

Abstract
Background:
Immunosuppressive medications are widely used for the prevention of allograft rejection in transplantation and graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Despite their clinical utility, these medications are accompanied by multiple off-target effects, some of which may be mediated by their effects on mitochondria.
Methods:
We examined the effect of commonly used immunosuppressive reagents, mycophenolate mofetil (MMF), cyclosporine A (CsA), rapamycin, and tacrolimus on mitochondrial function in human T-cells. T-cells were cultured in the presence of immunosuppressive medications in a range of therapeutic doses. After incubation, mitochondrial membrane potential, reactive oxygen species (ROS) production, and apoptotic cell death were measured by flow cytometry after staining with DiOC6, MitoSOX Red, and Annexin V and 7-AAD, respectively. Increases in cytosolic cytochrome c were demonstrated by Western blot. T-cell basal oxygen consumption rates were measured using a Seahorse bioanalyzer.
Results:
T-cells demonstrated significant levels of mitochondrial depolarization after treatment with therapeutic levels of MMF but not after treatment with CsA, tacrolimus, or rapamycin. Only MMF induced T-cell ROS production and induced significant levels of apoptotic cell death that were associated with increased levels of cytosolic cytochrome c. MMF decreased T-cell basal oxygen consumption within its therapeutic range, and CsA demonstrated a trend toward this result.
Conclusions:
The impairment of mitochondrial function by commonly used immunosuppressive reagents may impair T-cell differentiation and function by decreasing energy production, producing toxic ROS, and inducing apoptotic cell death.
Keywords: Mitochondria, Immunosuppression, T-cells, Membrane potential, Reactive oxygen species, Oxygen consumption rate
Introduction
Despite numerous advancements in medical therapies, organ transplantation remains the gold standard therapy for end-stage organ disease. However, the acceptance of foreign tissue requires the use of medications that suppress T-cell responses.1,2 It is increasingly recognized that cell metabolism plays a critical role in T-cell activation, differentiation, and function.3–6 T-cells rely primarily on mitochondrial oxidative phosphorylation pathways to generate ATP, although once activated, they also increase their utilization of glycolysis, a process referred to as the “Warburg effect.”7 Mitochondrial ATP production, Ca2+ uptake, and oxidative phosphorylation8 all increase during T-cell activation and mitochondria accumulate at the immune synapse during antigen recognition.9
With current immunosuppression regimens, the median survival of solid organ transplants continues to improve, with 1-y survival exceeding 90% for many organs.10–12 The long-term need for immunosuppression raises the concern of accumulating toxicities related to the use of immunosuppressive medications. It has been demonstrated that some of these toxicities are due to the effects of these medications on mitochondrial function. For example, calcineurin inhibitors have been shown to cause mitochondrial dysfunction in kidney tubule cells, potentially explaining the nephrotoxicity commonly caused by these medications.13,14 Kidney tubule cells treated with cyclosporine A (CsA) show mitochondrial swelling, cytochrome c release, and decreased mitochondrial membrane potential, processes associated with apoptotic cell death.15 Mitochondrial dysfunction in endothelial cells caused by CsA may also be responsible for the development of hypertension associated with this medication.16 Furthermore, treatment of monocytes with mycophenolate mofetil (MMF) and rapamycin has been shown to induce apoptosis.17,18 The effects of these medications on mitochondria could help explain their toxicities in various tissues.
Mitochondria are intimately tied to T-cell immune function.9 However, little is known about the effect of immunosuppressive medications on T-cell mitochondrial function. The aim of this study was therefore to investigate how commonly used immunosuppressive medications affect the function of T-cell mitochondria, focusing on drug levels within clinically targeted pharmacologic ranges.
Materials and methods
Materials
Jurkat cells, an immortalized line of human CD4+ T-lymphocyte cells, were obtained from the American Tissue Culture Collection (TIB-152, Manassas, VA) and were maintained in a 5% CO2 atmosphere at 37°C in complete Roswell Park Memorial Institute (RPMI) media containing 10% heat-inactivated fetal bovine serum (Mediatech Inc, Manassas, VA), penicillin/streptomycin (100 U/mL each), 0.05 mM beta-mercaptoethanol, 2 mM L-glutamine, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, and 10 mM HEPES (all obtained from Life Technologies, Carlsbad, CA). CsA, MMF, tacrolimus, and rapamycin were obtained from Cayman Chemical (Ann Arbor, MI, items #12088, #13988, #10007965, #13346). Stock solutions of CsA (5 mM), tacrolimus (5 mM), rapamycin (5 mM), and MMF (20 mM) were prepared in DMSO, aliquoted and stored at −20°C. MitoSOX Red and MitoTracker Green fluorescent probes were obtained from Life Technologies. 7-Amino-actinomycin D (7-AAD), annexin V, and 10 X annexin V staining buffer were obtained from BD Biosciences (Franklin Lakes, NJ). Human tumor necrosis factor alpha was from Enzo Life Sciences (Farmingdale, NY). Tween-20 was from SigmaeAldrich (St. Louis, MO).
Flow cytometry
Jurkat cells were plated at a density of 500,000 cells/mL in complete RPMI medium. CsA was added to make final concentrations of 125, 250, and 500 nM. Final concentrations of MMF were 3, 10, and 30 μM. Final concentrations of rapamycin were 5, 20, and 40 nM. Final concentrations of tacrolimus were 6, 18, and 36 nM. Experimental concentrations were calculated using commonly targeted blood levels of these medications to include therapeutic and supratherapeutic levels. Targeted blood levels of tacrolimus after transplant range from 5 to 15 ng/mL, with higher levels being targeted soon after transplant and tapering slowly over time. This range was used to calculate the molarity of the drug in solution, which was used in the experimental conditions. The 6 nM and 18 nM conditions represent the lower and upper limits of this therapeutic range, and 36 nM represents a supratherapeutic level. The same method was used to determine the experimental conditions for CsA and MMF. After 24 h of incubation at 37°C, cells were harvested and assayed for mitochondrial membrane potential, reactive oxygen species (ROS) production, apoptosis, as described below. Cell staining was quantified using a BD LSRFortessa flow cytometer from BD Biosciences. Data were analyzed using FlowJo software (Ashland, OR). Cells treated with 50 ng/mL human tumor necrosis factor alpha for 24 h were used as a positive control for gating for ROS production, apoptosis, and mitochondrial membrane potential assays.
Mitochondrial membrane potential
Mitochondrial membrane potential was assessed using the DiOC6 fluorescent probe. Harvested Jurkat cells were resuspended in 50 nM staining solution and incubated for 15 min at 37°C. Cells were then washed and resuspended in PBS.
ROS production
Superoxide production was quantified using MitoSOX Red. Harvested Jurkat cells were resuspended in 50 nM MitoSOX Red staining solution. Cells were incubated at room temperature protected from light for 30 min. Cells were then washed and resuspended in PBS.
Apoptosis
Apoptosis was assessed by dual staining with annexin V and 7-AAD using the PE Annexin V Apoptosis Detection Kit I (#559763, BD Biosciences) at 4°C for 30 min.
Oxygen consumption
Jurkat cells were harvested and plated in 24-well plates in complete RPMI media and treated with immunosuppressive reagents. The choice of which immunosuppressive reagents to test was determined using data obtained in flow cytometry experiments. Those reagents with positive flow cytometry results were further assessed using the Seahorse Bio-analyzer. After 24 h incubation with CsA, cells were recounted and 75–150,000 cells per well were replated in glucose-free RPMI media with 5 mM galactose on poly-D lysine-coated 24-well plates. Cells were incubated with MMF for 4 h and replated as mentioned previously. A Seahorse XFe24 Bioanalyzer (Seahorse Bioscience, Billerica, MA) was used to measure basal oxygen consumption rates (OCRs) as previously described.19,20
Cytochrome c western blot
Jurkat cells were plated in complete 1 RPMI and 0, 3, 10, and 30 μM MMF was added to each treatment group flask. Cells were then incubated overnight at 37°C in humidified air and 5% CO2. After 24 h, cells were collected and mitochondrial and cytosolic cellular fractions were separated using a cytochrome c release apoptosis assay kit after manufacturer’s instructions (Abcam Cambridge, United Kingdom, product #65311). Protein concentration was measured using a Bradford assay, and 20 μg of protein was separated by electrophoresis on “Any kD” polyacrylamide gels (Bio-Rad, Hercules, CA). Proteins were then transferred to nitrocellulose membrane. The membrane was blocked for 1 h in 1% fish gelatin and then incubated overnight with a 1:200 dilution of cytochrome c antibody provided with the cytochrome release kit. The membrane was washed 3 times in PBS supplemented with 0.1% (v/v) Tween-20 (PBS-T). The membrane was then incubated for 1 h with HRP-conjugated goat anti-rabbit antibody (Jackson ImmunoResearch Laboratories, Inc, West Grove, PA) at a 1:5000 dilution in PBS-T, and then washed 3 more times in PBS-T and imaged using the Li-Cor Odyssey Imaging System (Lincoln, NE). Western blot membranes were stripped of antibodies using Restore Plus Western Blot Stripping Buffer (Thermo-Fisher Scientific) before reprobing with anti-GAPDH antibody (1:10,000 dilution in PBS-T, Abcam, #ab181602), followed by HRP-conjugated goat anti-rabbit antibody (1:5000 in PBS-T).
Statistical analysis
Flow cytometry experiments were performed in quadruplicate. Seahorse assays were performed in triplicate. Individual results are shown in graphical form, and the median and range of each experimental condition are also indicated. Data were analyzed using GraphPad Prism software (La Jolla, CA). Comparisons between experimental and control groups and between each experimental group were performed by two-tailed unpaired Student’s t-test, and P values ≤ 0.05 were considered significant. P values were adjusted for multiple comparisons using the Bonferroni correction. P values significant after correction are marked with an asterisk.
Results
The mitochondrial membrane potential generated by proton pumps is essential to the process of oxidative phosphorylation and is a key indicator of cell health. We used DiOC6, a fluorescent dye that accumulates primarily in mitochondria with active membrane potentials, to measure mitochondrial membrane depolarization using a flow cytometric assay. We observed that MMF caused a decrease in mitochondrial membrane potential in a concentration-dependent manner at therapeutic (10 μM-3 μM) and supratherapeutic (30 μM) levels (Fig. 1A and B). CsA caused a trend toward decreased mitochondrial membrane potential (P = 0.01 and P = 0.04 at 125 nm and 250 nm, respectively, when compared to vehicle control), which were not significant after correction for multiple comparisons. In contrast, neither rapamycin nor tacrolimus affected mitochondrial membrane potential at the doses tested.
Fig. 1 –
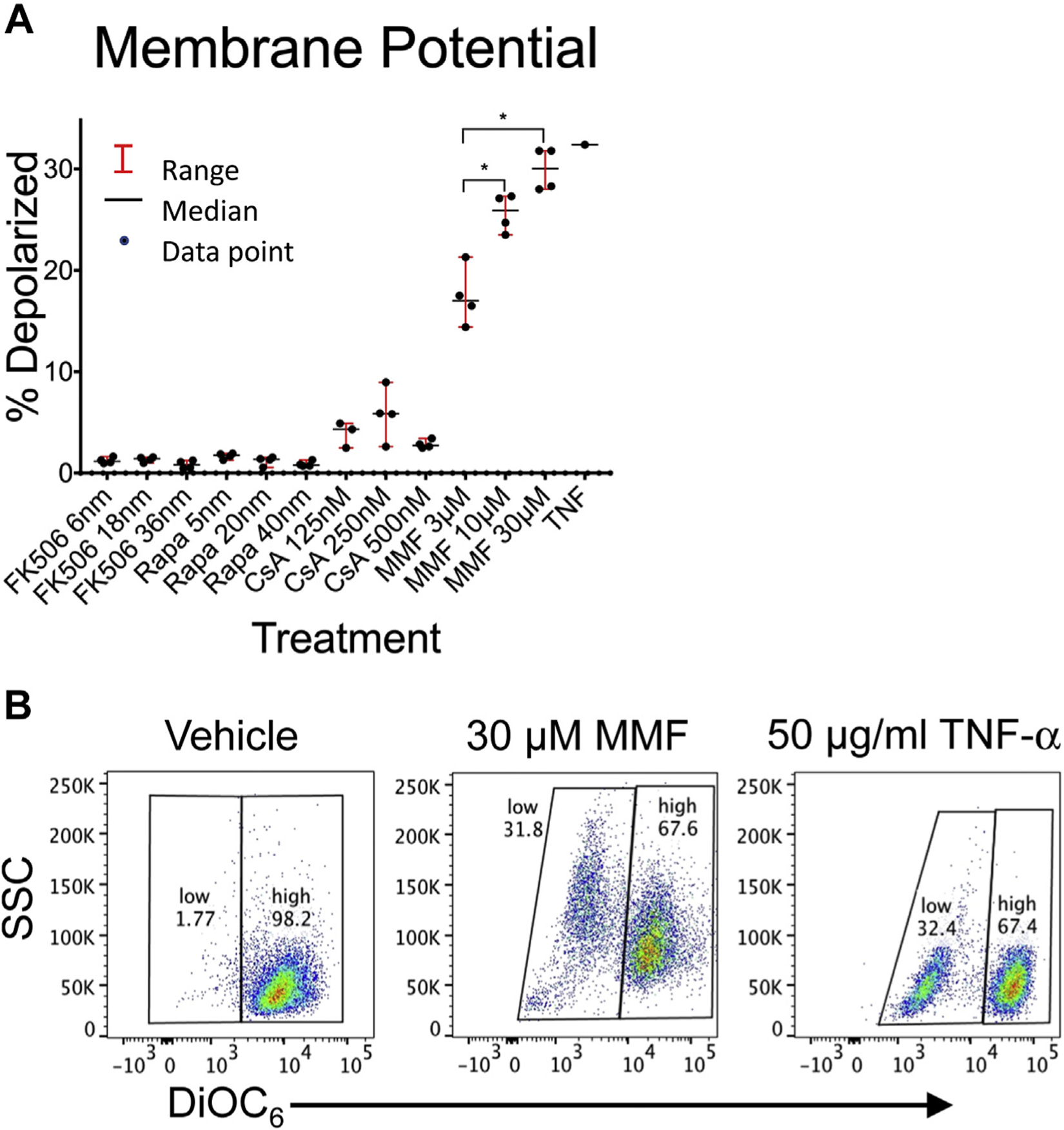
Effect of immunosuppression reagents on T-cell mitochondrial membrane potential. Jurkat cells were incubated for 24 h with therapeutic and supratherapeutic levels of tacrolimus (FK506), rapamycin (Rapa), cyclosporine A (CsA), and mycophenolate mofetil (MMF). After 24-h incubation, cells were stained with DiOC6 and analyzed by flow cytometry for mitochondrial membrane depolarization as assessed by decreased DiOC6 fluorescence. (A) The percentage of cells with reduced DiOC6 staining is shown for each drug and concentration in quadruplicate. The median and range of each data set is also shown. (B) Representative flow cytometry after incubation with vehicle, 30 μM MMF, and TNF-α control. Comparisons between treatment and vehicle control groups and between each treatment groups were performed using a two-tailed unpaired Student’s t-test. *P-values were adjusted for multiple comparisons using the Bonferroni correction with a standard significant P value of 0.05. Significant P-values after correction are marked with an asterisk. TNF-α = tumor necrosis factor alpha.
We next measured the production of mitochondrial ROS, which include superoxide and hydrogen peroxide molecules, as an indicator of mitochondrial oxidative stress, damage, and dysfunction after treatment with immunosuppressive reagents. We used MitoSOX Red to assess ROS production in a flow cytometric assay. We found that MMF caused a significant increase in ROS production both within and above its therapeutic range (Fig. 2A and B). CsA, rapamycin, and tacrolimus had no effect on ROS production at the concentrations and over the time period (24 h) tested.
Fig. 2 –
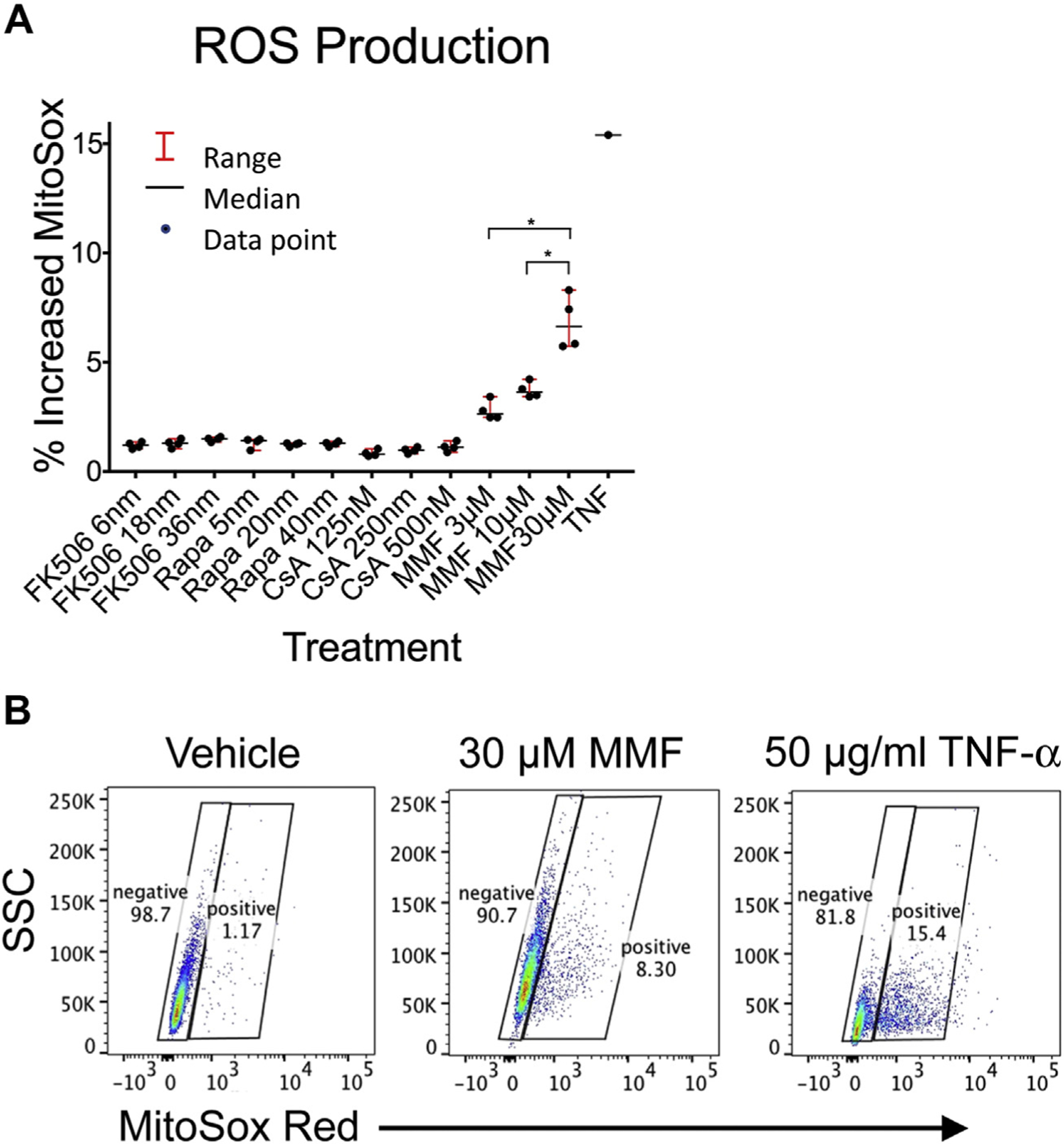
Effect of immunosuppression reagents on T-cell reactive oxygen species (ROS) production. Jurkat cells were incubated for 24 h with therapeutic and supratherapeutic levels of tacrolimus (FK506), rapamycin (Rapa), cyclosporine A (CsA), and mycophenolate mofetil (MMF). After 24-h incubation, cells were stained with MitoSOX Red and analyzed by flow cytometry for ROS production as assessed by increased MitoSOX fluorescence. (A) The percentage of cells with increased MitoSOX staining is shown for each drug and concentration in quadruplicate. The median and range are also indicated. (B) Representative flow cytometry after incubation with vehicle control, 30 μM MMF, and TNF-α control. Comparisons between treatment and vehicle control groups and between each treatment group were performed using a two-tailed unpaired Student’s t-test. *P-values were adjusted for multiple comparisons using the Bonferroni correction with a standard significant P value of 0.05. Significant P-values after correction are marked with an asterisk. TNF-α = tumor necrosis factor alpha.
Mitochondrial depolarization can be associated with the opening of mitochondrial permeability transition pores, allowing cytochrome c release into the cytoplasm and the initiation of the proteolytic caspase cascade that ultimately results in apoptotic cell death. MMF treatment of Jurkat cells demonstrated a concentration-dependent increase in apoptosis (Fig. 3A and B). The association of the apoptotic T-cell death with mitochondrial dysfunction was supported by Western blot demonstrating an increase in cytosolic cytochrome c (Fig. 4). None of the other medications tested had significant effects on apoptosis at the dosages and incubation time tested.
Fig. 3 –
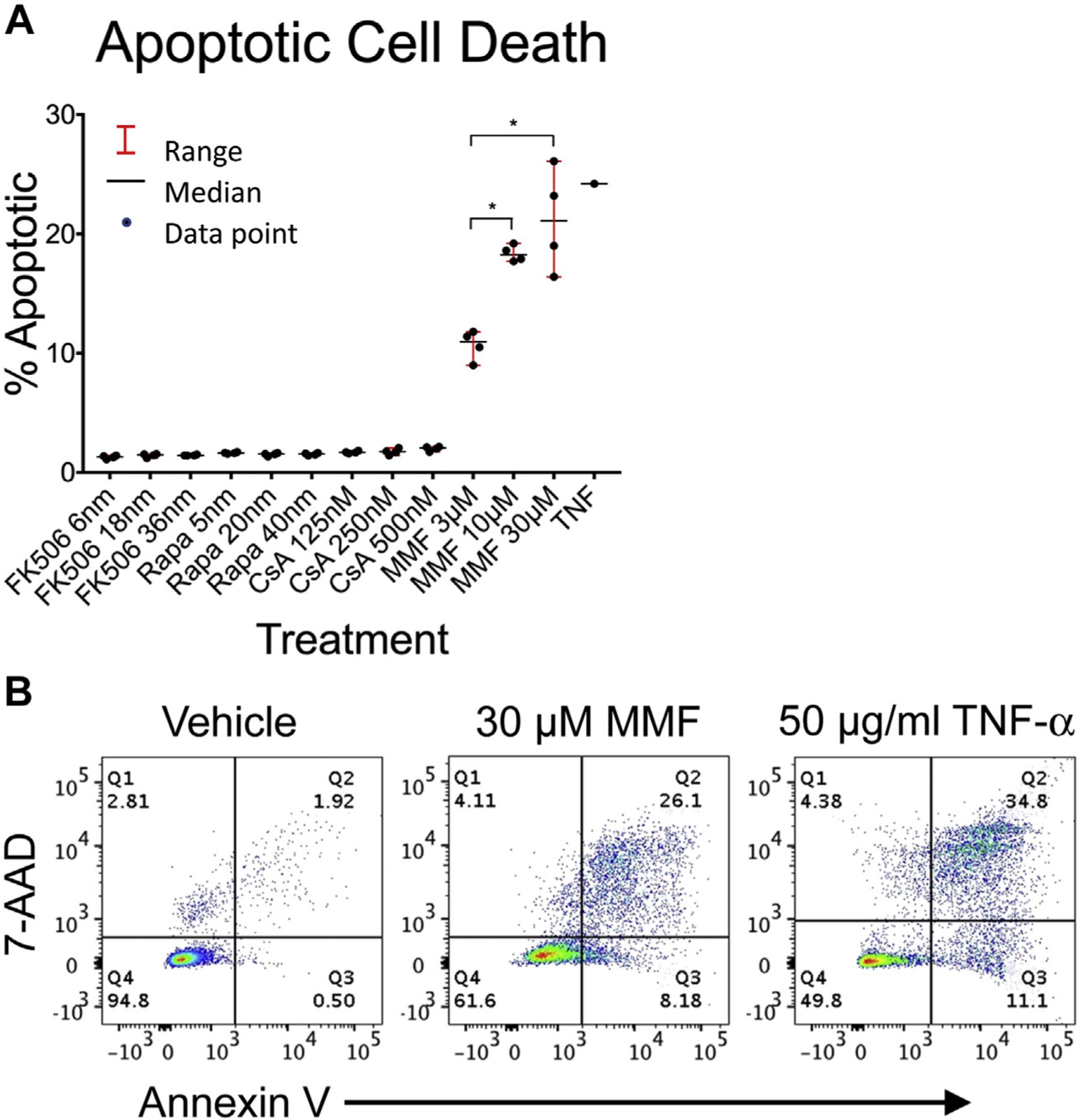
Effect of immunosuppression reagents on apoptotic T-cell death. Jurkat cells were incubated for 24 h with therapeutic and supratherapeutic levels of tacrolimus (FK506), rapamycin (Rapa), cyclosporine A (CsA), and mycophenolate mofetil (MMF). After 24-h incubation, cells were stained with Annexin V and 7-AAD and analyzed by flow cytometry for apoptotic cell death defined as 7-AAD+/Annexin V+ cells. (A) The percentage of apoptotic cells is shown for each drug and concentration in quadruplicate. The median and range for each treatment group are also shown. (B) Representative flow cytometry after incubation with 30 μM MMF, vehicle control, and TNF-α–epositive control. Comparisons between treatment and vehicle control groups and between each treatment groups were performed using a two-tailed unpaired Student’s t-test. *P-values were adjusted for multiple comparisons using the Bonferroni correction with a standard significant P value of 0.05. Significant P-values after correction are marked with an asterisk. TNF-α = tumor necrosis factor alpha.
Fig. 4 –
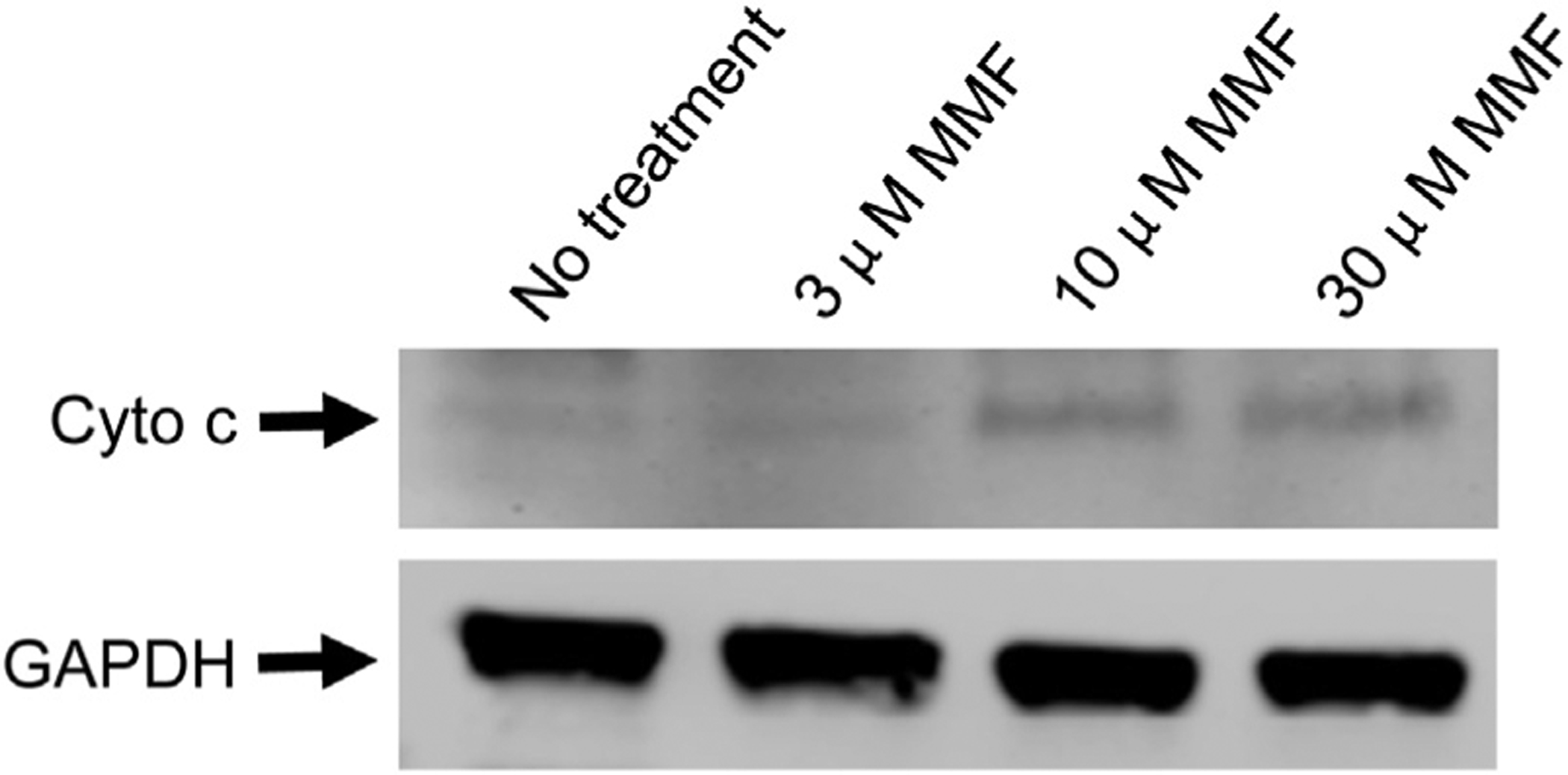
Treatment of T-cells with mycophenolate mofetil (MMF) results in increased cytosolic cytochrome c. Jurkat cells were treated for 24 h with increasing concentrations of MMF and their cytoplasmic fractions were isolated. Western blot analysis was then performed for cytochrome c and GAPDH (loading control).
We next sought to investigate the functional impact of the two immunosuppressive medications that caused mitochondrial membrane depolarization, MMF and CsA, on mitochondrial metabolic function. For this purpose, we measured T-cell basal oxygen consumption using a Seahorse bioanalyzer. Incubation with CsA for 24 h caused a trend toward decreased basal oxygen consumption (Fig. 5A). Incubation of T-cells with MMF for 4 h caused a significant reduction in basal oxygen consumption at the lowest dose tested (Fig. 5B).
Fig. 5 –
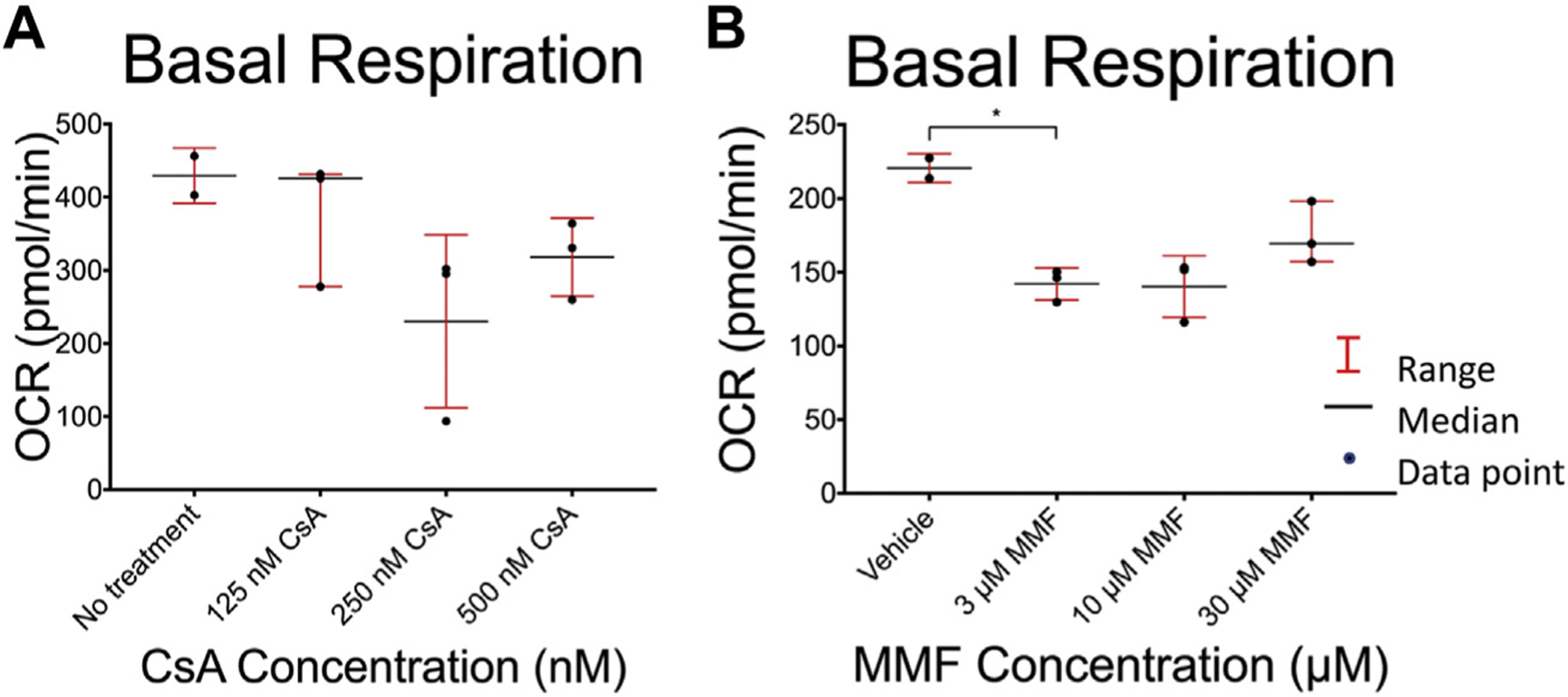
Effect of immunosuppression reagents on T-cell basal oxygen consumption rate (OCR). Jurkat cells OCR was measured after 4 h of treatment with mycophenolate mofetil (MMF) and 24 h of treatment with cyclosporine A (CsA) at the concentrations shown. After 24 h, cells were recounted and plated on poly-D lysine–treated Seahorse plates in glucose-free RPMI media with 5 mM galactose. Cells incubated for 4 h were immediately replated. A Seahorse bioanalyzer was then used to measure basal OCR following treatment with (A) CsA and (B) MMF. Comparisons between treatment and vehicle control groups and between each treatment group were performed using a two-tailed unpaired Student’s t-test. *P-values were adjusted for multiple comparisons using the Bonferroni correction with a standard significant P value of 0.05. Significant P-values after correction are marked with an asterisk.
Discussion
Immunosuppressive medications commonly used in organ transplantation have previously been shown to cause dysfunction in endothelial cells and monocytes, providing a potential mechanism for some of their known toxicities. Although the side effects of these medications and their relationship to mitochondrial function are well documented, these medications specifically target T-cells, which are reliant on mitochondrial function to supply the energy needed to produce their effector functions. Here we demonstrated that MMF causes a loss of mitochondrial membrane potential and increases ROS production associated with increases in cytoplasmic cytochrome c levels and the induction of apoptotic cell death. In comparison, CsA had a nonsignificant trend toward decreased mitochondrial membrane potential, whereas rapamycin and tacrolimus demonstrated no effect on mitochondrial function within their therapeutic dose ranges.
MMF has previously been shown to decrease glucose up-take in human T-cells and inhibit glycolysis, leading to decreased intracellular ATP content and apoptosis.21 MMF has also been shown to cause preferential apoptosis of activated murine T-cells in vivo.22 We have extended these observations to show a concentration-dependent effect of MMF on ROS production and membrane potential, demonstrating these effects both within and above the therapeutic range.
In contrast to the toxic effects of MMF, CsA did not cause apoptotic cell death. This is in line with previous findings that CsA inhibits mitochondrial transition pore formation, which allows the release of mitochondrial cytochrome c into the cytoplasm, a key step in the initiation of apoptosis.16 Previous studies using 20-fold higher levels of CsA (10 μM) have shown that at these levels CsA promotes apoptosis in Jurkat cells in association with mitochondrial dysfunction.23 These differences may be a consequence of CsA dosage: A dose-dependent effect of CsA has been previously demonstrated such that nanomolar concentrations decrease mitochondria transition pore opening, whereas micromolar concentrations lead to mitochondrial membrane depolarization, cytochrome c translocation to the cytosol, and apoptosis.24 Overall, our results indicate that therapeutic doses of CsA may cause mitochondrial stress without causing apoptosis. Previous studies have shown that tacrolimus causes increased apoptosis through ROS generation and decreased membrane potential.25 However, these studies were performed using tacrolimus levels approximately 1000-fold higher than those used in our experiments. In another study by Migita et al.,26 tacrolimus at lower concentrations of 20–200 ng/mL did not affect mitochondrial function. We would attribute the differences in our results between CsA and tacrolimus, both calcineurin inhibitors, to biochemical differences between the molecules themselves. Calcineurin is widely expressed throughout the body, and so the potential of target effects of these medications are numerous. Accordingly, CsA and tacrolimus have been shown to have different effects in a wide variety of tissues despite their similar mechanisms of action. These include vascular smooth muscle, osteoblasts, pancreatic beta cells, and others.17,27–29 These differences are reflected in the slightly different side effect profiles between CsA and tacrolimus, and so one might expect that their effects in our study may also differ slightly.29,30 With regard to rapamycin, we have confirmed previous studies that rapamycin, at similar concentrations to those used in the present study, does not cause apoptosis in Jurkat cells.31
We chose to work with Jurkat cells because, as CD4+ cells they play a critical role in the acute cellular rejection most common among transplant recipients. As such, we felt that the CD4+ subset was an appropriate starting point from which to investigate the effects of these medications on T-cells and their immune function more specifically. Indeed, there has been some study into the effects of these medications on different T-cellsubsets. Calcineurin inhibitors have been shown to decrease the amount of regulatory T-cells in the early posteliver transplant period.32 Similarly, tacrolimus has been shown to inhibit clonal expansion of naïve T-cells and development of cytotoxic and memory T-cells. In contrast, CsA treatment results in increased development of memory cells but has no effect on the development of effector cytotoxic responses.33,34 MMF has also been shown to reduce the amount of circulating CD4+, CD8+, and CD16+ lymphocytes.35 These results indicate that these medications exert a more specific effect on T-cells than general toxicity; however, these studies focused only on the absolute count and immune responsiveness of these T-cell subsets and did not evaluate their mitochondrial function in any way. And so, we feel that a more detailed analysis of the mitochondrial function of these T-cell subsets is warranted, in addition to further testing on human T-cells.
In summary, we have shown that MMF causes dose-dependent mitochondrial dysfunction and apoptosis within and above its therapeutic range. In contrast, calcineurin inhibitors and rapamycin cause little to no mitochondrial dysfunction or apoptosis at clinically relevant doses. This research provides insight into the interconnected metabolic and immune pathways in T-cells by demonstrating that common immunosuppressive drugs can cause mitochondrial dysfunction and affect mitochondrial metabolism in human T-cells. We believe these effects may contribute to the immunosuppressive effect of these medications by interfering with critical mitochondrial functions required for T-cell activation. Although these results demonstrate different degrees of mitochondrial dysfunction caused by these medications, we believe that this is a potential explanation for any clinical data suggesting superiority of one regimen over another. Therefore, we would not advocate for a particular regimen over another based on these data alone. Further research should seek to continue to enumerate the effects of these medications by focusing on primary cell lines and on specific T-cell subsets.
Acknowledgment
This project was supported by funding from the NIH/NIAID (AI101263) to T.B. Authors’ contributions: A.N. conceived and designed the analysis, collected the data, performed the data analysis, and wrote the paper; M.S. performed the data analysis and wrote the paper; T.L. collected the data and performed the data analysis; M.Z. collected the data and performed the data analysis; L.L. collected the data and performed the data analysis; J.M. conceived and designed the analysis; T.B. conceived and designed the analysis, performed the data analysis, and wrote the paper.
Footnotes
Disclosure
The authors reported no proprietary or commercial interest in any product mentioned or concept discussed in this article.
REFERENCES
- 1.Bamoulid J, Staeck O, Halleck F, et al. The need for minimization strategies: current problems of immunosuppression. Transpl Int. 2015;28:891–900. [DOI] [PubMed] [Google Scholar]
- 2.Chinen J, Buckley RH. Transplantation immunology: solid organ and bone marrow. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S324–S335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van der Windt GJW, Pearce EL. Metabolic switching and fuel choice during T cell differentiation and memory development. Immunol Rev. 2012;249:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sack MN. T cells: mitochondrial fidelity and metabolic agility control immune cell fate and function. J Clin Invest. 2018;128:3651–3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leavy O. T cells: mitochondria and T cell activation. Nat Rev Immunol. 2013;13:224. [DOI] [PubMed] [Google Scholar]
- 6.Chao T, Wang H, Ho PC. Mitochondrial control and Guidance of cellular activities of T cells. Front Immunol. 2017;8:473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buck MD, O’Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. 2015;212:1345–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davide FG, Dostert C, Brenner D. Reactive oxygen species: involvement in T cell signaling and metabolism. Cell Press Rev. 2018;1455:1–14. [DOI] [PubMed] [Google Scholar]
- 9.Frauwirth KA, Riley JL, Harris MH, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769–777. [DOI] [PubMed] [Google Scholar]
- 10.Gubser PM, Bantug GR, Razik L, et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol. 2013;14:1064–1072. [DOI] [PubMed] [Google Scholar]
- 11.Quintana A, Schwindling C, Wenning AS, et al. T cell activation requires mitochondrial translocation to the immunological synapse. Proc Natl Acad Sci USA. 2007;104:14418–14423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ledderose C, Bao Y, Lidicky M, et al. Mitochondria are gate-keepers of T cell function by producing the ATP that drives purigenic signaling. J Biol Chem. 2014;289:25936–25945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.United States Department of Health and Human Services. Kaplan-Meier Graft Survival Rates for Transplants Performed: 2008–2015. 2018. Available at: https://optn.transplant.hrsa.gov. Washington, DC. [Google Scholar]
- 14.Illsinger S, Janzen N, Lucke T, et al. Cyclosporine A: impact on mitochondrial function in endothelial cells. Clin Transplant. 2011;25:584–593. [DOI] [PubMed] [Google Scholar]
- 15.Illsinger S, Goken C, Brockmann M, et al. Effect of tracolimus on energy metabolism in human umbilical endothelial cells. Ann Transplant. 2011;16:68–75. [DOI] [PubMed] [Google Scholar]
- 16.Xiao Z, Shan J, Li C, et al. Mechanisms of cyclosporine-induced renal cell apoptosis: a systematic review. Am J Nephrol. 2013;37:30–40. [DOI] [PubMed] [Google Scholar]
- 17.Kędzierska K, Sporniak-Tutak K, Kolasa A, et al. The effect of immunosuppressive therapy on renal cell apoptosis in native rat kidneys. Histol Histopathol. 2015;30:105–116. [DOI] [PubMed] [Google Scholar]
- 18.De Arriba G, Calvino M, Benito S, Parra T. Cyclosporine A-induced apoptosis in renal tubular cells is related to oxidative damage and mitochondrial fission. Toxicol Lett. 2013;218:30–38. [DOI] [PubMed] [Google Scholar]
- 19.Nowak M, Tardivel S, Nguyen-Khoa T, et al. Mycophenolate mofetil and rapamycin induce apoptosis in the human monocytic U937 cell line through two different pathways. J Cell Biochem. 2017;118:3480–3487. [DOI] [PubMed] [Google Scholar]
- 20.Takebe N, Cheng X, Fandy TE, et al. IMP dehydrogenase inhibitor mycophenolate mofetil induces caspase-dependent apoptosis and cell cycle inhibition in multiple myeloma cells. Mol Cancer Ther. 2006;5:457–466. [DOI] [PubMed] [Google Scholar]
- 21.Luz AL, Rooney JP, Kubik LL, Gonzalez CP, Song DH, Meyer JN. Mitochondrial morphology and fundamental parameters of the mitochondrial respiratory chain are altered in Caenorhabditis elegans strains deficient in mitochondrial dynamics and homeostasis processes. PLoS One. 2015;10:e0130940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luz AL, Smith LL, Rooney JP, Meyer JN. Seahorse Xfe 24 extracellular flux analyzer-based analysis of cellular respiration in Caenorhabditis elegans. Curr Protoc Toxicol. 2015;66, 25.7.1–25.7.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernandez-Ramos AA, Marchetti-Laurent C, Poindessous V, et al. A comprehensive characterization of the impact of mycophenolic acid of the metabolism of Jurkat T cells. Sci Rep. 2017;7:10550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi SJN, Lee HK, Kim NH, Chung SY. Mycophenolic acid mediated mitochondrial membrane potential transition change lead to T lymphocyte apoptosis. J Korean Surg Soc. 2011;81:235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao J, Sana R, Calder V, et al. Mitochondrial permeability transition pore in inflammatory apoptosis of human conjunctival epithelial cells and T cells: effects of cyclosporine A. Cornea. 2013;54:4717–4733. [DOI] [PubMed] [Google Scholar]
- 26.Bustamante J, Lopes EC, Garcia M, et al. Disruption of mitochondrial membrane potential during apoptosis induced by PSC 833 and CsA in multidrug-resistant lymphoid leukemia. Toxicol Appl Pharmacol. 2004;199:44–51. [DOI] [PubMed] [Google Scholar]
- 27.Martin-Fernandez M, Rupert M, Montero M, de la Piedra C. Effects of cyclosporine, tacrolimus, and rapamycin on osteoblasts. Transplant Proc. 2017;49:2219–2224. [DOI] [PubMed] [Google Scholar]
- 28.Chakkera HA, Kudva Y, Kaplan B. Calcineurin inhibitors: pharmacologic mechanisms impacting both insulin resistance and insulin Secretion leading to glucose dysregulation and diabetes Mellitus. Clin Pharmacol Ther. 2017;101:114–120. [DOI] [PubMed] [Google Scholar]
- 29.Penninga L, Penninga EI, Moller CH, Iverson M, Steinbruchel DA, Gluud C. Tacrolimus versus cyclosporin as primaryimmunosuppression for lung transplant recipients. Cochrane Database Syst Rev. 2013: CD008817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Helmschrott M, Rivinius R, Ruhparwar A, et al. Advantageous effects of immunosuppression with tacrolimus in comparison with cyclosporine A regarding renal function in patients after heart transplantation. Drug Des Devel Ther. 2015;9:1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tao Z, Jones E, Goodisman J, Souid A. Quantitative measure of cytotoxicity of anticancer drugs and other agents. Anal Biochem. 2008;381:43–52. [DOI] [PubMed] [Google Scholar]
- 32.Kim HY, Cho ML, Jhun JY, et al. The imbalance of T helper 17/regulatory T cells and memory B cells during the early post-transplantation period in peripheral blood of living donor liver transplantation recipients under calcineurin inhibitor-based immunosuppression. Immunology. 2013;138:124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koenen HJ, Michielsen EC, Verstappen J, Fasse E, Joosten I. Superior T-cell suppression by rapamycin and FK506 over rapamycin and cyclosporine A because of abrogated cytotoxic T-lymphocyte induction, impaired memory responses, and persistent apoptosis. Transplantation. 2003;75:1581–1590. [DOI] [PubMed] [Google Scholar]
- 34.Tsuda K, Yamanaka K, Kitagawa H, et al. Calcineurin inhibitors suppress cytokine production from memory T cells and differentiation of naïve T cells into cytokine-producing mature T cells. PLoS One. 2012;7:e31465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bravo Soto JA, Esteban de la Rosa RJ, Luna del Castillo JD, et al. Effect of mycophenolate mofetil regimen on peripheral blood lymphocyte subsets in kidney transplant recipients. Transplant Proc. 2003;35:1355–1359. [DOI] [PubMed] [Google Scholar]