

Abstract
Detection of double-stranded RNAs (dsRNAs) is a central mechanism of innate immune defense in many organisms. We here discuss several families of dsRNA-binding proteins involved in mammalian antiviral innate immunity. These include RIG-I-like receptors, protein kinase R, oligoadenylate synthases, adenosine deaminases acting on RNA, RNA interference systems, and other proteins containing dsRNA-binding domains and helicase domains. Studies suggest that their functions are highly interdependent and that their interdependence could offer keys to understanding the complex regulatory mechanisms for cellular dsRNA homeostasis and antiviral immunity. This review aims to highlight their interconnectivity, as well as their commonalities and differences in their dsRNA recognition mechanisms.
Keywords: dsRNA, RIG-I-like receptor, protein kinase R, oligoadenylate synthase, dsRNA-dependent adenosine deaminase, RNA interference
INTRODUCTION
Detection of foreign nucleic acids is a central mechanism for innate immune defense in most organisms studied to date. Accumulating evidence suggests that the innate immune system can be stimulated not only by pathogen-derived nucleic acids but also by host-derived nucleic acids that signal pathologic cellular states. Such self-derived danger signals include altered structure, location, or loss of interaction partners of cellular DNA or RNA. One of the best-characterized foreign features of RNA is the double-stranded structure. Double-stranded RNAs (dsRNAs) often accumulate during viral infection as a result of viral RNA replication, but they are also present in uninfected cells to varying degrees. Much progress has been made in our understanding of how the innate immune system distinguishes between cellular dsRNA versus viral dsRNAs in normal physiological conditions, and how and when such discrimination breaks down, causing self dsRNA– triggered sterile inflammation. These investigations led to the identification of new sources of viral and cellular dsRNAs and a previously unappreciated network of dsRNA-binding proteins that regulate innate immunity. These proteins include classical innate immune molecules, such as RIG-I-like receptors (RLRs), protein kinase R (PKR), and oligoadenylate synthases (OASes), but also other dsRNA-binding proteins previously known to function in cellular dsRNA homeostasis without a direct connection to innate immunity.
In this review, we discuss several families of cellular dsRNA–binding proteins involved in antiviral innate immunity (Figure 1a, Table 1). We focus on the structural and biochemical mechanisms of how these proteins recognize dsRNAs and how the specific dsRNA binding mode allows discrimination between self and nonself dsRNAs, and between normal and altered self dsRNAs. We also discuss how these proteins influence each other’s functions in the context of antiviral immunity and cellular dsRNA biology. We begin by describing general structural principles of dsRNA and dsRNA-protein interactions, and several important points to consider in investigating dsRNA-binding proteins.
Figure 1.

dsRNA-binding proteins in innate immunity. (a) Network of dsRNA sensors and modulators in innate immunity. The diagram below shows their interdependence. Blue arrows and red inhibition lines indicate reported stimulatory and inhibitory relationships, respectively. (b) Structures of dsRNA and dsDNA, which display A-form and B-form double helixes, respectively. (c) Watson-Crick base pair interactions, with edges facing major versus minor grooves indicated. Note that relative locations of hydrogen bond donors and acceptors differ for all four nucleotides in the major groove but are degenerate in the minor groove. Abbreviations: ADAR, adenosine deaminase acting on RNA; PKR, protein kinase R; RLR, RIG-I-like receptor.
Table 1.
dsRNA-binding proteins
| dsRNA-binding proteins |
dsRNA-binding domains |
Best-known functions | Effect on other dsRNA-binding proteins |
Disease implication |
|---|---|---|---|---|
| RLRs | Helicase domain, C-terminal domain | Antiviral signal activation | Induce PKR, OASes, and ADARs through interferon signaling | Aicardi-Goutieres syndrome, Singleton-Merten syndrome, systemic lupus erythematosus |
| PKR | dsRBDs | Translational suppression | Amplifies RLR signaling | Unknown |
| OASes, RNase L | NTase domains | Translational suppression | Amplify RLR signaling | Unknown |
| ADARs | dsRBDs | RNA editing and resolving dsRNA structure | Suppress RLRs, PKR, OASes; assist Dicer | Aicardi-Goutieres syndrome |
| Dicer, Drosha | PAZ, dsRBDs, RNase III, helicase domain | Processing dsRNAs | Inhibit RLRs | Age-related macular degeneration, polyglutamine diseases |
| PACT, TRBP | dsRBDs | Regulate PKR, Dicer, and RLRs | Inhibit PKR, promote Dicer and RLRs | Dystonia |
| Helicases (besides RLRs and Dicer) | Helicase domains and others | Regulate dsRNA-dependent immunity | Unknown | Unknown |
Abbreviations: ADAR, adenosine deaminases acting on RNA; dsRBD, dsRNA-binding domain; NTase, nucleotide transferase; OASes, oligoadenylate synthases; PAZ, Piwi Argonaute and Zwille; PKR, protein kinase R; RLR, RIG-I-like receptor; RNase, ribonuclease.
GENERAL STRUCTURAL PRINCIPLES OF dsRNA AND dsRNA-PROTEIN INTERACTIONS
The structure of dsRNA deviates significantly from that of dsDNA (Figure 1b). dsRNA displays an A-form duplex with deep and narrow major grooves and shallow and wider minor grooves (Figure 1b). dsDNA, on the other hand, forms the B-form duplex, with wide major grooves and narrow minor grooves (1, 2). As such, the major groove of dsDNA is accessible for protein secondary structure, while that of dsRNA is generally not (1, 2). Since the major groove, not the minor groove, displays edges of bases distinct in all four nucleotides (3) (Figure 1c), proteins interacting with the dsDNA major groove have the potential to discriminate among the four nucleotides and bind DNA with high sequence specificity (4). By contrast, dsRNA-binding proteins primarily interact with the RNA backbone and the minor groove and are thus largely sequence independent. While dsRNA sequence can indirectly affect protein binding to a certain extent (5), sequence dependence of dsRNA-binding proteins is generally limited.
CHALLENGES IN STUDYING dsRNA-PROTEIN INTERACTIONS
It is important to recognize common challenges and misconceptions in studying dsRNA-protein interactions. First and foremost, RNA affinity should not be equated to functional relevance. While some of the dsRNA-binding proteins in this review have a significantly higher affinity for dsRNA, many also bind other types of nucleic acids with comparable affinity through non-specific electrostatic interactions. As such, efforts to identify ligands solely based on affinity are inappropriate for these proteins. This was particularly the case for MDA5, an RLR that recognizes dsRNA. MDA5 forms filaments and activates antiviral signaling pathways only upon dsRNA binding (6, 7), but it also binds single-stranded RNA (ssRNA) with equivalent affinity (6). In addition to the high-affinity interaction with ssRNAs, which are abundant in cells, highly cooperative filament formation of MDA5 disfavors enrichment of stimulatory dsRNAs by MDA5 pull-down or by other affinity-based methods (8). By contrast, a conformation-specific method directly utilizing MDA5 filament formation enabled selective isolation of its dsRNA ligands (8).
Another challenge relates to the method of RNA preparation and common presence of RNA by-products or multiple conformers. In vitro transcription with T7 phage RNA polymerase is one of the most common methods for preparing RNAs. But it is known to generate a variety of unintended by-products, one of which is dsRNA formed by intended sense RNA and fully complementary antisense RNA (9). This can lead to misdefining RNA specificity for a protein that is in fact dsRNA specific (10). Even if the RNA of the intended sequence can be obtained without the by-products, whether the prepared RNA adopts the desired RNA structure is of another general concern. For example, hairpin RNAs can open up and pair with the complementary regions in trans rather than in cis, leading to longer dimeric or multimeric dsRNA formation with distinct biological activities. In fact, such dimer formation by hairpin RNAs has caused confusion as to the RNA specificity of PKR, a dsRNA-dependent kinase (11, 12). In many cases, native gel purification, rather than denaturing gel purification, and careful diagnostic analyses are required to determine the precise identity of the RNAs under investigation.
Additionally, several biochemical and molecular biology tools that are well established for ssRNA and dsDNA are often unsuitable for or difficult to apply to dsRNA. For example, protein-RNA UV cross-linking approaches used for isolating in vivo ligands are often known to be inefficient for dsRNA-binding proteins (13, 14), although successful cases do exist (15, 16). The inefficient cross-linking is likely due to limited contacts between protein side chains and dsRNA bases. Reverse transcription is another example that is difficult to apply to dsRNA. Even with heat denaturation during primer binding, duplex structure can be restored during reverse transcription and can block the activity of the reverse transcriptase, which generally lacks the strand-displacement activity. Knocking down dsRNA by small interfering RNA (siRNA) is also challenging, as the actions of siRNAs require accessibility of the complementary region in the target RNA. Finally, RNase treatment is frequently used to examine whether certain biological events are dependent on RNAs. However, dsRNAs, unlike ssRNAs, are more resistant to RNases, possibly requiring different choices of RNase treatment conditions to examine dsRNA dependence.
The challenges described above, some more serious than others, demand careful examination of dsRNA-protein interactions with the understanding that dsRNAs have different physicochemical properties than ssRNA and dsDNA, and that most molecular biology tools are developed for ssRNA/dsDNA and may not be suitable for dsRNA. With these considerations in mind, we discuss seven families of dsRNA-binding proteins at the interface of innate immunity and cellular dsRNA biology (Figure 1a). We apologize in advance for the studies omitted in this review due to the limited space. More extensive reviews for each family of proteins are available elsewhere.
RIG-I-LIKE RECEPTORS
RLRs consist of RIG-I, MDA5, and LGP2 and are innate immune receptors that recognize viral dsRNAs during infection. Upon viral dsRNA binding, RIG-I and MDA5 activate the common downstream adaptor molecule MAVS. Activated MAVS then recruits multiple signaling molecules, including TRAFs, TBK1, and IRF3/7, leading to the transcriptional upregulation of type I interferons and other proinflammatory cytokines (17, 18). Unlike RIG-I and MDA5, LGP2 does not directly activate MAVS but is thought to positively and negatively regulate MDA5 and RIG-I (19–21), respectively, although additional regulatory activities were also reported (22). Despite sharing the same downstream signaling pathways, RIG-I and MDA5 play nonredundant roles by recognizing largely distinct groups of viral RNAs (17, 18). RIG-I recognizes dsRNA structure in a manner dependent on the dsRNA end structure, in particular the blunt end and 5′-triphosphate group (5′ppp) (10). 5′ppp is present in all nascent transcripts but is generally removed from cellular RNAs through normal 5′ processing (e.g., endoribonuclease cleavage and 7-methyl guanosine cap addition) in the nucleus. By contrast, many (but not all) viral RNAs do not undergo 5′ processing and harbor 5′ppp. When these viral RNAs form dsRNAs with blunt ends, as in copy-back defective interfering particles or in genome panhandles, they can robustly activate RIG-I signaling. By contrast, dsRNA recognition by MDA5 is independent of 5′ppp or dsRNA end structure but instead requires a long (generally kb) duplex stem, as in the replication intermediates of picornaviruses. Note that the dsRNA length threshold for MDA5 is determined by filament assembly and disassembly kinetics and thus can change depending on the level of MDA5 and dsRNA (23, 24).
Detailed structural and biochemical studies revealed how individual RLRs differentially recognize dsRNAs and carry out their respective antiviral functions. We first describe the molecular mechanism of RIG-I, and then those of MDA5 and LGP2. In the absence of viral RNA, RIG-I is in the autorepressed state wherein the signaling domain, the N-terminal tandem CARD (2CARD), is prevented from activating MAVS (25). The autorepressed 2CARD is liberated when dsRNA binds the helicase and C-terminal domains (Hel-CTD) of RIG-I (26, 27), although ATP binding was also proposed to be important (25, 28). Multiple studies showed that a release of 2CARD is not sufficient for MAVS activation; it additionally requires a tetramerization of 2CARD (29). At least two non–mutually exclusive mechanisms have been proposed to stimulate 2CARD tetramerization. First, while RIG-I binds to the end of dsRNA as a monomer (to sense 5′ppp and blunt ends), it assembles a filamentous oligomer on longer dsRNAs through ATP-driven translocation (30–33) (Figure 2a). RIG-I filament formation in turn promotes the 2CARD tetramerization through a proximity-induced mechanism (30) (Figure 2a). Second, K63-linked polyubiquitin (K63-Ubn), synthesized by the E3 ligase TRIM25 and RIPLET, also plays an important role in tetramerization of RIG-I 2CARD by wrapping around the core tetramer structure of 2CARD (34, 35) (Figure 2a). The 2CARD tetramer then acts as a helical template to nucleate the MAVS filament formation (36), which in turn serves as a signaling platform to recruit downstream signaling molecules (36, 37).
Figure 2.
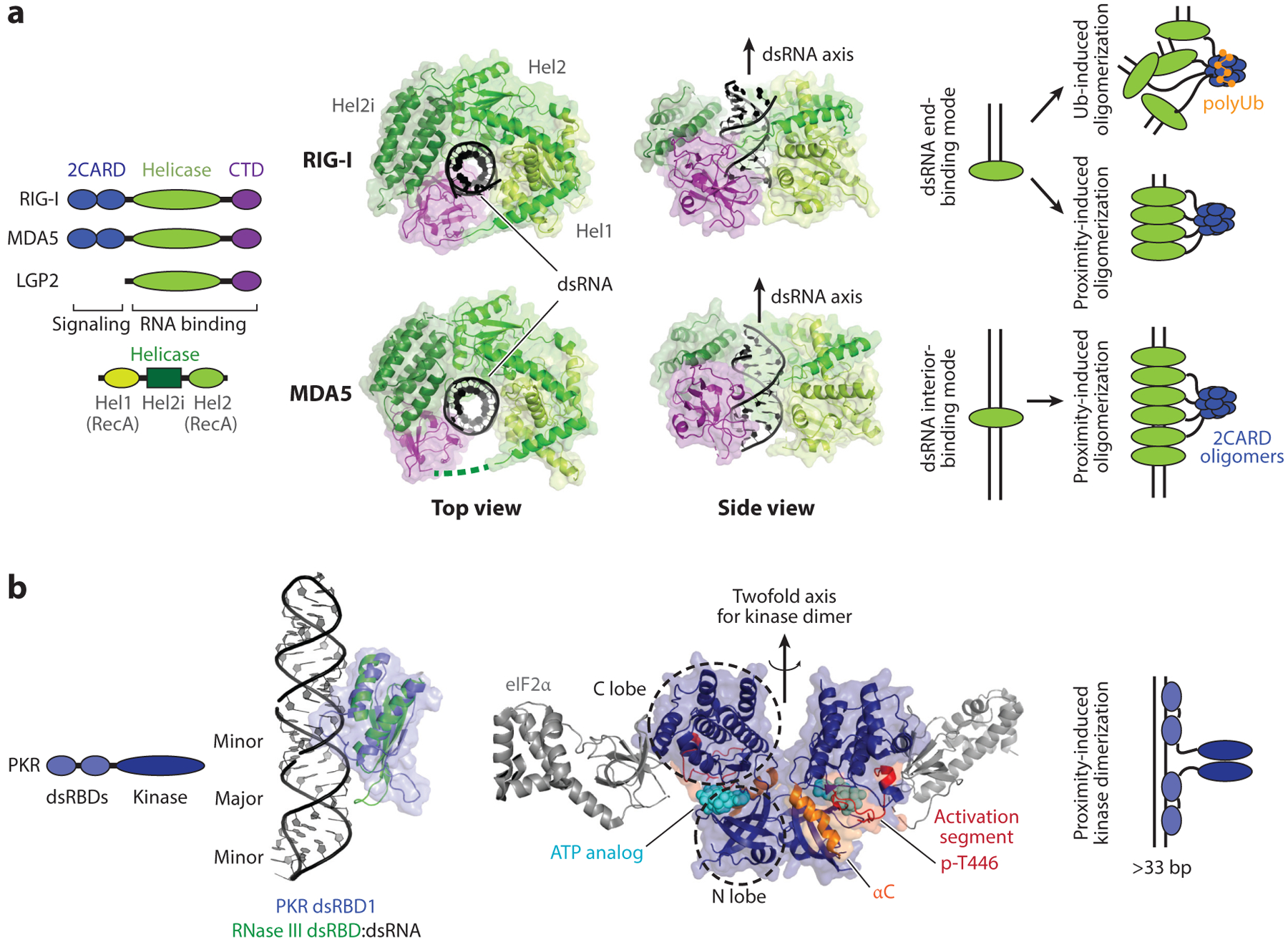
RLR and PKR. (a) Domain architectures and structures of human RIG-I and MDA5 in complex with dsRNA (PDB: 3TMI and 4GL2). RIG-I binds the dsRNA end as a monomer (as shown in the crystal structure), but on long dsRNA, it assembles into filamentous oligomers. Filament formation of RIG-I on dsRNA promotes 2CARD tetramerization through a proximity-induced mechanism. The K63-linked polyubiquitin chain also promotes 2CARD tetramerization. In contrast to RIG-I, MDA5 only binds the dsRNA interior, not the end, and its filament formation is obligatory. As with RIG-I, 2CARD tetramerization (with or without the K63-linked polyubiquitin chain) is required for antiviral signal activation. (b) Domain architecture and structures of PKR. The structure of PKR dsRBDs in complex with dsRNA is not available, but studies suggest that PKR dsRBDs bind dsRNA in a manner similar to other canonical dsRBDs. The RNA-free structure of PKR dsRBD1 (blue) (PDB: 1QU6) is overlaid on the structure of Aquifex aeolicus RNase III in complex with dsRNA (green) (PDB: 2EZ6). On the right is the structure of the PKR kinase domain (dark blue) in the active dimeric state and in complex with eIF2α (gray) (PDB: 2A19). Analogous to RLRs, PKR utilizes dsRNA binding and the proximity-induced mechanism to dimerize and activate the kinase domain. Abbreviations: CTD, C-terminal domain; dsRBD, dsRNA-binding domain; PKR, protein kinase R; RLR, RIG-I-like receptor; Ub, ubiquitin.
MDA5 has a similar domain architecture and signaling mechanism as RIG-I. MDA5 2CARD activates MAVS by forming an analogous oligomer, which in turn nucleates the MAVS filament formation. MDA5 also binds dsRNA using Hel-CTD and forms filaments along the length of dsRNA (Figure 2a). Unlike RIG-I, however, MDA5 does not bind dsRNA ends, and filament formation is obligatory for stable dsRNA binding and signal activation (6, 23). Furthermore, the MDA5 filament undergoes end disassembly during ATP hydrolysis (23), which results in a selective destabilization of the MDA5 filament on short dsRNA, and preferential accumulation of MDA5 on long dsRNA (23). This leads to selective recognition of long dsRNAs generated during viral replication and robust discrimination against short cellular dsRNAs during normal physiological conditions (24, 38).
The third RLR member, LGP2, contains the helicase domain and CTD but lacks 2CARD and is thus unable to activate MAVS. Structural studies, however, suggest that LGP2 binds dsRNA in a manner similar to RIG-I (39). It preferentially binds dsRNA ends (albeit in a 5′ppp-independent manner), and at the same time forms filamentous oligomers upon ATP hydrolysis (39–41). Previous studies suggest that LGP2 sensitizes MDA5 to dsRNA ligands and facilitates MDA5 filament formation by assisting the otherwise slow and inefficient nucleation process (42). Considering the high dsRNA affinity of LGP2 and its structural similarity to MDA5, LGP2 may serve as a nucleator for the MDA5 filament assembly, although direct interaction between LGP2 and MDA5 has not been demonstrated.
In all cases of RLRs, they recognize dsRNA structure and additional features, such as 5′ppp and dsRNA length, for robust discrimination between self and nonself dsRNAs. Despite these multilayered discriminatory mechanisms, accumulating evidence suggests that RLRs misrecognize self RNAs under various pathologic conditions. In mice, a single point mutation (G821S) in MDA5 leads to constitutive activation of its signaling activity and development of a multiple-organ autoimmune disease (43). In humans, mutations in at least seven different residues of MDA5 were shown to cause a broad spectrum of autoinflammatory diseases, such as Aicardi-Goutières syndrome and systemic lupus erythematosus (44–46). While some studies have suggested the possibility of ligand-independent activation of MDA5 (47), others have reported the role of endogenous dsRNAs, in particular those formed by inverted repeat Alu elements (IR-Alus), in constitutive activation of MDA5 (8). Similar IR-Alu-mediated activation was observed for wild-type MDA5 under the condition where the dsRNA-modifying enzyme ADAR1 (adenosine deaminase acting on RNA 1) is deficient (discussed below) (8, 48). Recent studies showed that a chemotherapeutic agent, 5-aza-2′-deoxycytidine, also activates MDA5 by upregulating endogenous retrovirus dsRNAs, leading to sterile inflammation that is beneficial for the subsequent cancer immunotherapies (49, 50). A more recent study showed that mitochondrial dsRNAs accumulated under pathologic conditions can be released to the cytosol and function as an additional source of endogenous ligands for MDA5 (51).
Several point mutations in RIG-I were also found to constitutively activate antiviral signaling, causing another autoinflammatory disease, Singleton-Merten syndrome (52, 53). Misrecognition of endogenous RNAs was also proposed to underlie aberrant activation of RIG-I, but the identity of the endogenous RNA ligand is less clear. Wild-type RIG-I can also be activated by ionizing radiation (IR) cancer therapy (54). IR was proposed to induce mislocalization of U1 and U2 small nuclear RNAs (snRNAs) and subsequent misrecognition by RIG-I. However, the precise nature of the RIG-I-stimulatory elements in U1/U2 snRNAs is yet unclear, as U1 and U2 snRNAs harbor tri-methyl-guanosine cap, not 5′ppp. In the context of breast cancer cells, RIG-I was found to be activated by 5′ppp-harboring transcripts of RNA polymerase III (RNAP III). It was proposed that cancer cell–stromal cell interactions upregulate RNAP III activity, leading to abnormal accumulation of RNAP III transcripts, such as RNA7SL1, without corresponding upregulation of its protein partner that normally shields 5′ppp (55, 56). Similar unshielding of 5′ppp-containing RNAP III transcripts was proposed for the activation of RIG-I during infection with herpes simplex virus 1, Epstein-Barr virus, and influenza A virus (57). Additionally, multiple other conditions were also shown to generate altered self RNAs, by the action of RNase L (58, 59) or inaction of the RNA exosome (RNase) component, SKIV2L (60), thereby triggering RIG-I signaling. In most cases, however, the proposed RNA ligands are not known to harbor canonical RIG-I-stimulatory elements, i.e., 5′ppp and blunt-ended dsRNA structure. Mechanistic details of how these RNAs stimulate RIG-I remain to be further investigated.
RLRs can also function beyond antiviral signaling pathways and impact the functions of other dsRNA-binding proteins. RIG-I and MDA5 were found to remodel dsRNA-protein complexes (dsRNPs) in an ATP-dependent manner (61). This remodeling activity can result in displacement of viral proteins from dsRNAs, exposing otherwise sequestered dsRNA to the host immune system, such as RLRs and PKR (61). In line with this observation, RIG-I and MDA5 were shown to have a signaling-independent antiviral activity against a broad range of viruses (61–64). Whether LGP2 displays a similar dsRNP-remodeling activity is as yet unclear, but recent studies have also suggested LGP2 activities beyond antiviral immunity. LGP2 inhibits Dicer-dependent processing of pre-microRNA (miRNA) and long dsRNA, thereby suppressing biogenesis of miRNA and siRNA, respectively (65, 66). These observations together suggest that RLRs have multiple functions in antiviral immunity and host dsRNA biology beyond simply activating the antiviral signaling pathway.
PROTEIN KINASE R
PKR is a dsRNA-dependent protein kinase and is transcriptionally upregulated by interferon. PKR normally exists in the latent form and is activated by dsRNA binding (67). One of the best-characterized targets of PKR is eIF2α, the α subunit of the eukaryotic translational initiation factor eIF2. eIF2 delivers the initiator tRNA to the small ribosomal subunit and is essential for cap-dependent translation. Phosphorylation of eIF2α prevents recycling of eIF2 and suppresses cap-dependent translational initiation. Thus, activation of PKR leads to the global shutdown of protein synthesis and inhibition of viral replication, cell cycle progression, and cell growth.
PKR consists of two tandem repeats of dsRNA-binding domains (dsRBDs) and a kinase domain (68, 69) (Figure 2b). dsRBDs are one of the most common dsRNA-binding protein motifs. Multiple structures of dsRBDs with and without dsRNA are available (5, 70) that show that dsRBDs have a conserved protein core structure and a common dsRNA-binding mode. dsRBD binds to one face of dsRNA primarily through phosphate and ribose backbones of two adjacent minor grooves (Figure 2b). Consistent with the backbone-binding mode, most dsRBDs, including PKR dsRBDs, interact with dsRNA in a sequence-independent manner (71). Studies showed that while the first dsRBD acts as the primary dsRNA binder on short dsRNAs (72, 73), both dsRBDs are required for high-affinity interaction with longer dsRNAs and dsRNA-dependent activation of the kinase domain (see below) (73, 74).
Earlier studies suggested that PKR exists in the autorepressed state in the absence of dsRNA, and dsRNA binding releases autorepression, thereby activating the kinase activity (75, 76). The second dsRBD was proposed to mediate autorepression by directly binding to the kinase domain. However, other studies have argued against the autorepression model and supported the role of dsRNA-induced dimerization as a mechanism for PKR activation. For example, biophysical analysis showed that latent PKR is in the extended monomeric conformation (77), inconsistent with the folded autoinhibited conformational state. Deletion of dsRBDs does not constitutively activate PKR, while replacement of dsRBDs by an artificial dimerizing domain does (78). The minimal dsRNA length (~33 bp) required for PKR activation also coincides with the minimal length stably recruiting two PKR molecules (74). Additionally, an excess amount of stimulatory dsRNA, which promotes monomeric PKR binding but suppresses PKR dimerization, was found to inhibit PKR activity (79).
Structures of the kinase domain further showed a detailed picture of the dimerization-induced activation of PKR (Figure 2b). Although the structure of inactive, monomeric PKR is not available, the structure of the active, dimeric PKR kinase domain suggested that the dimerization of the N-terminal lobe of the kinase domain likely induces conformational changes in the αC helix and the activation segments in a manner similar to other well-characterized kinases (69, 80) (Figure 2b). The structure also showed how the autophosphorylation of Thr446 in the activation segment further helps stabilize the active site conformation (80, 81) (Figure 2b). Altogether, these observations support the model where dsRNA binding brings two or more PKR molecules in close proximity to allow dimerization of the kinase domain (Figure 2b). Once dimerized, the kinase domain autophosphorylates in cis, which further stabilizes the dimeric structure even in the absence of dsRNA and fully activates kinase activity (78).
Accumulating evidence suggests that PKR can tolerate a certain level of structural irregularities (e.g., mismatches, bulges) in dsRNA (79) and can be activated by cellular dsRNAs under various physiological conditions. For example, in the absence of ADAR1, cellular dsRNAs formed by unmodified IR-Alu were proposed to activate PKR (48). Similarly, breakdown of the nuclear membrane during mitosis and consequent exposure of an excess amount of nuclear IR-Alus were also proposed to activate PKR (82). Additional sources of endogenous ligands for PKR include cellular small nucleolar RNAs (snoRNAs) (83) and dsRNAs formed by mitochondrial sense and antisense transcripts (84), although mechanistic details of how these RNAs access cytosolic PKR remain unclear. Intriguingly, RNAs with short stem loops were also shown to activate PKR in a 5′ppp-dependent manner (85). This observation raises the possibility that PKR could be activated by a broader range of cellular RNAs, for example 5′ppp-containing RNAP III transcripts, as proposed for RIG-I (55, 57).
Innate immune functions of PKR appear to be complex. Several studies showed that PKR enhances the antiviral signaling activities of RLRs against certain viruses (86–88). Multiple, mutually nonexclusive mechanisms have been proposed. In one mechanism, PKR phosphorylates molecules involved in the signaling pathway, such as IkB (89), thereby altering their activities and synergizing with RLR signaling. However, it remains controversial whether PKR directly phosphorylates those signaling molecules or whether the effect is indirect (90, 91). Another proposal is that PKR amplifies RLR signaling by inducing formation of stress granules, the cytosolic nonmembranous granules formed upon dsRNA stimulation and enriched for RLRs and other signaling molecules (88, 92). However, there are cases where RIG-I and MDA5 are recruited to stress granules without activating antiviral signaling (93), suggesting that stress granule localization is not sufficient. PKR was also found to promote IFN-α/β production by stabilizing their mRNAs, rather than by upregulating their transcription (86), although the underlying mechanism for this observation is also unclear. Finally, PKR was proposed to exert its impact on RLR through translational suppression (94, 95). This proposal is supported by the observation that chemical inhibition of protein synthesis by cycloheximide also amplifies RLR signaling. All in all, PKR clearly plays an important role in antiviral immunity and cellular dsRNA biology, not only by linking dsRNA detection to translational inhibition but also by stimulating the antiviral signaling pathways.
OLIGOADENYLATE SYNTHASES AND RNase L
OASes are a family of interferon-inducible enzymes that synthesize 2′−5′ phosphodiester–linked oligoadenylates (2–5An) upon binding to dsRNA (96, 97). 2–5An function as a second messenger activating latent endoribonuclease RNase L by inducing its homodimerization or oligomerization (98). Activated RNase L cleaves unpaired RNA with minimal sequence specificity, targeting a wide range of RNAs, including rRNAs, tRNAs, other noncoding RNAs, and mRNAs, from both viruses and host. Accordingly, activation of the OAS–RNase L system leads to global suppression of protein synthesis, inhibition of cell growth, and induction of cell death, effects analogous to those of PKR, but more rapid and possibly with a broader impact (96, 97).
There are four isoforms of OASes in humans (Figure 3a). OAS1–3 are enzymatically active, while OASL is not. OAS1, 2, and 3 respectively harbor one, two, and three tandem repeats of a nucleotidyl transferase (NTase) domain that belongs to the family of template-independent nucleotide polymerases (96). Structural studies showed that OAS NTases bind one side of dsRNA (99, 100) in a manner analogous to that of dsRBDs (Figure 3a). Unlike dsRBDs, however, OAS NTases undergo a conformational change upon dsRNA binding, which activates its enzymatic activity. More specifically, dsRNA binding organizes the three conserved aspartic acids in the active site for coordinating the magnesium ions required for catalysis (99). The mechanism by which OASes couple dsRNA binding to 2–5An synthesis is similar to that of cGAS (Figure 3a), a cytosolic DNA sensor that catalyzes synthesis of 2′−3′ cGAMP upon dsDNA binding (101, 102). This suggests a conserved mechanism for double-stranded nucleic acid recognition and antiviral signaling.
Figure 3.

OASes and ADARs. (a) Domain architectures and structures of human OAS1–3 and OASL. The structure of OAS1 in complex with dsRNA (PDB: 4IG8). The OAS1 structure is also superposed onto cGAS in complex with dsDNA (PDB: 6CT9) and OAS3 (first pseudo-NTase domain) in complex with dsRNA (PDB: 4S3N). The second UbL domain of OASL (green, PDB: 1WH3) is superposed onto the structure of Ub (dark blue) on the right. (b) Schematic of adenosine-to-inosine modification and domain architectures of human ADAR1–3. The structure of dsRBD1–2 of ADAR2 in complex with dsRNA (PDB: 2L3J). Structures of ADAR1 ZBD1 in complex with dsRNA in the Z-form (PDB: 2GXB) and the deaminase domain of ADAR2 (PDB: 5HP2) are also shown on the right. Abbreviations: ADAR, adenosine deaminase acting on RNA 1; dsRBD, dsRNA-binding domain; NTase, nucleotidyl transferase; OAS, oligoadenylate synthase; PDB, Protein Data Bank; Ub, ubiquitin; ZBD, Z-DNA-binding domain.
OAS2 and OAS3 have two and three copies of NTase domains, but only one domain is catalytically active; the other domains (pseudo-NTase) lack the catalytic triad in the active site (103, 104). The pseudo-NTase domains, however, appear to have important functions. For example, the first pseudo-NTase domain in OAS3 binds dsRNA using the same binding mode as the catalytically active NTase domain of OAS1 (105, 106) (Figure 3a). It is possible that the second pseudo-NTase domain of OAS3 also binds dsRNA, thereby increasing dsRNA affinity of OAS3. Intriguingly, OAS3 responds to dsRNA more efficiently than OAS1 and OAS2 and serves as the primary upstream receptor for RNase L during viral infection (107). OAS3 was also found to preferentially recognize long (>50 bp) dsRNAs, perhaps reflecting linear arrangements of the three NTase domains along dsRNA (103).
OASL, an enzymatically inactive member of the OAS family, was also shown to display broad antiviral activity (108). OASL contains a single pseudo-NTase (Figure 3a), which also appears to bind dsRNA in a manner similar to OAS1 (109). Following the pseudo-NTase domain are the two tandem repeats of ubiquitin-like domains (UbLs, Figure 3a) (110). Both pseudo-NTase and UbLs are required for the observed antiviral activity of OASL (111). A previous study suggested that UbLs of OASL can functionally substitute K63-Ubn in inducing RIG-I 2CARD tetramerization (112), although the nature of this interaction remains to be further characterized. In mice, Oasl1 and Oasl2 are related to human OASL, with 70% and 50% sequence identity, respectively. While Oasl2 is necessary for efficient RIG-I signaling, as with human OASL (112), Oasl1 binds the 5′ untranslated region (UTR) of IRF7 mRNA, inhibits its translation, and negatively regulates antiviral immunity (113). This observation raises the possibility that human OASL plays a more complex role than simply assisting RIG-I.
Accumulating evidence suggests that the OAS–RNase L system has virus-independent activities. RNase L was found to regulate the steady-state mRNA levels of certain genes involved in cell growth, proliferation, and differentiation (114–116). Whether this basal activity of RNase L requires 2–5An and upstream OAS enzymes is unclear. The lack of dsRNA modification by ADAR1 (see the next section) was also shown to activate RNase L (117), causing cytotoxicity in an RNase L–dependent manner. In this case, RNase L activation is most likely mediated by upstream OASes (in particular OAS3) and their activation by unmodified cellular dsRNAs (e.g., IR-Alus). Intriguingly, OAS2 was found to be involved in normal developmental process (118), and OAS1 was proposed to recognize a wide range of ssRNAs beyond the canonical dsRNAs (119). Although more detailed analysis is required to understand the potential role of cellular RNAs in the reported functions of OASes/RNaseL beyond antiviral immunity, they certainly add a level of complexity in dsRNA biology and antiviral immunity.
ADENOSINE DEAMINASES ACTING ON RNA
ADARs are a class of enzymes that modify adenosines (A) to inosines (I) in dsRNAs (120–122) (Figure 3b). Since inosine is recognized as guanosine by reverse transcriptase and ribosomes, ADARs can be mutagenic when the modification occurs in coding regions. In noncoding duplexed regions, A-to-I modification replaces the A:U base pair by the weaker A:I base pair interaction (Figure 3b), resulting in weakening or partial melting of the dsRNA structure (120–122). Both the mutagenic activity and the duplex-melting activity have a profound impact on dsRNA biology and innate immunity.
Humans have three ADAR proteins, ADAR1–3, all of which have tandem repeats of RNA-binding domains [Z-DNA-binding domain (ZBD), dsRBD, and/or arginine-rich domain] followed by the deaminase domain (Figure 3b). ZBDs and arginine-rich domains (R-domains) are specific to ADAR1 and ADAR3, respectively, while dsRBD repeats are common in all three ADAR proteins. While dsRBDs are thought to bind dsRNAs with little or no sequence specificity, structures of ADAR2 dsRBDs with a hairpin dsRNA (Figure 3b) suggested that ADAR2 dsRBDs can recognize limited sequence information both through the minor groove contact and through indirect sensing of structural irregularities embedded within the hairpin dsRNA (5, 123). ZBDs of ADAR1 are also known to bind dsRNA in a sequence-dependent manner, displaying higher affinity for purine-pyrimidine repeat sequences that readily form left-handed Z-forms (124) (Figure 3b). The R-domain, on the other hand, appears to allow sequence- and structure-independent electrostatic interaction with nucleic acids (125).
The deaminase domain also has an intrinsic affinity for dsRNA with limited sequence preference. The crystal structures of ADAR2 showed that the deaminase domain binds dsRNA with a highly localized distortion in RNA, causing flipping of the target adenosine base out of the RNA duplex and its insertion into the active site (126, 127) (Figure 3b). While the mechanism for base flipping appears somewhat analogous to those of DNA methyltransferases (MTases) (128), ADAR2 and the DNA MTases flip their target bases through minor and major grooves, respectively, reflecting their differences in dsRNA- and dsDNA-binding modes. As the sequence nearby the target site would likely affect the efficiency of target base flipping, the deaminase domain, in addition to the other RNA-binding domains, was proposed to influence the site selectivity of ADARs (127).
ADAR1–3 have largely distinct groups of dsRNA substrates and expression patterns (129). ADAR1 primarily modifies dsRNAs in noncoding regions, such as ~300-bp IR-Alus in introns and 3′ UTRs of mRNAs (129, 130). By contrast, ADAR2 is responsible for modifying small hair-pins in coding regions (129). ADAR1 and ADAR2 also have largely distinct expression patterns; ADAR1 is ubiquitously expressed, while ADAR2 is restricted to the central nervous system. The observed difference in their target sites, however, appears to reflect their intrinsic difference in RNA specificity rather than expression patterns. In contrast to ADAR1 and ADAR2, ADAR3 lacks enzymatic activity despite harboring catalytic residues, and was proposed to function primarily as an inhibitor of ADAR2 (129, 131).
Consistent with the distinct modification targets and expression patterns, ADAR1 and ADAR2 have different biological functions. ADAR2 knockout causes postnatal seizures and early death in mouse, but this phenotype can be rescued by a single A-to-G substitution in one of the ADAR2 target sites in the Gria2 gene (132). By contrast, ADAR1 deletion is embryonically lethal (133–135). ADAR1 has two isoforms, constitutively expressed p110 in the nucleus and interferon-inducible p150, that are in both the cytosol and the nucleus (Figure 3b). Similar embryonic lethality was observed with selective deletion of p150 (136) or mice with a catalytically deficient knock-in mutation (137). The embryonic lethality of ADAR1 knockout in mouse was largely rescued by double knockout of MDA5 or MAVS, but not RIG-I (136–138). In humans, loss-of-function mutations in ADAR1 cause Aicardi-Goutières syndrome (139), the inflammatory disease that can also be caused by gain-of-function mutations in MDA5 (44). Further analysis showed that lack of A-to-I modification leads to stabilization of cellular dsRNAs (in particular IR-Alus), which in turn activates MDA5 (8), PKR (48), and OASes/RNase L (117). Consistent with the role of ADAR1 in suppressing these dsRNA-dependent antiviral molecules, ADAR1 has been shown to be a broadly acting proviral factor (108, 122). However, ADAR1 can also display antiviral activities against a subset of viruses or under certain circumstances, likely by directly modifying viral RNAs and accumulating deleterious mutations in the viral genomes (140, 141).
Besides the canonical antiviral pathways, ADAR1 also participates in the RNA interference (RNAi) pathway (142). ADAR1 was shown to modify certain precursors of miRNA and siRNA, thereby impairing their proper maturation (143) or altering their target mRNAs (144). However, more recent studies found that ADAR1, in particular the p110 isoform, can also have a positive effect on Dicer activity (145). ADAR1 forms a complex with Dicer and stimulates its activity independent of the deaminase activity (145). It was proposed that ADAR1 carries out different functions depending on the oligomeric state and interaction partners (145); the full picture remains to be further examined.
Thus, the primary functions of ADAR1 appear to regulate other dsRNA-binding proteins in the host innate immunity and the RNAi system. Whether ADARs are reciprocally regulated by other cellular dsRNA-binding proteins is yet unclear. While DHX9 and Staufen 1 were shown to modulate the metabolism of IR-Alus, the primary targets of ADAR1, the level of A-to-I modification of IR-Alus was found to be largely unaffected by DHX9 and Staufen 1 (146, 147). Future studies aimed at identifying regulatory factors for ADAR1 would be important.
RNA INTERFERENCE PATHWAY
Drosha, Dicer, and Argonautes are conserved RNases involved in RNAi pathways. Drosha and Dicer are responsible for biogenesis of miRNA and siRNA, while Argonaute proteins silence mRNAs harboring the sequence complementary to the miRNA or siRNA (148–150). miRNAs are synthesized by RNAP II as primary transcripts (pri-miRNAs). Drosha processes pri-miRNAs to ~70-nt-long pre-miRNAs in the nucleus (148, 150). Pre-miRNAs are exported to the cytosol and processed by Dicer, producing mature miRNAs harboring an ~21-bp duplexed region with 2-nt 3′ overhangs at both ends. Dicer can also process exogenously introduced long dsRNAs, such as viral replication intermediates, to generate siRNAs (148, 150). Mature miRNA and siRNA duplexes are then loaded onto Argonaute proteins, at which point the guide strand is selected, while the passenger strand is degraded. The resultant complex of Argonaute and guide-strand RNA, termed the RNA-induced silencing complex (RISC), binds to a target mRNA for gene silencing. Humans have four Argonaute proteins (Ago1–4), of which only Ago2 has nuclease activity to slice target mRNAs. However, all four Ago proteins can silence target genes by recruiting effector molecules for translational suppression, deadenylation or mRNA degradation (149).
Previous structural and biochemical analyses of Drosha, Dicer, and Argonautes offer detailed pictures of how these enzymes function. Drosha and Dicer have dsRNA-specific RNase III domains and share similar structural organization and dsRNA recognition mechanisms (151–153) (Figure 4a). Both enzymes utilize the PAZ domain to fix one end of pri-miRNA/pre-miRNA and position dsRNA for accurate cleavage by the RNase III domains (Figure 4a). Structural analysis of Dicer revealed that the PAZ domain interacts with dsRNA in at least two conformations representing the preslicing and slicing modes (153, 154) (Figure 4a). Additional accessory dsRBDs, both within Drosha/Dicer and from their interaction partners (e.g., DGCR8 and TRBP, respectively), were proposed to further facilitate dsRNA binding and dsRNA positioning. Dicer also contains the helicase domain homologous to those of RLRs (Figure 4a). The structures of human and Drosophila Dicers suggest that the helicase domain can also interact with dsRNA in at least two distinct modes (Figure 4a), one similarly to RLR (155) and the other distinct from RLR (153). The latter was observed in the preslicing conformation. How the helicase domain interacts with dsRNA in the slicing mode and whether it adopts the conformation similar to RLR remain to be investigated. Unlike Drosha and Dicer, Argonautes have an RNase H–like domain, rather than RNase III, and display vastly different domain architectures. Structures showed that Ago2 binds guide RNA through the phosphate backbone and extends the guide RNA to facilitate target mRNA recognition (149).
Figure 4.

Drosha/Dicer/Ago2 and PACT/TRBP. (a) Domain architectures and structures of human Drosha, Dicer, and Ago2. The structures of human Dicer (PDB: 5ZAL and 4NHA) in complex with dsRNA showed two distinct conformations representing the preslicing mode (where dsRNA is far from the slicing center) and the slicing mode (where dsRNA contacts the slicing center). dsRNA in the slicing mode was modeled based on the structure of Dicer platform-PAZ in complex with dsRNA (PDB:4NHA). Note that in the preslicing conformation, the helicase domain (green) interacts with dsRNA in a manner distinct from the RLR helicase domains. By contrast, the structure of Drosophila Dicer-2 (PDB: 6BU9, right) shows that the Dicer helicase domain partially encircles dsRNA, similarly to the RLR helicase domains. (b) Domain architectures and structures of PACT and TRBP. The structures of canonical dsRBD1–2 of TRBP in complex with dsRNA (PDB: 5N8M) and noncanonical dsRBD3 in complex with Dicer Hel2i (PDB: 4WYQ). The overlay of the two structures on the right shows that noncanonical dsRBD3 is structurally similar to canonical dsRBDs. Abbreviations: dsRBD, dsRNA-binding domain; RLR, RIG-I-like receptor.
Accumulating evidence suggests that Drosha and Dicer can function beyond biogenesis of siRNAs/miRNAs (156). For example, Drosha has been shown to regulate mRNA stability and processing of a select group of RNAs (157). Dicer was shown to process the Huntingtin mRNA when the CAG repeat sequence expands and forms a stable duplex hairpin (158). Dicer was also shown to cleave Alu RNAs and prevent excessive accumulation of Alu RNAs that can trigger NLRP3-mediated inflammation (159, 160). Interestingly, the immune-stimulatory RNAs in this case were reported to be individual Alu RNAs synthesized by RNAP III, rather than IR-Alus synthesized by RNAP II. Whether Dicer can also process IR-Alus and perturb the dynamics of other dsRNA-binding proteins, such as MDA5, PKR, OASes, and ADARs, remains unknown.
How does the RNAi system affect viral replication and antiviral immunity? While the importance of RNAi in antiviral defense has been widely documented in plants and invertebrates (161), its potential antiviral function in mammals has been controversial. On the one hand, Dicer-mediated cleavage of viral dsRNAs could lead to suppression of viral replication by knocking down viral genes. On the other hand, it could also suppress dsRNA-dependent antiviral immunity by eliminating dsRNA ligands for RLR, PKR, and OASes. Multiple lines of evidence have suggested that RNAi and the mammalian antiviral signaling pathways are mutually inhibitory (65, 66, 162). Interferon signaling blocks RISC by ADP-ribosylation, while several interferon-stimulated genes (ISGs) were found to be targeted by RISC (162). LGP2, a CARD-less RLR and an ISG, was also found to inhibit Dicer-dependent processing of pre-miRNA and long dsRNA, thereby suppressing miRNA/siRNA biogenesis (65, 66).
Other studies, however, suggest that Drosha, Dicer, and Argonaute proteins can exert antiviral activities in both RISC-dependent and -independent manners. It was initially thought that Dicer and Ago2 had antiviral functions in only a few undifferentiated tissues where interferon response was minimal or lacking (163, 164); a more recent study has suggested that Ago2 has antiviral activity in a wider range of cell types, including differentiated and interferon-competent cells (165). Furthermore, Drosha was also shown to suppress several positive-strand viruses by binding structural elements in viral RNAs and blocking actions of the RNA-dependent RNAP (166). These studies together offer multiple mechanisms by which RNAi machineries can exert antiviral activity. However, future studies are required to fully understand how these antiviral activities of RNAi interplay with mutually suppressive interferon signaling.
PACT AND TRBP
PACT and TRBP are paralogous dsRNA-binding proteins involved in many branches of dsRNA biology, including functions of RLRs, PKR, and Dicer (167). Both PACT and TRBP consist of three tandem repeats of dsRBDs (Figure 4b). As described for PKR and ADARs, dsRBDs have a conserved structure and dsRNA-binding mode. In PACT and TRBP, the first two (dsRBD1 and 2) are canonical dsRBDs with demonstrated dsRNA-binding activity (similar to those of PKR and ADAR1), while the third domain (dsRBD3) is a noncanonical dsRBD that has the canonical dsRBD structure but lacks dsRNA-binding activity (167) (Figure 4b). Instead, dsRBD3s of PACT and TRBP are involved in a broad range of protein-protein interactions. They were shown to form homodimers as well as heterodimers with each other, other dsRBDs (70, 167–169), and a helicase domain (170) (Figure 4b). To make matters more complex, dsRBD1 and 2 of PACT and TRBP were also proposed to interact with other proteins in dsRNA-dependent and -independent manners (171). As such, despite having a simple repeat of dsRBDs, PACT and TRBP are involved in multiple aspects of dsRNA biology through various protein-protein interactions as well as protein-dsRNA interactions.
One of the first reported functions of PACT and TRBP is to modulate the PKR activity. Earlier studies proposed that PACT directly binds PKR and activates the PKR kinase activity in the absence of dsRNA (172–174), while TRBP inhibits PKR by directly binding to PKR and/or PACT (175–177). A homozygous missense mutation (P222L) in PACT was shown to cause hyperactivation of PKR and early-onset dystonia DYT16 (178), supporting the role of PACT in PKR modulation. However, the exact nature of the PKR-modulatory activity of PACT (and the impact of the mutation P222L) has been controversial, with more recent studies supporting the role of PACT as a negative regulator of PKR, rather than a positive regulator. Mice deficient in the PACT homolog, RAX, display developmental and reproductive defects, and these defects were rescued by double knockout of PKR or expression of a kinase-dead mutant of PKR (i.e., a dominant negative mutant) (179). These observations indicate that the primary function of PACT is to suppress the aberrant activation of PKR. The PKR-suppressive role of PACT has also been observed in the context of HIV infection (180) and the deficiency of the splicing factor TIA1/TIAL1 (181). The reason for the discrepancy between the earlier and more recent studies is yet unclear—it might reflect the context-dependent role of PACT (e.g., cell stress state, posttranslational modifications) (180, 182, 183) or the accidental mutation in the PACT construct reported to be widely used in the earlier studies (167, 182). More detailed understanding of the structural and biochemical mechanism is required to fully define the role of PACT and TRBP in PKR regulation.
PACT and TRBP were also proposed to be involved in biogenesis of miRNAs and siRNAs. They interact directly with Dicer and fine-tune its RNA cleavage sites (167, 170, 184–186). Detailed structural and biochemical studies revealed that TRBP utilizes dsRBD3 to bind the helicase domain of Dicer (in particular the Hel2i subdomain, Figure 4b) and promotes Dicer activity (170, 184). Although a similar binding mode was proposed for PACT, PACT and TRBP differentially affect Dicer substrate specificity, cleavage kinetics and accuracy, isomer production, strand selection, and RISC assembly (184, 185, 187–189). The structural basis for the differential effects of PACT and TRBP remains to be further examined.
PACT and TRBP were also found to regulate RLRs. Earlier studies suggested that PACT activates RIG-I and MDA5 through a mechanism independent of PKR and Dicer (190–194). TRBP, on the other hand, was proposed to interact with LGP2, assisting MDA5 detection of cardioviruses (195). Despite having a helicase domain (including Hel2i) closely related to that of Dicer, PACT was proposed to bind RIG-I CTD, not Hel2i, and to stimulate the ATPase activity of RIG-I (190). However, in an apparent contradiction to these findings, more recent studies have shown that PACT is specifically involved in MDA5-mediated antiviral signaling, not RIG-I-mediated signaling (196, 197). It was also proposed that PACT interacts with LGP2 in a dsRNA-dependent manner and that this interaction is required for PACT-mediated MDA5 stimulation (197).
All in all, it is clear that PACT and TRBP function as a hub for dsRNA biology, not only by sharing common dsRNA substrates but also by interacting with various dsRNA-binding proteins in the host. However, the specific roles of PACT and TRBP in functions of PKR, Dicer, and RLRs remain controversial, and their molecular mechanisms unclear. These limitations likely reflect the complex network of interactions among dsRNA-binding proteins, and the challenges in dissecting mechanisms that connect many of these dsRNA-binding proteins.
HELICASES (BESIDES RLRs AND DICER)
Helicases are involved in nearly all nucleic acid biology, from DNA replication, transcription, and translation to dsRNA-dependent innate immunity. While the term helicase implies duplex-unwinding activity, many proteins in the helicase superfamily do not display such activity (198, 199). In fact, helicases are classified based on not duplex-unwinding activity but the presence of conserved sequence motifs (198, 199). Helicases often contain one or two RecA-like domains, and utilize the interface of two RecA-like domains (either within a polypeptide or in an oligomer) to bind ATP and cognate nucleic acids. Nucleic acid binds one side of the interface of the RecA-like domains, which in turn organizes the active site on the other side for ATP binding and/or hydrolysis (198, 199). The ATP binding or hydrolysis, in turn, triggers additional conformational changes in the helicase protein, often resulting in remodeling of the bound nucleic acid or other neighboring proteins.
A small group of helicases recognize dsRNAs and display dsRNA-dependent ATPase activity without duplex unwinding; they are named dsRNA-activated ATPases (DRAs) (200). They include RLRs and Dicer, which commonly share the two RecA-like domains and an insertion domain (Hel2i) between the two. DRAs belong to helicase superfamily 2, one of the largest families of helicases. Recent studies, however, suggest that several other helicases in superfamily 2 are also involved in sensing foreign dsRNAs and regulating cellular dsRNA homeostasis.
One of these helicases is DHX9 (also known as RHA), a highly abundant and multifunctional RNA/DNA helicase involved in DNA replication, transcription, and posttranscriptional and translational regulation (201). It contains two tandem repeats of dsRBDs followed by the DExH-motif helicase domain, and it displays an ATP-driven dsRNA-unwinding activity (201). Earlier studies showed that DHX9 is packaged into HIV-1 virions and is a host factor necessary for HIV-1 replication (202). More recently, DHX9 was shown to play key roles in cellular dsRNA homeostasis by binding IR-Alus and cooperating with ADAR1 in resolving cellular dsRNA structures (146). In apparent contradiction to its role in dsRNA unwinding and resolution, DHX9 was also proposed to function as a dsRNA sensor upstream of MAVS (203) and the Nlrp9b inflammasome (204). The interplay between the activities of DHX9 in resolving dsRNA and sensing dsRNA is yet unclear.
Several other helicases in superfamily 2 have also been proposed to activate antiviral and inflammatory signaling pathways in response to foreign dsRNAs. For example, DDX60 is an interferon-inducible helicase and was found to perform antiviral functions in at least two distinct manners (108, 205, 206). DDX60 assists RLRs in sensing foreign dsRNAs in a tissue-dependent manner (205, 206), but it also degrades viral RNAs, thereby conferring RLR- and interferon-independent antiviral activities (206). However, a more recent study failed to confirm the antiviral function of DDX60 against a broad range of viruses (207), demanding more detailed studies to understand the precise role of DDX60. Additional examples of helicases reported to function in innate immune signaling in response to dsRNA include DHX15 (208), DHX33 (209, 210), DHX29 (211, 212), DDX3 (213, 214), and the complex of DDX1-DDX21-DHX36 (215). Some helicases, such as DDX17, were shown to function as antiviral effectors independent of immune signaling pathways (16), analogous to the reported effector functions of RLRs (61) and DDX60 (206). The antiviral activity of DDX17, however, appears dependent on the virus; DDX17 inhibits bunyavirus replication (16) and promotes replication of HIV-1 (216) and influenza A (217). Although more detailed studies are required to mechanistically define antiviral functions of many of these helicases, these studies place helicases as additional factors that further increase the complexity of the dsRNA biology network and antiviral immunity.
CONCLUDING REMARKS
We discussed seven families of dsRNA-binding proteins involved in mammalian antiviral innate immunity. The main goal was not necessarily to cover each family in depth, but rather to highlight their commonality, differences, and interdependence in their functions. Structures of many of these proteins show that they commonly recognize phosphate backbones and minor grooves of dsRNA, the binding mode largely incompatible with a high degree of sequence dependence. They also utilize several common mechanisms to couple dsRNA binding to their respective downstream functions. These include dsRNA-mediated, proximity-induced oligomerization (RLRs and PKR); dsRNA-mediated allosteric regulation (OASes and helicases); and simple recruitment of effector domains to dsRNA (ADARs, Drosha, and Dicer).
Their functional interdependence, however, appears more complex and is further confounded by seemingly conflicting reports in some of these cases (Figure 1a, Table 1). On the one hand, their functions are inevitably linked through the shared dsRNA substrate—both pathogen-derived and cellular dsRNAs. In particular, various sources of cellular dsRNAs have been identified to play increasingly important roles in human dsRNA biology and pathology, including Alu retroelements, triple-repeat expansions, and mitochondrial dsRNAs. However, it is also clear that the interdependence of these dsRNA-binding proteins goes far beyond simply sharing dsRNA substrates and is further defined by protein-protein interactions. Much remains to be investigated to structurally and biochemically define their interactions and interdependence. Such system-level understanding of cellular dsRNA biology is likely required for comprehensive understanding of dsRNA-mediated antiviral immunity and regulation.
ACKNOWLEDGMENTS
The author acknowledges Burroughs Wellcome Fund and NIH grants (R01AI106912 and R01AI111784).
Footnotes
DISCLOSURE STATEMENT
The author is not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Neidle S 2008. Principles of Nucleic Acid Structure. London: Academic [Google Scholar]
- 2.Gesteland RF, Cech TR, Atkins JF. 1999. The RNA World. Cold Spring Harbor, NY: Cold Spring Harb. Lab. Press; 2nd ed. [Google Scholar]
- 3.Seeman NC, Rosenberg JM, Rich A. 1976. Sequence-specific recognition of double helical nucleic acids by proteins. PNAS 73:804–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rohs R, Jin X, West SM, Joshi R, Honig B, Mann RS. 2010. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem 79:233–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masliah G, Barraud P, Allain FH. 2013. RNA recognition by double-stranded RNA binding domains: a matter of shape and sequence. Cell Mol. Life Sci 70:1875–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peisley A, Lin C, Bin W, Orme-Johnson M, Liu M, et al. 2011. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. PNAS 108:21010–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berke IC, Modis Y. 2012. MDA5 cooperatively forms dimers and ATP-sensitive filaments upon binding double-stranded RNA. EMBO J. 7:1714–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmad S, Mu X, Yang F, Greenwald E, Park JW, et al. 2018. Breaching self-tolerance to Alu duplex RNA underlies MDA5-mediated inflammation. Cell 172:797–810 [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes a method to identify RNA ligand for MDA5 using protein conformation instead of affinity.
- 9.Mu X, Greenwald E, Ahmad S, Hur S. 2018. An origin of the immunogenicity of in vitro transcribed RNA. Nucleic Acids Res. 46:5239–49 [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes a common dsRNA by-product of in vitro transcription.
- 10.Schlee M, Roth A, Hornung V, Hagmann CA, Wimmenauer V, et al. 2009. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 31:25–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heinicke LA, Wong CJ, Lary J, Nallagatla SR, Diegelman-Parente A, et al. 2009. RNA dimerization promotes PKR dimerization and activation. J. Mol. Biol 390:319–38 [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes a common phenomenon of hairpin RNA dimerization and its impact on PKR activity analysis.
- 12.Hull CM, Bevilacqua PC. 2016. Discriminating self and non-self by RNA: roles for RNA structure, misfolding, and modification in regulating the innate immune sensor PKR. Acc. Chem. Res 49:1242–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu ZR, Wilkie AM, Clemens MJ, Smith CW. 1996. Detection of double-stranded RNA-protein interactions by methylene blue-mediated photo-crosslinking. RNA 2:611–21 [PMC free article] [PubMed] [Google Scholar]
- 14.Ricci EP, Kucukural A, Cenik C, Mercier BC, Singh G, et al. 2014. Staufen1 senses overall transcript secondary structure to regulate translation. Nat. Struct. Mol. Biol 21:26–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sugimoto Y, Vigilante A, Darbo E, Zirra A, Militti C, et al. 2015. hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature 519:491–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moy RH, Cole BS, Yasunaga A, Gold B, Shankarling G, et al. 2014. Stem-loop recognition by DDX17 facilitates miRNA processing and antiviral defense. Cell 158:764–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu B, Hur S. 2015. How RIG-I like receptors activate MAVS. Curr. Opin. Virol 12:91–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kato H, Takahasi K, Fujita T. 2011. RIG-I-like receptors: cytoplasmic sensors for non-self RNA. Immunol. Rev 243:91–98 [DOI] [PubMed] [Google Scholar]
- 19.Venkataraman T, Valdes M, Elsby R, Kakuta S, Caceres G, et al. 2007. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J. Immunol 178:6444–55 [DOI] [PubMed] [Google Scholar]
- 20.Childs KS, Randall RE, Goodbourn S. 2013. LGP2 plays a critical role in sensitizing mda-5 to activation by double-stranded RNA. PLOS ONE 8:e64202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Satoh T, Kato H, Kumagain Y, Yoneyama M, Sato S, et al. 2010. LGP2 is a positive regulator of RIG-I– and MDA5-mediated antiviral responses. PNAS 107:1512–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parisien JP, Lenoir JJ, Mandhana R, Rodriguez KR, Qian K, et al. 2018. RNA sensor LGP2 inhibits TRAF ubiquitin ligase to negatively regulate innate immune signaling. EMBO Rep. 19:e45176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peisley A, Jo M, Lin C, Wu B, Orme-Johnson M, et al. 2012. Kinetic mechanism for viral dsRNA length discrimination by MDA5 filament. PNAS 109:E3340–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feng Q, Hato SV, Langereis MA, Zoll J, Virgen-Slane R, et al. 2012. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep. 29:1187–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kowalinski E, Lunardi T, McCarthy AA, Louber J, Brunel J, et al. 2011. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 147:423–35 [DOI] [PubMed] [Google Scholar]
- 26.Luo D, Kohlway A, Vela A, Pyle AM. 2012. Visualizing the determinants of viral RNA recognition by innate immune sensor RIG-I. Structure 20:1983–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang F, Ramanathan A, Miller MT, Tang G-Q, Gale M Jr., et al. 2011. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature 479:423–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rawling DC, Fitzgerald ME, Pyle AM. 2015. Establishing the role of ATP for the function of the RIG-I innate immune sensor. eLife 4:e03931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sohn J, Hur S. 2016. Filament assemblies in foreign nucleic acid sensors. Curr. Opin. Struct. Biol 37:134–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peisley A, Wu B, Yao H, Walz T, Hur S. 2013. RIG-I forms signaling-competent filaments in an ATP-dependent, ubiquitin-independent manner. Mol. Cell 51:573–83 [DOI] [PubMed] [Google Scholar]
- 31.Patel JR, Jain A, Chou YY, Baum A, Ha T, Garcia-Sastre A. 2013. ATPase-driven oligomerization of RIG-I on RNA allows optimal activation of type-I interferon. EMBO Rep. 14:780–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Myong S, Cui S, Cornish PV, Kirchhofer A, Gack MU, et al. 2009. Cytosolic viral sensor RIG-I is a 5′-triphosphate–dependent translocase on double-stranded RNA. Science 323:1070–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Devarkar SC, Schweibenz B, Wang C, Marcotrigiano J, Patel SS. 2018. RIG-I uses an ATPase-powered translocation-throttling mechanism for kinetic proofreading of RNAs and oligomerization. Mol. Cell 72:355–68.e354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang X, Kinch LN, Brautigam CA, Chen X, Du F, et al. 2012. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 36:959–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peisley A, Wu B, Xu H, Chen ZJ, Hur S. 2014. Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature 509:110–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu B, Peisley A, Tetrault D, Li Z, Egelman EH, et al. 2014. Molecular imprinting as a signal activation mechanism of the viral RNA sensor RIG-I. Mol. Cell 55:511–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. 2011. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146:448–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Triantafilou K, Vakakis E, Kar S, Richer E, Evans GL, Triantafilou M. 2012. Visualisation of direct interaction of MDA5 and the dsRNA replicative intermediate form of positive strand RNA viruses. J. Cell Sci 125:4761–69 [DOI] [PubMed] [Google Scholar]
- 39.Uchikawa E, Lethier M, Malet H, Brunel J, Gerlier D, Cusack S. 2016. Structural analysis of dsRNA binding to anti-viral pattern recognition receptors LGP2 and MDA5. Mol. Cell 62:586–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li X, Ranjith-Kumar CT, Brooks MT, Dharmaiah S, Herr AB, et al. 2009. The RIG-I-like receptor LGP2 recognizes the termini of double-stranded RNA. J. Biol. Chem 284:13881–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bruns AM, Pollpeter D, Hadizadeh N, Myong S, Marko JF, Horvath CM. 2013. ATP hydrolysis enhances RNA recognition and antiviral signal transduction by the innate immune sensor, laboratory of genetics and physiology 2 (LGP2). J. Biol. Chem 288:938–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bruns AM, Leser GP, Lamb RA, Horvath CM. 2014. The innate immune sensor LGP2 activates antiviral signaling by regulating MDA5-RNA interaction and filament assembly. Mol. Cell 55:771–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Funabiki M, Kato H, Miyachi Y, Toki H, Motegi H, et al. 2014. Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity 40:199–212 [DOI] [PubMed] [Google Scholar]
- 44.Rice GI, del Toro Duany Y, Jenkinson EM, Forte GM, Anderson BH, et al. 2014. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat. Genet 46:503–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Eyck L, De Somer L, Pombal D, Bornschein S, Frans G, et al. 2015. IFIH1 mutation causes systemic lupus erythematosus with selective IgA deficiency. Arthritis Rheumatol. 67:1592–97 [DOI] [PubMed] [Google Scholar]
- 46.Bursztejn AC, Briggs TA, del Toro Duany Y, Anderson BH, O’Sullivan J, et al. 2015. Unusual cutaneous features associated with a heterozygous gain-of-function mutation in IFIH1: overlap between Aicardi-Goutières and Singleton-Merten syndromes. Br. J. Dermatol 173:1505–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oda H, Nakagawa A, Abe J, Awaya T, Funabiki M, et al. 2014. Aicardi-Goutières syndrome is caused by IFIH1 mutations. Am. J. Hum. Genet 95:121–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chung H, Calis JJA, Wu X, Sun T, Yu Y, et al. 2018. Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell 172:811–24.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, et al. 2015. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 162:974–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, et al. 2015. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 162:961–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, et al. 2018. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560:238–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jang M-A, Kim EK, Now H, Nguyen NTH, Kim W-J, et al. 2015. Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome. Am. J. Hum. Genet 96:266–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ferreira CR, Crow YJ, Gahl WA, Goldbach-Mansky R, Hur S, et al. 2018. DDX58 and classic Singleton-Merten syndrome. J. Clin. Immnol In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ranoa DR, Parekh AD, Pitroda SP, Huang X, Darga T, et al. 2016. Cancer therapies activate RIG-I-like receptor pathway through endogenous non-coding RNAs. Oncotarget 7:26496–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nabet BY, Qiu Y, Shabason JE, Wu TJ, Yoon T, et al. 2017. Exosome RNA unshielding couples stromal activation to pattern recognition receptor signaling in cancer. Cell 170:352–66.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, et al. 2014. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 159:499–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chiang JJ, Sparrer KMJ, van Gent M, Lassig C, Huang T, et al. 2018. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nat. Immunol 19:53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Malathi K, Dong B, Gale M Jr., Silverman RH. 2007. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448:816–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Malathi K, Saito T, Crochet N, Barton DJ, Gale M Jr., Silverman RH. 2010. RNase L releases a small RNA from HCV RNA that refolds into a potent PAMP. RNA 16:2108–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eckard SC, Rice GI, Fabre A, Badens C, Gray EE, et al. 2014. The SKIV2L RNA exosome limits activation of the RIG-I-like receptors. Nat. Immunol 15:839–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yao H, Dittmann M, Peisley A, Hoffmann H-H, Gilmore RH, et al. 2015. ATP-dependent effector-like functions of RIG-I-like receptors. Mol. Cell 58:541–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sato S, Li K, Kameyama K, Hayashi T, Ishida Y, et al. 2015. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 42:123–32 [DOI] [PubMed] [Google Scholar]
- 63.Weber M, Sediri H, Felgenhauer U, Binzen I, Banfer S, et al. 2015. Influenza virus adaptation PB2– 627K modulates nucleocapsid inhibition by the pathogen sensor RIG-I. Cell Host Microbe 17:309–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Benitez AA, Panis M, Xue J, Varble A, Shim JV, et al. 2015. In vivo RNAi screening identifies MDA5 as a significant contributor to the cellular defense against influenza A virus. Cell Rep. 11:1714–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.van der Veen AG, Maillard PV, Schmidt JM, Lee SA, Deddouche-Grass S, et al. 2018. The RIG-I-like receptor LGP2 inhibits Dicer-dependent processing of long double-stranded RNA and blocks RNA interference in mammalian cells. EMBO J. 37:e97479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Takahashi T, Nakano Y, Onomoto K, Murakami F, Komori C, et al. 2018. LGP2 virus sensor regulates gene expression network mediated by TRBP-bound microRNAs. Nucleic Acids Res. 46:9134–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pfeller CK, Li Z, George CX, Samuel CE. 2011. Protein kinase PKR and RNA adenosine deaminase ADAR1: new roles for old players as modulators of the interferon response. Curr. Opin. Immunol 23:573–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nanduri S, Carpick BW, Yang Y, Williams BR, Qin J. 1998. Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J. 17:5458–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dey M, Cao C, Dar AC, Tamura T, Ozato K, et al. 2005. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2α substrate recognition. Cell 122:901–13 [DOI] [PubMed] [Google Scholar]
- 70.Gleghorn ML, Maquat LE. 2014. ‘Black sheep’ that don’t leave the double-stranded RNA-binding domain fold. Trends Biochem. Sci 39:328–40 [DOI] [PMC free article] [PubMed] [Google Scholar]; This review provides a comprehensive overview of dsRBD structures and functions.
- 71.Bevilacqua PC, Cech TR. 1996. Minor-groove recognition of double-stranded RNA by the double-stranded RNA-binding domain from the RNA-activated protein kinase PKR. Biochemistry 35:9983–94 [DOI] [PubMed] [Google Scholar]
- 72.Kim I, Liu CW, Puglisi JD. 2006. Specific recognition of HIV TAR RNA by the dsRNA binding domains (dsRBD1–dsRBD2) of PKR. J. Mol. Biol 358:430–42 [DOI] [PubMed] [Google Scholar]
- 73.Ucci JW, Kobayashi Y, Choi G, Alexandrescu AT, Cole JL. 2007. Mechanism of interaction of the double-stranded RNA (dsRNA) binding domain of protein kinase R with short dsRNA sequences. Biochemistry 46:55–65 [DOI] [PubMed] [Google Scholar]
- 74.Husain B, Mukerji I, Cole JL. 2012. Analysis of high-affinity binding of protein kinase R to double-stranded RNA. Biochemistry 51:8764–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gelev V, Aktas H, Marintchev A, Ito T, Frueh D, et al. 2006. Mapping of the auto-inhibitory interactions of protein kinase R by nuclear magnetic resonance. J. Mol. Biol 364:352–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nanduri S, Rahman F, Williams BR, Qin J. 2000. A dynamically tuned double-stranded RNA binding mechanism for the activation of antiviral kinase PKR. EMBO J. 19:5567–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McKenna SA, Lindhout DA, Kim I, Liu CW, Gelev VM, et al. 2007. Molecular framework for the activation of RNA-dependent protein kinase. J. Biol. Chem 282:11474–86 [DOI] [PubMed] [Google Scholar]
- 78.Dey M, Mann BR, Anshu A, Mannan MA. 2014. Activation of protein kinase PKR requires dimerization-induced cis-phosphorylation within the activation loop. J. Biol. Chem 289:5747–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Husain B, Hesler S, Cole JL. 2015. Regulation of PKR by RNA: formation of active and inactive dimers. Biochemistry 54:6663–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dar AC, Dever TE, Sicheri F. 2005. Higher-order substrate recognition of eIF2α by the RNA-dependent protein kinase PKR. Cell 122:887–900 [DOI] [PubMed] [Google Scholar]
- 81.Li FZ, Li SW, Wang Z, Shen YQ, Zhang TC, Yang X. 2013. Structure of the kinase domain of human RNA-dependent protein kinase with K296R mutation reveals a face-to-face dimer. Chinese Sci. Bull 58:998–1002 [Google Scholar]
- 82.Kim Y, Lee JH, Park JE, Cho J, Yi H, Kim VN. 2014. PKR is activated by cellular dsRNAs during mitosis and acts as a mitotic regulator. Genes Dev. 28:1310–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Youssef OA, Safran SA, Nakamura T, Nix DA, Hotamisligil GS, Bass BL. 2015. Potential role for snoRNAs in PKR activation during metabolic stress. PNAS 112:5023–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim Y, Park J, Kim S, Kim M, Kang MG, et al. 2018. PKR senses nuclear and mitochondrial signals by interacting with endogenous double-stranded RNAs. Mol. Cell 81:1051–63.e6 [DOI] [PubMed] [Google Scholar]
- 85.Nallagatla SR, Hwang J, Toroney R, Zheng X, Cameron CE, Bevilacqua PC. 2007. 5′-Triphosphate-dependent activation of PKR by RNAs with short stem-loops. Science 318:1455–58 [DOI] [PubMed] [Google Scholar]
- 86.Schulz O, Pichlmair A, Rehwinkel J, Rogers NC, Scheuner D, et al. 2010. Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe 7:354–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pham AM, Santa Maria FG, Lahiri T, Friedman E, Marie IJ, Levy DE. 2016. PKR transduces MDA5-dependent signals for type I IFN induction. PLOS Pathog. 12:e1005489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Oh SW, Onomoto K, Wakimoto M, Onoguchi K, Ishidate F, et al. 2016. Leader-containing uncapped viral transcript activates RIG-I in antiviral stress granules. PLOS Pathog. 12:e1005444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zamanian-Daryoush M, Mogensen TH, DiDonato JA, Williams BR. 2000. NF-κB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-κB-inducing kinase and IκB kinase. Mol. Cell. Biol 20:1278–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sadler AJ, Williams BR. 2007. Structure and function of the protein kinase R. Curr. Top. Microbiol. Immunol 316:253–92 [DOI] [PubMed] [Google Scholar]
- 91.Ishii T, Kwon H, Hiscott J, Mosialos G, Koromilas AE. 2001. Activation of the I kappa B alpha kinase (IKK) complex by double-stranded RNA-binding defective and catalytic inactive mutants of the interferon-inducible protein kinase PKR. Oncogene 20:1900–12 [DOI] [PubMed] [Google Scholar]
- 92.Onomoto K, Jogi M, Yoo JS, Narita R, Morimoto S, et al. 2013. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLOS ONE 7:e43031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Langereis MA, Feng Q, van Kuppeveld FJ. 2013. MDA5 localizes to stress granules, but this localization is not required for the induction of type I interferon. J. Virol 87:6314–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.McAllister CS, Taghavi N, Samuel CE. 2012. Protein kinase PKR amplification of interferon βinduction occurs through initiation factor eIF-2α-mediated translational control. J. Biol. Chem 287:36384–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dalet A, Arguello RJ, Combes A, Spinelli L, Jaeger S, et al. 2017. Protein synthesis inhibition and GADD34 control IFN-β heterogeneous expression in response to dsRNA. EMBO J. 36:761–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kristiansen H, Gad HH, Eskildsen-Larsen S, Despres P, Hartmann R. 2011. The oligoadenylate synthetase family: an ancient protein family with multiple antiviral activities. J. Interferon Cytokine Res 31:41–47 [DOI] [PubMed] [Google Scholar]
- 97.Hovanessian AG, Justesen J. 2007. The human 2′−5′ oligoadenylate synthetase family: unique interferon-inducible enzymes catalyzing 2′−5′ instead of 3′−5′ phosphodiester bond formation. Biochimie 89:779–88 [DOI] [PubMed] [Google Scholar]
- 98.Han Y, Donovan J, Rath S, Whitney G, Chitrakar A, Korennykh A. 2014. Structure of human RNase L reveals the basis for regulated RNA decay in the IFN response. Science 343:1244–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Donovan J, Dufner M, Korennykh A. 2013. Structural basis for cytosolic double-stranded RNA surveillance by human oligoadenylate synthetase 1. PNAS 110:1652–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hartmann R, Justesen J, Sarkar SN, Sen GC, Yee VC. 2003. Crystal structure of the 2′-specific and double-stranded RNA-activated interferon-induced antiviral protein ′5′-oligoadenylate synthetase. Mol. Cell 12:1173–85 [DOI] [PubMed] [Google Scholar]
- 101.Hornung V, Hartmann R, Ablasser A, Hopfner KP. 2014. OAS proteins and cGAS: unifying concepts in sensing and responding to cytosolic nucleic acids. Nat. Rev. Immunol 14:521–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhou W, Whiteley AT, de Oliveira Mann CC, Morehouse BR, Nowak RP, et al. 2018. Structure of the human cGAS-DNA complex reveals enhanced control of immune surveillance. Cell 174:300–11.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Donovan J, Whitney G, Rath S, Korennykh A. 2015. Structural mechanism of sensing long dsRNA via a noncatalytic domain in human oligoadenylate synthetase 3. PNAS 112:3949–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sarkar SN, Ghosh A, Wang HW, Sung SS, Sen GC. 1999. The nature of the catalytic domain of 2 −5′-oligoadenylate synthetases. J. Biol. Chem 274:25535–42 [DOI] [PubMed] [Google Scholar]
- 105.Ilson DH, Torrence PF, Vilcek J. 1986. Two molecular weight forms of human 2′,5′-oligoadenylate synthetase have different activation requirements. J. Interferon Res 6:5–12 [DOI] [PubMed] [Google Scholar]
- 106.Ibsen MS, Gad HH, Thavachelvam K, Boesen T, Despres P, Hartmann R. 2014. The 2′−5′-oligoadenylate synthetase 3 enzyme potently synthesizes the 2′−5′-oligoadenylates required for RNase L activation. J. Virol 88:14222–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li Y, Banerjee S, Wang Y, Goldstein SA, Dong B, et al. 2016. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. PNAS 113:2241–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, et al. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ibsen MS, Gad HH, Andersen LL, Hornung V, Julkunen I, et al. 2015. Structural and functional analysis reveals that human OASL binds dsRNA to enhance RIG-I signaling. Nucleic Acids Res. 43:5236–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hartmann R, Olsen HS, Widder S, Jorgensen R, Justesen J. 1998. p59 OASL, a 2′−5′ oligoadenylate synthetase like protein: a novel human gene related to the 2′−5′ oligoadenylate synthetase family. Nucleic Acids Res. 26:4121–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Marques J, Anwar J, Eskildsen-Larsen S, Rebouillat D, Paludan SR, et al. 2008. The p59 oligoadenylate synthetase-like protein possesses antiviral activity that requires the C-terminal ubiquitin-like domain. J. Gen. Virol 89:2767–72 [DOI] [PubMed] [Google Scholar]
- 112.Zhu J, Zhang Y, Ghosh A, Cuevas RA, Forero A, et al. 2014. Antiviral activity of human OASL protein is mediated by enhancing signaling of the RIG-I RNA sensor. Immunity 40:936–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lee MS, Kim B, Oh GT, Kim YJ. 2013. OASL1 inhibits translation of the type I interferon-regulating transcription factor IRF7. Nat. Immunol 14:346–55 [DOI] [PubMed] [Google Scholar]
- 114.Al-Ahmadi W, Al-Haj L, Al-Mohanna FA, Silverman RH, Khabar KS. 2009. RNase L downmodulation of the RNA-binding protein, HuR, and cellular growth. Oncogene 28:1782–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Brennan-Laun SE, Li XL, Ezelle HJ, Venkataraman T, Blackshear PJ, et al. 2014. RNase L attenuates mitogen-stimulated gene expression via transcriptional and post-transcriptional mechanisms to limit the proliferative response. J. Biol. Chem 289:33629–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fabre O, Salehzada T, Lambert K, Boo Seok Y, Zhou A, et al. 2012. RNase L controls terminal adipocyte differentiation, lipids storage and insulin sensitivity via CHOP10 mRNA regulation. Cell Death Differ. 19:1470–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li Y, Banerjee S, Goldstein SA, Dong B, Gaughan C, et al. 2017. Ribonuclease L mediates the cell-lethal phenotype of double-stranded RNA editing enzyme ADAR1 deficiency in a human cell line. eLife 6:e25687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Oakes SR, Gallego-Ortega D, Stanford PM, Junankar S, Au WWY, et al. 2017. A mutation in the viral sensor 2′−5′-oligoadenylate synthetase 2 causes failure of lactation. PLOS Genet. 13:e1007072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hartmann R, Norby PL, Martensen PM, Jorgensen P, James MC, et al. 1998. Activation of 2 −5′ oligoadenylate synthetase by single-stranded and double-stranded RNA aptamers. J. Biol. Chem 273:3236–46 [DOI] [PubMed] [Google Scholar]
- 120.Walkley CR, Li JB. 2017. Rewriting the transcriptome: adenosine-to-inosine RNA editing by ADARs. Genome Biol. 18:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Slotkin W, Nishikura K. 2013. Adenosine-to-inosine RNA editing and human disease. Genome Med. 5:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.George CX, John L, Samuel CE. 2014. An RNA editor, adenosine deaminase acting on double-stranded RNA (ADAR1). J. Interferon Cytokine Res 34:437–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Stefl R, Oberstrass FC, Hood JL, Jourdan M, Zimmermann M, et al. 2010. The solution structure of the ADAR2 dsRBM-RNA complex reveals a sequence-specific readout of the minor groove. Cell 143:225–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Placido D, Brown BA 2nd, Lowenhaupt K, Rich A, Athanasiadis A. 2007. A left-handed RNA double helix bound by the Z alpha domain of the RNA-editing enzyme ADAR1. Structure 15:395–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chen CX, Cho DS, Wang Q, Lai F, Carter KC, Nishikura K. 2000. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 6:755–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Thomas JM, Beal PA. 2017. How do ADARs bind RNA? New protein-RNA structures illuminate substrate recognition by the RNA editing ADARs. BioEssays 39:1600187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Matthews MM, Thomas JM, Zheng Y, Tran K, Phelps KJ, et al. 2016. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat. Struct. Mol. Biol 23:426–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Klimasauskas S, Kumar S, Roberts RJ, Cheng X. 1994. HhaI methyltransferase flips its target base out of the DNA helix. Cell 76:357–69 [DOI] [PubMed] [Google Scholar]
- 129.Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, et al. 2017. Dynamic landscape and regulation of RNA editing in mammals. Nature 550:249–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hundley HA, Bass BL. 2010. ADAR editing in double-stranded UTRs and other noncoding RNA sequences. Trends Biochem. Sci 35:377–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Oakes E, Anderson A, Cohen-Gadol A, Hundley HA. 2017. Adenosine deaminase that acts on RNA 3 (ADAR3) binding to glutamate receptor subunit B pre-mRNA inhibits RNA editing in glioblastoma. J. Biol. Chem 292:4326–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Higuchi M, Maas S, Single FN, Hartner J, Rozov A, et al. 2000. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406:78–81 [DOI] [PubMed] [Google Scholar]
- 133.Hartner JC, Schmittwolf C, Kispert A, Muller AM, Higuchi M, Seeburg PH. 2004. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J. Biol. Chem 279:4894–902 [DOI] [PubMed] [Google Scholar]
- 134.Hartner JC, Walkley CR, Lu J, Orkin SH. 2008. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat. Immunol 10:109–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hauenstein A, Zhang L, Wu H. 2015. The hierarchical structural architecture of inflammasomes, supramolecular inflammatory machines. Curr. Opin. Struct. Biol 31:75–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. 2015. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 43:933–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, et al. 2015. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349:1115–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, et al. 2014. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 9:1482–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, et al. 2010. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat. Genet 44:1243–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ward SV, George CX, Welch MJ, Liou LY, Hahm B, et al. 2011. RNA editing enzyme adenosine deaminase is a restriction factor for controlling measles virus replication that also is required for embryogenesis. PNAS 108:331–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Biswas N, Wang T, Ding M, Tumne A, Chen Y, et al. 2012. ADAR1 is a novel multi targeted anti-HIV-1 cellular protein. Virology 422:265–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nishikura K 2016. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol 17:83–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, et al. 2006. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol 13:13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. 2007. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 315:1137–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ota H, Sakurai M, Gupta R, Valente L, Wulff BE, et al. 2013. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell 153:575–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Aktas T, Avsar Ilik I, Maticzka D, Bhardwaj V, Pessoa Rodrigues C, et al. 2017. DHX9 suppresses RNA processing defects originating from the Alu invasion of the human genome. Nature 544:115–19 [DOI] [PubMed] [Google Scholar]
- 147.Elbarbary RA, Li W, Tian B, Maquat LE. 2013. STAU1 binding 3′ UTR IRAlus complements nuclear retention to protect cells from PKR-mediated translational shutdown. Genes Dev. 27:1495–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wilson RC, Doudna JA. 2013. Molecular mechanisms of RNA interference. Annu. Rev. Biophys 42:217–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sheu-Gruttadauria J, MacRae IJ. 2017. Structural foundations of RNA silencing by Argonaute. J. Mol. Biol 429:2619–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kim VN, Han J, Siomi MC. 2009. Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol 10:126–39 [DOI] [PubMed] [Google Scholar]
- 151.Kwon SC, Nguyen TA, Choi YG, Jo MH, Hohng S, et al. 2016. Structure of human DROSHA. Cell 164:81–90 [DOI] [PubMed] [Google Scholar]
- 152.Macrae IJ, Li F, Zhou K, Cande WZ, Doudna JA. 2006. Structure of Dicer and mechanistic implications for RNAi. Cold Spring Harb. Symp. Quant. Biol 71:73–80 [DOI] [PubMed] [Google Scholar]
- 153.Liu Z, Wang J, Cheng H, Ke X, Sun L, et al. 2018. Cryo-EM structure of human Dicer and its complexes with a pre-miRNA substrate. Cell 173:1191–203.e12. Erratum. 2018. Cell 173:1549–50 [DOI] [PubMed] [Google Scholar]
- 154.Tian Y, Simanshu DK, Ma JB, Park JE, Heo I, et al. 2014. A phosphate-binding pocket within the platform-PAZ-connector helix cassette of human Dicer. Mol. Cell 53:606–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sinha NK, Iwasa J, Shen PS, Bass BL. 2018. Dicer uses distinct modules for recognizing dsRNA termini. Science 359:329–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Burger K, Gullerova M. 2015. Swiss army knives: non-canonical functions of nuclear Drosha and Dicer. Nat. Rev. Mol. Cell Biol 16:417–30 [DOI] [PubMed] [Google Scholar]
- 157.Lee D, Shin C. 2018. Emerging roles of DROSHA beyond primary microRNA processing. RNA Biol. 15:186–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Banez-Coronel M, Porta S, Kagerbauer B, Mateu-Huertas E, Pantano L, et al. 2012. A pathogenic mechanism in Huntington’s disease involves small CAG-repeated RNAs with neurotoxic activity. PLOS Genet. 8:e1002481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kaneko H, Dridi S, Tarallo V, Gelfand BD, Fowler BJ, et al. 2011. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 471:325–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, et al. 2012. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 149:847–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ding SW. 2010. RNA-based antiviral immunity. Nat. Rev. Immunol 10:632–44 [DOI] [PubMed] [Google Scholar]
- 162.Seo GJ, Kincaid RP, Phanaksri T, Burke JM, Cox JE, et al. 2013. Reciprocal inhibition between intracellular antiviral signaling and the RNAi machinery in mammalian cells. Cell Host Microbe 14:435–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Maillard PV, Ciaudo C, Marchais A, Li Y, Jay F, et al. 2013. Antiviral RNA interference in mammalian cells. Science 342:235–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Li Y, Lu J, Han Y, Fan X, Ding SW. 2013. RNA interference functions as an antiviral immunity mechanism in mammals. Science 342:231–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Li Y, Basavappa M, Lu J, Dong S, Cronkite DA, et al. 2016. Induction and suppression of antiviral RNA interference by influenza A virus in mammalian cells. Nat. Microbiol 2:16250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Aguado LC, Schmid S, May J, Sabin LR, Panis M, et al. 2017. RNase III nucleases from diverse kingdoms serve as antiviral effectors. Nature 547:114–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Heyam A, Lagos D, Plevin M. 2015. Dissecting the roles of TRBP and PACT in double-stranded RNA recognition and processing of noncoding RNAs. Wiley Interdiscip. Rev. RNA 6:271–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jakob L, Treiber T, Treiber N, Gust A, Kramm K, et al. 2016. Structural and functional insights into the fly microRNA biogenesis factor Loquacious. RNA 22:383–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Heyam A, Coupland CE, Degut C, Haley RA, Baxter NJ, et al. 2017. Conserved asymmetry under-pins homodimerization of Dicer-associated double-stranded RNA-binding proteins. Nucleic Acids Res. 45:12577–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wilson RC, Tambe A, Kidwell MA, Noland CL, Schneider CP, Doudna JA. 2015. Dicer-TRBP complex formation ensures accurate mammalian microRNA biogenesis. Mol. Cell 57:397–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Chukwurah E, Willingham V, Singh M, Castillo-Azofeifa D, Patel RC. 2018. Contribution of the two dsRBM motifs to the double-stranded RNA binding and protein interactions of PACT. J. Cell Biochem 119:3598–607 [DOI] [PubMed] [Google Scholar]
- 172.Patel RC, Sen GC. 1998. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 17:4379–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Peters GA, Dickerman B, Sen GC. 2009. Biochemical analysis of PKR activation by PACT. Biochemistry 48:7441–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Singh M, Patel RC. 2012. Increased interaction between PACT molecules in response to stress signals is required for PKR activation. J. Cell Biochem 113:2754–64 [DOI] [PubMed] [Google Scholar]
- 175.Park H, Davies MV, Langland JO, Chang HW, Nam YS, et al. 1994. TAR RNA-binding protein is an inhibitor of the interferon-induced protein kinase PKR. PNAS 91:4713–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Chukwurah E, Patel RC. 2018. Stress-induced TRBP phosphorylation enhances its interaction with PKR to regulate cellular survival. Sci. Rep 8:1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Laraki G, Clerzius G, Daher A, Melendez-Pena C, Daniels S, Gatignol A. 2008. Interactions between the double-stranded RNA-binding proteins TRBP and PACT define the Medipal domain that mediates protein-protein interactions. RNA Biol. 5:92–103 [DOI] [PubMed] [Google Scholar]
- 178.Vaughn LS, Bragg DC, Sharma N, Camargos S, Cardoso F, Patel RC. 2015. Altered activation of protein kinase PKR and enhanced apoptosis in dystonia cells carrying a mutation in PKR activator protein PACT. J. Biol. Chem 290:22543–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Dickerman BK, White CL, Kessler PM, Sadler AJ, Williams BR, Sen GC. 2015. The protein activator of protein kinase R, PACT/RAX, negatively regulates protein kinase R during mouse anterior pituitary development. FEBS J. 282:4766–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Clerzius G, Shaw E, Daher A, Burugu S, Gelinas JF, et al. 2013. The PKR activator, PACT, becomes a PKR inhibitor during HIV-1 replication. Retrovirology 10:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Meyer C, Garzia A, Mazzola M, Gerstberger S, Molina H, Tuschl T. 2018. The TIA1 RNA-binding protein family regulates EIF2AK2-mediated stress response and cell cycle progression. Mol. Cell 69:622–35.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Daher A, Laraki G, Singh M, Melendez-Pena CE, Bannwarth S, et al. 2009. TRBP control of PACT-induced phosphorylation of protein kinase R is reversed by stress. Mol. Cell Biol 29:254–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Singh M, Castillo D, Patel CV, Patel RC. 2011. Stress-induced phosphorylation of PACT reduces its interaction with TRBP and leads to PKR activation. Biochemistry 50:4550–60 [DOI] [PubMed] [Google Scholar]
- 184.Lee HY, Zhou K, Smith AM, Noland CL, Doudna JA. 2013. Differential roles of human Dicer-binding proteins TRBP and PACT in small RNA processing. Nucleic Acids Res. 41:6568–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Lee Y, Hur I, Park SY, Kim YK, Suh MR, Kim VN. 2006. The role of PACT in the RNA silencing pathway. EMBO J. 25:522–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kok KH, Ng MH, Ching YP, Jin DY. 2007. Human TRBP and PACT directly interact with each other and associate with Dicer to facilitate the production of small interfering RNA. J. Biol. Chem 282:17649–57 [DOI] [PubMed] [Google Scholar]
- 187.Fukunaga R, Han BW, Hung JH, Xu J, Weng Z, Zamore PD. 2012. Dicer partner proteins tune the length of mature miRNAs in flies and mammals. Cell 151:533–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Kim Y, Yeo J, Lee JH, Cho J, Seo D, et al. 2014. Deletion of human tarbp2 reveals cellular microRNA targets and cell-cycle function of TRBP. Cell Rep. 9:1061–74 [DOI] [PubMed] [Google Scholar]
- 189.Noland CL, Doudna JA. 2013. Multiple sensors ensure guide strand selection in human RNAi pathways. RNA 19:639–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kok KH, Lui PY, Ng MH, Siu KL, Au SW, Jin DY. 2011. The double-stranded RNA-binding protein PACT functions as a cellular activator of RIG-I to facilitate innate antiviral response. Cell Host Microbe 9:299–309 [DOI] [PubMed] [Google Scholar]
- 191.Ho TH, Kew C, Lui PY, Chan CP, Satoh T, et al. 2016. PACT- and RIG-I-dependent activation of type I interferon production by a defective interfering RNA derived from measles virus vaccine. J. Virol 90:1557–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Luthra P, Ramanan P, Mire CE, Weisend C, Tsuda Y, et al. 2013. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe 14:74–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ding Z, Fang L, Yuan S, Zhao L, Wang X, et al. 2017. The nucleocapsid proteins of mouse hepatitis virus and severe acute respiratory syndrome coronavirus share the same IFN-beta antagonizing mechanism: attenuation of PACT-mediated RIG-I/MDA5 activation. Oncotarget 8:49655–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Siu KL, Yeung ML, Kok KH, Yuen KS, Kew C, et al. 2014. Middle East respiratory syndrome coronavirus 4a protein is a double-stranded RNA-binding protein that suppresses PACT-induced activation of RIG-I and MDA5 in the innate antiviral response. J. Virol 88:4866–76 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 195.Komuro A, Homma Y, Negoro T, Barber GN, Horvath CM. 2016. The TAR-RNA binding protein is required for immunoresponses triggered by Cardiovirus infection. Biochem. Biophys. Res. Commun 480:187–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Lui PY, Wong LR, Ho TH, Au SWN, Chan CP, et al. 2017. PACT facilitates RNA-induced activation of MDA5 by promoting MDA5 oligomerization. J. Immunol 199:1846–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Miyamoto M, Komuro A. 2017. PACT is required for MDA5-mediated immunoresponses triggered by Cardiovirus infection via interaction with LGP2. Biochem. Biophys. Res. Commun 494:227–33 [DOI] [PubMed] [Google Scholar]
- 198.Singleton MR, Dillingham MS, Wigley DB. 2007. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem 76:23–50 [DOI] [PubMed] [Google Scholar]
- 199.Putnam AA, Jankowsky E. 2013. DEAD-box helicases as integrators of RNA, nucleotide and protein binding. Biochim. Biophys. Acta 1829:884–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Luo D, Kohlway A, Pyle AM. 2013. Duplex RNA activated ATPases (DRAs): platforms for RNA sensing, signaling and processing. RNA Biol 10:111–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lee T, Pelletier J. 2016. The biology of DHX9 and its potential as a therapeutic target. Oncotarget 7:42716–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, et al. 2008. Identification of host proteins required for HIV infection through a functional genomic screen. Science 319:921–26 [DOI] [PubMed] [Google Scholar]
- 203.Zhang Z, Yuan B, Lu N, Facchinetti V, Liu YJ. 2011. DHX9 pairs with IPS-1 to sense double-stranded RNA in myeloid dendritic cells. J. Immunol 187:4501–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Zhu S, Ding S, Wang P, Wei Z, Pan W, et al. 2017. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 546:667–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Miyashita M, Oshiumi H, Matsumoto M, Seya T. 2011. DDX60, a DEXD/H box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Mol. Cell. Biol 31:3802–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Oshiumi H, Miyashita M, Okamoto M, Morioka Y, Okabe M, et al. 2015. DDX60 is involved in RIG-I-dependent and independent antiviral responses, and its function is attenuated by virus-induced EGFR activation. Cell Rep. 11:1193–207 [DOI] [PubMed] [Google Scholar]
- 207.Goubau D, van der Veen AG, Chakravarty P, Lin R, Rogers N, et al. 2015. Mouse superkiller-2-like helicase DDX60 is dispensable for type I IFN induction and immunity to multiple viruses. Eur. J. Immunol 45:3386–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Wang P, Zhu S, Yang L, Cui S, Pan W, et al. 2015. Nlrp6 regulates intestinal antiviral innate immunity. Science 350:826–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Mitoma H, Hanabuchi S, Kim T, Bao M, Zhang Z, et al. 2013. The DHX33 RNA helicase senses cytosolic RNA and activates the NLRP3 inflammasome. Immunity 39:123–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Liu Y, Lu N, Yuan B, Weng L, Wang F, et al. 2014. The interaction between the helicase DHX33 and IPS-1 as a novel pathway to sense double-stranded RNA and RNA viruses in myeloid dendritic cells. Cell Mol. Immunol 11:49–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Sugimoto N, Mitoma H, Kim T, Hanabuchi S, Liu YJ. 2014. Helicase proteins DHX29 and RIG-I cosense cytosolic nucleic acids in the human airway system. PNAS 111:7747–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zhu Q, Tan P, Li Y, Lin M, Li C, et al. 2018. DHX29 functions as an RNA co-sensor for MDA5-mediated EMCV-specific antiviral immunity. PLOS Pathog. 14:e1006886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Gringhuis SI, Hertoghs N, Kaptein TM, Zijlstra-Willems EM, Sarrami-Forooshani R, et al. 2017. HIV-1 blocks the signaling adaptor MAVS to evade antiviral host defense after sensing of abortive HIV-1 RNA by the host helicase DDX3. Nat. Immunol 18:225–35 [DOI] [PubMed] [Google Scholar]
- 214.Gu L, Fullam A, McCormack N, Hohn Y, Schroder M. 2017. DDX3 directly regulates TRAF3 ubiquitination and acts as a scaffold to co-ordinate assembly of signalling complexes downstream from MAVS. Biochem. J 474:571–87 [DOI] [PubMed] [Google Scholar]
- 215.Zhang Z, Kim T, Bao M, Facchinetti V, Jung SY, et al. 2011. DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity 34:866–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Sithole N, Williams C, Vaughan A, Lever A. 2015. The roles of DEAD box helicases in the life cycle of HIV-1. Lancet 385(Suppl. 1):S89. [DOI] [PubMed] [Google Scholar]
- 217.Bortz E, Westera L, Maamary J, Steel J, Albrecht RA, et al. 2011. Host- and strain-specific regulation of influenza virus polymerase activity by interacting cellular proteins. mBio 2:e00151–11 [DOI] [PMC free article] [PubMed] [Google Scholar]