

Abstract
Chemotherapeutics that self-assemble into colloids have limited efficacy above their critical aggregation concentration due to their inability to penetrate intact plasma membranes. Even when colloid uptake is promoted, issues with colloid escape from the endolysosomal pathway persist. By stabilizing acid-responsive lapatinib colloids through coaggregation with fulvestrant, and inclusion of transferrin, we demonstrate colloid internalization by cancer cells, where subsequent lapatinib ionization leads to endosomal leakage and increased cytotoxicity. These results demonstrate a strategy for triggered drug release from stable colloidal aggregates.
Graphical Abstract
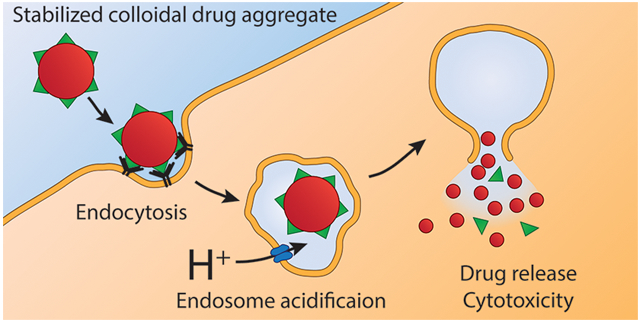
Some small-molecule drugs spontaneously self-assemble under aqueous conditions to form colloidal aggregates.1-3 The formation of these colloids is governed by a critical aggregation concentration (CAC) and generally occurs when water is added to a solution of drug in a water-miscible organic solvent.4 Although colloidal drug aggregates are best known for causing false hits in early drug discovery,5-7 recent efforts have aimed to stabilize these drug-rich particles for delivery.8-10
Development of stable colloidal drug aggregates has paradoxically created a new problem: colloid-associated drug does not permeate the cell membrane to interact with intracellular targets.11,12 For colloid-bound drugs, drug release can be achieved by disrupting the particles to yield drug monomers.12,13 Colloid dissolution has traditionally been accomplished by adding detergent; however, this strategy is more useful in vitro than in vivo, where toxic excipients are dose-limited.
To enhance colloid cytotoxicity without adding an exogenous reagent, we sought to exploit an endogenous stimulus to trigger drug release from the stable colloids. One such stimulus is the acidic environment of the endosomes and lysosomes of cells.14 Fortuitously, the formation of some colloidal drug aggregates is pH-dependent, with the colloidal form of weakly basic drugs dissolving at low pH.15-17 This behavior creates an opportunity for colloids that are stable outside the cell to be disrupted intracellularly by the acidic endolysosomal pathway.18-21 Colloidal drug aggregates stabilized with a targeting antibody have been shown to enter cancer cells via endocytosis,22 suggesting that acidification can trigger release.
To test this mechanism, we designed a stable, targeted colloidal formulation containing the ionizable chemotherapeutic lapatinib, and demonstrated acid-triggered release. We found that the cytotoxicity of the targeted formulation was amplified compared to nontargeted controls due to colloid endocytosis and subsequent endosomal escape.
■ RESULTS AND DISCUSSION
Results.
We began by identifying colloid-forming drugs that could respond to acidic conditions by measuring the critical aggregation concentration (CAC) of several aggregators as a function of pH (Table 1). Critical aggregation concentrations were calculated by plotting the scattering intensity (indicative of colloid number and size) from dynamic light scattering (DLS) versus concentration (Tables S1, S2 and Figure S1).1,23 Weakly basic aggregators, such as clotrimazole and lapatinib, had pH-dependent CACs, which should enable colloid disruption as a function of pH; conversely, aggregators with pKa values well below our pH range, such as fulvestrant and sorafenib, did not dissolve on acidification.
Table 1.
Colloid-Forming Drugs with pKa’s > 5 Show pH-Dependent Critical Aggregation Concentrations (CACs) between Physiological pH 7.4 and Endosomal pH 5.5a
| Drug name | Structure and protonation site | Critical aggregation concentration (μM) |
pKa of DH+ | ||
|---|---|---|---|---|---|
| pH 7.4 | pH 5.5 | ||||
| pH-responsive | Lapatinib | 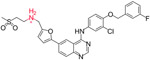 |
1.2 ± 0.3 | 17 ± 3 | 7.2 |
| Clotrimazole | 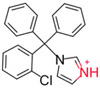 |
24 ± 1 | ~100 | 6.62 | |
| Nilotinib | 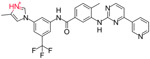 |
4.4 ± 1.4 | 15 ± 5 | 6.3 | |
| Pazopanib | 20 ± 4 | ~100 | 5.07 | ||
| Unresponsive | Sorafenib | 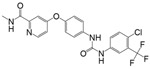 |
5.9 ± 0.5 | 2.4 ± 1.1 | 2.03 |
| Fulvestrant | 1.4 ± 0.4 | 1.6 ± 0.0 | −0.88 | ||
CACs were measured in pH-adjusted phosphate buffered saline (PBS) by dynamic light scattering as described in the Supporting Information (n = 3, mean ± 95% CI) and pKa’s were obtained from DrugBank.ca.
We worked to develop a stable colloidal formulation of lapatinib due to its potency and robust response to acidic pH. Although lapatinib forms colloidal aggregates on its own, they are only transiently stable and precipitate within a few hours (Figure S2). We screened excipients to stabilize colloidal lapatinib against precipitation (Figures S2 and S3)8,24-26 and found that coaggregation with fulvestrant, another colloid-forming drug, resulted in stable, spherical colloids, approximately 200 nm diameter (Figure S4) with slight positive charge (Figure S5). We found that a 1:3 molar ratio of lapatinib/fulvestrant was best stabilized against precipitation by excipients such as the graft copolymer poly(D,L-lactide-co-2-methyl-2-carboxytrimethylene carbonate)-graft-poly(ethylene glycol) (PLAC-PEG, Figures 1 and S6) and the protein transferrin (Figures S7 and S8). We also confirmed that these colloids are stable in cell culture media (Figure S9).
Figure 1.

Colloidal lapatinib is stabilized by coaggregation with fulvestrant and polymeric surfactant. Colloidal drug aggregates were formulated with 50 μM total drug, and 0.01 mg mL−1 poly(D,L-lactide-co-2-methyl-2-carboxytrimethylene carbonate)-graft-poly(ethylene glycol) (PLAC-PEG) in phosphate buffered saline. A 1:3 mol ratio of lapatinib/fulvestrant was selected for its stability against (A) growth and (B) precipitation as measured by dynamic light scattering. (C) Fluorescence measurement (λex = 340 nm, λem = 450 nm) confirmed the presence of lapatinib in these co-colloids (n = 3, mean ± SD).
We investigated the response of the stable colloids to acidic conditions. Colloids were formulated in PBS and subsequently acidified, mimicking the pH within the endolysosomal pathway. To determine whether the lapatinib/fulvestrant co-colloids changed as a function of pH, we measured colloid size and scattering intensity by DLS (Figure S10). We observed relatively little change in colloid size after acidification. Next, we centrifuged the suspension to pellet the colloids, and then quantified free drug in the supernatant after ensuring that no colloids were present. The amount of released lapatinib increased with increasing acidity (Figure 2A), whereas the release of the nonionizable fulvestrant was unaffected by pH (Figure 2B). Importantly, lapatinib release could be triggered at any time after formulation, and rapidly reached an equilibrium that was sustained for 24 h (Figure 2C) whereas fulvestrant remained unresponsive to acid (Figure 2D). The amount of drug released from the colloids remained similar from both 50 μM to 200 μM colloidal aggregates (Figure S11). Additionally, similar trends were observed (Figure S12) in PBS containing 10% (v/v) fetal bovine serum (FBS), demonstrating a similar response in serum-containing media. We also observed an increased baseline level of drug release in serum-containing media, which is consistent with previous reports that show how proteins increase the CAC of colloidal aggregators.10,27 These results suggest that the stable lapatinib colloids will respond to the acid stimulus in the endolysosomal pathway.
Figure 2.

Lapatinib release from stable lapatinib-fulvestrant co-colloids is triggered by acidic conditions. Colloids were formulated with 12.5 μM lapatinib, 37.5 μM fulvestrant, and 0.01 mg mL−1 poly(D,L-lactide-co-2-methyl-2-carboxytrimethylene carbonate)-graft-poly(ethylene glycol) (PLAC-PEG) in PBS. (A) Lapatinib is released from the co-colloids with decreasing pH, whereas (B) fulvestrant is retained within the colloids. (C) Lapatinib release from co-colloids is triggered by acidification to pH 5.5 (colored arrows), whereas (D) fulvestrant release is unaffected (n ≥ 3, mean ± SD, one-way ANOVA with Tukey’s posthoc test, ns p > 0.05, ****p < 0.0001 compared to initial release at pH 7.4).
We then investigated the endocytosis of our stable, acid-responsive colloidal drug aggregates. To measure uptake, we incorporated a hydrophobically modified BODIPY dye which is fluorescent only when incorporated within the colloid.2 These colloids appeared as punctate structures within MDA-MB-231-H2N cells (Figure 3, Figure S13). Although we observed minimal uptake of the PLAC-PEG stabilized colloids, we observed that transferrin-stabilized colloids were taken up to a much greater extent (Figure 3C). This result held for both lapatinib/fulvestrant colloids and colloids of fulvestrant alone, and aligns with previous research demonstrating that stabilizers that interact specifically with cells can elicit endocytosis.22
Figure 3.

Decoration of colloidal drug aggregates with transferrin enhances their endocytosis. MDA-MB-231-H2N cells were treated with colloids (Table S3) with the addition of 500 nM hydrophobic BODIPY dye. Images: cells were treated with lapatinib and fulvestrant colloids stabilized with either (A) transferrin or (B) poly(D,L-lactide-co-2-methyl-2-carboxytrimethylene carbonate)-graft-poly(ethylene glycol) (PLAC-PEG) for 1 h followed by fresh media for 23 h. The cells were labeled with CellMask Green (1×, 5 min) and imaged under live cell conditions. Transferrin-stabilized colloids were visualized as punctate structures inside the cells whereas PLAC-PEG-stabilized colloids were not observed inside the cells. (C) Cells were treated for 3 h, and fluorescence of the BODIPY dye inside the cells was then analyzed by flow cytometry (n = 3 biological replicates, mean ± SD, two-way ANOVA with Tukey’s posthoc test, ***p < 0.001, ****p < 0.0001 compared to all other groups).
We next examined the in vitro toxicity of acid-responsive, stable lapatinib/fulvestrant colloids against lapatinib-sensitive, HER2-overexpressing MDA-MB-231-H2N cells. The transferrin-stabilized lapatinib/fulvestrant colloids were substantially more toxic than any other formulation, including PLAC-PEG stabilized lapatinib/fulvestrant colloids that were minimally endocytosed (Figure 4A).
Figure 4.

Lapatinib/fulvestrant colloids are cytotoxic after endocytosis and subsequent endosome disruption. Colloidal drug aggregates were formulated as described in Table S3, with the addition of 2 μM 7-aminoactinomycin D (7-AAD) to monitor endosome disruption in those experiments. Colloids composed of 50 μM lapatinib and 150 μM fulvestrant and stabilized with transferrin resulted in greater (A) cytotoxicity and (B) endosomal disruption (measured by amount of nuclear dye) than either co-colloids stabilized with PLAC-PEG or colloids of fulvestrant alone (150 μM fulvestrant plus stabilizer) (n = 9 biological replicates and separate colloid formulations for the toxicity experiment and n = 3 for the endosome disruption experiment, mean ± SD, two-way ANOVA with Tukey’s posthoc test, *p < 0.05, **p < 0.01, ****p < 0.0001). (C) Schematic describing how the membrane-impermeant nucleic acid stain 7-AAD was used to test for endosomal escape. (D) Nuclear 7-AAD fluorescence (red) is visible after treatment with lapatinib-fulvestrant colloids stabilized with transferrin, demonstrating its endosomal escape. (E) Permeabilized cells accumulate 7-AAD (red) in their nuclei as expected, whereas (F) live cells treated with 7-AAD alone do not. The presence of cells in each region of interest was verified using the transmission channel prior to capturing these images.
To determine the mechanism of the increased toxicity of endocytosed lapatinib-containing colloids versus nonendocytosed colloids, we investigated the ability of colloidal lapatinib to disrupt the endosomes using a fluorescence dequenching assay.28,29 Incorporating the membrane impermeant nuclear stain 7-aminoactinomycin D (7-AAD) into the colloids allowed us to measure its fluorescence in MDA-MB-231-H2N cells. The fluorescence of 7-AAD intensifies on DNA binding. Therefore, 7-AAD staining should only be observed in the nucleus following both colloid endocytosis and subsequent endosomal escape. Lapatinib/fulvestrant-transferrin colloids resulted in higher nuclear 7-AAD fluorescence than fulvestrant-transferrin colloids and PLAC-PEG stabilized formulations (Figures 4B-D and S14). Viability of the cells remained unchanged after 3 h of treatment, indicating that drug toxicity did not interfere with this assay (Figure S15).
An alternative explanation to endosome disruption is simple diffusion of lapatinib from the endosomes, which we probed with a membrane permeability assay. The transmembrane diffusion of lapatinib was reduced when the colloids were acidified (Figure S16), likely because the concentration of free, uncharged lapatinib decreased with increasing acidity (Figure S17).
Notwithstanding the importance of transferrin for cellular uptake, we wondered about its stability in serum as displacement of the macromolecular coronas by serum proteins can affect circulating nanoparticles.30 To test whether serum proteins could displace the transferrin stabilizer, we formulated colloidal drug aggregates stabilized with fluorophore-labeled transferrin and then pelleted the colloids by centrifugation. Quantifying the fraction of transferrin remaining in the supernatant, we found that most of the transferrin was displaced after 6 h of incubation in 10% serum (Figure S18).
Discussion.
We show that one can trigger the release of weakly basic colloid-forming drugs from stable colloidal drug aggregates by acidification of the medium. By exploiting the local acidity in the endolysosomal pathway, we demonstrate triggered drug release and its consequent cytotoxicity, thereby overcoming a key challenge of colloidal stability, and hence inactivity, after cell uptake. Thus, whereas previous research has shown that colloidal aggregation can cause false negative hits in cytotoxicity assays,11,12 this effect can be overcome by protonation of the colloidal drug and subsequent endosome disruption.
Interestingly, we observed that the amount of lapatinib released was less than its CAC at a comparable pH. We attribute this behavior to the fulvestrant, which remains in the colloidal state and acts as a sink for lapatinib. This hypothesis is supported by studies on the phase behavior of co-colloids that show a reduction in effective CAC when multiple drugs coexist in the colloid.31-33 This physical interaction between lapatinib and fulvestrant suggests that they mix to form co-colloids and explains how fulvestrant stabilizes lapatinib against precipitation.
We found that endocytosis of the acid-responsive lapatinib/fulvestrant colloids greatly enhances their cytotoxicity due to increased drug transport from the endosomes into the cytosol. This endosomal escape could occur by one of two mechanisms: either the increased concentration of free lapatinib enhances the amount of lapatinib diffusion across the endosomal membrane or the weakly basic lapatinib disrupts the integrity of the endosomes through osmotic pressure effects.28,34,35 As lapatinib is slow to cross membranes under acidic conditions, the enhanced diffusion mechanism is unlikely. Transport of ionizable drugs, such as protonated (cationic) lapatinib at reduced pH, diffuse more slowly through the lipid membranes.36,37 This prediction was supported by our estimate of free, uncharged lapatinib as a function of pH. With respect to the proton-sponge mechanism, the 7-AAD fluorescence quenching assay suggests that acid-responsive colloidal drug aggregates can disrupt endosomes, leading to drug leakage into the cytosol and ultimately enhancing drug cytotoxicity. This type of endosomal escape strategy has been exploited to deliver therapeutics that are unable to escape the endosomes themselves.35,38,39 We have adapted this approach to deliver a new type of cargo: colloidal drug aggregates. Furthermore, we avoid the use of acid-responsive but pharmacologically inert excipients by using a drug that is naturally acid-responsive. Conceivably, such pharmacologically active drug colloids could be used instead of the traditionally used inert carriers to deliver proteins or nucleic acids.
Notwithstanding these results, colloidal drug aggregates are admittedly in dynamic equilibrium with free drug, and a small amount of that free drug may diffuse across lipid membranes. As a result, the cells that are highly sensitive to lapatinib may be killed even in the absence of colloid endocytosis. Another consequence of this behavior is that drug diffusion out of the endolysosomal pathway could slowly occur simultaneously with the proton sponge effect.
Although our results demonstrate that endosomal escape of lapatinib is enhanced by the acidic microenvironment, quantifying endosomal escape remains a challenge for the field, with many methods relying on inferences drawn from models.29 Importantly, we show endosomal escape in live cells through a membrane disruption mechanism. Furthermore, we show that only colloidal drugs that are inherently acid-responsive can have their release triggered by acidic conditions whereas those that are unresponsive to acid cannot.
To be useful in vivo, there are still hurdles to overcome in designing these colloidal formulations. For example, many nanomaterials, including colloidal drug aggregates, can be solubilized by serum proteins,10,40-42 which inherently limits the in vivo utility of targeting the tumor. Thus, this issue will need to be considered during the development of colloids for disease treatment.
Conclusions.
Recent attempts to exploit colloidal drug aggregates have been hindered, until now, by the inability to control drug release. We demonstrate acid-triggered release from stable colloidal drug aggregates, endosomal disruption, and enhanced cytotoxicity. We demonstrate the selective, stimulus-responsive release of drugs from colloidal aggregates, thereby laying the framework for controlled release from a colloid that is inactive until its target is reached.
■ METHODS
Materials.
Lapatinib and sorafenib were purchased from MedChemExpress. Nilotinib and pazopanib were purchased from Cedarlane. Fulvestrant was purchased from SelleckChem. Poly(D,L-lactide-co-2-methyl-2-carboxy-trimethylene carbonate)-graft-poly(ethylene glycol) (PLAC-PEG) was synthesized by ring-opening polymerization using a pyrenebutanol initiator to a molecular weight of 12 kDa and conjugated with an average of three 10 kDa PEG chains/backbone as previously described.43 Ultrapure polysorbate 80 (UP80) was purchased from NOF America Corporation. Clotrimazole, norethindrone, dimethyl sulfoxide (DMSO), transferrin, EDTA, dodecane, lecithin, poly(acrylic acid) (PAA), methylcelluose, and hydroxypropylmethylcellulose (HPMC) were purchased from Sigma-Aldrich. Transferrin-Alexa Fluor 488 conjugate, RPMI 1640 cell culture medium, penicillin-streptomycin solution, trypsin-EDTA solution, Hank’s balanced salt solution, PrestoBlue cell viability reagent, CholEsteryl BODIPY 542/563 C11, CellMask Green 1000× solution, and 7-AAD were purchased from Thermo Fisher Scientific. Fetal bovine serum and Dulbecco’s phosphate buffered saline were purchased from Wisent Bio Products. Ultrapure Congo red was purchased from Enzo Life Sciences. HPLC grade acetonitrile and methanol were purchased from Caledon Laboratories. Mass spectrometry grade formic acid was purchased from Fluka.
Colloidal Drug Aggregate Formulation.
Colloidal drug aggregates were formulated as described previously.8 Briefly, the colloids form spontaneously when water is added to drug that is dissolved in a water-miscible organic solvent (usually DMSO). Final colloid suspensions were typically made at a 1 mL scale with 1% (v/v) final DMSO concentration. First, solutions of drug, polymer, and dye were prepared in 10 μL of DMSO at 100× the final concentration. For colloids formulated under serum-free conditions, 890 μL of double distilled water was then added, followed by 100 μL of 10× PBS (for experiments without cells) or 10× RPMI 1640 (for cell experiments). For colloids formulated under 10% (v/v) serum conditions, 800 μL of double distilled water was added, followed by 90 μL of 10× PBS or 10× media, and finally 100 μL of FBS. Transferrin stabilized colloids were prepared by supplementing the water added with a small amount of 5 mg mL−1 transferrin in PBS. When the pH of the colloid solution needed to be adjusted, the amount of water added was reduced by 10 μL, and 10 μL of aqueous citric acid was added as the last formulation step. The concentration of citric acid used depended on the desired final pH. In PBS, the concentration was 0.12 M for pH 6.5, 0.3 M for pH 5.5, 0.35 M for pH 5.2, and 0.5 M for pH 4.5. In PBS with 10% (v/v) FBS, the concentration was 0.15 M for pH 6.5, 0.35 M for pH 5.5, and 0.6 M for pH 4.5. For time-based studies, the colloid suspensions were incubated at 37 °C between time points.
Characterization by Dynamic Light Scattering.
Colloid diameter, polydispersity index (dispersity), and scattering intensity were measured by dynamic light scattering (DLS) using a DynaPro Plate Reader II (Wyatt Technologies) that was optimized by the manufacturer for detection of colloidal aggregates (i.e., 100–1000 nm particles). The instrument was configured with a 60 mW 830 nM laser and detector angle of 158°. A 100 μL sample of each formulation was pipetted into a 96-well plate and measured with 20 acquisitions per sample.
Characterization of Zeta Potential.
Colloids were prepared as described above, but with a single addition of 0.1 mM KCl solution instead of water and buffer. Zeta potential was immediately assessed using a Malvern Nano-ZS instrument (Malvern Panalytical).
Fluorescent Intensity Characterization.
Colloid suspensions were prepared as described above. A 100 μL sample of each formulation was pipetted into a 96-well plate. Fluorescence was measured using a Tecan Infinite Pro 200 plate reader.
Characterization by Transmission Electron Microscopy (TEM).
Colloidal formulations (5 μL) were deposited onto freshly glow-discharged 400 mesh carbon coated copper TEM grids (Ted Pella, Inc.) and allowed to adhere for 5 min. Excess liquid was removed, followed by a quick wash with 5 μL of water. Grids were then imaged on a LEO 912B Energy Filtered transmission electron microscope operating at 120 kV.
Assessment of Colloid-Bound Transferrin.
Colloid formulations were prepared as described above with transferrin-Alexa Fluor 488 conjugate as the stabilizer. At predetermined time points during incubation at 37 °C, formulations were centrifuged at 16,000g for 1 h, followed by withdrawal of 100 μL of supernatant for measurement. The amount of transferrin remaining in the supernatant was measured using fluorimetry (λex = 488 nm, λem = 530 nm). The fraction of transferrin bound to the colloid was calculated with eq 1.
| (1) |
Assessment of Drug Release.
Drug release was measured by centrifuging to pellet the colloids and then by quantifying the drug in the supernatant. Colloid formulations were centrifuged at 16,000g for 1 h, followed by withdrawal of 100 μL for quantification. Another 100 μL was withdrawn for DLS analysis to confirm that the colloids had completely settled out of solution. Noncentrifuged colloid suspensions were used as controls.
Membrane Permeability Assay.
Supported artificial lipid membranes were prepared according to the manufacturer’s instructions (EMD Millipore cat# MATRNPS50). Briefly, a 1% (w/w) solution of egg lecithin in dodecane was prepared by sonicating until the solution was no longer cloudy. A 5 μL aliquot of this phospholipid solution was dropped by pipet into each well of the mesh plate, which wet the membrane and caused it to become translucent. Receiver solution was prepared as 1% (v/v) DMSO and 10% (v/v) FBS in PBS, and 340 μL was pipetted into each well of the receiver plate. Lapatinib and fulvestrant solutions ranging from 0 to 5 μM for constructing the standard curve were prepared in identical media and pipetted into separate wells of the receiver plate. Colloids were prepared in PBS containing 10% (v/v) FBS as described above, and 150 μL was pipetted into the mesh plate wells corresponding to the receiver solution in the receiver plate. The mesh plate and receiver plate were gently mated, resulting in the receiver solution wetting the bottom of the phospholipid membrane. A plate cover was added, and the assembly edges were sealed with parafilm to prevent evaporation. The sealed assembly was then incubated at 37 °C for 6 h. Finally, the stack was carefully disassembled and a 200 μL sample of receiver and standard curve solutions was withdrawn for drug quantification as described below. Each donor and receiver well were tested with a pH strip to verify the integrity of the membrane. A separate test with Trypan blue verified that the membranes were impermeable to this colloidal dye.
Drug Concentration Quantification.
Drug concentration in samples from drug release and membrane permeability assays was quantified by high pressure liquid chromatography coupled with tandem mass spectrometry (HPLC-MS-MS). Protein was precipitated from samples in serum containing media by spiking with 10 μL of formic acid and adding acetonitrile to a final volume of 1 mL. The precipitated samples were then centrifuged at 16,000 g for 5 min to pellet the proteins. The drug-containing supernatant was then diluted in methanol such that the final drug concentration was less than 100 ng mL−1. During the final dilution, internal standards (nilotinib for lapatinib and norethindrone for fulvestrant) were added to a final concentration of 25 ng/mL each. Standard curves were prepared in a similar way, by diluting 100 mM solutions of drug in DMSO with methanol to final concentrations of 100, 75, 50, 25, 10, 5, 2.5, and 1 ng mL−1, each with 25 ng mL−1 of internal standard.
Cell Culture.
MDA-MB-231-H2N cells were a generous gift from R. Kerbel (Sunnybrook Research Institute, Toronto, ON, Canada). Cells were maintained in a humidified incubator at 37 °C with 5% atmospheric CO2. Cells were grown in 75 cm2 tissue culture flasks with 10 mL of RPMI 1640 supplemented with 10% FBS, 10 UI/mL penicillin, and 10 μg mL−1 streptomycin. Cells were passaged twice per week with a typical subculture ratio of 1:16.
Cell Viability Experiments.
After passaging, cell suspensions were diluted into fresh media and 200 μL was pipetted into each well of a 96-well plate. Two thousand cells per well were plated and allowed to adhere overnight. Then, the media was withdrawn and replaced with treatment formulations (prepared as described above). Cells were incubated during the experiment in a humidified incubator at 37 °C with 5% atmospheric CO2. For treatments lasting less than 3 days, the treatment solutions were removed after the prescribed time, cells were washed with fresh media, and 200 μL of fresh media was added. Cell viability was assessed 3 days after commencing treatment using the PrestoBlue viability assay according to the manufacturer’s protocol. Cell viability is reported as a percentage of the vehicle (DMSO with no drug or excipient) control.
Confocal Imaging of Treated Cells.
Cells were seeded at approximately 2 × 105 cells per well in 8-chamber tissue culture treated glass coverslips and allowed to adhere overnight. Treatments were prepared as described above and 300 μL applied to each well. Cells were incubated during the experiment in a humidified incubator at 37 °C with 5% (v/v) atmospheric CO2. The treatment solutions were removed after the prescribed time, cells washed with fresh media, and 300 μL of fresh media added. For fixed cells, 4% (w/w) aqueous paraformaldehyde was applied for 10 min, followed by staining (when appropriate), and finally blank PBS. For live cell conditions, cells were washed with fresh media, stained, and imaged under Hank’s balanced salt solution. Cells were imaged on an Olympus FV1000 inverted confocal microscope using a 1.42 NA 60× oil immersion lens (Olympus PLAPON 60XO). Laser and detector settings were held constant between different treatment conditions.
Flow Cytometry.
Cells were seeded at approximately 2 × 105 cells per well in 24-well plates and allowed to adhere overnight. Treatments were prepared as described above, and 500 μL was applied to each well. Cells were incubated during the experiment in a humidified incubator at 37 °C with 5% (v/v) atmospheric CO2. For treatments lasting less than 3 h, the treatment solutions were removed after the prescribed time, cells were washed with fresh media, and 500 μL of fresh media was added. Three hours after treatment initiation, cells were washed three times with media, detached using 500 μL of accutase solution, spiked with 500 μL of media, then centrifuged at 400g to pellet the cells. Cells were then resuspended in cold flow buffer (PBS supplemented with 2% (v/v) FBS and 2 mM EDTA) and kept on ice until measurement. The flow buffer was supplemented with 7-AAD at 2 μg mL−1 as a vital stain, except for blank controls and in the endosome escape assay where 7-AAD had already been added. Cell fluorescence was quantified using a BD Accuri C6 flow cytometer with excitation wavelength of 488 nm and emission filters of 585/40 nm (BODIPY colloid dye) and >670 nm (7-AAD). Data were analyzed using the BD Accuri C6 Plus software and reported as the fluorescence of the live cell fraction (gated using scattering and 7-AAD) averaged between three biological replicates.
Supplementary Material
■ ACKNOWLEDGMENTS
This work was supported by U.S. National Institutes of General Medical Sciences (Grant GM71630 to B.K.S and M.S.S, and by R35GM122481to B.K.S.) and the Natural Sciences and Engineering Research Council (NSERC Discovery Grant to M.S.S). E.N.D and A.N.G are thankful for the support of Queen Elizabeth II Graduate Scholarships in Science & Technology and NSERC postgraduate scholarships, respectively. A.N.G. was also supported by an NSERC CREATE in M3 scholarship. We thank M. Forbes from the University of Toronto Advanced Instrumentation for Molecular Structure lab for assistance with mass spectrometry, J. Logie for synthesis of the PLAC-PEG polymer, N. Coombs from the University of Toronto Centre for Nanostructure Imaging for assistance with TEM imaging, P. Gilbert and S. Davoudi for use of their flow cytometer, and members of the Shoichet laboratories for their thoughtful comments.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschem-bio.9b00247.
Data on CAC calculation, colloid characterization, and supporting confocal images (PDF)
The authors declare the following competing financial interest(s): We have submitted a provisional patent on the topic of acid-responsive colloidal drug aggregates.
■ REFERENCES
- (1).Coan KED, and Shoichet BK (2008) Stoichiometry and Physical Chemistry of Promiscuous Aggregate-Based Inhibitors. J. Am. Chem. Soc 130, 9606–9612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Seidler J, McGovern SL, Doman TN, and Shoichet BK (2003) Identification and Prediction of Promiscuous Aggregating Inhibitors among Known Drugs. J. Med. Chem 46, 4477–4486. [DOI] [PubMed] [Google Scholar]
- (3).Doak AK, Wille H, Prusiner SB, and Shoichet BK (2010) Colloid Formation by Drugs in Simulated Intestinal Fluid. J. Med. Chem 53, 4259–4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Brick MC, Palmer HJ, and Whitesides TH (2003) Formation of Colloidal Dispersions of Organic Materials in Aqueous Media by Solvent Shifting. Langmuir 19, 6367–6380. [Google Scholar]
- (5).Coan KED, Maltby DA, Burlingame AL, and Shoichet BK (2009) Promiscuous Aggregate-Based Inhibitors Promote Enzyme Unfolding. J. Med. Chem 52, 2067–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).McGovern SL, Caselli E, Grigorieff N, and Shoichet BK (2002) A Common Mechanism Underlying Promiscuous Inhibitors from Virtual and High-Throughput Screening. J. Med. Chem 45, 1712–1722. [DOI] [PubMed] [Google Scholar]
- (7).Ganesh AN, Donders EN, Shoichet BK, and Shoichet MS (2018) Colloidal aggregation: From screening nuisance to formulation nuance. Nano Today 19, 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ganesh AN, Logie J, McLaughlin CK, Barthel BL, Koch TH, Shoichet BK, and Shoichet MS (2017) Leveraging Colloidal Aggregation for Drug-Rich Nanoparticle Formulations. Mol. Pharmaceutics 14, 1852–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Shamay Y, Shah J, Işık M, Mizrachi A, Leibold J, Tschaharganeh DF, Roxbury D, Budhathoki-Uprety J, Nawaly K, Sugarman JL, Baut E, Neiman MR, Dacek M, Ganesh KS, Johnson DC, Sridharan R, Chu KL, Rajasekhar VK, Lowe SW, Chodera JD, and Heller DA (2018) Quantitative self-assembly prediction yields targeted nanomedicines. Nat. Mater 17, 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ganesh AN, Aman A, Logie J, Barthel BL, Cogan P, Alawar R, Koch TH, Shoichet BK, and Shoichet MS (2019) Colloidal Drug Aggregate Stability in High Serum Conditions and Pharmacokinetic Consequence. ACS Chem. Biol 14, 751–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Owen SC, Doak AK, Ganesh AN, Nedyalkova L, McLaughlin CK, Shoichet BK, and Shoichet MS (2014) Colloidal Drug Formulations Can Explain “Bell-Shaped” Concentration–Response Curves. ACS Chem. Biol 9, 777–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Owen SC, Doak AK, Wassam P, Shoichet MS, and Shoichet BK (2012) Colloidal Aggregation Affects the Efficacy of Anticancer Drugs in Cell Culture. ACS Chem. Biol 7, 1429–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).McLaughlin CK, Duan D, Ganesh AN, Torosyan H, Shoichet BK, and Shoichet MS (2016) Stable Colloidal Drug Aggregates Catch and Release Active Enzymes. ACS Chem. Biol 11 , 992–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Mindell JA (2012) Lysosomal Acidification Mechanisms. Annu. Rev. Physiol 74, 69–86. [DOI] [PubMed] [Google Scholar]
- (15).Indulkar AS, Box KJ, Taylor R, Ruiz R, and Taylor LS (2015) pH-Dependent Liquid–Liquid Phase Separation of Highly Supersaturated Solutions of Weakly Basic Drugs. Mol. Pharmaceutics 12, 2365–2377. [DOI] [PubMed] [Google Scholar]
- (16).Frenkel YV, Clark AD, Das K, Wang Y-H, Lewi PJ, Janssen PAJ, and Arnold E (2005) Concentration and pH Dependent Aggregation of Hydrophobic Drug Molecules and Relevance to Oral Bioavailability. J. Med. Chem 48, 1974–1983. [DOI] [PubMed] [Google Scholar]
- (17).Sugihara H, and Taylor LS (2018) Evaluation of Pazopanib Phase Behavior Following pH-Induced Supersaturation. Mol. Pharmaceutics 15, 1690–1699. [DOI] [PubMed] [Google Scholar]
- (18).Ivanov AI, Ed. (2008) Exocytosis and endocytosis, Humana Press, Totowa, NJ. [Google Scholar]
- (19).Canton I, and Battaglia G (2012) Endocytosis at the nanoscale. Chem. Soc. Rev 41, 2718. [DOI] [PubMed] [Google Scholar]
- (20).Doherty GJ, and McMahon HT (2009) Mechanisms of Endocytosis. Annu. Rev. Biochem 78, 857–902. [DOI] [PubMed] [Google Scholar]
- (21).Wang J, MacEwan SR, and Chilkoti A (2017) Quantitative Mapping of the Spatial Distribution of Nanoparticles in Endo-Lysosomes by Local pH. Nano Lett. 17, 1226–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ganesh AN, McLaughlin CK, Duan D, Shoichet BK, and Shoichet MS (2017) A New Spin on Antibody–Drug Conjugates: Trastuzumab-Fulvestrant Colloidal Drug Aggregates Target HER2-Positive Cells. ACS Appl. Mater. Interfaces 9, 12195–12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ilevbare GA, and Taylor LS (2013) Liquid–Liquid Phase Separation in Highly Supersaturated Aqueous Solutions of Poorly Water-Soluble Drugs: Implications for Solubility Enhancing Formulations. Cryst. Growth Des 13, 1497–1509. [Google Scholar]
- (24).Tadros TF, Ed. (2007) Colloid stability: the role of surface forces, Wiley-VCH Verlag, Weinheim. [Google Scholar]
- (25).D’Addio SM, and Prud’homme RK (2011) Controlling drug nanoparticle formation by rapid precipitation. Adv. Drug Delivery Rev 63, 417–426. [DOI] [PubMed] [Google Scholar]
- (26).Bakshi RP, Tatham LM, Savage AC, Tripathi AK, Mlambo G, Ippolito MM, Nenortas E, Rannard SP, Owen A, and Shapiro TA (2018) Long-acting injectable atovaquone nanomedicines for malaria prophylaxis. Nat. Commun 9, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Indulkar AS, Mo H, Gao Y, Raina SA, Zhang GGZ, and Taylor LS (2017) Impact of Micellar Surfactant on Supersaturation and Insight into Solubilization Mechanisms in Supersaturated Solutions of Atazanavir. Pharm. Res 34, 1276–1295. [DOI] [PubMed] [Google Scholar]
- (28).Vermeulen LMP, Brans T, Samal SK, Dubruel P, Demeester J, De Smedt SC, Remaut K, and Braeckmans K (2018) Endosomal Size and Membrane Leakiness Influence Proton Sponge-Based Rupture of Endosomal Vesicles. ACS Nano 12, 2332–2345. [DOI] [PubMed] [Google Scholar]
- (29).Martens TF, Remaut K, Demeester J, De Smedt SC, and Braeckmans K (2014) Intracellular delivery of nanomaterials: How to catch endosomal escape in the act. Nano Today 9, 344–364. [Google Scholar]
- (30).Ke PC, Lin S, Parak WJ, Davis TP, and Caruso F (2017) A Decade of the Protein Corona. ACS Nano 11, 11773–11776. [DOI] [PubMed] [Google Scholar]
- (31).Trasi NS, and Taylor LS (2015) Thermodynamics of Highly Supersaturated Aqueous Solutions of Poorly Water-Soluble Drugs—Impact of a Second Drug on the Solution Phase Behavior and Implications for Combination Products. J. Pharm. Sci 104, 2583–2593. [DOI] [PubMed] [Google Scholar]
- (32).Knapik-Kowalczuk J, Tu W, Chmiel K, Rams-Baron M, and Paluch M (2018) Co-Stabilization of Amorphous Pharmaceuticals—The Case of Nifedipine and Nimodipine. Mol. Pharmaceutics 15, 2455–2465. [DOI] [PubMed] [Google Scholar]
- (33).Feng BY, and Shoichet BK (2006) Synergy and Antagonism of Promiscuous Inhibition in Multiple-Compound Mixtures. J. Med. Chem 49, 2151–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Benjaminsen RV, Mattebjerg MA, Henriksen JR, Moghimi SM, and Andresen TL (2013) The Possible “Proton Sponge ” Effect of Polyethylenimine (PEI) Does Not Include Change in Lysosomal pH. Mol. Ther 21, 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Varkouhi AK, Scholte M, Storm G, and Haisma HJ (2011) Endosomal escape pathways for delivery of biologicals. J. Controlled Release 151 , 220–228. [DOI] [PubMed] [Google Scholar]
- (36).Amidon GL, Lennernäs H, Shah VP, and Crison JR (1995) A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of in Vitro Drug Product Dissolution and in Vivo Bioavailability. Pharm. Res 12, 413–420. [DOI] [PubMed] [Google Scholar]
- (37).Mahoney BP, Raghunand N, Baggett B, and Gillies RJ (2003) Tumor acidity, ion trapping and chemotherapeutics. Biochem. Pharmacol 66, 1207–1218. [DOI] [PubMed] [Google Scholar]
- (38).Smith SA, Selby LI, Johnston APR, and Such GK (2019) The Endosomal Escape of Nanoparticles: Toward More Efficient Cellular Delivery. Bioconjugate Chem. 30, 263–272. [DOI] [PubMed] [Google Scholar]
- (39).Cullis PR, and Hope MJ (2017) Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol. Ther 25, 1467–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Sun X, Wang G, Zhang H, Hu S, Liu X, Tang J, and Shen Y (2018) The Blood Clearance Kinetics and Pathway of Polymeric Micelles in Cancer Drug Delivery. ACS Nano 12, 6179–6192. [DOI] [PubMed] [Google Scholar]
- (41).Talelli M, Barz M, Rijcken CJF, Kiessling F, Hennink WE, and Lammers T (2015) Core-crosslinked polymeric micelles: Principles, preparation, biomedical applications and clinical translation. Nano Today 10, 93–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Svenson S (2014) What nanomedicine in the clinic right now really forms nanoparticles?: What nanomedicine really forms nanoparticles. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol 6, 125–135. [DOI] [PubMed] [Google Scholar]
- (43).Logie J, Owen SC, McLaughlin CK, and Shoichet MS (2014) PEG-Graft Density Controls Polymeric Nanoparticle Micelle Stability. Chem. Mater 26, 2847–2855. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.