

Abstract
Eukaryotic translation initiation factor 4E (eIF4E) binds the m7GTP cap structure at the 5′-end of mRNAs, stimulating the translation of proteins implicated in cancer cell growth and metastasis. eIF4E is a notoriously challenging target, and most of the reported inhibitors are negatively charged guanine analogues with negligible cell permeability. To overcome these challenges, we envisioned a covalent targeting strategy. As there are no cysteines near the eIF4E cap binding site, we developed a covalent docking approach focused on lysine. Taking advantage of a “make-on-demand” virtual library, we used covalent docking to identify arylsulfonyl fluorides that target a noncatalytic lysine (Lys162) in eIF4E. Guided by cocrystal structures, we elaborated arylsulfonyl fluoride 2 to 12, which to our knowledge is the first covalent eIF4E inhibitor with cellular activity. In addition to providing a new tool for acutely inactivating eIF4E in cells, our computational approach may offer a general strategy for developing selective lysine-targeted covalent ligands.
Recent studies by academic and industrial laboratories have catalyzed renewed interest in chemical probes and drugs with a covalent mechanism of action.1,2 Most targeted covalent drugs act by modifying cysteine residues, which are potent nucleophiles. Cysteines are relatively rare in the proteome, and consequently, they are often not present in ligand binding sites. An alternative covalent strategy involves targeting lysine.3,4 Although lysine is more prevalent than cysteine, it is also much less nucleophilic. These attributes make lysine a challenging target for covalent inhibitor design and raise concerns about the selectivity of lysine-targeted probes.5,6 Consequently, most lysine-targeted inhibitors were designed by starting with a potent non-covalent inhibitor and appending an appropriately positioned electrophile.4,7 A general computational screening approach for the direct identification of lysine-targeted ligands would enable a covalent targeting strategy for the multitude of proteins that lack a ligandable cysteine.
Cap-dependent eukaryotic translation initiation factor 4E (eIF4E), which binds the 5′-m7GTP cap of cellular mRNAs, exemplifies the potential of lysine-directed covalent inhibitors. Molecules that bind and occlude the cap binding site of eIF4E are attractive as potential anticancer leads and tools for studying cap-dependent translation initiation.8,9 Although a small molecule has been reported to block eIF4E binding to eIF4G,10 there are few published inhibitors that bind in the m7GTP pocket, and most are nucleotide analogues resembling m7GTP.11,12 These inhibitors bind eIF4E reversibly and are negatively charged; removing the negative charge results in a drastic loss in binding affinity. Not surprisingly, these inhibitors are inactive in cells (or weakly active as prodrugs),13 likely because of a lack of membrane permeability. With these challenges in mind, we were motivated to pursue a covalent inhibition strategy. Without a cysteine near the cap binding site, proximal lysines emerged as potentially attractive nucleophiles. In particular, the paralogue-specific Lys162 in the eIF4E cap binding site (replaced by Ile in eIF4E2 and Val in eIF4E3) directly hydrogen-bonds with the β-phosphate of m7GTP.14,15
To exploit Lys162 as a nucleophile, we built a virtual make-on-demand library of arylsulfonyl fluorides—which are precedented as electrophiles for lysine modification4,7,16—based on building blocks and syntheses developed at Enamine.17,18 A variety of computational docking methods have been reported to identify covalent ligands.19-22 Our strategy builds upon the DOCKovalent method, which targets cysteines and serines with large libraries of electrophiles,23 adapting it to the greater conformational flexibility of lysine residues. As in DOCKovalent, library molecules are scored by their physical complementarity to the binding site in poses that position an electrophile to react with the nucleophile. In contrast to our previous study, we were faced with a flexible lysine side chain, which is challenging to model accurately in docking. We therefore followed initial docking geometries with minimization of the best-ranked molecules using molecular mechanics and then by further MM/GBSA rescoring.24,25 This not only used a higher level of theory in evaluating the docked molecules but better sampled and evaluated the possible lysine conformations. Before this approach was applied to eIF4E, it was tested for the ability to reproduce the geometries of 16 protein–ligand complexes with covalent lysine-targeted ligands. The pose reproduction rate here was 69%, with average root-mean-square deviations of 1.8 Å when we docked to predicted lysine rotamers (Figure S1 and Table S1), suggesting that the method might be useful prospectively.
Accordingly, we docked 88 186 make-on-demand18 arylsulfonyl fluorides against the X-ray structure of the eIF4E cap binding site. The top-ranked 219 molecules (0.24%), as evaluated by DOCKovalent and refined by minimization and rescoring, were inspected for interactions with key residues, such as Trp56, Trp102, and Glu103, and for internal ligand strain, which DOCKovalent treats only approximately.23 Seven compounds were prioritized, purchased, and assayed for covalent binding to eIF4E by protein mass spectrometry (Figure 1a and Table S2). At 100 μM, two arylsulfonyl fluoride docking hits (2 and 3) substantially labeled eIF4E after incubation for 3 h (Table S2), with compound 2 showing the highest labeling specificity as determined by competition with m7GTP (Figure 1b). Modeling suggested the presence of a deep lipophilic pocket adjacent to compound 2 in the cap binding site (Figure 1c), whose occupation might improve the potency. We targeted this pocket by designing a second virtual library of 2239 new compounds, each of which contains a variable hydrophobic substituent appended to the aniline of 2. These analogues were docked and modeled into the site. Five high-ranking compounds were synthesized and purchased (Figure S2). Compounds 8 and 9 showed improved potency (Figure 2a,b), labeling eIF4E to 68% and 41%, respectively, after treatment with 30 μM compound for 3 h. LC–MS/MS analysis confirmed K162 as the major site of modification by compound 9, although modification of K206 (adjacent to the m7GTP pocket) was also detected (Figure S3).
Figure 1.

Covalent docking to eIF4E Lys162. (a) Arylsulfonyl fluorides 1–7 prioritized by covalent docking. (b) eIF4E (1 μM) was treated with compound 2 (100 μM) with or without m7GTP (100 μM). At the indicated time points, eIF4E labeling by 2 was quantified by LC–MS. (c) Docked pose of compound 2 covalently bound to Lys162 (green) of eIF4E (docked to PDB ID 4DT6; pink side chains, gray surface).
Figure 2.

Structure-guided optimization of covalent eIF4E inhibitors. (a) Secondary docking hits 8 and 9. (b) WT and K162R eIF4E were treated with 8 and 9, followed by LC–MS analysis. (c) Cocrystal structure of 9/eIF4E (PDB ID 6U09). Electron density (2Fo – Fc) is shown at a contour level of 1σ. (d) Overlay of compound 9/eIF4E (cyan) with the ligand from PDB ID 4DUM (yellow), which guided the design of compounds 10–12 (e). (f) eIF4E was treated with 9–12 for 1 h at pH 8.1 (left) or 15 min at pH 7 (right), followed by LC–MS analysis. (g) Kinetic parameters for modification of eIF4E by compounds 10–12 (pH 8.1).
To support further optimization, we determined the cocrystal structure of compound 9 bound to eIF4E at 1.79 Å resolution (PDB ID 6U09; Figure 2c). This structure confirmed the binding mode anticipated by docking, with the isoquinolone core interacting with eIF4E in a manner similar to the guanine of m7GTP (Figure S4). As designed, the 4-pyridylmethyl substituent of 9 fits into the hydrophobic pocket, with the pyridine nitrogen accepting a hydrogen bond from the hydroxyl of Ser92. Continuous electron density between Lys162 and the sulfonyl group was observed, consistent with covalent bond formation. A similar binding mode for compound 8 was also confirmed by a second cocrystal structure at 1.96 Å resolution, in this case with the 4-cyanobenzyl substituent occupying the hydrophobic pocket (PDB ID 6U06; Figure S4).
An overlay of the eIF4E/9 complex with a previously reported eIF4E inhibitor that has nanomolar binding affinity but lacks cellular activity12 suggested two further directions for optimization. By analogy to this inhibitor (Figure 2d), we first added a 2-Cl substituent to the pyridine to afford compound 10 (Figure 2e), which increased the potency by 50-fold relative to compound 9 in a 1 h labeling experiment (Figure 2f, left). Second, as suggested by the overlay (Figure 2d), we replaced the isoquinolone core with a quinazolinone bearing an exocyclic amine (compounds 11 and 12; Figure 2e), which was designed to form a second hydrogen bond with Glu103 (Figure S5). Consistent with the importance of this interaction, the IC50 improved 157-fold and 581-fold for 11 and 12, respectively, in a 15 min labeling reaction performed at neutral pH (Figure 2f, right). To better describe the efficiency of covalent bond formation, we measured two kinetic parameters, kinact and Ki. Despite sharing the same electrophile, kinact for 11 and 12 was improved 40- to 50-fold relative to that for 10, whereas Ki was only slightly altered (Figures 2g and S6). These surprising results suggest that the exocyclic amine in 11 and 12 exerts a stronger effect on the rate of covalent bond formation than on the reversible binding affinity, perhaps by better orienting the reversibly bound arylsulfonyl fluoride for nucleophilic attack by Lys162. Compound 12 showed reduced labeling of K162R eIF4E (relative to 8 and 9) and undetectable labeling of S92H eIF4E (Figures S7 and S8), consistent with a precise binding orientation in the eIF4E pocket and proximity-accelerated modification of Lys162. Because both eIF4E2 and eIF4E3 lack the equivalent of Ser92 and Lys162, these widely expressed eIF4E paralogues are predicted to be resistant to compound 12.
Given its high kinact/Ki value (0.33 μM−1 min−1), we prioritized compound 12 for cellular efficacy experiments. After treatment of Jurkat cells with increasing concentrations of 12 (and 9–11 at 10 μM), cell lysates were prepared, and endogenous eIF4E was enriched using m7GTP-agarose beads. Encouragingly, Western blot analysis of cell lysates prior to affinity enrichment with m7GTP-agarose revealed a dose-dependent shift of eIF4E (25 kDa) to a form with a higher molecular weight (MW), likely due to covalent modification by compound 12 (Figure 3a, “lysates”). Consistent with this interpretation, analysis of the m7GTP pulldown samples (Figure 3a, “elution”) revealed a dose-dependent decrease in bound eIF4E, with the higher-MW form detected solely in the supernatant. Notably, compounds 9 and 10 were less active toward eIF4E in cells versus compound 12, consistent with their reduced potency against the purified protein (Figure 3a).
Figure 3.
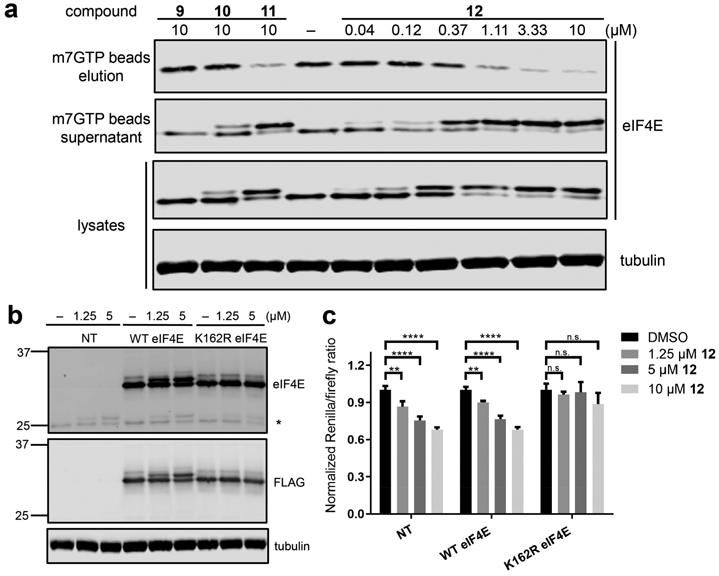
Covalent inactivation of eIF4E in cells. (a) Jurkat cells were treated with 9–12. After 3 h, cell lysates were prepared, and an aliquot of each sample was analyzed by Western blotting for eIF4E and tubulin (“lysates”). Remaining lysate samples were enriched with m7GTP-agarose beads, and the bound (“elution”) and unbound (“supernatant”) fractions were analyzed by Western blotting. (b) HEK293T cells stably overexpressing WT or K162R FLAG-eIF4E or nontransduced cells (NT) were treated with 12 for 30 min. Cell lysates were prepared and analyzed by Western blotting (* denotes endogenous eIF4E). (c) Stable cell lines from (b) were treated with 12 prior to transfection with a bicistronic plasmid comprising a cap-dependent cistron (Renilla luciferase) followed by a cap-independent cistron (firefly luciferase). Cells were incubated with 12 or DMSO for 6 h, after which the Renilla and firefly luciferase activities were measured (Figure S9). Data are means ± SEM (n = 3). **, P < 0.01; ****, P < 0.0001.
To further characterize the cellular effects of compound 12, we generated stable HEK293T cell lines overexpressing FLAG-tagged wild-type (WT) and K162R eIF4E. As expected, treatment of these cells with compound 12 (1.25 and 5 μM) induced a MW shift for WT eIF4E but not K162R eIF4E, providing additional support for covalent modification of Lys162 (Figure 3b). Parallel experiments in cells transfected with a bicistronic dual-luciferase reporter26 revealed inhibition of cap-dependent but not cap-independent translation, as shown by a decrease in the Renilla/firefly luciferase activity ratio (Figures 3c and S9). Consistent with an on-target mechanism of action (despite the possibility of covalent off-target reactions), compound 12 inhibited cap-dependent translation in nontransduced cells and cells overexpressing WT eIF4E but not in cells overexpressing the K162R mutant. Thus, overexpression of an eIF4E mutant lacking Lys162 confers resistance to compound 12 in the cap-dependent translation assay, providing genetic evidence that covalent modification of eIF4E underlies its inhibitory effects. We note that after a longer incubation period (24 h), the extent of eIF4E modification by 12 was slightly reduced (Figure S10), likely reflecting decomposition of the arylsulfonyl fluoride27 and resynthesis of unmodified eIF4E. We assessed the chemical stability of compound 12 under various buffer conditions at 37 °C and observed both hydrolysis and glutathione-mediated reduction (t1/2 = 3–30 min; Figure S11), potentially explaining why 12 did not modify 100% of endogenous or overexpressed eIF4E in HEK293T cells. Nevertheless, compound 12 represents the first example of a lysine-targeted eIF4E inhibitor with cellular activity. Its high kinact/Ki compensates for its instability, enabling rapid modification of intracellular eIF4E before its depletion from the culture medium.
Three features of this study merit emphasis. First, the docking strategy for covalent lysine inhibitors may find wide application to other challenging protein targets, especially those lacking a ligandable cysteine. Second, there is much interest in a recent 100- to 1000-fold increase in accessible chemical space via make-on-demand libraries. 17 This study suggests that this space may be expanded substantially by identifying new chemotypes (e.g., arylsulfonyl fluorides) that fit within the reaction schemes and building blocks underlying the make-on-demand approach.28,29 Finally, compound 12 together with the resistant mutant allele (K162R) provides a new chemical biology toolkit for acute inactivation of eIF4E and elucidation of its complex cellular roles in translation control.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Ono Pharma Foundation (J.T.), Pfizer (J.T.), NIH NIGMS (R35 GM122481, B.K.S.), NIH NCI (F31CA214028, A.C.), and the China Scholarship Council (201409110014, X.W.). We acknowledge support from Advanced Light Source beamline 8.3.1. We thank Xi Liu for help with data processing, Yurii Moroz and Andriy Yakymenko (Enamine) for help building the virtual library, and Haoyuan Wang and Keely Oltion for helpful discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b10377.
Detailed experimental procedures, crystallographic statistics and collection parameters, and synthesis and spectral characterization of compounds (PDF)
The authors declare the following competing financial interest(s): J.T. is a cofounder and shareholder of Global Blood Therapeutics, Principia Biopharma, Kezar Life Sciences, and Cedilla Therapeutics.
Contributor Information
Xiaobo Wan, Department of Pharmaceutical Chemistry and Department of Cellular and Molecular Pharmacology, University of California, San Francisco, California 94158, United States.
Tangpo Yang, Department of Cellular and Molecular Pharmacology, University of California, San Francisco, California 94158, United States.
Adolfo Cuesta, Department of Cellular and Molecular Pharmacology, University of California, San Francisco, California 94158, United States.
Xiaming Pang, Department of Urology, University of California, San Francisco, California 94158, United States.
Trent E. Balius, Department of Pharmaceutical Chemistry, University of California, San Francisco, California 94158, United States
John Irwin, Department of Pharmaceutical Chemistry, University of California, San Francisco, California 94158, United States.
Brian K. Shoichet, Department of Pharmaceutical Chemistry, University of California, San Francisco, California 94158, United States.
Jack Taunton, Department of Cellular and Molecular Pharmacology, University of California, San Francisco, California 94158, United State.
REFERENCES
- (1).Lagoutte R; Patouret R; Winssinger N Covalent inhibitors: an opportunity for rational target selectivity. Curr. Opin. Chem. Biol 2017, 39, 54–63. [DOI] [PubMed] [Google Scholar]
- (2).Baillie TA Targeted Covalent Inhibitors for Drug Design. Angew. Chem., Int. Ed 2016, 55 (43), 13408–13421. [DOI] [PubMed] [Google Scholar]
- (3).Pettinger J; Jones K; Cheeseman MD Lysine-Targeting Covalent Inhibitors. Angew. Chem., Int. Ed 2017, 56 (48), 15200–15209. [DOI] [PubMed] [Google Scholar]
- (4).Cuesta A; Taunton J Lysine-Targeted Inhibitors and Chemoproteomic Probes. Annu. Rev. Biochem 2019, 88, 365–381. [DOI] [PubMed] [Google Scholar]
- (5).Hacker SM; Backus KM; Lazear MR; Forli S; Correia BE; Cravatt BF Global profiling of lysine reactivity and ligandability in the human proteome. Nat. Chem 2017, 9 (12), 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ward CC; Kleinman JI; Nomura DK NHS-Esters As Versatile Reactivity-Based Probes for Mapping Proteome-Wide Ligandable Hotspots. ACS Chem. Biol 2017, 12 (6), 1478–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Cuesta A; Wan X; Burlingame AL; Taunton J Ligand Conformational Bias Drives Enantioselective Modification of a Surface-Exposed Lysine on Hsp90. J. Am. Chem. Soc 2020, 142, 3392–3400. [DOI] [PubMed] [Google Scholar]
- (8).Truitt ML; Conn CS; Shi Z; Pang X; Tokuyasu T; Coady AM; Seo Y; Barna M; Ruggero D Differential Requirements for eIF4E Dose in Normal Development and Cancer. Cell 2015, 162 (1), 59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Siddiqui N; Sonenberg N Signalling to eIF4E in cancer. Biochem. Soc. Trans 2015, 43 (5), 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Moerke NJ; Aktas H; Chen H; Cantel S; Reibarkh MY; Fahmy A; Gross JD; Degterev A; Yuan J; Chorev M; Halperin JA; Wagner G Small-Molecule Inhibition of the Interaction between the Translation Initiation Factors eIF4E and eIF4G. Cell 2007, 128 (2), 257–267. [DOI] [PubMed] [Google Scholar]
- (11).Ghosh P; Park C; Peterson MS; Bitterman PB; Polunovsky VA; Wagner CR Synthesis and evaluation of potential inhibitors of eIF4E cap binding to 7-methyl GTP. Bioorg. Med. Chem. Lett 2005, 15 (8), 2177–2180. [DOI] [PubMed] [Google Scholar]
- (12).Chen X; Kopecky DJ; Mihalic J; Jeffries S; Min X; Heath J; Deignan J; Lai S; Fu Z; Guimaraes C; Shen S; Li S; Johnstone S; Thibault S; Xu H; Cardozo M; Shen W; Walker N; Kayser F; Wang Z Structure-Guided Design, Synthesis, and Evaluation of Guanine-Derived Inhibitors of the eIF4E mRNA-Cap Interaction. J. Med. Chem 2012, 55 (8), 3837–3851. [DOI] [PubMed] [Google Scholar]
- (13).Ghosh B; Benyumov AO; Ghosh P; Jia Y; Avdulov S; Dahlberg PS; Peterson M; Smith K; Polunovsky VA; Bitterman PB; Wagner CR Nontoxic Chemical Interdiction of the Epithelial-to-Mesenchymal Transition by Targeting Cap-Dependent Translation. ACS Chem. Biol 2009, 4 (5), 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Marcotrigiano J; Gingras A-C; Sonenberg N; Burley SK Cocrystal Structure of the Messenger RNA 5′ Cap-Binding Protein (eIF4E) Bound to 7-methyl-GDP. Cell 1997, 89 (6), 951–961. [DOI] [PubMed] [Google Scholar]
- (15).Tomoo K; Shen X; Okabe K; Nozoe Y; Fukuhara S; Morino S; Ishida T; Taniguchi T; Hasegawa H; Terashima A; Sasaki M; Katsuya Y; Kitamura K; Miyoshi H; Ishikawa M; Miura K.-i. Crystal structures of 7-methylguanosine 5′-triphosphate (m7GTP)- and P1-7-methylguanosine-P3-adenosine-5′,5′-triphosphate (m7GpppA)-bound human full-length eukaryotic initiation factor 4E: biological importance of the C-terminal flexible region. Biochem. J 2002, 362 (3), 539–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Colman RF Affinity labeling of purine nucleotide sites in proteins. Annu. Rev. Biochem 1983, 52 (1), 67–91. [DOI] [PubMed] [Google Scholar]
- (17).Lyu J; Wang S; Balius TE; Singh I; Levit A; Moroz YS; O’Meara MJ; Che T; Algaa E; Tolmachova K; Tolmachev AA; Shoichet BK; Roth BL; Irwin JJ Ultra-large library docking for discovering new chemotypes. Nature 2019, 566 (7743), 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Tolmachova KA; Moroz YS; Konovets A; Platonov MO; Vasylchenko OV; Borysko P; Zozulya S; Gryniukova A; Bogolubsky AV; Pipko S; Mykhailiuk PK; Brovarets VS; Grygorenko OO (Chlorosulfonyl)benzenesulfonyl Fluorides—Versatile Building Blocks for Combinatorial Chemistry: Design, Synthesis and Evaluation of a Covalent Inhibitor Library. ACS Comb. Sci 2018, 20 (11), 672–680. [DOI] [PubMed] [Google Scholar]
- (19).Bianco G; Forli S; Goodsell DS; Olson AJ Covalent docking using autodock: Two-point attractor and flexible side chain methods. Protein Sci. 2016, 25 (1), 295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Katritch V; Byrd CM; Tseitin V; Dai D; Raush E; Totrov M; Abagyan R; Jordan R; Hruby DE Discovery of small molecule inhibitors of ubiquitin-like poxvirus proteinase I7L using homology modeling and covalent docking approaches. J. Comput.- Aided Mol. Des 2007, 21 (10), 549–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Scholz C; Knorr S; Hamacher K; Schmidt B DOCKTITE-a highly versatile step-by-step workflow for covalent docking and virtual screening in the molecular operating environment. J. Chem. Inf. Model 2015, 55 (2), 398–406. [DOI] [PubMed] [Google Scholar]
- (22).Scarpino A; Ferenczy GG; Keserű GM Comparative Evaluation of Covalent Docking Tools. J. Chem. Inf. Model 2018, 58 (7), 1441–1458. [DOI] [PubMed] [Google Scholar]
- (23).London N; Miller RM; Krishnan S; Uchida K; Irwin JJ; Eidam O; Gibold L; Cimermančič P; Bonnet R; Shoichet BK; Taunton J Covalent docking of large libraries for the discovery of chemical probes. Nat. Chem. Biol 2014, 10, 1066–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Del Rio A; Sgobba M; Parenti MD; Degliesposti G; Forestiero R; Percivalle C; Conte PF; Freccero M; Rastelli G A computational workflow for the design of irreversible inhibitors of protein kinases. J. Comput.-Aided Mol. Des 2010, 24 (3), 183–194. [DOI] [PubMed] [Google Scholar]
- (25).Genheden S; Ryde U The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discovery 2015, 10 (5), 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Poulin F; Gingras A-C; Olsen H; Chevalier S; Sonenberg N 4E-BP3, a New Member of the Eukaryotic Initiation Factor 4E-binding Protein Family. J. Biol. Chem 1998, 273 (22), 14002–14007. [DOI] [PubMed] [Google Scholar]
- (27).Gambini L; Baggio C; Udompholkul P; Jossart J; Salem AF; Perry JJP; Pellecchia M Covalent Inhibitors of Protein-Protein Interactions Targeting Lysine, Tyrosine, or Histidine Residues. J. Med. Chem 2019, 62 (11), 5616–5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Liu Z; Li J; Li S; Li G; Sharpless KB; Wu P SuFEx Click Chemistry Enabled Late-Stage Drug Functionalization. J. Am. Chem. Soc 2018, 140 (8), 2919–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Laudadio G; Bartolomeu A. d. A.; Verwijlen LMHM; Cao Y; de Oliveira KT; Noël T Sulfonyl Fluoride Synthesis through Electrochemical Oxidative Coupling of Thiols and Potassium Fluoride. J. Am. Chem. Soc 2019, 141 (30), 11832–11836. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.