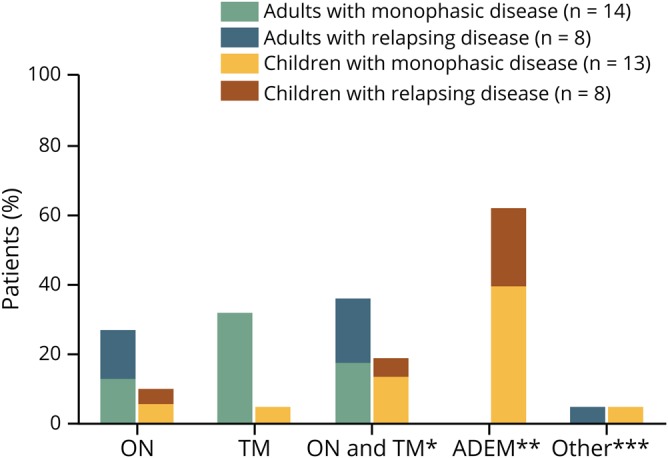
*6/12 patients fulfilling Wingerchuk 2015 diagnostic criteria for NMOSD. **Relapsing ADEM group only consists of patients with ADEM-ON (ADEM followed by (recurrent) optic neuritis). ***Including 1 adult with a relapsing encephalitis (double syndrome MOG-IgG/NMDA-R) and 1 child with a progressive ‘leukodystrophy-like’ disease. ADEM = acute disseminated encephalomyelitis; MOG-IgG = myelin oligodendrocyte glycoprotein antibody; MOGAD = MOG-IgG-associated disease; ON = optic neuritis; TM = transverse myelitis.
Furthermore, among the MOG-IgG–seropositive patients, 1 patient was diagnosed with a progressive leukodystrophy-like disorder with a severe regression of cognitive functions (figure 1). This patient deceased 6 years after the start of first symptoms. Another MOG-IgG–seropositive patient presented with a recurrent encephalitis. In this patient, NMDA receptor (NMDAR) antibodies were found additionally to the positive MOG-IgG. This patient was diagnosed with a double syndrome (figure 1) because besides typical anti-NMDAR encephalitis features, this patients also had clinical and radiologic findings compatible with a demyelinating disorder, including diplopia, visual impairment, urge incontinence, and demyelinating lesions on MRI.
Finally, 1 MOG-IgG–seropositive patient diagnosed with monophasic ADEM also had transient AQP4-IgG; he twice had a weak positive CBA test result for AQP4-IgG, i.e., during onset of disease and in the first follow-up sample 5 years later. In the following sample, 1 year later, he became AQP4-IgG seronegative. Because the MOG-IgG remained detectable and because of his clinical phenotype, which was more compatible with MOGAD than AQP4-IgG–seropositive NMOSD, this patient was only included in the MOG-IgG–seropositive patient group.
Patient characteristics AQP4-IgG–seropositive patients
The majority of AQP4-IgG–seropositive patients were female (86%), and 67% of patients had a relapsing disease course (table 1). Within a median follow-up of 65 months (range: 6–312 months), all AQP4-IgG–seropositive patients were diagnosed with NMOSD (all fulfilling the Wingerchuk 2015 diagnostic criteria).
HLA association in MOG-IgG–seropositive patients
In MOG-IgG–seropositive patients, a lower proportion of HLA-A*68 (0.0% vs 9.2%, OR 0.11, 95% CI, 0.007–1.838, p = 0.0299) and a higher proportion of HLA-B*08 (41.9% vs 25.6%, OR 2.11, 95% CI, 1.154–3.842, p = 0.0217) were observed compared with controls. However, these results did not remain significant after statistical correction for multiple testing (table 2). No significant HLA-A, -B, -C, -DR, and -DQ associations were found in MOG-IgG–seropositive patients (table e-1, links.lww.com/NXI/A214).
Table 2.
HLA allele frequencies in MOG-IgG–seropositive patients and controlsa

Because of the heterogeneity in the MOG-IgG–seropositive patient group, different subanalyses were performed. A subanalysis with exclusion of MOG-IgG–seropositive patients with above described rare MOGAD presentations (n = 3; progressive leukodystrophy-like disorder, double syndrome with NMDAR encephalitis, and ADEM with coexisting transient weak positive AQP4-IgG) showed comparable results, with, in addition, an increased frequency of HLA-DRB1*15 (40.0% vs 24.2%, OR 2.11, 95% CI, 1.127–3.949, p = 0.0260). However, this association did not remain significant after statistical correction for multiple testing.
A subanalysis on age group, comparing MOG-IgG–seropositive children (n = 21) with MOG-IgG–seropositive adults (n = 22), showed no significant differences nor tendencies.
Further subanalyses in MOG-IgG–seropositive patients on disease onset phenotype (opticospinal, n = 28 vs non-opticospinal, n = 15), disease course (monophasic, n = 27 vs multiphasic, n = 16), and final diagnosis (ADEM, n = 13 vs non-ADEM, n = 30) showed no significant differences nor tendencies for the HLA-A*01, -B*08, -DRB1*03 haplotype, which is the most relevant haplotype in the European population (see below).
HLA association in AQP4-IgG–seropositive patients
Table 3 shows significant HLA associations in the AQP4-IgG–seropositive patients compared with controls. After correction for multiple testing, a strong positive association for HLA-A*01 (61.9% vs 33.7%, OR 3.16, 95% CI, 1.707–5.863, pc = 0.0045), HLA-B*08 (61.9% vs 25.6%, OR 4.66, 95% CI, 2.513–8.643, pc < 0.0001), and HLA-DRB1*03 (51.2% vs 27.6%, OR 2.75, 95% CI, 1.495–5.042, pc = 0.0199) remained significant (figure 2), suggestive of a role of this conserved HLA-A*01, -B*08, -DRB1*03 haplotype. In 16/42 (38%) of patients, this haplotype was present. All HLA allelic frequencies of AQP4-IgG–seropositive patients are displayed in table e-2 (links.lww.com/NXI/A214).
Table 3.
HLA allele frequencies in AQP4-IgG–seropositive patients and controlsa

Figure 2. Forest plot of significant HLA associations in AQP4-IgG–seropositive patients*.

*After correction for multiple testing (Sidak method).
Discussion
We found clear differences in HLA Class I and II alleles in Dutch MOG-IgG– and AQP4-IgG–seropositive patients with European ancestry. In AQP4-IgG–seropositive patients, we found a strong positive association with HLA-A*01, -B*08, and -DRB1*03. In contrast, we did not find any evidence for a particular HLA association in MOG-IgG–seropositive patients.
These findings are relevant for a further distinction between MOGAD from AQP4-IgG–positive NMOSD. Where MOG-IgG was first found to represent a subset of patients with AQP4-IgG–negative NMOSD, nowadays MOG-IgG is consistently identified in a broad spectrum of acquired demyelinating syndromes, which extend beyond the NMOSD phenotype.1–4,12 Previous studies have identified some clinical and imaging features that can distinguish MOGAD from AQP4-IgG–positive NMOSD; however, in several patients, the clinical and radiologic phenotype still overlaps.12,15–18 Nevertheless, the immunopathogenic differences in these diseases, i.e., an astrocytopathy in AQP4-IgG–positive NMOSD and an oligodendrocytopathy in MOGAD, suggest 2 distinctive autoimmune disorders.12 The present findings further support these differences on the basis of the immunogenetic features and therefore once again emphasize the probable pathophysiologic difference between MOG-IgG– and AQP4-IgG–related diseases.
Although HLA associations in AQP4-IgG–mediated NMOSD have previously been studied in a few ethnic populations worldwide, this study was the first investigating the Dutch population with European ancestry. Our finding of HLA-DRB1*03 as risk allele in AQP4-IgG–mediated disorders confirms and extends the results previously obtained in patients with NMOSD from different non-Asian populations.19–24 Also, the recent large genome-wide association study from Estrada et al.25 found HLA-DRB1*03:01 as the most significant HLA allele in patients with NMOSD from Texas compared with controls of European ancestry, which was driven by the presence of AQP4-IgG. HLA-DRB1*03 has not been found as risk allele in 2 studies with European26 and Arabic27 patients with NMOSD. However, these 2 studies had smaller proportions of AQP4-IgG–seropositive patients among their included patients with NMOSD (respectively, 61% and 49%, compared with 73–100% in the other mentioned cohorts), what could explain the absence of a significant association with HLA-DRB1*03. An association with HLA-DPB1*0501 has previously been described only in Asian patients with AQP4-IgG–positive NMOSD28–30 and was not found in our Dutch patients with European ancestry.
In addition, we found a strong correlation with HLA-A*01 (OR 3.16) and HLA-B*08 (OR 4.66): 2 HLA Class I genes. These associations have never been described in the previous literature, possibly as most previous studies only focused on the HLA Class II alleles. AQP4-IgG has been established as pathogenic autoantigen, directly targeting the AQP4 water channel on the foot of the astrocytes and causing complement-mediated necrosis, secondary demyelination, inflammation, and subsequently neuronal death.9,10,31 Therefore, the expectation of HLA Class II associations in this presumed primary B cell–mediated disorder is imaginable. However, also HLA Class I genes can play a key or additional role in initiating or maintaining autoimmune responses, primarily by triggering a CD8+ cytotoxic T-cell response.32 Our found associations with HLA Class I alleles could indicate involvement of such T-cell responses in the pathogenesis of AQP4-IgG–mediated NMOSD. Nonetheless, HLA-A*01 and HLA-B*08 are also known to form an ancestral European haplotype together with (among others) HLA-DRB1*03 due to linkage.33 Whether there is an independent contribution of HLA-A*01 and HLA-B*08 in AQP4-IgG–mediated NMOSD or whether these HLA Class I associations are only a result of linkage with DRB1*03 remains to be established.
Of interest, this ancestral European haplotype including HLA-DRB1*03 has also been linked to several other autoimmune diseases, such as SLE, Sjögren syndrome, myasthenia gravis, Graves disease, and celiac disease.32,33 This is in accordance with the clinical observation in AQP4-IgG–seropositive patients that around 25% of these patients have coexisting autoimmune disorders as listed above.34–38 In our AQP4-IgG–seropositive patients 19% also had an accompanying autoimmune disease.
To our best knowledge, this was the first study worldwide investigating HLA associations in MOGAD. Of interest, we found no evidence for particular HLA associations in our MOG-IgG–seropositive patients. This finding is striking because one could expect a contribution of HLA in the pathophysiology of MOGAD due to the presence of the autoantibodies. However, in contrast to AQP4-IgG, the pathogenicity of MOG-IgG has not fully been elucidated.15,39 MOG-IgG targets the CNS-specific MOG protein, which is expressed on the outer surface of the myelin sheaths and plasma membrane of oligodendrocytes.4,40,41 Although the MOG protein only represents a minor component of myelin (0.05%),41 its location makes it highly immunogenic4 and is an imaginable target for autoantibodies in a demyelinating disease. However, although in vitro studies have demonstrated that human MOG-IgG activates complement and cellular-dependent cytotoxicity, in vivo rodent models could not confirm the human MOG-IgG to cause demyelination in these animals.15,42–45 Nevertheless, there are various possible explanations for the human MOG-IgG not being pathogenic in rodents, most importantly because human MOG-IgG simply does not recognize the rodent MOG protein.39 Additionally to these immunologic studies, histopathologic findings from MOG-IgG–seropositive patients show activated complement at sites of ongoing demyelinating.39,46,47 This again supports a humoral immune pathogenesis and a potential pathogenic activity of human MOG-IgG.
Importantly, our negative result for HLA associations in MOGAD does not directly reject the assumption that these disorders are primary B cell-mediated due to the potential T cell-independent B cell activation by antibody-inducing signals from, for example, dendritic cells, macrophages, and granulocytes.48 An alternative explanation may be that many different HLA Class II/peptide complexes are able to trigger CD4+ T cells to provide help to the B cells resulting in IgG antibodies in all patients irrespective of their HLA-DR type.
One could hypothesize that the distinctive clinical phenotypes within the MOGAD could have a different underlying pathogenesis with potentially a different immunogenetic background. The broad spectrum of MOGAD is also variable with age at onset. In children, ADEM is more prevalent compared with adults. In adults, most patients have ON, (LE)TM, or both (either simultaneously or subsequently).49 Despite these apparent phenotypical differences, we did not find differences in HLA allele frequencies when comparing subgroups regarding age (child vs adult), disease course (mono- vs multiphasic), and clinical presentation (opticospinal vs non-opticospinal and ADEM vs non-ADEM), neither after exclusion of rare MOGAD presentations. This again supports our main finding regarding the absence of HLA association in MOGAD, with also the absence of HLA association in distinctive MOGAD subgroups.
In general, confounding due to linkage with neighboring HLA and non-HLA genes is a possible limitation when studying HLA associations. Furthermore, as described before, we should be reminiscent of the ethnic variation of HLA associations within the same disease. We therefore only included patients with European ancestry in our study. A specific limitation of our study is the relatively small sample size, which is inherent to the rarity of both antibody-mediated diseases. However, our AQP4-IgG–seropositive patient group was very homogeneous, and therefore, we do not expect that the relatively low number has influenced the results in this patient group. In contrast, our MOG-IgG–seropositive patient group was more heterogeneous regarding clinical phenotype and age at onset, as a result of the broad spectrum in MOGAD. Because of the small sample size, only limited subanalyses were possible to overcome this issue.
In conclusion, we show no evidence for HLA association in MOGAD in the Dutch population with European ancestry. Whereas in AQP4-IgG–seropositive patients, the newly found association with HLA-A*01 and HLA-B*08, together with the confirmation of the earlier described association with HLA-DRB1*03, is suggestive of a role of this haplotype in the etiology of these AQP4-IgG–mediated diseases in patients with European ancestry. These findings are suggestive of a different immunogenetic background and therefore once again emphasize MOGAD and AQP4-IgG–positive NMOSD to be distinctive demyelinating disorders of the CNS.
Similar studies need to be conducted in MOGAD, preferably with international collaboration for larger sample sizes. Furthermore, it would be interesting to investigate the independent risk of the found HLA Class I and II alleles to AQP4-IgG–positive NMOSD.
Acknowledgment
The authors thank the patients and their families for their participation in this study. Rogier Q. Hintzen is deceased.
Glossary
- ADEM
acute disseminated encephalomyelitis
- AQP4
aquaporin-4
- CBA
cell-based assay
- HLA
human leukocyte antigen
- LETM
longitudinally extensive transverse myelitis
- MOG
myelin oligodendrocyte glycoprotein
- MOGAD
myelin oligodendrocyte glycoprotein–associated disease
- NMDAR
NMDA receptor
- NMOSD
neuromyelitis optica spectrum disorder
- ON
optic neuritis
- pc
p after correction
- SSO
sequence-specific oligonucleotide
- TM
transverse myelitis
Appendix. Authors
Study funding
The study was supported by the Dutch MS research Foundation. This study was not industry sponsored.
Disclosure
A.L. Bruijstens, Y.Y.M. Wong, D.E. van Pelt, P.J.E. van der Linden, G.W. Haasnoot, R.Q. Hintzen, F.H.J. Claas, R.F. Neuteboom, and B.H.A. Wokke report no disclosures relevant to the manuscript. Go to Neurology.org/NN for full disclosures.
References
- 1.Ketelslegers IA, Van Pelt DE, Bryde S, et al. . Anti-MOG antibodies plead against MS diagnosis in an Acquired Demyelinating Syndromes cohort. Mult Scler 2015;21:1513–1520. [DOI] [PubMed] [Google Scholar]
- 2.Rostasy K, Mader S, Schanda K, et al. . Anti-myelin oligodendrocyte glycoprotein antibodies in pediatric patients with optic neuritis. Arch Neurol 2012;69:752–756. [DOI] [PubMed] [Google Scholar]
- 3.de Mol CL, Wong Y, van Pelt ED, et al. . The clinical spectrum and incidence of anti-MOG-associated acquired demyelinating syndromes in children and adults. Mult Scler Epub 2019 May 16. [DOI] [PMC free article] [PubMed]
- 4.Reindl M, Di Pauli F, Rostasy K, Berger T. The spectrum of MOG autoantibody-associated demyelinating diseases. Nat Rev Neurol 2013;9:455–461. [DOI] [PubMed] [Google Scholar]
- 5.Wingerchuk DM, Banwell B, Bennett JL, et al. . International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015;85:177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lennon VA, Wingerchuk DM, Kryzer TJ, et al. . A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004;364:2106–2112. [DOI] [PubMed] [Google Scholar]
- 7.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 2005;202:473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ketelslegers IA, Modderman PW, Vennegoor A, Killestein J, Hamann D, Hintzen RQ. Antibodies against aquaporin-4 in neuromyelitis optica: distinction between recurrent and monophasic patients. Mult Scler 2011;17:1527–1530. [DOI] [PubMed] [Google Scholar]
- 9.Jarius S, Wildemann B. AQP4 antibodies in neuromyelitis optica: diagnostic and pathogenetic relevance. Nat Rev Neurol 2010;6:383–392. [DOI] [PubMed] [Google Scholar]
- 10.Parratt JD, Prineas JW. Neuromyelitis optica: a demyelinating disease characterized by acute destruction and regeneration of perivascular astrocytes. Mult Scler 2010;16:1156–1172. [DOI] [PubMed] [Google Scholar]
- 11.Fang L, Kang X, Wang Z, et al. . Myelin oligodendrocyte glycoprotein-IgG contributes to oligodendrocytopathy in the presence of complement, distinct from astrocytopathy induced by AQP4-IgG. Neurosci Bull 2019;35:853–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pittock SJ, Lucchinetti CF. Neuromyelitis optica and the evolving spectrum of autoimmune aquaporin-4 channelopathies: a decade later. Ann N Y Acad Sci 2016;1366:20–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong YYM, Hacohen Y, Armangue T, et al. . Paediatric acute disseminated encephalomyelitis followed by optic neuritis: disease course, treatment response and outcome. Eur J Neurol 2018;25:782–786. [DOI] [PubMed] [Google Scholar]
- 14.Verduyn W, Doxiadis II, Anholts J, et al. . Biotinylated DRB sequence-specific oligonucleotides. Comparison to serologic HLA-DR typing of organ donors in eurotransplant. Hum Immunol 1993;37:59–67. [DOI] [PubMed] [Google Scholar]
- 15.Reindl M, Waters P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat Rev Neurol 2019;15:89–102. [DOI] [PubMed] [Google Scholar]
- 16.van Pelt ED, Wong YY, Ketelslegers IA, Hamann D, Hintzen RQ. Neuromyelitis optica spectrum disorders: comparison of clinical and magnetic resonance imaging characteristics of AQP4-IgG versus MOG-IgG seropositive cases in The Netherlands. Eur J Neurol 2016;23:580–587. [DOI] [PubMed] [Google Scholar]
- 17.Sato DK, Callegaro D, Lana-Peixoto MA, et al. . Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology 2014;82:474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitley J, Woodhall M, Waters P, et al. . Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology 2012;79:1273–1277. [DOI] [PubMed] [Google Scholar]
- 19.Blanco Y, Ercilla-Gonzalez G, Llufriu S, et al. . [HLA-DRB1 typing in Caucasians patients with neuromyelitis optica] HLA-DRB1 en pacientes caucasicos con neuromielitis optica. Rev Neurol 2011;53:146–152. [PubMed] [Google Scholar]
- 20.Zephir H, Fajardy I, Outteryck O, et al. . Is neuromyelitis optica associated with human leukocyte antigen?. Mult Scler 2009;15:571–579. [DOI] [PubMed] [Google Scholar]
- 21.Deschamps R, Paturel L, Jeannin S, et al. . Different HLA class II (DRB1 and DQB1) alleles determine either susceptibility or resistance to NMO and multiple sclerosis among the French Afro-Caribbean population. Mult Scler 2011;17:24–31. [DOI] [PubMed] [Google Scholar]
- 22.Brum DG, Barreira AA, dos Santos AC, et al. . HLA-DRB association in neuromyelitis optica is different from that observed in multiple sclerosis. Mult Scler 2010;16:21–29. [DOI] [PubMed] [Google Scholar]
- 23.Alvarenga MP, Fernandez O, Leyva L, et al. . The HLA DRB1*03:01 allele is associated with NMO regardless of the NMO-IgG status in Brazilian patients from Rio de Janeiro. J Neuroimmunol 2017;310:1–7. [DOI] [PubMed] [Google Scholar]
- 24.Alonso VR, de Jesus Flores Rivera J, Garci YR, et al. . Neuromyelitis optica (NMO IgG+) and genetic susceptibility, potential ethnic influences. Cent Nerv Syst Agents Med Chem 2018;18:4–7. [DOI] [PubMed] [Google Scholar]
- 25.Estrada K, Whelan CW, Zhao F, et al. . A whole-genome sequence study identifies genetic risk factors for neuromyelitis optica. Nat Commun 2018;9:1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Asgari N, Nielsen C, Stenager E, Kyvik KO, Lillevang ST. HLA, PTPN22 and PD-1 associations as markers of autoimmunity in neuromyelitis optica. Mult Scler 2012;18:23–30. [DOI] [PubMed] [Google Scholar]
- 27.Brill L, Mandel M, Karussis D, et al. . Increased occurrence of anti-AQP4 seropositivity and unique HLA Class II associations with neuromyelitis optica (NMO), among Muslim Arabs in Israel. J Neuroimmunol 2016;293:65–70. [DOI] [PubMed] [Google Scholar]
- 28.Matsushita T, Matsuoka T, Isobe N, et al. . Association of the HLA-DPB1*0501 allele with anti-aquaporin-4 antibody positivity in Japanese patients with idiopathic central nervous system demyelinating disorders. Tissue Antigens 2009;73:171–176. [DOI] [PubMed] [Google Scholar]
- 29.Wang H, Dai Y, Qiu W, et al. . HLA-DPB1 0501 is associated with susceptibility to anti-aquaporin-4 antibodies positive neuromyelitis optica in southern Han Chinese. J Neuroimmunol 2011;233:181–184. [DOI] [PubMed] [Google Scholar]
- 30.Yoshimura S, Isobe N, Matsushita T, et al. . Distinct genetic and infectious profiles in Japanese neuromyelitis optica patients according to anti-aquaporin 4 antibody status. J Neurol Neurosurg Psychiatry 2013;84:29–34. [DOI] [PubMed] [Google Scholar]
- 31.Lucchinetti CF, Guo Y, Popescu BF, Fujihara K, Itoyama Y, Misu T. The pathology of an autoimmune astrocytopathy: lessons learned from neuromyelitis optica. Brain Pathol 2014;24:83–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gough SC, Simmonds MJ. The HLA region and autoimmune disease: associations and mechanisms of action. Curr Genomics 2007;8:453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gambino CM, Aiello A, Accardi G, Caruso C, Candore G. Autoimmune diseases and 8.1 ancestral haplotype: an update. HLA 2018;92:137–143. [DOI] [PubMed] [Google Scholar]
- 34.Pittock SJ, Lennon VA, de Seze J, et al. . Neuromyelitis optica and non organ-specific autoimmunity. Arch Neurol 2008;65:78–83. [DOI] [PubMed] [Google Scholar]
- 35.Jarius S, Ruprecht K, Wildemann B, et al. . Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation 2012;9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wandinger KP, Stangel M, Witte T, et al. . Autoantibodies against aquaporin-4 in patients with neuropsychiatric systemic lupus erythematosus and primary Sjogren's syndrome. Arthritis Rheum 2010;62:1198–1200. [DOI] [PubMed] [Google Scholar]
- 37.Jarius S, Jacob S, Waters P, Jacob A, Littleton E, Vincent A. Neuromyelitis optica in patients with gluten sensitivity associated with antibodies to aquaporin-4. J Neurol Neurosurg Psychiatry 2008;79:1084. [DOI] [PubMed] [Google Scholar]
- 38.Jarius S, Paul F, Franciotta D, et al. . Neuromyelitis optica spectrum disorders in patients with myasthenia gravis: ten new aquaporin-4 antibody positive cases and a review of the literature. Mult Scler 2012;18:1135–1143. [DOI] [PubMed] [Google Scholar]
- 39.Peschl P, Bradl M, Hoftberger R, Berger T, Reindl M. Myelin oligodendrocyte glycoprotein: deciphering a target in inflammatory demyelinating diseases. Front Immunol 2017;8:529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brunner C, Lassmann H, Waehneldt TV, Matthieu JM, Linington C. Differential ultrastructural localization of myelin basic protein, myelin/oligodendroglial glycoprotein, and 2',3'-cyclic nucleotide 3'-phosphodiesterase in the CNS of adult rats. J Neurochem 1989;52:296–304. [DOI] [PubMed] [Google Scholar]
- 41.Hemmer B, Archelos JJ, Hartung HP. New concepts in the immunopathogenesis of multiple sclerosis. Nat Rev Neurosci 2002;3:291–301. [DOI] [PubMed] [Google Scholar]
- 42.Peschl P, Schanda K, Zeka B, et al. . Human antibodies against the myelin oligodendrocyte glycoprotein can cause complement-dependent demyelination. J Neuroinflammation 2017;14:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou D, Srivastava R, Nessler S, et al. . Identification of a pathogenic antibody response to native myelin oligodendrocyte glycoprotein in multiple sclerosis. Proc Natl Acad Sci U S A 2006;103:19057–19062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mader S, Gredler V, Schanda K, et al. . Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflammation 2011;8:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bettelli E, Baeten D, Jager A, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T and B cells cooperate to induce a Devic-like disease in mice. J Clin Invest 2006;116:2393–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spadaro M, Gerdes LA, Mayer MC, et al. . Histopathology and clinical course of MOG-antibody-associated encephalomyelitis. Ann Clin Transl Neurol 2015;2:295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang JJ, Jaunmuktane Z, Mummery C, Brandner S, Leary S, Trip SA. Inflammatory demyelination without astrocyte loss in MOG antibody-positive NMOSD. Neurology 2016;87:229–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cerutti A, Puga I, Cols M. New helping friends for B cells. Eur J Immunol 2012;42:1956–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen L, Chen C, Zhong X, et al. . Different features between pediatric-onset and adult-onset patients who are seropositive for MOG-IgG: a multicenter study in South China. J Neuroimmunol 2018;321:83–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw data used in this article are available at the Erasmus University Medical Center and could be shared by request from a qualified investigator, maintaining anonymization of the individual patients.