

Abstract
We tested the hypothesis that P2X4 purinergic receptor (P2X4) exacerbates ischemic acute kidney injury (AKI) by promoting renal tubular inflammation after ischemia and reperfusion (IR). Supporting this, P2X4 deficient (KO) mice were protected against ischemic AKI with significantly attenuated renal tubular necrosis, inflammation and apoptosis when compared to P2X4 wild type (WT) mice subjected to renal IR. Furthermore, WT mice treated with a P2X4 allosteric agonist ivermectin had exacerbated renal IR injury whereas P2X4 WT mice treated with a selective P2X4 antagonist (5-BDBD) were protected against ischemic AKI. Mechanistically, induction of kidney NLRP3 inflammasome signaling after renal IR was significantly attenuated in P2X4 KO mice. A P2 agonist ATPγS increased NLRP3 inflammasome signaling (NLRP3 and caspase 1 induction and IL-1β processing) in isolated renal proximal tubule cells from WT mice whereas these increases were absent in renal proximal tubules isolated from P2X4 KO mice. Moreover, 5-BDBD attenuated ATPγS induced NLRP3 inflammasome induction in renal proximal tubules from WT mice. Finally, a P2X4 agonist ivermectin induced NLRP3 inflammasome and pro-inflammatory cytokines in cultured human proximal tubule cells. Taken together, our studies suggest that renal proximal tubular P2X4 activation exacerbates ischemic AKI and promotes NLRP3 inflammasome signaling.
Keywords: Apoptosis, inflammation, inflammasome, ischemia and reperfusion injury, necrosis, neutrophil
Introduction
Acute kidney injury (AKI) is a major health care challenge that cost more than $10 billion per year in the US (1, 2). Unfortunately, despite many decades of intense research, there is no effective preventive measure or therapy for AKI (3, 4). Renal ischemia and reperfusion (IR) injury is a frequent cause of clinical AKI in patients undergoing cardiac, vascular or liver transplant surgeries with ~50–80% post-operative risk for ischemic AKI (5, 6). Clinical AKI frequently leads to extra-renal systemic complication resulting in hepatic, pulmonary and gut dysfunction as well as sepsis contributing to its very high mortality (3).
Renal IR injury results in proximal tubular necrosis followed by subsequent inflammatory response and apoptotic tubular cell death (7–9). Inflammatory response after renal IR is a complex process orchestrated by induction and release of pro-inflammatory cytokines and chemokines by renal tubular cells that attract influx of inflammatory leukocytes including neutrophils, T-cells and macrophages to the site of injury (10–12). Therefore, limiting the inflammatory response would be an attractive target for therapy in clinical ischemic AKI. Damage Associated Molecular Pattern (DAMP) ligands released by dying or dead renal cells orchestrate renal tubular inflammatory response and cell death after IR (13–15). Indeed, multiple DAMPs have been identified including ATP, HMGB-1, uric acid and mitochondrial DNA that activate pathways to induce and potentiate renal cell death and a sterile inflammatory process via innate immune system activation after IR (16).
ATP is one of the DAMPs that rapidly initiates tissue inflammation and immune cell infiltration (17, 18). Since the intracellular ATP concentration is far greater than extracellular levels, impairment of membrane integrity after IR results in rapid release of intracellular ATP into the extracellular space (19–21). Extracellular ATP binds to 2 distinct membrane anchored P2 purinergic receptor subtypes –ligand-gated ionotropic P2X receptors (P2X1–7) and metabotropic G-protein coupled P2Y receptors (P2Y1,2,4,6,11–14) (19, 22). The P2X receptors allow influx and efflux of cations (Na+, K+ and Ca++) upon activation and are involved in a variety of pathological responses including inflammation, tissue damage and the graft-versus-host disease (19, 20). Of P2X receptor subtypes, P2X7 receptor subtype has received most scientific attention as it exacerbates a variety of kidney pathology including ischemic AKI, septic AKI and diabetic nephropathy (19, 23, 24). P2X4 receptor is closely related to P2X7 receptors in terms of amino acid sequence, channel physiology, chromosomal location and tissue distribution (25). Although P2X7 receptor is scantly expressed in normal kidney tissue (and upregulated after tissue injury and inflammation), P2X4 receptors are widely expressed in normal kidney cells including renal proximal tubules (19, 26). Furthermore, P2X4 receptor is one of the most sensitive purinergic receptors activated by nanomolar extracellular ATP (20, 25). In contrast, P2X7 receptor has low affinity for extracellular ATP and requires mM ATP concentration for its activation (19).
Unlike the better-characterized role for P2X7 (19, 21, 23), the role for P2X4 in renal pathology and injury remains unclear. In a mouse model of ureteral obstruction induced renal inflammation, P2X4 deletion exacerbated renal fibrosis suggesting a protective and anti-fibrotic role for kidney P2X4 (27). However, P2X4 activation may play a detrimental role in diabetic nephropathy by promoting renal tubular inflammation (28). Since the effect P2X4 activation in ischemic AKI has never been studied, we examined in this study the role for P2X4 in renal IR injury. We tested the hypothesis that P2X4 plays a detrimental role in ischemic AKI by promoting renal tubular inflammation with subsequent exacerbation of renal tubular death.
Materials and Methods
P2X4 and P2X7 mRNA expressions in P2X4 wild type and P2X4 deficient mice.
Because P2X4 and P2X7 have similar channel pharmacology as well as chromosomal location and some studies reported heteromeric co-expression of P2X4 and P2X7 (29, 30), we measured kidney P2X4 and P2X7 mRNA expressions in P2X4 WT and P2X4 KO mice with RT-PCR as described previously (31, 32). Primer design was based on published GenBank sequences. We also measured GAPDH mRNA expression to control for RNA loading.
Kidney P2X4 staining utilizing P2X4 reporter mice
Kidneys from P2X4 tdTomato BAC transgenic mice (B6N.Cg-Tg(P2rx4-tdTomato)1Khakh/j, Jax Labs) were fixed with 4% paraformaldehyde, dehydrated with 30% sucrose, frozen in O.C.T (Tissue-Tek, Torrance, CA) and cryosectioned (5 μm). Kidney sections were permeabilized and stained with green fluorescent PHA lectin (proximal tubule specific marker, Thermo Fisher Scientific, Waltham, MA) for 1h at RT, 1:100 dilution in 1% BSA-PBS. After washes, kidney slides were counterstained with DAPI to visualize cell nuclei, mounted with Vectashield (Vector, Burlingame, CA) mounting media, and imaged with IX71 Olympus fluorescent microscope.
Renal IR injury in mice
After Columbia University Institutional Animal Care and Use Committee approval, 8 to 10-week-old male P2X4 wild type (WT), P2X4 Heterozygous (HTZ) or P2X4 deficient (KO) mice (33) on a C57BL/6 background weighing 20–25g were anesthetized with pentobarbital i.p. (Sigma, St Louis, MO: 50 mg/kg body weight or to effect) and subjected to right nephrectomy and 30 min left renal ischemia as described previously (34, 35). Some mice then received a P2X4 allosteric receptor agonist Ivermectin (10 mg/kg, i.p.) 30 min before 20 min renal ischemia or a selective P2X4 antagonist (5-BDBD [5-(3-Bromophenyl)-1,3-dihydro-2H-benzofuro[3,2-e]-1,4-diazepin-2-on], 1 mg/kg, i.p.) 30 min before 30 min renal ischemia. Sham-operated animals underwent anesthesia followed by laparotomy, right nephrectomy, bowel manipulations and wound closure without renal ischemia. Body temperature of all mice were sustained at ~37°C using a surgical heating pad during surgery as well as during recovery from anesthesia. For pain management, all mice received 0.5–1 mg/kg s.c. buprenorphine SR prior to surgery.
Detection of renal injury after IR
Twenty-four hrs after renal IR injury or sham surgery, we measured plasma BUN and creatinine using an enzymatic blood urea nitrogen (BUN) and creatinine reagent kits (Thermo Fisher Scientific, MA). This creatinine measurement method limits the interferences from mouse plasma chromagens known to occur in the Jaffe method. We also performed qRT-PCR for kidney NGAL mRNA from mice subjected to sham-surgery or to renal IR injury. NGAL is an early and sensitive marker of renal tubular injury (36).
Histological detection of kidney injury
Twenty-four hrs after renal IR injury or sham surgery, kidney hematoxylin-eosin (H&E) sections were blindly assessed using a grading scale of kidney necrotic IR injury to the proximal tubules (0–4, Renal Injury Score) as outlined by Jablonski et al. (37) as described previously. Briefly, the renal pathologist was blinded to the experimental conditions. Deidentified slides were H&E-stained coronal cross-sections of bivalved whole kidney showing full-thickness cortex and medulla. The cortical and medullary parenchyma was evaluated in its entirety in all the microscopic fields covering the entire slide to generate the Jablonski score.
Determination of plasma IL-1β in mice
We measured plasma IL-1β levels in P2X4 WT, P2X4 HTZ and P2X4 KO mice subjected to sham-surgery or renal IR injury using commercially available ELISA kits (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions.
Detection of kidney apoptosis
Twenty four hrs after sham surgery or renal IR injury, TUNEL staining detected fragmented DNA as described (38) using a commercially available kit (Roche, Indianapolis, IN). Apoptotic TUNEL positive cells were quantified in 5–7 randomly chosen 200X microscope images fields in the corticomedullary junction and results were expressed as apoptotic cells counted per 200X field.
Detection of kidney neutrophil infiltration
Kidney neutrophil infiltration after IR injury or sham surgery was detected with immunohistochemistry using rat anti-mouse Ly6G monoclonal antibody (Thermo Fisher Scientific, Inc., Pittsburgh, PA) as described (32, 39). Primary IgG2a antibody (MCA1212, AbD Serotec, Raleigh, NC) was utilized as a negative isotype control (data not shown). Quantification of kidney infiltrating neutrophils was performed using 5–7 randomly chosen 200X microscope image fields (corticomedullary junction for kidney neutrophils) and results were expressed as neutrophils counted per 200X field.
Kidney proximal tubule cell culture
Immortalized human proximal tubular cell line (HK-2; American Type Culture Collection, Manassas, VA) was kept in low glucose DMEM/F12 medium supplemented with 10% FBS at 37°C in a humidified 5% CO2 atmosphere as described (40, 41). Mouse kidney proximal tubules were isolated using Percoll density gradient separation as described previously (42) and maintained in high glucose DMEM/Ham’s F12 medium plus 10% FBS, 2 mM L-glutamine, and 10 mM HEPES. After 24 hrs of serum deprivation, confluent cultured proximal tubule cells were treated with 500 μM ATPγS or with 10 μM ivermectin for 6 hrs or 24 hrs to measure mRNA or protein expressions respectively. Some cells were pretreated with a P2X4 receptor antagonist 10 μM 5-BDBD 30 min before ATPγS treatment.
Immunoblotting
We performed NLRP3, IL-1β and caspase 1 immunoblotting as described previously (43–45). Primary antibodies that recognize NLRP3, pro- and cleaved IL-1β and pro- and cleaved caspase 1 were from Cell Signaling Technology (Danvers, MA).
Q-RTPCR for pro-inflammatory cytokine and chemokine and inflammasome pathway associated mRNA expression
Renal inflammation after IR was also assessed by measuring pro-inflammatory mRNA markers including IL-6, IL-8, intercellular adhesion molecule-1 (ICAM-1), monocyte chemo-attractive protein-1 (MCP-1), macrophage inflammatory protein-2 (MIP-2), and tumor necrosis factor-α (TNF-α) and inflammasome mRNA markers including IL-1β, IL-18 and NLRP3 using quantitative RT-PCR as described previously with primers listed in Table 1 (31, 32). Primer design was based on published GenBank sequences. We measured GAPDH mRNA expression to confirm equal RNA loading.
Table 1.
Primers used in quantitative reverse transcription polymerase chain reactions to amplify mouse cDNAs based on published GenBank sequences. Annealing temperatures used for each primer are also provided.
| Primers | Sequence (Sense/Antisense) | Annealing Temp (°C) |
|---|---|---|
| mouse TNF-α | 5’-TACTGAACTTCGGGGTGATTGGTCC-3’ | 65 ℃ |
| 5’-CAGCCTTGTCCCTTGAAGAGAACC-3’ | ||
| mouse MCP-1 | 5’-ACCTGCTGCTACTCATTCAC-3’ | 60 ℃ |
| 5’-TTGAGGTGGTTGTGGAAAAG-3’ | ||
| mouse MIP-2 | 5’-CCAAGGGTTGACTTCAAGAAC-3’ | 60 ℃ |
| 5’-AGCGAGGCACATCAGGTACG-3’ | ||
| mouse KC | 5’-CAATGAGCTGCGCTGTCAGTG-3’ | 60 ℃ |
| 5’-CTTGGGGACACCTTTTAGCATC-3’ | ||
| mouse IL-6 | 5’-CCGGAGAGGAGACTTCACAG-3’ | 62 ℃ |
| 5’-GGAAATTGGGGTAGGAAGGA-3’ | ||
| mouse ICAM-1 | 5’-TGTTTCCTGCCTCTGAAGC-3’ | 60 ℃ |
| 5’-CTTCGTTTGTGATCCTCCG-3’ | ||
| mouse NGAL | 5’-CACCACGGACTACAACCAGTTCGC-3’ | 66 ℃ |
| 5’-TCAGTTGTCAATGCATTGGTCGGTG-3’ | ||
| mouse P2X4R | 5’-TCGTGTGGGAAAAGGGCTAC-3’ | 66 ℃ |
| 5’-TCCAGTCCCAATTCCACTGC-3’ | ||
| mouse P2X7R | 5’-TGTGTGCATTGACTTGCTCA-3’ | 60 ℃ |
| 5’-CTTGCAGACTTTTCCCAAGC-3’ | ||
| mouse NLRP3 | 5’-CATGGCTGTGTGGATCTTTG-3’ | 66 ℃ |
| 5’-GAGATGCGGGAGAGATATCC-3’ | ||
| mouse IL-1β | 5’-CTGAAAGCTCTCCACCTC-3’ | 56 ℃ |
| 5’-TGCTGATGTACCAGTTGGGG-3’ | ||
| mouse IL-18 | 5’-TCTTGCGTCAACTTCAAGGA-3’ | 60 ℃ |
| 5’-GGGTTCACTGGCACTTTGAT-3’ | ||
| human MCP-1 | 5’-AGCAAGTGTCCCAAAGAAGC-3’ | 64 ℃ |
| 5’-CTCAAAACATCCCAGGGGTA-3’ | ||
| human MIP-2 | 5’-CTTGCCAGCTCTCCTCCTC-3’ | 64 ℃ |
| 5’-GCTTTCTGCCCATTCTTGAG-3’ | ||
| human IL-8 | 5’-TCTGCAGCTCTGTGTGAAGG-3’ | 64 ℃ |
| 5’-ATTGCATCTGGCAACCCTAC-3’ | ||
| human IL-6 | 5’-AAAGAGGCACTGGCAGAAAA-3’ | 64 ℃ |
| 5’-CATGCTACATTTGCCGAAGA-3’ | ||
| human NLRP3 | 5’-CCGAAGTGGGGTTCAGATAA-3’ | 62 ℃ |
| 5’-GATGAGACGCAGTCGTGTGT-3’ | ||
| human IL-1β | 5’-CATTGCTCAAGTGTCTGAAGC-3’ | 62 ℃ |
| 5’-CATGGCCACAACAACTGACG-3’ | ||
| GAPDH | 5’-ACCACAGTCCATGCCATCAC-3’ | 65 ℃ |
| 5’-CACCACCCTGTTGCTGTAGCC-3’ | ||
Statistical analysis
Data were analyzed with Student’s t-test, one-way ANOVA plus Tukey’s post hoc multiple comparison test or Mann–Whitney nonparametric U test to analyze renal injury scores. All data are expressed throughout the text as means ± SEM.
Results
Kidney P2X4 and P2X7 receptor mRNA expression mice
We measured kidney mRNA expression for P2X4 and P2X7 since previous studies suggested heteromeric co-expression of P2X4 and P2X7 (30, 46). Figure 1A shows lack of P2X4 mRNA in P2X4 KO mice kidneys whereas the expression of P2X7 mRNA was similar between P2X4 WT and P2X4 KO mice (N=5–9). P2X4 HTZ mice had ~60% reduction in kidney P2X4 mRNA with equivalent P2X7 mRNA expression.
Figure 1. Confirmation of kidney P2X4 deletion in P2X4 deficient mice and examination of P2X4 expression in kidney epithelia.
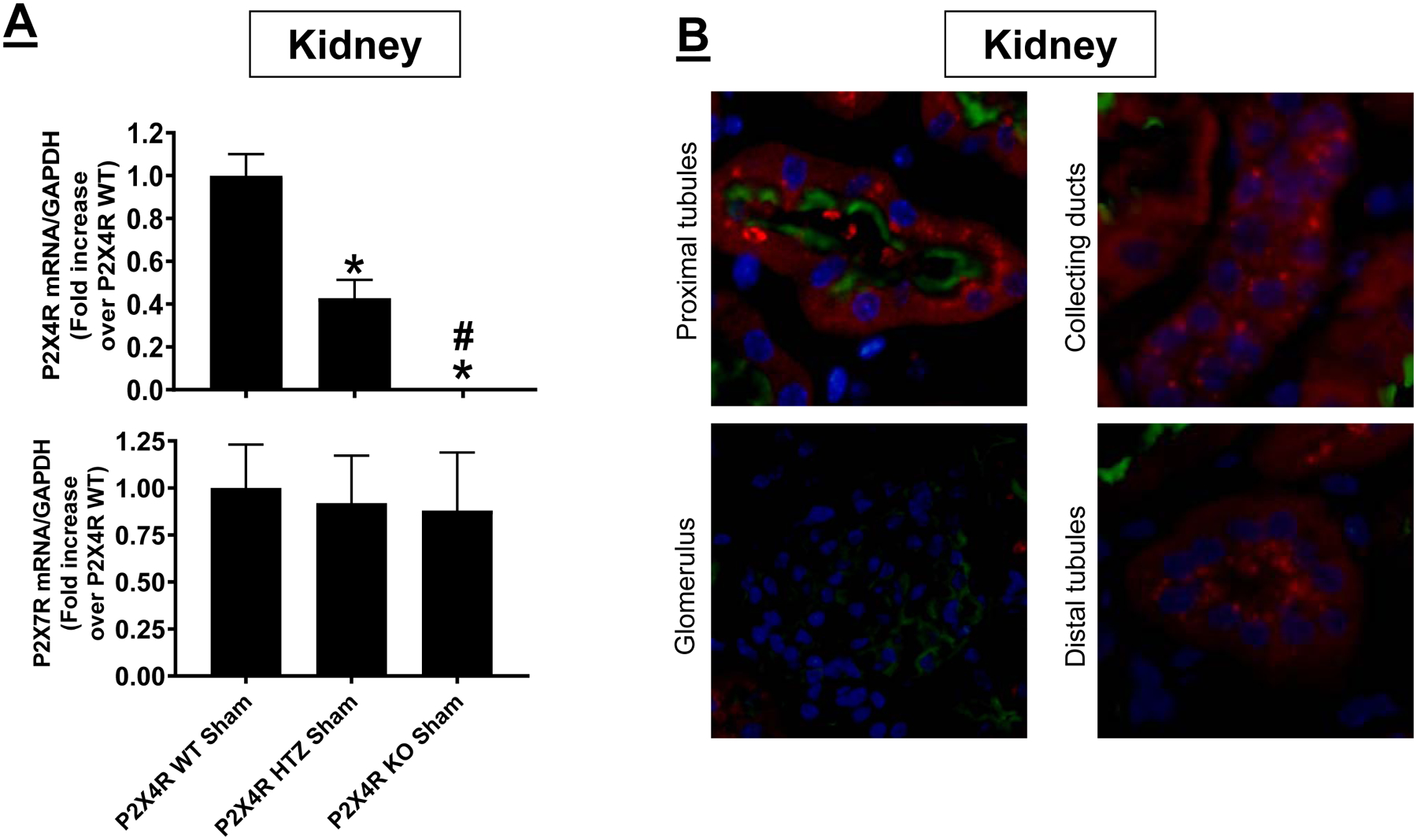
A) Representative gel images and band intensity quantifications of P2X4 mRNA normalized to GAPDH from RT-PCR reactions in kidney tissues of P2X4 wild type (WT), P2X4 heterozygous (HTZ) and P2X4 deficient (KO) mice. For statistical analysis, one-way ANOVA with plus Tukey’s post hoc multiple comparison test was used to detect significant changes. *P<0.05 vs. P2X4 WT mice (N=9) and #P<0.05 vs. P2X4 HTZ mice (N=5). Error bars represent 1 SEM. B) To determine P2X4 expression in kidney epithelia, we detected P2X4 protein expression in various segments of the kidney utilizing the P2X4 tdTomato reporter mice. We show that P2X4 is expressed in proximal tubules (stained with Phaseolus vulgaris leucoagglutinin lectin to identify the brush border of proximal tubules, green), collecting ducts and distal tubule epithelia but was absent in glomeruli (Representative of 7 kidney sections).
We also detected P2X4 protein expression in various segments of the kidney utilizing the P2X4 tdTomato reporter mice (47). Figure 1B (representative of 7 kidney slides) shows that P2X4 expression was evident in proximal tubules stained with Phaseolus vulgaris leucoagglutinin lectin to identify the brush border of proximal tubules (green), collecting duct and distal tubule epithelia but was absent in glomeruli. Renal IR injury did not increase P2X4 expression in tubular and ductal epithelia (data not shown).
P2X4 deficiency protects against ischemic AKI
Plasma creatinine and BUN values were similar between P2X4 WT, P2X4 HTZ and P2X4 KO mice subjected to sham-operation (Figure 2). P2X4 WT mice subjected to 30 min renal IR had significantly higher plasma BUN and creatinine as well as kidney NGAL mRNA (N=5–10) compared to sham-operated mice (N=4–5). We show here that mice lacking P2X4 were protected against ischemic AKI compared to P2X4 WT mice as demonstrated by reduced plasma BUN and creatinine as well as kidney NGAL mRNA expression (N=5–10). P2X4 HTZ mice subjected to renal IR also were protected against AKI when compared to P2X4 WT mice but the attenuation of renal injury was less than that of P2X4 KO mice.
Figure 2. P2X4 deficiency protects against ischemic AKI in mice.
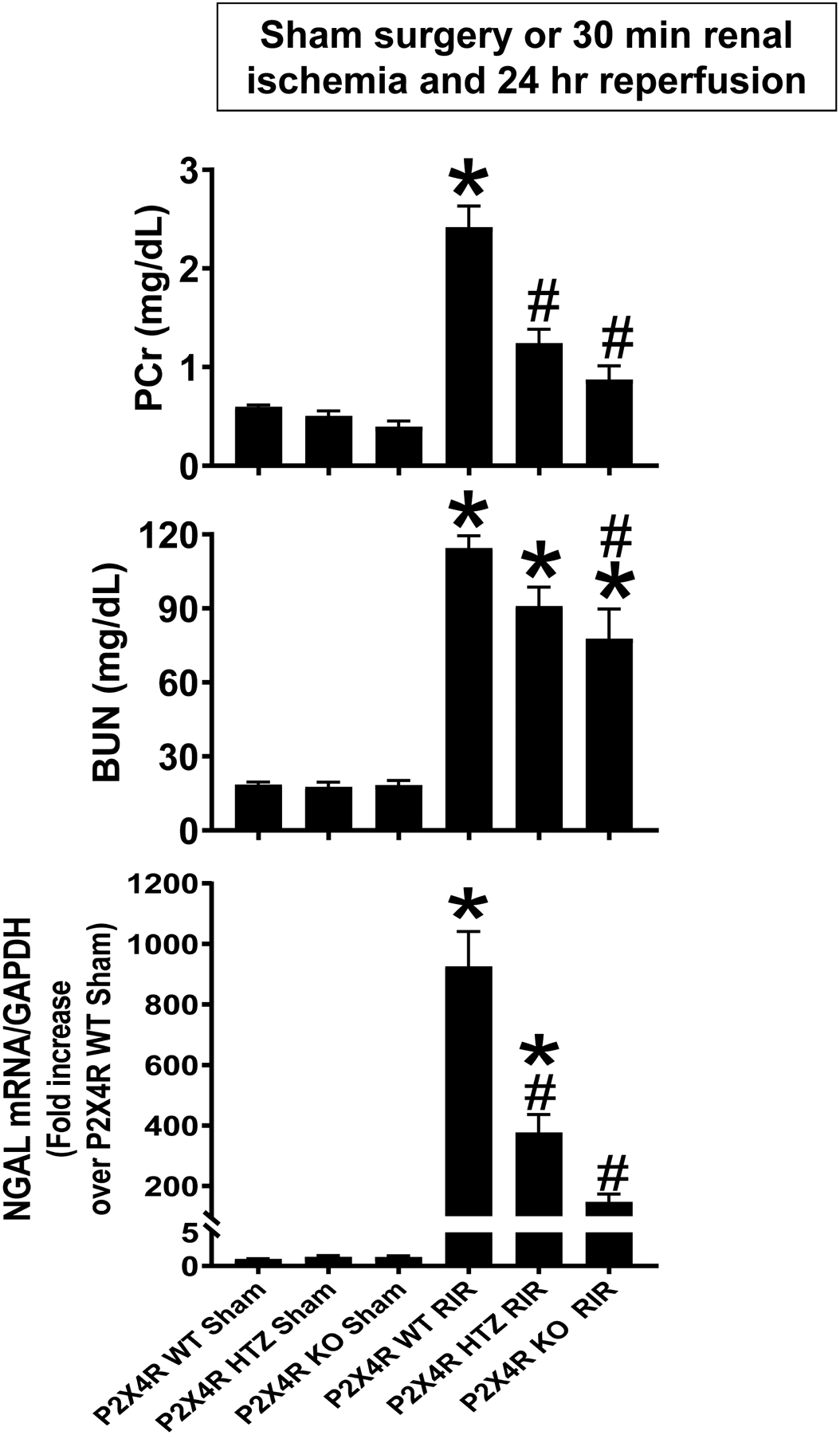
P2X4 wild type (WT), P2X4 heterozygous (HTZ) and P2X4 deficient (KO) mice were subjected to sham-surgery (N=4–5) or to 30 min renal ischemia and 24 hr reperfusion (IR, N=5–10). Plasma BUN and creatinine as well as kidney NGAL mRNA were measured. For statistical analysis, the one-way ANOVA plus Tukey’s post hoc multiple comparison test wase used to detect significant changes. *P<0.05 vs. P2X4 WT mice subjected to sham surgery. #P<0.05 vs. P2X4 subjected to renal IR. Error bars represent 1 SEM.
P2X4 deficiency reduces renal tubular necrosis after ischemic AKI
Figure 3A shows representative kidney H&E images from P2X4 WT, P2X4 HTZ and P2X4 KO mice subjected to sham surgery or 30 min renal ischemia and 24 hr reperfusion (magnification 200X, N=3–5). P2X4 WT mice subjected to renal IR showed severe tubular necrosis and proteinaceous casts as well as increased tubular dilatation and congestion (Figure 3A). P2X4 deficiency in P2X4 HTZ or P2X4 KO mice led to decreased renal tubular necrosis, congestion and cast formation compared to P2X4 WT mice subjected to renal IR. Kidneys from P2X4 KO mice had significantly reduced renal tubular injury score compared to P2X4 WT mice after IR (Figure 3B).
Figure 3. P2X4 deficiency reduces renal tubular necrosis after ischemic AKI.
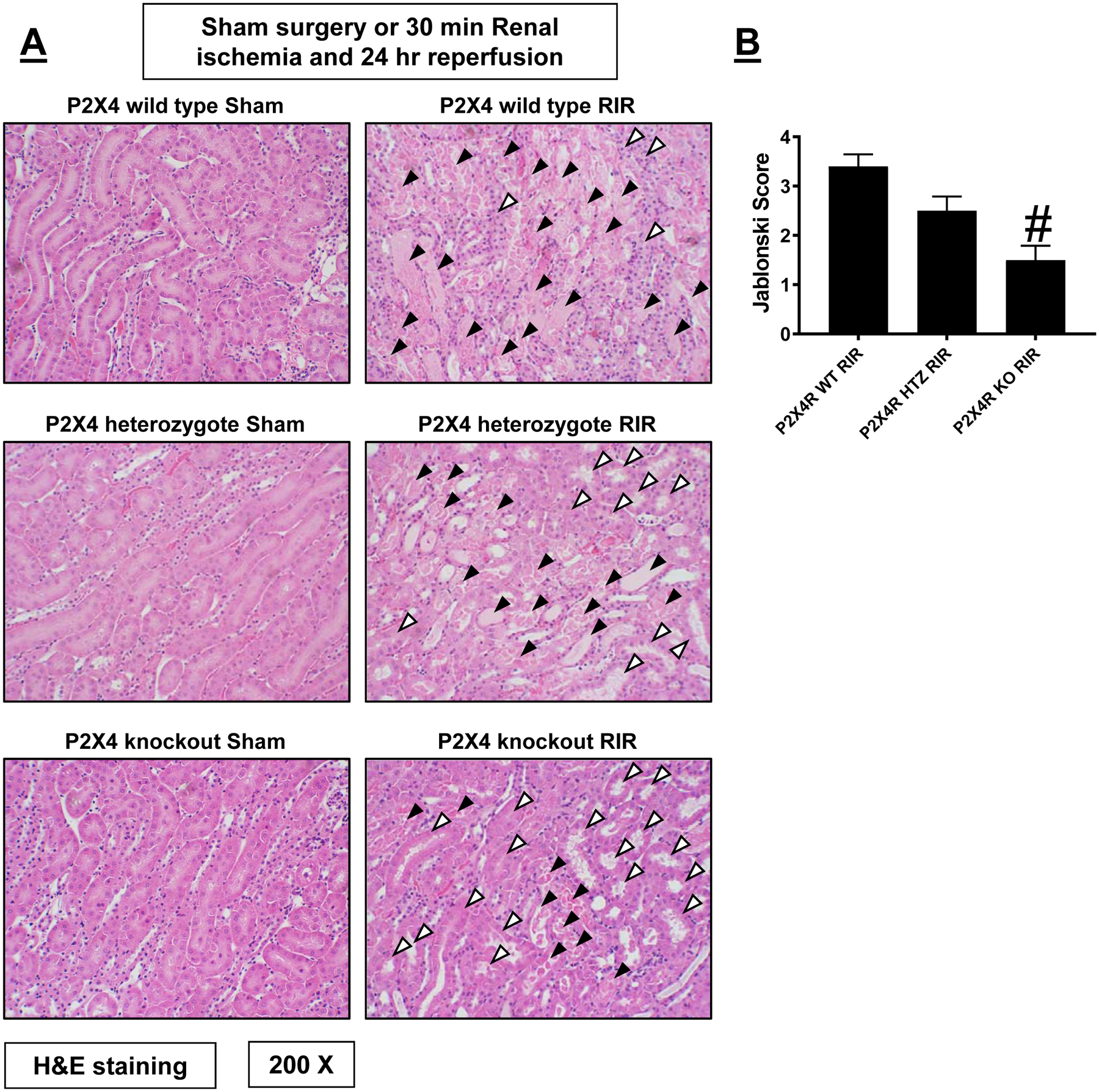
Representative H&E images (from 3–5 experiments) of kidneys from P2X4 wild type (WT), P2X4 heterozygous (HTZ) and P2X4 deficient (KO) mice subjected to sham surgery or to 30 min renal ischemia and 24 hrs reperfusion (IR, magnification 200X). Renal injury scores assessing the degree of renal tubular necrosis is also shown here (scale: 0–4) 24 hrs after renal IR (N=3–5). Filled arrowheads indicate damaged tubules (tubular necrosis, proteinaceous casts, tubular dilatation, and congestion) and blank arrowheads indicate normal or uninjured renal tubules after IR. For statistical analysis, the Mann–Whitney nonparametric test was used to detect significant changes. #P<0.05 vs. P2X4 WT mice subjected to renal IR injury. Error bars represent 1 SEM.
P2X4 deficiency attenuates kidney apoptosis after ischemic AKI
Figure 4A shows representative TUNEL staining images indicative of renal apoptosis and Figure 4B shows counts of TUNEL positive kidney cells (B) from P2X4 WT, P2X4 HTZ and P2X4 KO mice subjected to sham surgery (N=3) or to 30 min renal IR (N=5–7, magnification 200X). Many TUNEL (fragmented DNA) positive cells were detected suggestive of renal tubular apoptosis in the kidneys from P2X4 WT mice subjected to renal IR injury. TUNEL positive kidney cell counts were significantly reduced in P2X4 KO or P2X4 HTZ subjected to renal IR injury.
Figure 4. P2X4 deficiency attenuates kidney apoptosis after ischemic AKI.
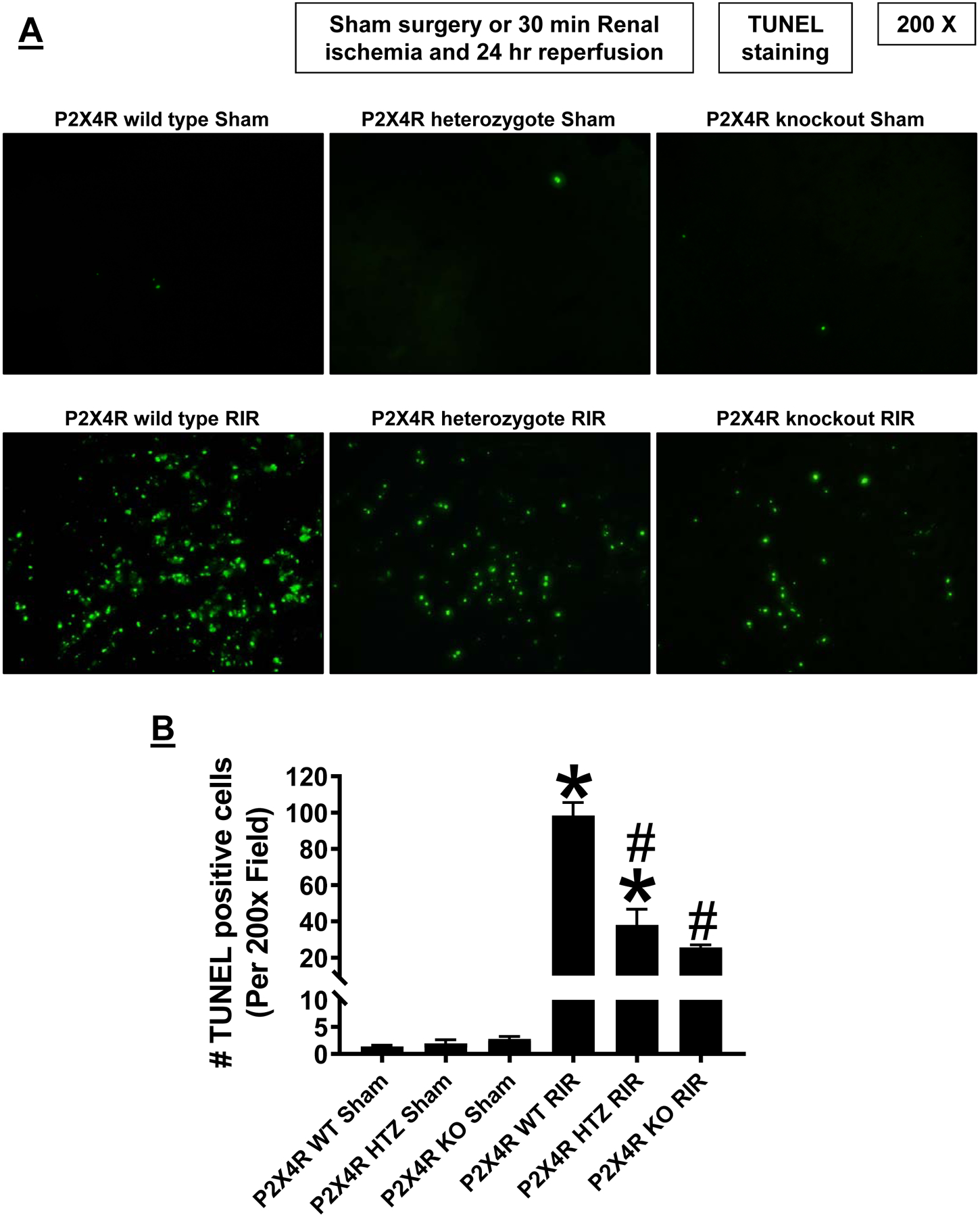
A. Representative images of terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling (TUNEL) staining indicative of renal tubular apoptosis and counts of TUNEL positive kidney cells (B) in the kidneys of P2X4 wild type (WT), P2X4 heterozygous (HTZ) and P2X4 deficient (KO) mice subjected to sham-surgery (N=3) or to 30 min renal ischemia and 24 hr reperfusion (IR, N=5–7 magnification 200X). *P<0.05 vs. P2X4 WT mice subjected to sham surgery. #P<0.05 vs. P2X4 WT mice subjected to renal IR. Error bars represent 1 SEM. For statistical analysis, the one-way ANOVA plus Tukey’s post hoc multiple comparison test was used to detect significant changes.
P2X4 deficiency reduces kidney neutrophil infiltration after ischemic AKI
Figure 5A shows representative immunohistochemistry images and Figure 5B shows counts of infiltrating kidney neutrophils in the kidneys of P2X4 WT, P2X4 HTZ and P2X4 KO mice subjected to sham surgery (N=3) or to 30 min renal IR (N=5–7, magnification 200X). Kidney neutrophil infiltration increased significantly in P2X4 WT mice subjected to renal IR. P2X4 deficiency in P2X4 HTZ or P2X4 KO mice resulted in significantly attenuated kidney neutrophil infiltration after renal IR compared to P2X4 WT mice.
Figure 5. P2X4 deficiency decreases kidney neutrophil infiltration after ischemic AKI.

Representative images of immunohistochemistry for neutrophils (A, dark brown) and counts of infiltrating kidney neutrophils (B) in the kidneys of P2X4 wild type (WT), P2X4 heterozygous (HTZ) and P2X4 deficient (KO) mice subjected to sham-surgery (N=3) or to 30 min renal ischemia and 24 hr reperfusion (IR, N=5–7, magnification 200X). For statistical analysis, the one-way ANOVA plus Tukey’s post hoc multiple comparison test was used to detect significant changes.*P<0.05 vs. P2X4 WT mice subjected to sham surgery. #P<0.05 vs. P2X4 WT mice subjected to renal IR. $ P<0.05 vs. P2X4 HTZ mice subjected to renal IR. Error bars represent 1 SEM.
P2X4 deficiency downregulates pro-inflammatory chemokine and cytokine induction after ischemic AKI
Figure 6 shows fold increases in pro-inflammatory mRNAs normalized to GAPDH for each indicated mRNA (N=5–9). Ischemic AKI increased all pro-inflammatory genes measured in P2X4 WT mice. P2X4 deficiency reduced the expression of neutrophil and macrophage attracting chemokines KC, MIP-2 and MCP-1 after renal IR compared to P2X4 WT mice. Moreover, ICAM-1, IL-6 as well as TNF-α induction was attenuated in P2X4 KO mice. These decreases were more pronounced in P2X4 KO mice when compared to P2X4 HTZ mice.
Figure 6. P2X4 deficiency attenuates kidney pro-inflammatory chemokine/cytokine induction after ischemic AKI.
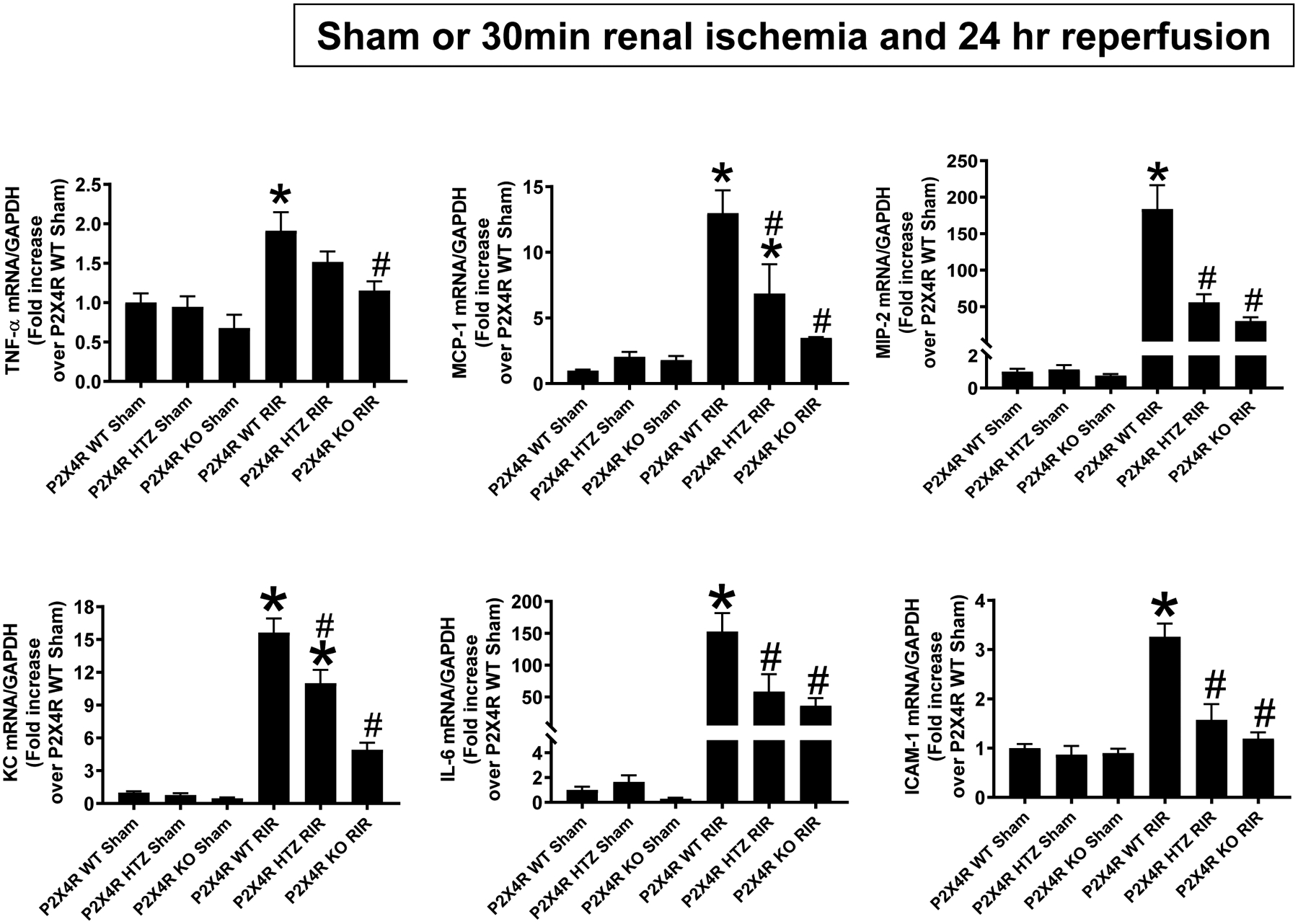
With quantitative RT-PCR, we measured the expression of pro-inflammatory cytokine and chemokine mRNAs [keratinocyte-derived cytokine (KC), monocyte chemoattractive protein-1 (MCP-1), macrophage inflammatory protein-2 (MIP-2), tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6) and intercellular adhesion molecule-1 (ICAM-1)] in the kidneys of P2X4 wild type (WT), P2X4 heterozygous (HTZ) and P2X4 deficient (KO) mice 24 hr after sham-surgery or 30 min renal ischemia (IR). Fold increases in pro-inflammatory mRNAs normalized to GAPDH from quantitative RT-PCR reactions for each indicated mRNA (N=4–6) are shown. For statistical analysis, the one-way ANOVA plus Tukey’s post hoc multiple comparison test wase used to detect significant changes. *P < 0.05 vs. P2X4 WT sham-operated mice. #P < 0.05 vs. P2X4 WT mice subjected to renal IR injury. Error bars represent 1 SEM.
P2X4 antagonist 5-BDBD protects against renal IR injury in mice
Since global P2X4 deficiency may lead to compensatory changes and may not reflect the true physiology of acute receptor deficiency, we complemented the P2X4 gene deletion studies with studies utilizing a specific antagonist (5-BDBD) for mouse P2X4. Sham-operated P2X4 WT mice treated with either vehicle (Intralipid 20%, N=3) or with 1 mg/kg 5-BDBD (N=3) had no detectable renal injury measured by plasma creatinine, BUN and kidney NGAL mRNA (Figure 7A), renal tubular necrosis (Figures 7B and C), renal tubular apoptosis (Figures 7D and E), neutrophil infiltration (Figures 7F and G) and kidney cytokine mRNA induction (Figure 7H). Vehicle-treated P2X4 WT mice subjected to 30 min renal IR (N=5) had significant renal injury, renal tubular necrosis, apoptosis, neutrophil infiltration and kidney cytokine mRNA induction compared to sham-operated mice. Consistent with findings in P2X4 deficient mice, P2X4 WT mice pretreated with 5-BDBD and subjected to renal IR had significantly attenuated renal injury, renal tubular necrosis, apoptosis, neutrophil infiltration and kidney cytokine mRNA induction compared to vehicle-treated P2X4 WT mice subjected to renal IR injury (N=6).
Figure 7. P2X4 antagonism with 5-BDBD protects against renal IR injury in mice.
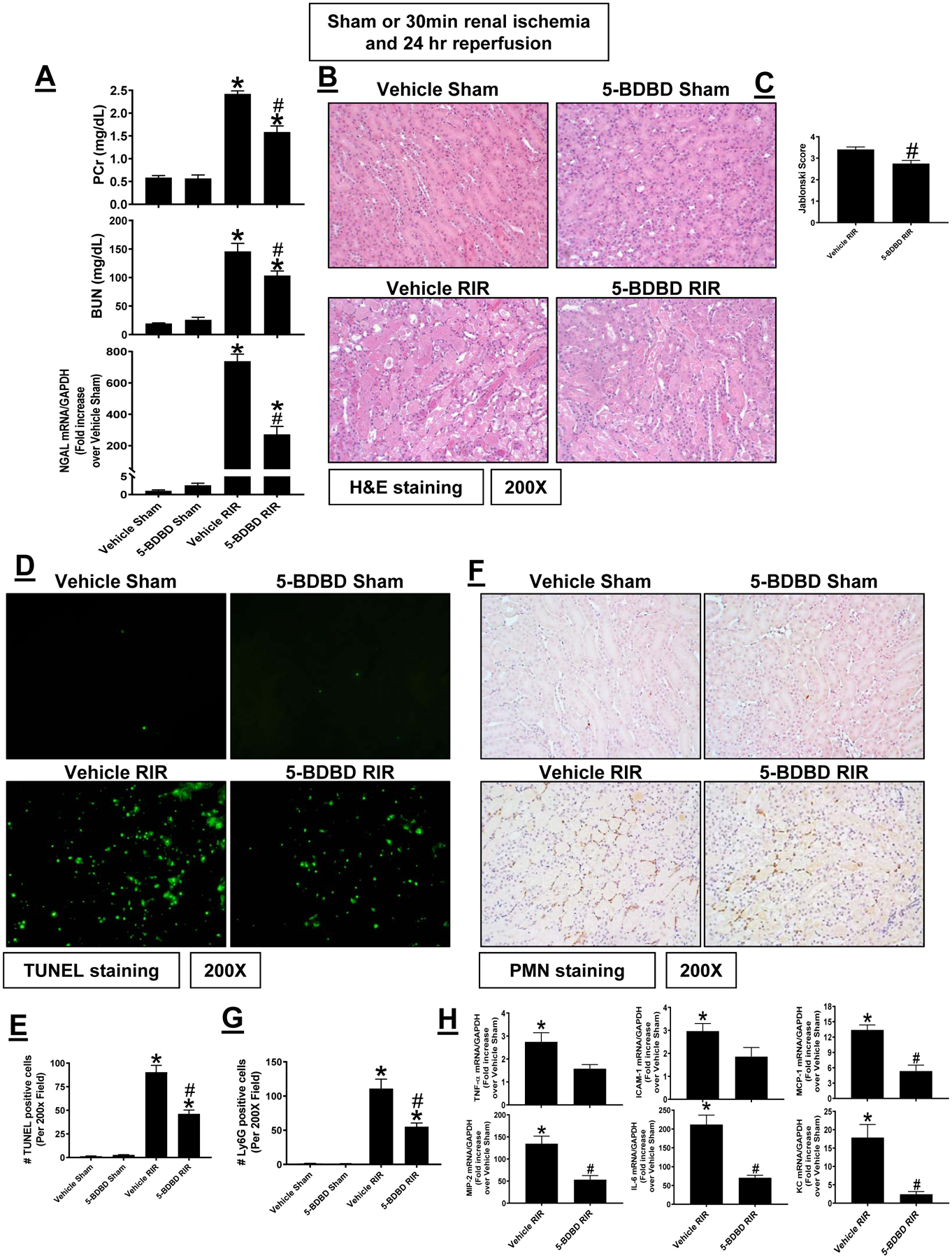
Wild type mice were treated with vehicle (intralipid 20%) or with 5-BDBD (a selective P2X4 antagonist, 1 mg/kg 30 min before surgery) and subjected to sham-surgery or to 30 min renal ischemia and 24 hr reperfusion (IR, N=5–6). 5-BDBD treatment reduced renal injury measured by plasma creatinine, BUN and kidney NGAL mRNA expression (A), renal tubular necrosis (B and C), renal tubular apoptosis (D and E), neutrophil infiltration (F and G) and kidney cytokine mRNA induction (H). TUNEL and neutrophil immunohistochemistry show 200X magnification images. For RTPCR, fold increases in pro-inflammatory mRNAs normalized to GAPDH from quantitative RT-PCR reactions for each indicated mRNA are shown. *P < 0.05 vs. P2X4 WT sham-operated mice. #P < 0.05 vs. P2X4 WT mice subjected to renal IR injury. Error bars represent 1 SEM.
P2X4 allosteric agonist Ivermectin exacerbates ischemic AKI in mice
Since P2X4 deletion or antagonist protected against ischemic AKI, we next tested whether acute P2X4 activation exacerbates renal IR injury in mice. Sham-operated P2X4 WT mice treated with either vehicle (Intralipid 20%) or with a P2X4 allosteric agonist Ivermectin (10 mg/kg) 30 min before sham surgery had no detectable renal injury measured by plasma creatinine, BUN and NGAL (data not shown), renal tubular necrosis (Figures 8B and C), renal tubular apoptosis (Figures 8D and E), neutrophil infiltration (Figures 8F and G) and kidney cytokine mRNA induction (Figure 8H). Vehicle-treated P2X4 WT mice subjected to 20 min. renal IR had moderate renal injury with mild renal tubular necrosis, apoptosis, neutrophil infiltration and kidney cytokine mRNA induction (Figure 8, N=4). Consistent with the hypothesis that P2X4 activation is detrimental for renal IR, P2X4 WT mice treated with Ivermectin and subjected to mild 20 min renal IR suffered increased renal injury, renal tubular necrosis, apoptosis, neutrophil infiltration and kidney cytokine mRNA induction compared to vehicle treated P2X4 WT mice subjected to 20 min renal IR injury (N=4–5).
Figure 8. P2X4 allosteric agonist Ivermectin exacerbates ischemic AKI in mice.
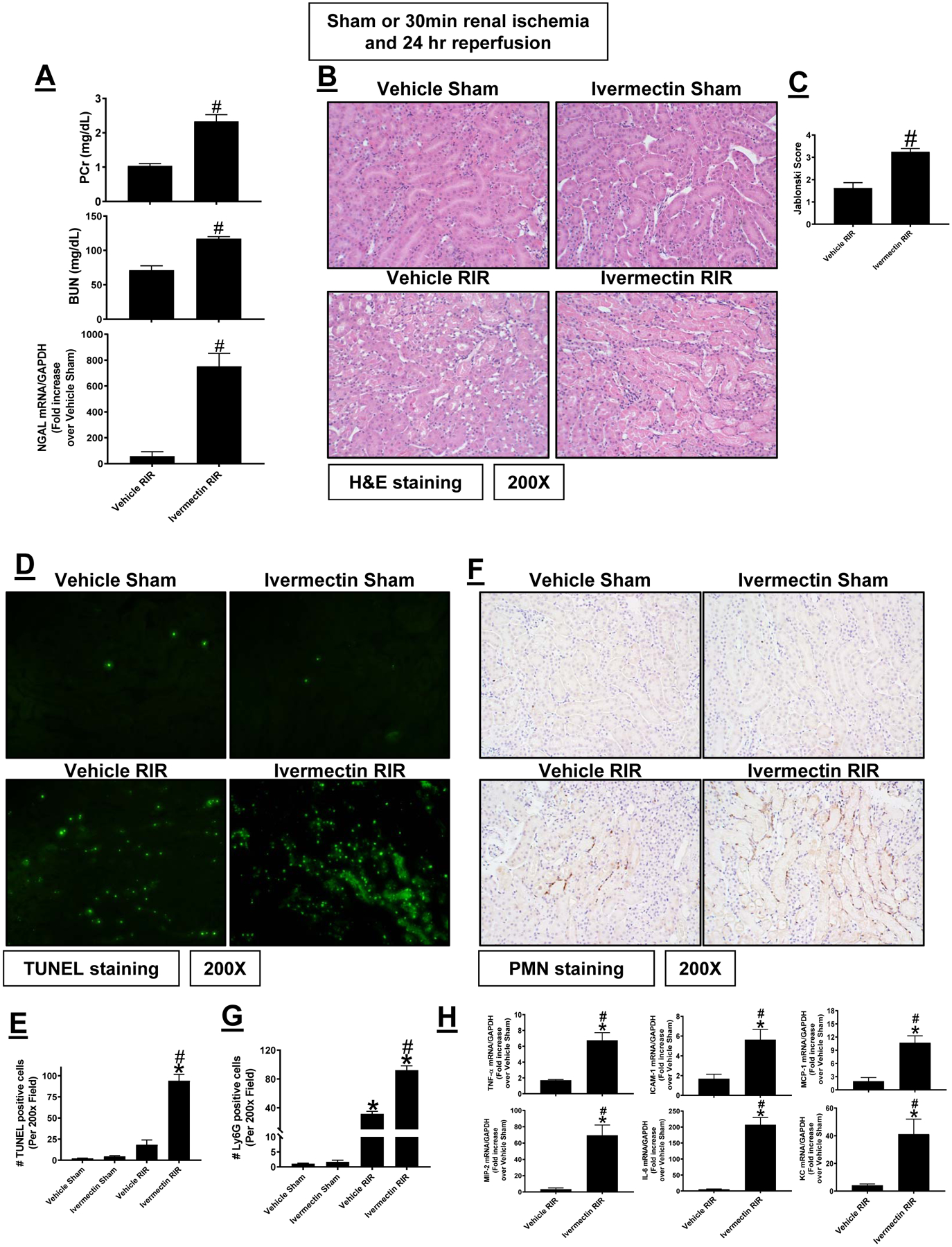
Wild type mice were treated with vehicle (intralipid 20%) or with ivermectin (a P2X4 allosteric agonist, 10 mg/kg 30 min before surgery) and subjected to sham-surgery or to 20 min renal ischemia and 24 hr reperfusion (IR, N=4–5). Ivermectin treatment increased renal injury measured by plasma creatinine, BUN and kidney NGAL mRNA (A), renal tubular necrosis (B and C), renal tubular apoptosis (D and E), neutrophil infiltration (F and G) and kidney cytokine mRNA induction (H). TUNEL and neutrophil immunohistochemistry show 200X magnification images. For RTPCR, fold increases in pro-inflammatory mRNAs normalized to GAPDH from quantitative RT-PCR reactions for each indicated mRNA are shown. *P < 0.05 vs. P2X4 WT sham-operated mice. #P < 0.05 vs. P2X4 WT mice subjected to renal IR injury. Error bars represent 1 SEM.
Induction of renal tubular NLP3 inflammasome signaling with P2X4 activation
P2X4 signaling induces NLRP3 inflammasome signaling in diabetes and bladder injury models (28, 48) and NLRP3 inflammasome is critical for exacerbation of ischemic AKI (49–51). Kidneys from P2X4 KO mice subjected to 30 min renal IR had significantly attenuated induction of NLRP3, IL-1β and IL-18 mRNA after renal IR compared to P2X4 WT mice supporting the hypothesis that P2X4 directly induces kidney NLRP3 inflammasome signaling (Figure 9A, N=5–9). Furthermore, kidneys from P2X4 KO mice had significantly reduced induction of NLRP3, pro- as well as cleaved forms of caspase 1 and IL-1β protein expression after renal IR compared to P2X4 WT mice (Figure 9B, N=4–5). Consistent with these findings, plasma IL-1β levels were significantly attenuated in P2X4 KO mice when compared to P2X4 WT mice subjected to renal IR (Figure 9C, N=5–7). These results suggest that P2X4 signaling upregulates kidney NLRP3 inflammation pathway perhaps leading to increased renal tubular inflammatory response and renal cell death after IR.
Figure 9. P2X4-mediated induction of renal tubular NLP3 inflammasome signaling.
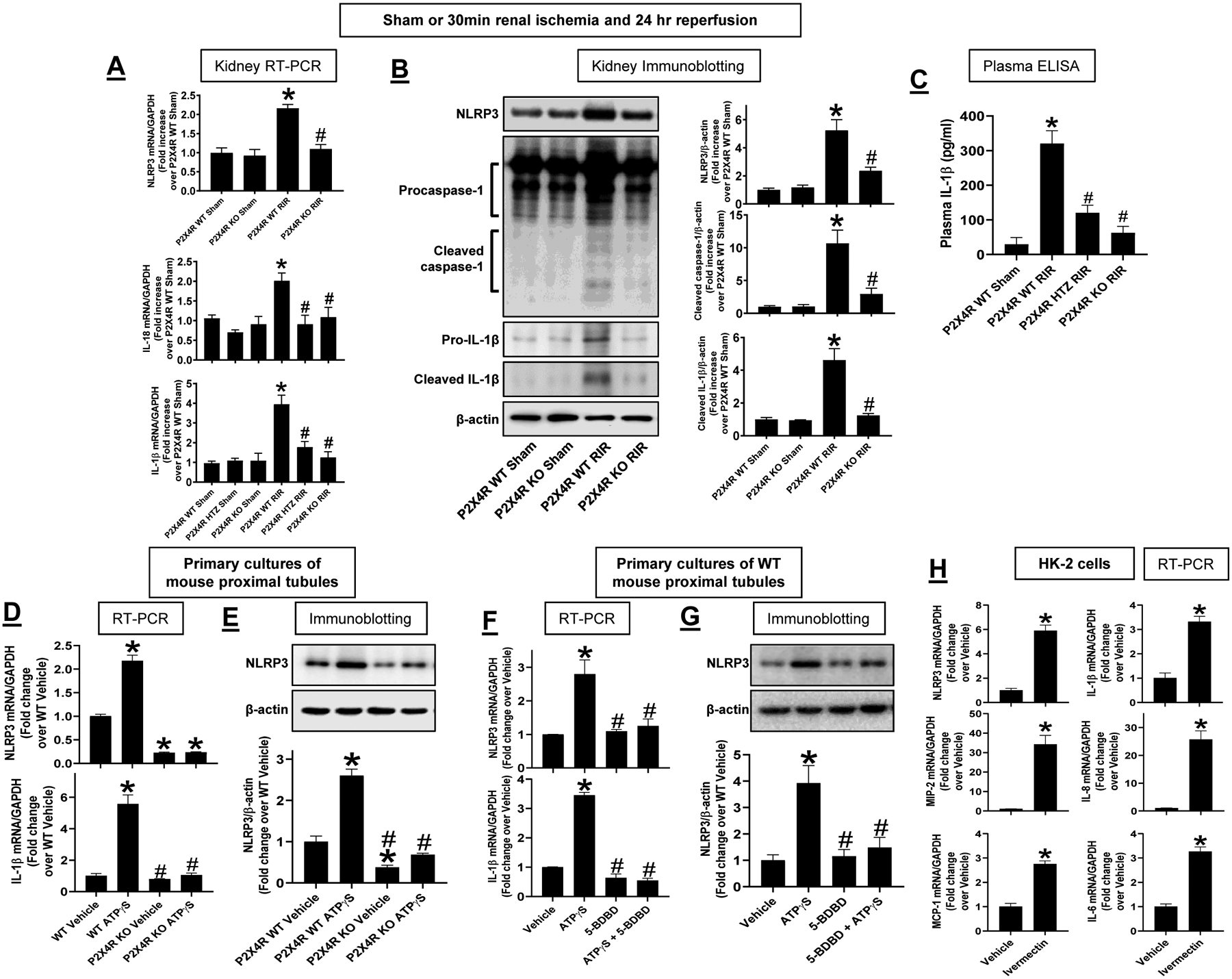
A. With RT-PCR, we measured the expression of NLRP3, IL-1β and IL-18 mRNAs in the kidneys of P2X4 wild type (WT), P2X4 heterozygous (HTZ) and P2X4 deficient (KO) mice 24 hr after sham-surgery or 30 min renal ischemia (IR). Fold increases in mRNAs normalized to GAPDH from quantitative RT-PCR reactions for each indicated mRNA (N=5–9) are shown. B. Representative immunoblotting images for NLRP3, pro- and cleaved caspase 1 and pro- and cleaved IL-1β (top) and band intensity quantifications normalized to β-actin (N=4–5, bottom) in the kidneys of P2X4 WT, P2X4 HTZ and P2X4 KO mice 24 hr after sham-surgery or 30 min renal ischemia. C. Plasma IL-1β measured by ELISA in P2X4 WT mice subjected to sham surgery and P2X4 WT, P2X4 HTZ and P2X4 KO mice subjected to 30 m in renal ischemia and 24 hr reperfusion (N=5–7). D. RTPCR measurements of NLRP3 and IL-1β mRNA in primary cultures of renal proximal tubules from P2X4 WT and P2X4 KO mice treated with vehicle (DW) or with 500 μM ATPγS for 6 hr (N=3). Fold increases in mRNAs normalized to GAPDH from quantitative RT-PCR reactions for each indicated mRNA are shown. E. Representative immunoblotting images for NLRP3 (top) and band intensity quantifications normalized to β-actin (N=3, bottom) in primary cultures of renal proximal tubules from P2X4 WT and P2X4 KO mice treated with vehicle (vehicle) or with 500 μM ATPγS for 24 hr (N=3). F. RTPCR measurements of NLRP3 and IL-1β mRNA in primary cultures of renal proximal tubules from P2X4 WT mice treated with vehicle (DW) or with 500 μM ATPγS for 6 hr (N=3). To test for the effect of blocking P2X4, some cells were pretreated with 10 μM 5-BDBD 30 min before ATPγS treatment. Fold increases in mRNAs normalized to GAPDH from quantitative RT-PCR reactions for each indicated mRNA are shown. G. Representative immunoblotting images for NLRP3 (top) and band intensity quantifications normalized to β-actin (N=3, bottom) in primary cultures of renal proximal tubules from P2X4 WT mice treated with vehicle (DW) or with 500 μM ATPγS for 6 hr (N=3). Some cells were pretreated with 10 μM 5-BDBD 30 min before ATPγS treatment for 24 hr. H. RTPCR measurements of NLRP3 and IL-1β mRNA as well as pro-inflammatory cytokines (MIP-2, IL-8, IL-6 and MCP-1) in human renal proximal tubule (HK-2) cells treated with vehicle (DMSO) or with 10 μM an P2X4 allosteric agonist Ivermectin for 6 hr (N=6). Fold increases in mRNAs normalized to GAPDH from quantitative RT-PCR reactions for each indicated mRNA are shown.
To determine whether P2X4 activates renal proximal tubular NLRP3 inflammasome signaling, we isolated renal proximal tubule cells from P2X4 WT or P2X4 KO mice and treated the cells with a P2X4 agonist ATPγS (500 μM) for 6 hrs (Figure 9D) or 24 hrs (Figure 9E). Figure 9D shows significant induction of NLRP3 and IL-1β mRNA in primary cultures of P2X4 renal proximal tubule cells treated with ATPγS (N=3). In contrast, ATPγS failed to induce NLRP3 and IL-1β mRNA in renal proximal tubules cells from P2X4 KO mice. Similarly, ATPγS induced NLRP3 protein expression in P2X4 WT but not from P2X4 KO renal proximal tubule cells (Figure 9E, N=3). To complement the gene deletion studies, we treated primary cultures of renal proximal tubule cells with either vehicle or with 5-BDBD (10 μM), a specific P2X4 antagonist. Figure 9F and Figure 9G show that 5-BDBD significantly attenuated the ATPγS-mediated induction of NLRP3 and IL-1β mRNA and NLRP3 protein when compared to vehicle-treated renal proximal tubule cells. Furthermore, Ivermectin (10 μM) induced NLRP3 and IL-1β mRNA in human renal proximal tubule (HK-2) cells when compared to vehicle-treated cells (Figure 9H, N=6). In addition, ivermectin increased the expression of MIP-2, IL-8, MCP-1 and IL-6 mRNA expression in HK-2 cells consistent with P2X4-mediated exacerbation of renal tubular inflammation.
Discussion
In this study, we demonstrate that P2X4 has a detrimental role in renal IR injury by accentuating renal tubular necrosis, inflammation and apoptosis. Mice deficient in P2X4 and WT mice pretreated with a selective P2X4 antagonist had significantly reduced ischemic AKI with decreased renal tubular necrosis, inflammation and apoptosis. Conversely, an allosteric enhancer for P2X4 markedly increased renal dysfunction after IR with exacerbated renal tubular necrosis, inflammation and apoptosis. We also demonstrated that P2X4 directly induces NLRP3 inflammasome signaling in renal proximal tubule cells providing a potential mechanism for P2X4-mediared exacerbation of ischemic AKI.
There is no effective therapy or preventive measure for clinical AKI. Many promising potential therapies based on preclinical research failed to be effective clinically (52, 53). The main reason for lack of effective therapy for clinical AKI is the complex pathology it entails in a very heterogeneous patient population (54–56). Renal tubular cell death after IR results in several distinct pathways of cell death including necrosis and inflammation followed by renal tubular apoptosis (55). Therefore, it is critical that a renal protective strategy should be able to attenuate all or most of these cell injury pathways after IR.
Extracellular ATP released from dying or dead renal cells activate both P2X and P2Y purinergic receptors. Several P2 receptors are implicated in extracellular ATP-mediated inflammatory processes including P2Y2, P2X7 and P2X4 (19, 20). Of P2X receptor subtypes, the detrimental role for P2X7 in inflammatory response and ischemic cell death is widely studied (19, 23, 26). Although P2X4 and P2X7 have similar cellular effects, P2X7 expression is detected only under pathological condition and injured cells whereas P2X4 expression is detectable in normal physiological conditions (19, 26). Moreover, the role for P2X4 in ischemic renal cell death and inflammation is not known. We show here that deletion or antagonism of P2X4 decreases renal tubular necrosis, inflammation and apoptosis providing a scientific basis for modulating P2X4 to treat clinical AKI.
The mechanisms by which P2X4 activation leads to increased renal tubular cell death after IR remain unclear. Recent studies implicated P2X4-mediated activation of NOD-like receptor protein 3 (NLRP3) inflammasome signaling in several models including a diabetic nephropathy model of kidney injury and a bladder inflammation model (28, 48). We show in this study that P2X4 activation in cultured renal proximal tubule cells induces mRNA and protein expression of proteins assembled to form NLRP3 inflammasome, a critical component of the innate immune system. The NLRP3 inflammasome is a large (>700kDa) intracellular multiprotein complex that translates danger-associated molecular pattern molecule signaling and is critically important for the activation of inflammatory responses as well as programmed pro-inflammatory cell death pathway referred to as pyroptosis (57, 58). Upon activation, NLRP3 protein complex oligomerizes and recruits the apoptosis-associated speck-like protein containing a caspase recruiting domain (ASC) and caspase 1 to form the inflammasome with subsequent caspase 1 activation. Inflammasome activation and assembly promote maturation, proteolytic cleavage and secretion of pro-inflammatory cytokines IL-1β and IL-18 by activated caspase 1 of the inflammasome component. Active caspase 1 also drives pyroptosis in addition to cleaving to release active IL-1β (59).
We show that renal IR injury caused significant induction of NLRP3 inflammasome components including induction of renal tubular NLRP3 expression as well as upregulation of renal IL-1β and caspase 1. Consistent with the role of P2X4-mediated induction of NLRP3 inflammasome signaling, P2X4 KO mice as well as P2X4 WT mice treated with 5-BDBD had significantly reduced induction of NLRP3 inflammasome signaling and reduced inflammatory response (less cytokine generation and renal tubular neutrophil infiltration). Suggesting a critical role of NLRP3 inflammasome signaling in ischemic AKI, NLRP3 KO mice suffer reduced kidney injury after IR (50, 51). Indeed, deleting or inhibiting NLRP3 inflammasome formation attenuates renal tubular inflammation and damage in mice after ureteral obstruction or after kidney ischemia and reperfusion injury (51, 60). Furthermore, pharmacological inhibition of NLRP3 signaling attenuates renal IR injury (61, 62). Finally, studies show divergent and tissue specific role for leukocyte and renal tubular epithelial NLRP3 signaling in AKI and recovery from AKI. Leukocyte-associated NLRP3 drives renal tubular apoptosis after IR whereas kidney NLRP3 regulates tissue-healing process after renal IR (49). Our findings suggest that P2X4 receptor activation during and after ischemic AKI induces renal tubular NLRP3 inflammasome signaling that may exacerbate ischemic AKI. P2X4-mediated NLPR3 inflammasome induction with subsequent cleavage and release of mature IL-1β intra-renally and to systemic circulation may drive systemic inflammatory response and remote organ dysfunction that occur after AKI (57).
The exact mechanism by which P2X4 activation leads to induction of NLRP3 signaling remains unknown. P2X4 receptor, typical of all P2X receptors, is a ligand-gated cation channel that opens with ATP-mediated activation (20, 25). The P2X4 has a high permeability to calcium leading to intracellular depolarization and activation of diverse calcium-dependent intracellular processes and enzymes (20, 24, 25). Indeed, intracellular calcium influx and subsequent depolarization are early hallmark events during necrosis, inflammation and apoptosis (63–65). Furthermore, influx of calcium may lead to pathological levels of intracellular reactive oxygen species generation (66, 67). Pathological intracellular calcium overload as well as reactive oxygen species generation leads to mitochondrial dysfunction and subsequent cell death (68, 69). Increased reactive oxygen species generation as well as calcium overload with mitochondrial dysfunction that may follow may have resulted in induction of NLRP3 inflammasome signaling.
Complicating the role of P2X4 and injury response further, not all effects of P2X4 activation are detrimental to cellular survival (20, 25, 27, 70, 71). Most studies support a detrimental role for P2X4 in cellular and tissue injury including the lung, heart, brain, kidney and central nervous system as well as several models of injury including sepsis, ischemia and inflammation (20, 25). However, protective role for P2X4 also has been suggested. Cardiac-specific overexpression of the P2X4R was associated with an enhanced survival after LAD ligation (71). P2X4 deletion is neuroprotective acutely but induces a depressive phenotype during recovery from ischemic stroke (72). Suggesting a protective role for renal P2X4, P2X4 deficient mice had exacerbated kidney fibrosis following unilateral ureteric obstruction (27). Furthermore, P2X4 receptor is required for neuroprotection via ischemic preconditioning (73). In addition, P2X4 receptors are protective against polymicrobial sepsis-induced organ injury and inflammation (74).
It appears that the balance between ATP and adenosine signaling is critical for ischemic and inflammatory tissue injury. ATP released by necrotic or dying cells is rapidly converted to adenosine via serial enzymatic actions of ecto-nucleoside-triphosphate-diphosphohydrolase1 (E-NTPDase1 or CD39) and ecto-5’-nucleotidase (CD73) (1, 2). Whereas ATP serves an pro-inflammatory and injurious molecule in many organs and cell types via activation of P2 receptors, extracellular adenosine attenuates kidney injury and inflammation via acting on P1 receptors in the kidney, heart and intestine (2, 5). Indeed, activation of A1, A2a or A2B P1 adenosine receptors protects against ischemic and inflammatory injury in the liver, kidney, intestine and heart (3, 5–7). Therefore, adenosine appears to counteract the detrimental effects of ATP indicating critical homeostatic balance between detrimental ATP signaling by P2 purinergic receptor activation and tissue protective adenosine signaling by P1 purinergic receptors.
Furthermore, modulation of purinergic signaling with exogenous P1 agonists as well as enzymes that promote P1 purinergic signaling can modulate many other biological as well as pathological processes including cardiovascular function, coagulation, inflammation and ischemic tissue injury (12–15). Interestingly, some of the tissue protective effects of P1 adenosine receptor signaling are mediated via activation of hypoxia inducible factor (HIF) transcription factors that appears to counterbalance the detrimental effects of ATP-mediated P2 purinergic receptor activation (18, 75). Indeed, adenosine produced from ATP degradation activates HIF transcription signaling to protect against ischemia, hypoxia and inflammatory response via P1 adenosine receptor activation (75, 76).
In summary, we demonstrate in this study that P2X4 receptor activation exacerbates ischemic AKI by potentiating renal tubular necrosis, inflammation and apoptosis. We hypothesize that ATP released by dying or dead renal cells after IR activates P2X4 leading to cellular depolarization due to rapid calcium influx (Figure 10). Subsequent NLRP3 inflammasome activation and induction result in IL-1β processing by activated caspase 1 would lead to renal tubular inflammatory and apoptotic cell death and systemic inflammatory response. Targeting P2X4 may provide a novel therapeutic approach to treat ischemic AKI. Further studies are needed to elucidate the detailed mechanistic link between P2X4 and NLRP3 inflammasome activation.
Figure 10. A proposed mechanism of P2X4 receptor-mediated exacerbation of ischemic AKI.
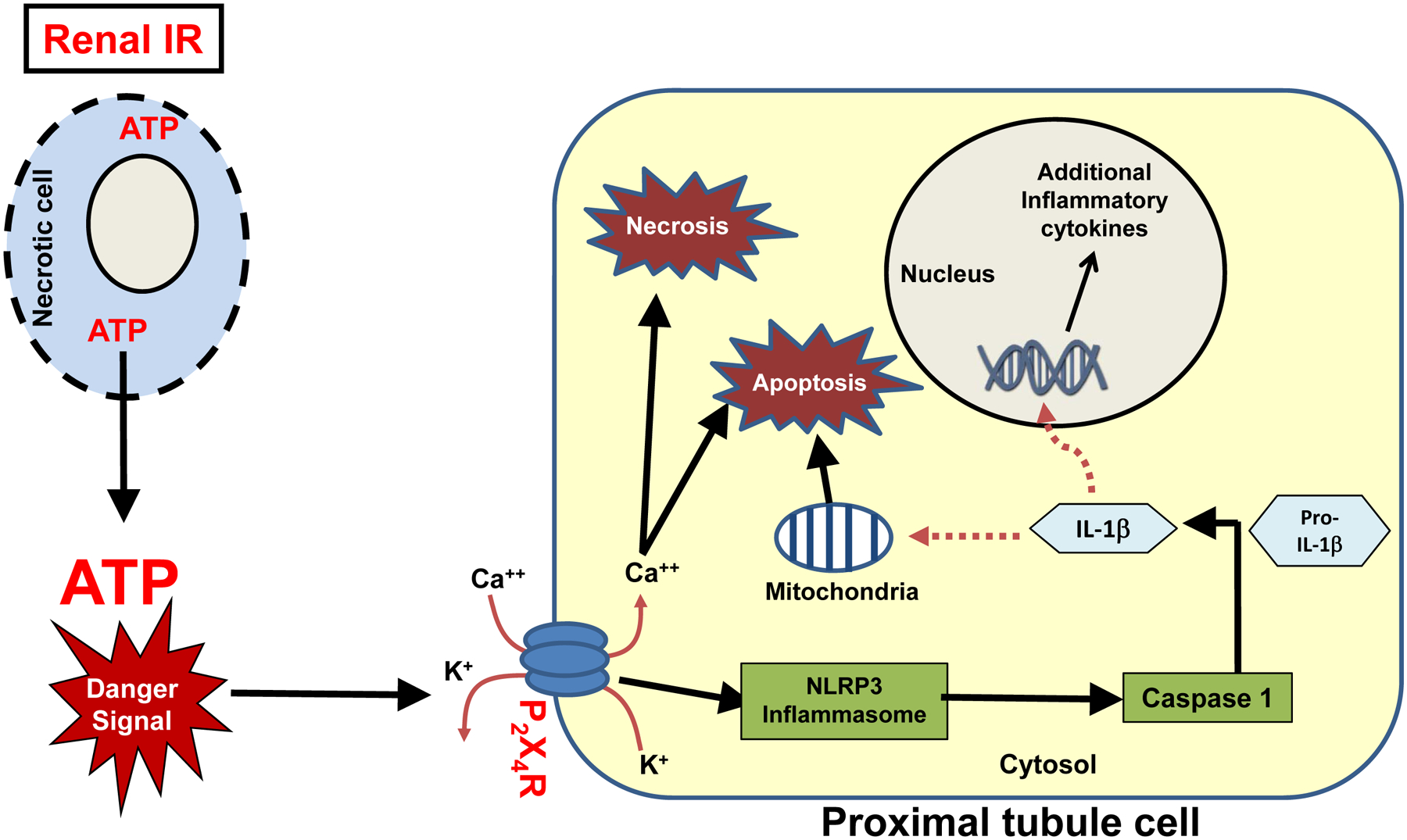
Our data show that P2X4 activation in renal proximal tubules induce NLRP3 inflammasome synthesis. Dying or necrotic renal cells release ATP to extracellular space. Opening of the P2X4 channels in response to extracellular ATP leads to K+ efflux and Ca++ influx potentially driving reactive oxygen species and NLRP3 inflammasome assembly. Subsequent caspase 1 activation, cleavage, and release of active IL-1β can induce additional renal tubular apoptosis and inflammation as well as systemic inflammatory response.
Acknowledgement:
This work was supported in part by Department of Anesthesiology, Columbia University and by NIH grants DK-109544 and DK-115694 to HTL and R01GM066189 and R01DK113790 to GH.
Nonstandard Abbreviations:
- AKI
acute kidney injury
- IR
ischemia and reperfusion
- DAMP
Damage Associated Molecular Pattern
- 5-BDBD
5-(3-Bromophenyl)-1,3-dihydro-2H-benzofuro[3,2-e]-1,4-diazepin-2-on
- BUN
blood urea nitrogen
- NGAL
neutrophil gelatinase-associated lipocalin
- ICAM-1
intercellular adhesion molecule-1
- MCP-1
monocyte chemo-attractive protein-1
- MIP-2
macrophage inflammatory protein-2
- TNF-α
tumor necrosis factor-α
- IL-6
Interleukin-6
- KC
keratinocyte chemoattractant
- NLRP3
nucleotide-binding domain (NOD)-like receptor protein 3
- TUNEL staining
terminal deoxynucleotidyl transferase dUTP nick end labeling staining
Footnotes
Disclosures: GH owns stock in Purine Pharmaceuticals, Inc., which has a patent license (62/532,619 and 62/562,770) for the use of P2X4 agonists in infections and sepsis.
References
- 1.Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, Robson SC, and Colgan SP (2003) Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. The Journal of experimental medicine 198, 783–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eltzschig HK, Weissmuller T, Mager A, and Eckle T (2006) Nucleotide metabolism and cell-cell interactions. Methods Mol Biol 341, 73–87 [DOI] [PubMed] [Google Scholar]
- 3.Yap SC, and Lee HT (2012) Adenosine and protection from acute kidney injury. Curr Opin Nephrol Hypertens 21, 24–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bauerle JD, Grenz A, Kim JH, Lee HT, and Eltzschig HK (2011) Adenosine generation and signaling during acute kidney injury. J Am Soc Nephrol 22, 14–20 [DOI] [PubMed] [Google Scholar]
- 5.Eltzschig HK, Bonney SK, and Eckle T (2013) Attenuating myocardial ischemia by targeting A2B adenosine receptors. Trends Mol Med 19, 345–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hart ML, Jacobi B, Schittenhelm J, Henn M, and Eltzschig HK (2009) Cutting Edge: A2B Adenosine receptor signaling provides potent protection during intestinal ischemia/reperfusion injury. J Immunol 182, 3965–3968 [DOI] [PubMed] [Google Scholar]
- 7.Han SJ, and Lee HT (2019) Mechanisms and therapeutic targets of ischemic acute kidney injury. Kidney Res Clin Pract 38, 427–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okusa MD, Linden J, Huang L, Rieger JM, Macdonald TL, and Huynh LP (2000) A(2A) adenosine receptor-mediated inhibition of renal injury and neutrophil adhesion. Am. J. Physiol Renal Physiol 279, F809–F818 [DOI] [PubMed] [Google Scholar]
- 9.Lee HT, Kim M, Joo JD, Gallos G, Chen JF, and Emala CW (2006) A3 adenosine receptor activation decreases mortality and renal and hepatic injury in murine septic peritonitis. Am. J. Physiol Regul. Integr. Comp Physiol 291, R959–R969 [DOI] [PubMed] [Google Scholar]
- 10.Gallos G, Ruyle TD, Emala CW, and Lee HT (2005) A1 adenosine receptor knockout mice exhibit increased mortality, renal dysfunction, and hepatic injury in murine septic peritonitis. Am. J. Physiol Renal Physiol 289, F369–F376 [DOI] [PubMed] [Google Scholar]
- 11.Lee HT, Gallos G, Nasr SH, and Emala CW (2004) A1 adenosine receptor activation inhibits inflammation, necrosis, and apoptosis after renal ischemia-reperfusion injury in mice. J. Am. Soc. Nephrol 15, 102–111 [DOI] [PubMed] [Google Scholar]
- 12.Koeppen M, Eckle T, and Eltzschig HK (2009) Selective deletion of the A1 adenosine receptor abolishes heart-rate slowing effects of intravascular adenosine in vivo. PloS one 4, e6784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eckle T, Kewley EM, Brodsky KS, Tak E, Bonney S, Gobel M, Anderson D, Glover LE, Riegel AK, Colgan SP, and Eltzschig HK (2014) Identification of hypoxia-inducible factor HIF-1A as transcriptional regulator of the A2B adenosine receptor during acute lung injury. J Immunol 192, 1249–1256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ehrentraut H, Clambey ET, McNamee EN, Brodsky KS, Ehrentraut SF, Poth JM, Riegel AK, Westrich JA, Colgan SP, and Eltzschig HK (2013) CD73+ regulatory T cells contribute to adenosine-mediated resolution of acute lung injury. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 27, 2207–2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hart ML, Kohler D, Eckle T, Kloor D, Stahl GL, and Eltzschig HK (2008) Direct treatment of mouse or human blood with soluble 5’-nucleotidase inhibits platelet aggregation. Arteriosclerosis, thrombosis, and vascular biology 28, 1477–1483 [DOI] [PubMed] [Google Scholar]
- 16.Colgan SP, and Eltzschig HK (2012) Adenosine and hypoxia-inducible factor signaling in intestinal injury and recovery. Annu Rev Physiol 74, 153–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eltzschig HK, Abdulla P, Hoffman E, Hamilton KE, Daniels D, Schonfeld C, Loffler M, Reyes G, Duszenko M, Karhausen J, Robinson A, Westerman KA, Coe IR, and Colgan SP (2005) HIF-1-dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J. Exp. Med 202, 1493–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JW, Koeppen M, Seo SW, Bowser JL, Yuan X, Li J, Sibilia M, Ambardekar AV, Zhang X, Eckle T, Yoo SH, and Eltzschig HK (2019) Transcription-independent Induction of ERBB1 through Hypoxia-inducible Factor 2A Provides Cardioprotection during Ischemia and Reperfusion. Anesthesiology [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menzies RI, Tam FW, Unwin RJ, and Bailey MA (2016) Purinergic signaling in kidney disease. Kidney Int [DOI] [PubMed] [Google Scholar]
- 20.Burnstock G (2016) P2X ion channel receptors and inflammation. Purinergic. Signal 12, 59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Solini A, Usuelli V, and Fiorina P (2015) The dark side of extracellular ATP in kidney diseases. J. Am. Soc. Nephrol 26, 1007–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jarvis MF, and Khakh BS (2009) ATP-gated P2X cation-channels. Neuropharmacology 56, 208–215 [DOI] [PubMed] [Google Scholar]
- 23.Rabadi MM, Kim M, Li H, Han SJ, Choi Y, D’Agati VD, and Lee HT (2017) ATP induces PAD4 in renal proximal tubule cells via P2X7 receptor activation to exacerbate ischemic AKI. Am J Physiol Renal Physiol, ajprenal 00364 02017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacobson KA, and Muller CE (2016) Medicinal chemistry of adenosine, P2Y and P2X receptors. Neuropharmacology 104, 31–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antonioli L, Blandizzi C, Fornai M, Pacher P, Lee HT, and Hasko G (2019) P2X4 receptors, immunity, and sepsis. Curr Opin Pharmacol 47, 65–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan Y, Bai J, Zhou X, Tang J, Jiang C, Tolbert E, Bayliss G, Gong R, Zhao TC, and Zhuang S (2015) P2X7 receptor inhibition protects against ischemic acute kidney injury in mice. Am. J. Physiol Cell Physiol 308, C463–C472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim MJ, Turner CM, Hewitt R, Smith J, Bhangal G, Pusey CD, Unwin RJ, and Tam FW (2014) Exaggerated renal fibrosis in P2X4 receptor-deficient mice following unilateral ureteric obstruction. Nephrol. Dial. Transplant 29, 1350–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen K, Zhang J, Zhang W, Zhang J, Yang J, Li K, and He Y (2013) ATP-P2X4 signaling mediates NLRP3 inflammasome activation: a novel pathway of diabetic nephropathy. Int J Biochem Cell Biol 45, 932–943 [DOI] [PubMed] [Google Scholar]
- 29.Hung SC, Choi CH, Said-Sadier N, Johnson L, Atanasova KR, Sellami H, Yilmaz O, and Ojcius DM (2013) P2X4 assembles with P2X7 and pannexin-1 in gingival epithelial cells and modulates ATP-induced reactive oxygen species production and inflammasome activation. PloS one 8, e70210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo C, Masin M, Qureshi OS, and Murrell-Lagnado RD (2007) Evidence for functional P2X4/P2X7 heteromeric receptors. Mol Pharmacol 72, 1447–1456 [DOI] [PubMed] [Google Scholar]
- 31.Park SW, Kim JY, Ham A, Brown KM, Kim M, D’Agati VD, and Lee HT (2012) A1 adenosine receptor allosteric enhancer PD-81723 protects against renal ischemia-reperfusion injury. Am. J. Physiol Renal Physiol 303, F721–F732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park SW, Kim M, Brown KM, D’Agati VD, and Lee HT (2012) Inhibition of sphingosine 1-phosphate receptor 2 protects against renal ischemia-reperfusion injury. J. Am. Soc. Nephrol 23, 266–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sim JA, Chaumont S, Jo J, Ulmann L, Young MT, Cho K, Buell G, North RA, and Rassendren F (2006) Altered hippocampal synaptic potentiation in P2X4 knock-out mice. J Neurosci 26, 9006–9009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim M, Park SW, Kim M, Chen SW, Gerthoffer WT, D’Agati VD, and Lee HT (2010) Selective Renal Over-Expression of Human Heat Shock Protein 27 Reduces Renal Ischemia-Reperfusion Injury in Mice. Am. J. Physiol Renal Physiol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee HT, Park SW, Kim M, Ham A, Anderson LJ, Brown KM, D’Agati VD, and Cox GN (2012) Interleukin-11 protects against renal ischemia and reperfusion injury. Am. J. Physiol Renal Physiol 303, F1216–F1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mishra J, Ma Q, Prada A, Mitsnefes M, Zahedi K, Yang J, Barasch J, and Devarajan P (2003) Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J. Am. Soc. Nephrol 14, 2534–2543 [DOI] [PubMed] [Google Scholar]
- 37.Jablonski P, Howden BO, Rae DA, Birrel CS, Marshall VC, and Tange J (1983) An experimental model for assessment of renal recovery from warm ischemia. Transplantation 35, 198–204 [DOI] [PubMed] [Google Scholar]
- 38.Park SW, Chen SW, Kim M, D’Agati VD, and Lee HT (2009) Human heat shock protein 27 overexpressing mice are protected against acute kidney injury after hepatic ischemia and reperfusion. Am. J. Physiol Renal Physiol 297, F885–F894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park SW, Kim M, Kim M, D’Agati VD, and Lee HT (2011) Sphingosine kinase 1 protects against renal ischemia-reperfusion injury in mice by sphingosine-1-phosphate1 receptor activation. Kidney Int 80, 1315–1327 [DOI] [PubMed] [Google Scholar]
- 40.Kim M, Kim M, Park SW, Pitson SM, and Lee HT (2010) Isoflurane protects human kidney proximal tubule cells against necrosis via sphingosine kinase and sphingosine-1-phosphate generation. Am. J. Nephrol 31, 353–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song JH, Kim M, Park SW, Chen SW, Pitson SM, and Lee HT (2010) Isoflurane via TGF-beta1 release increases caveolae formation and organizes sphingosine kinase signaling in renal proximal tubules. Am. J. Physiol Renal Physiol 298, F1041–F1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vinay P, Gougoux A, and Lemieux G (1981) Isolation of a pure suspension of rat proximal tubules. Am J Physiol 241, F403–F411 [DOI] [PubMed] [Google Scholar]
- 43.Lee HT, Kim JY, Kim M, Wang P, Tang L, Baroni S, D’Agati VD, and Desir GV (2013) Renalase protects against ischemic AKI. J Am Soc Nephrol 24, 445–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Joo JD, Kim M, Horst P, Kim J, D’Agati VD, Emala CW Sr., and Lee HT (2007) Acute and delayed renal protection against renal ischemia and reperfusion injury with A1 adenosine receptors. Am J Physiol Renal Physiol 293, F1847–F1857 [DOI] [PubMed] [Google Scholar]
- 45.Joo JD, Kim M, D’Agati VD, and Lee HT (2006) Ischemic preconditioning provides both acute and delayed protection against renal ischemia and reperfusion injury in mice. J. Am. Soc. Nephrol 17, 3115–3123 [DOI] [PubMed] [Google Scholar]
- 46.Fredholm BB, IJzerman AP, Jacobson KA, Linden J, and Muller CE (2011) International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors--an update. Pharmacol. Rev 63, 1–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu J, Bernstein AM, Wong A, Lu XH, Khoja S, Yang XW, Davies DL, Micevych P, Sofroniew MV, and Khakh BS (2016) P2X4 Receptor Reporter Mice: Sparse Brain Expression and Feeding-Related Presynaptic Facilitation in the Arcuate Nucleus. J Neurosci 36, 8902–8920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dunton CL, Purves JT, Hughes FM Jr., Jin H, and Nagatomi J (2018) Elevated hydrostatic pressure stimulates ATP release which mediates activation of the NLRP3 inflammasome via P2X4 in rat urothelial cells. Int Urol Nephrol 50, 1607–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bakker PJ, Butter LM, Claessen N, Teske GJ, Sutterwala FS, Florquin S, and Leemans JC (2014) A tissue-specific role for Nlrp3 in tubular epithelial repair after renal ischemia/reperfusion. Am J Pathol 184, 2013–2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim HJ, Lee DW, Ravichandran K, D OK, Akcay A, Nguyen Q, He Z, Jani A, Ljubanovic D, and Edelstein CL (2013) NLRP3 inflammasome knockout mice are protected against ischemic but not cisplatin-induced acute kidney injury. J Pharmacol Exp Ther 346, 465–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shigeoka AA, Mueller JL, Kambo A, Mathison JC, King AJ, Hall WF, Correia Jda S, Ulevitch RJ, Hoffman HM, and McKay DB (2010) An inflammasome-independent role for epithelial-expressed Nlrp3 in renal ischemia-reperfusion injury. J Immunol 185, 6277–6285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Molitoris BA (2014) Therapeutic translation in acute kidney injury: the epithelial/endothelial axis. J Clin Invest 124, 2355–2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Caestecker M, Humphreys BD, Liu KD, Fissell WH, Cerda J, Nolin TD, Askenazi D, Mour G, Harrell FE Jr., Pullen N, Okusa MD, Faubel S, and Group AAA (2015) Bridging Translation by Improving Preclinical Study Design in AKI. J Am Soc Nephrol 26, 2905–2916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elapavaluru S, and Kellum JA (2007) Why do patients die of acute kidney injury? Acta Clin. Belg. Suppl, 326–331 [DOI] [PubMed] [Google Scholar]
- 55.Ronco C, Bellomo R, and Kellum JA (2019) Acute kidney injury. Lancet 394, 1949–1964 [DOI] [PubMed] [Google Scholar]
- 56.Roy JP, and Devarajan P (2019) Acute Kidney Injury: Diagnosis and Management. Indian J Pediatr [DOI] [PubMed] [Google Scholar]
- 57.Chan AH, and Schroder K (2019) Inflammasome signaling and regulation of interleukin-1 family cytokines. The Journal of experimental medicine [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spinello A, Vecile E, Abbate A, Dobrina A, and Magistrato A (2019) How Can Interleukin-1 Receptor Antagonist Modulate Distinct Cell Death Pathways? J Chem Inf Model 59, 351–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Van Opdenbosch N, and Lamkanfi M (2019) Caspases in Cell Death, Inflammation, and Disease. Immunity 50, 1352–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vilaysane A, Chun J, Seamone ME, Wang W, Chin R, Hirota S, Li Y, Clark SA, Tschopp J, Trpkov K, Hemmelgarn BR, Beck PL, and Muruve DA (2010) The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J Am Soc Nephrol 21, 1732–1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nazir S, Gadi I, Al-Dabet MM, Elwakiel A, Kohli S, Ghosh S, Manoharan J, Ranjan S, Bock F, Braun-Dullaeus RC, Esmon CT, Huber TB, Camerer E, Dockendorff C, Griffin JH, Isermann B, and Shahzad K (2017) Cytoprotective activated protein C averts Nlrp3 inflammasome-induced ischemia-reperfusion injury via mTORC1 inhibition. Blood 130, 2664–2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tang TT, Lv LL, Pan MM, Wen Y, Wang B, Li ZL, Wu M, Wang FM, Crowley SD, and Liu BC (2018) Hydroxychloroquine attenuates renal ischemia/reperfusion injury by inhibiting cathepsin mediated NLRP3 inflammasome activation. Cell Death Dis 9, 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pefanis A, Ierino FL, Murphy JM, and Cowan PJ (2019) Regulated necrosis in kidney ischemia-reperfusion injury. Kidney Int 96, 291–301 [DOI] [PubMed] [Google Scholar]
- 64.Yuan X, Lee JW, Bowser JL, Neudecker V, Sridhar S, and Eltzschig HK (2017) Targeting Hypoxia Signaling for Perioperative Organ Injury. Anesth Analg [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Daemen MA, de Vries B, and Buurman WA (2002) Apoptosis and inflammation in renal reperfusion injury. Transplantation 73, 1693–1700 [DOI] [PubMed] [Google Scholar]
- 66.Frangogiannis NG (2007) Chemokines in ischemia and reperfusion. Thromb. Haemost 97, 738–747 [PubMed] [Google Scholar]
- 67.Torres M, and Forman HJ (2003) Redox signaling and the MAP kinase pathways. Biofactors 17, 287–296 [DOI] [PubMed] [Google Scholar]
- 68.Legrand M, Mik EG, Johannes T, Payen D, and Ince C (2008) Renal hypoxia and dysoxia after reperfusion of the ischemic kidney. Mol. Med 14, 502–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weinberg JM, Venkatachalam MA, Roeser NF, and Nissim I (2000) Mitochondrial dysfunction during hypoxia/reoxygenation and its correction by anaerobic metabolism of citric acid cycle intermediates. Proc. Natl. Acad. Sci. U. S. A 97, 2826–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang R, Beqiri D, Shen JB, Redden JM, Dodge-Kafka K, Jacobson KA, and Liang BT (2015) P2X4 receptor-eNOS signaling pathway in cardiac myocytes as a novel protective mechanism in heart failure. Comput Struct Biotechnol J 13, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang T, Shen JB, Yang R, Redden J, Dodge-Kafka K, Grady J, Jacobson KA, and Liang BT (2014) Novel protective role of endogenous cardiac myocyte P2X4 receptors in heart failure. Circ Heart Fail 7, 510–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Verma R, Cronin CG, Hudobenko J, Venna VR, McCullough LD, and Liang BT (2017) Deletion of the P2X4 receptor is neuroprotective acutely, but induces a depressive phenotype during recovery from ischemic stroke. Brain Behav Immun 66, 302–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ozaki T, Muramatsu R, Sasai M, Yamamoto M, Kubota Y, Fujinaka T, Yoshimine T, and Yamashita T (2016) The P2X4 receptor is required for neuroprotection via ischemic preconditioning. Sci Rep 6, 25893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Csoka B, Nemeth ZH, Szabo I, Davies DL, Varga ZV, Paloczi J, Falzoni S, Di Virgilio F, Muramatsu R, Yamashita T, Pacher P, and Hasko G (2018) Macrophage P2X4 receptors augment bacterial killing and protect against sepsis. JCI Insight 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Koeppen M, Lee JW, Seo SW, Brodsky KS, Kreth S, Yang IV, Buttrick PM, Eckle T, and Eltzschig HK (2018) Hypoxia-inducible factor 2-alpha-dependent induction of amphiregulin dampens myocardial ischemia-reperfusion injury. Nat Commun 9, 816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ju C, Colgan SP, and Eltzschig HK (2016) Hypoxia-inducible factors as molecular targets for liver diseases. J. Mol. Med. (Berl) [DOI] [PMC free article] [PubMed] [Google Scholar]