

Abstract
Genomic test results collected during the provision of medical care and stored in Electronic Health Record (EHR) systems represent an opportunity for clinical research into disease heterogeneity and clinical outcomes. In this paper, we evaluate the use of genomic test reports ordered for cancer patients in order to derive cancer subtypes and to identify biological pathways predictive of poor survival outcomes. A novel method is proposed to calculate patient similarity based on affected biological pathways rather than gene mutations. We demonstrate that this approach identifies subtypes of prognostic value and biological pathways linked to survival, with implications for precision treatment selection and a better understanding of the underlying disease. We also share lessons learned regarding the opportunities and challenges of secondary use of observational genomic data to conduct such research.
Keywords: Deep phenotyping, Computational cancer subtyping, Survival analysis, Secondary use of genomic data, Pathway analysis
1. Introduction
The vision of the Learning Healthcare System relies on the routine collection and ongoing use of clinical data to systematically extract medical knowledge and to apply it to patient care decisions [1]. Doing so at scale and in the context of Precision Oncology requires automated handling and integration of both clinical and genomic data, as well as effective approaches to mine data already acquired to date to enable retrospective knowledge generation. In practice, patients’ genomic test results are yet to be integrated into most EHR systems [2] and are thus underutilized for predicting clinical outcomes or optimizing patient care in a systematic and automated fashion.
To a large extent, bioinformatics research into cancer has relied on a limited set of publicly available datasets such as The Cancer Genome Atlas (TCGA) [3] and National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO) [4], which are clean, annotated, and multidimensional (containing multi-omics data). However, these datasets have several limitations. Clinical annotations are limited in scope and often manually curated, with curation relying on an ongoing directed effort rather than taking advantage of large-scale pre-existing passively collected data, such as the electronic health records data. These public datasets are decoupled from longitudinal clinical patient records, thus limiting our ability to leverage this data to study disease progression, response to treatment, or applications to inform clinical decisions. The curated snapshot data may harbor biases and may not be representative of the real-world population. Finally, many genomic variants are rare, and their identification would require a study of large cohorts, which is only possible to accomplish in the community at large in the clinical setting. For these reasons, the secondary use of clinical genomic test results represents a practical opportunity for expanding data acquisition for ongoing research, as well as for the development of genomic decision support systems.
On the other hand, genomic patient data routinely collected for oncology patients to support clinical decisions presents several challenges. First, it may be less comprehensive since only subsets of genes may be regularly tested in practice, and only results deemed clinically relevant at the time of interpretation are recorded and made available for downstream analysis. In addition, depending on the bioinformatics pipeline employed and inclusion of normal samples for filtering, the data may be biased by the inclusion of germline variants. Finally, genomic test reports may only be available in unstructured text format and are at least partially composed manually; extracting structured data from these reports is a laborious text mining task that may be error-prone and costly.
Cancer can be viewed as a disease of disrupted informational networks or pathways. In the seminal work on the landscapes of cancer, Vogelstein et al. (2013) showed that disruptions in 12 pathways governing cell fate, proliferation, and genome maintenance confer a selective growth advantage to cells and lead to tumorigenesis [5]. As a manifestation of cancer heterogeneity, patients may present with different mutated genes, but those genes may participate in the same biological process and therefore exert similar physiological effects and result in similar disease phenotypes. Elucidation of disrupted gene pathways from gene expression data is an area of active research in systems biology [6,7], and several gene pathway or network knowledge bases have been developed, including Reactome [8], KEGG [9], and Gene Ontology [10]. Zhao et al. [11] and Yang, Ge, and Zheng [12] have developed approaches to apply network topology knowledge to integrate gene expression data with somatic gene mutations on the network level. Kuijjer et al. [13] and Hofree et al. [14] demonstrated that grouping patients within cancer types based on somatic gene mutations alone mapped to altered pathways resulted in cancer subtypes of prognostic value. Altieri et al. [15] proposed a method applying a color coding technique to somatic mutation data overlaid on a protein-protein interaction network to identify a subnetwork of genes most predictive of survival within cancer-specific cohorts, while Fang and Gough [16] used Cox regression analysis with similar data to identify a subset of genes on a pan-cancer basis. All six teams used publicly available datasets from TCGA for their research. In the clinical setting, somatic mutations from cancer patient biopsies are routinely identified, particularly for more complex clinical cases. However, most often such tests are performed for a panel of genes (on the order of 500 genes) with known actionable associations with cancer, rather than the full exome. To our knowledge, this study is the first work demonstrating the application and utility of pathway analysis for cancer subtyping to real world data extracted from clinical cancer pathology reports.
In this research, we evaluate the use of pathology reports of genomic tumor sequencing and clinical data obtained from the electronic health records of the patients for mining cancer subtypes of prognostic value with the view to inform clinical care decisions as well as to derive a deeper understanding of cancer biology. We hypothesized that pathology test reports for a relatively large cancer panel would lend themselves to this approach despite certain limitations. These limitations include reported variants being limited to only cancer-associated genes, significant manual data curation that may not be generalizable to other datasets and may not scale for larger datasets, and potential biases introduced by filtering performed by bioinformatics pipelines. We combined biological pathway knowledge in the Reactome knowledgebase with somatic mutation data mined from clinical genomic tests, then applied unsupervised hierarchical clustering to identify clusters of patients with similarly mutated biological pathways. We obtained three sub-phenotypes of cancer patients across multiple cancer types that show significant differences in survival outcomes. We then identified biological pathways associated with worse survival outcomes and replicated a large subset of these pathways in a replication dataset.
Our key methodology contributions include an algorithm for patient similarity and disease subtyping in a pan-cancer cohort using a relatively small subset of somatic mutations mapped to affected biological pathways and a methodology to identify novel pan-cancer mechanisms implicated in worse survival outcomes based on differences among patient groups. This work demonstrates potential applications of the secondary use of genomic data obtained during routine provision of health care for research and clinical decision support.
2. Datasets
We analyzed a set of genomic test pathology reports ordered for 2906 patients of The NewYork Presbyterian Hospital (NYP), administered between March 7, 2011 and June 7, 2018. Test reports selected for analysis included 1414 reports for Columbia Combined Cancer Panel (CCCP), a panel of 467 cancer-associated genes analyzed with next-generation sequencing (NGS), with additional information on copy number variations (CNV). Samples were obtained during the provision of healthcare, sequenced and processed by the bioinformatics pipeline of the Columbia University Medical Center Laboratory of Personalized Genomic Medicine. Results were curated and reported in the genomic pathology reports finalized by a molecular pathologist, as previously reported [17]. Also included in this analysis were 366 test results from clinical cancer whole exome sequencing and transcriptome sequencing, with bioinformatics analysis and reporting performed as previously described [18]. A total of 10,791 gene variants, 1318 CNV events, and 196 gene fusions were automatically extracted from 1310 genomic test reports. Gene variants with a high probability of deleteriousness, as evaluated by CADD score [19] and MutPred [20], and all CNVs and fusions were retained. Genes were mapped to Reactome [8] pathways downloaded from reactome.org on April 13, 2019 (see Fig. 1).
Fig. 1.
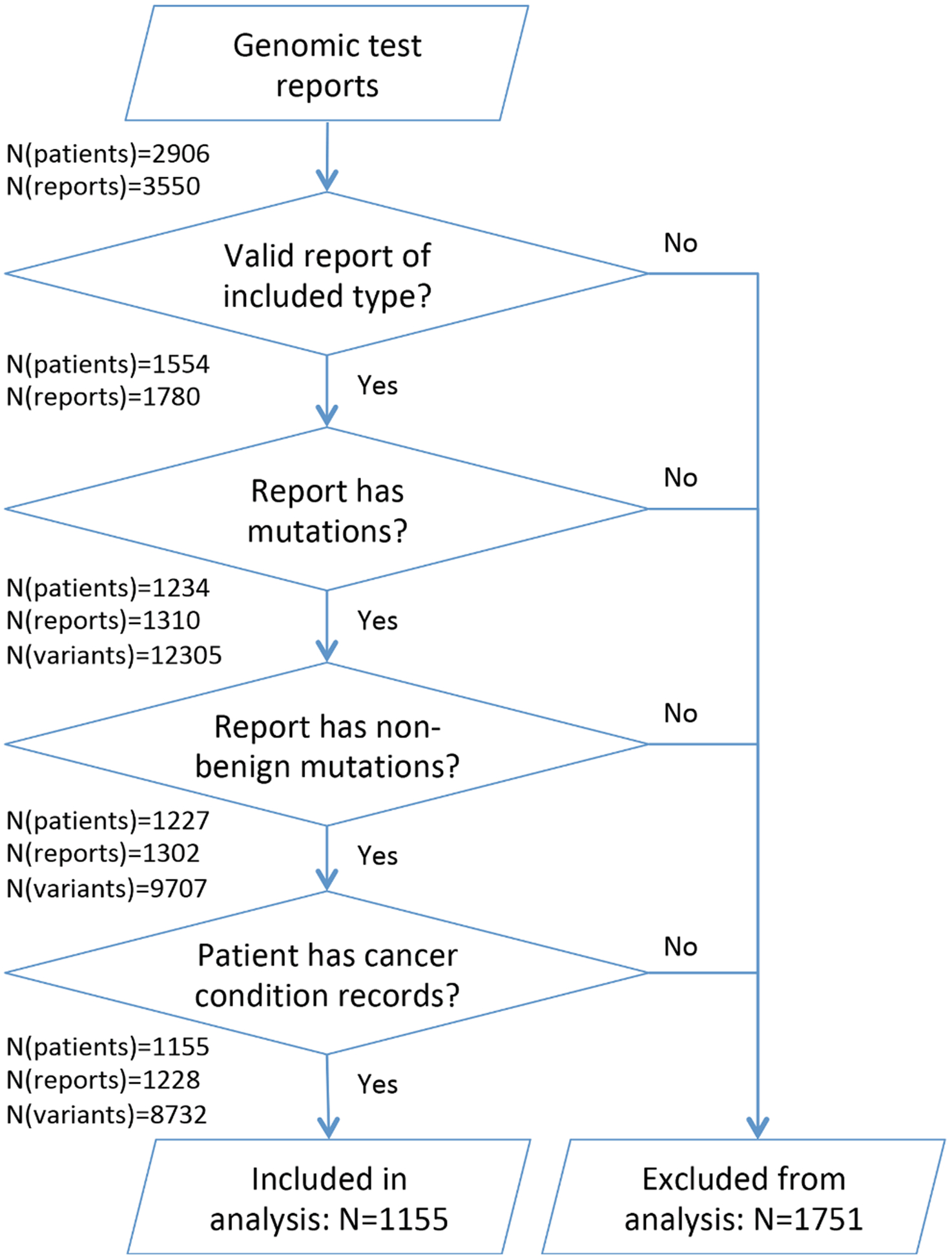
Obtaining patient cohort from genomic test reports and observational clinical data.
Clinical information for the same set of patients was obtained from Columbia University Irving Medical Center (CUIMC) Observational Health Data Sciences and Informatics (OHDSI) database containing data extracted from the NYP EHR. For 1554 patients that had a valid genomic test report, conditions and medications were extracted from OHDSI on July 30, 2018, and death dates were extracted on September 10, 2018. Clinical data represents 893,951 records of medical conditions for the period between November 17, 1985 and February 2, 2018 and 521,458 records of medications between October 18, 1996 and February 2, 2018. We mapped conditions to SNOMED-CT codes [21] representing all neoplastic diseases and retained for further analysis for 1155 patients diagnosed with a neoplastic disease. Primary cancer sites were obtained by mapping the SNOMED-CT disease codes to ICD-O-3 topology codes [22].
We obtained additional cancer-related data from the NYP Tumor Registry containing information for 38,776 NYP patients diagnosed with cancer between 1966 and 2017. This dataset included ICD-O-3 topological tumor classification, tumor grade, and dates of cancer diagnosis and death. Out of 1155 patients with genomic test results and a neoplastic disease diagnosis, 916 patients had a record in the Tumor Registry.
We performed validation of the proposed methodology and key findings on the integrated TCGA dataset of 3281 tumors across 12 tumor types along with key clinical data prepared and made available by Kandoth et al. – [23]. This dataset differs from the NYP dataset in a number of significant ways. Firstly, it does not apply manual selection or curation of significant genes and includes variants in 20,947 genes; however, it does not include gene fusions or CNVs. In contrast, the majority of our cases were derived from the Columbia cancer panel which reports on variants in only 467 genes and a smaller subset of cases derived from cancer whole exome and transcriptome sequencing (in total, 558 genes where reported between the two types of test reports), and these reported variants underwent manual curation. Since the validation dataset is not curated, it is significantly larger, with a total of 617,354 variants across 3281 tumor samples, compared to 12,305 variants for 2906 patients in the NYP dataset before pre-processing (8732 variants for 1155 patients after filtering). Finally, the validation dataset includes a different combination of primary tumor types and manually annotated verified clinical data such as survival, tumor type, and tumor stage.
3. Methods
The proposed analytical pipeline is outlined in Fig. 2 and consists of genomic and clinical data processing with subsequent analysis aimed at discovering cancer subtypes and predicting clinical outcomes.
Fig. 2.
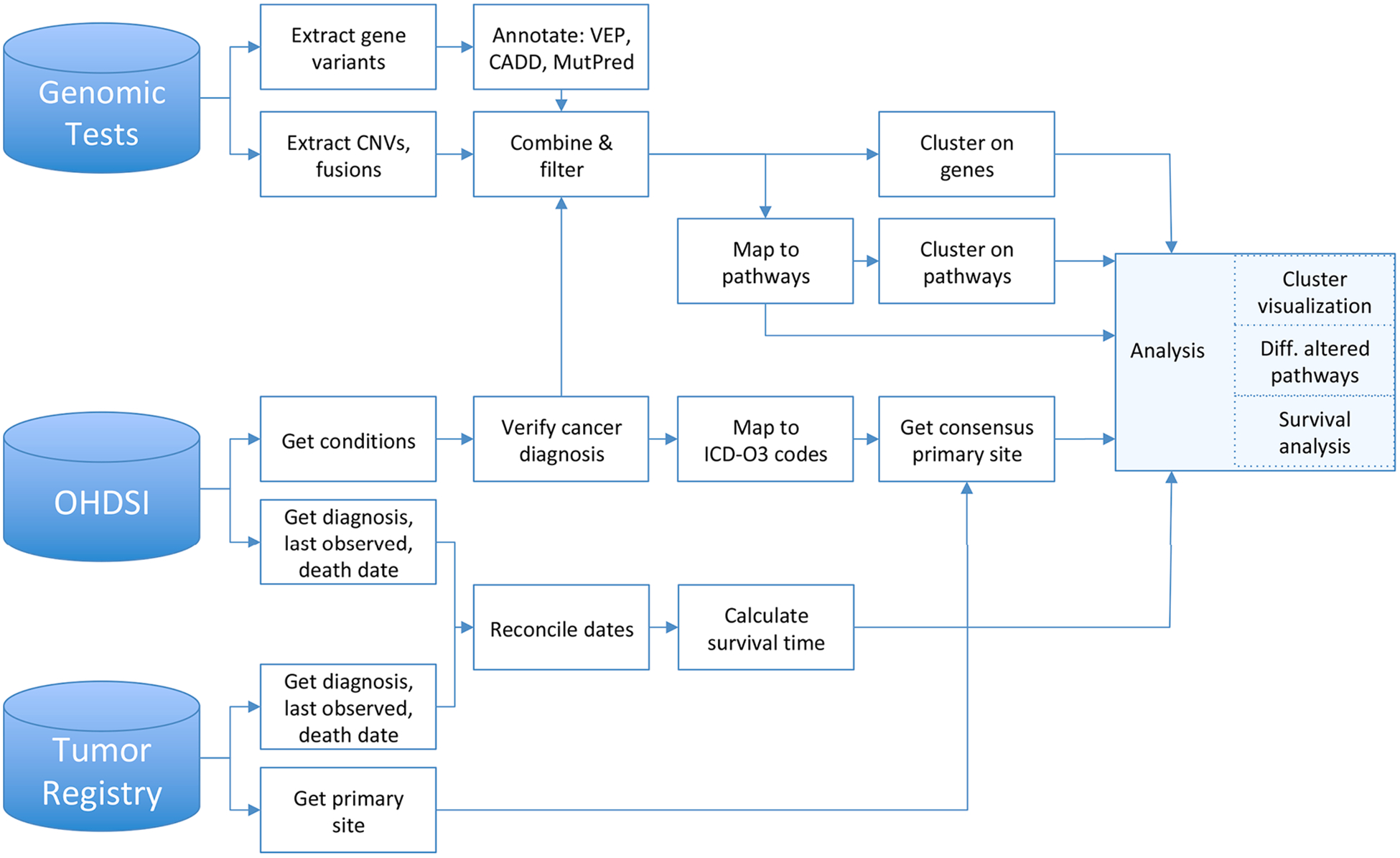
The data processing and analytical workflow.
3.1. Mining and classifying genomic test data
We extracted gene variants, gene amplifications, deletions, and fusions from the text of genomic test reports in R using regular expressions based on manually identified patterns. For example, gene variants were found by matching patterns like “Gene: SRSF2 Variant: NM_001195427 c.284C > A, p.P95H”, with small variations on punctuation. Detecting copy number variations and fusions required splitting text by sentences and phrases; gene name recognition; and identification of key phrase constructs like “presence of a LMNA-NTRK1 fusion”, “amplification of CDK4, MDM2, and TERT”, etc. Regular expressions used to extract gene variants, CNVs, and fusions are listed in Supplementary Table 1.
Variants in the reports had been classified and tiered by molecular pathologists by considering factors such as actionability, presence in targeted pathways, known roles in tumorigenesis, presence in population or cancer databases, predicted effect on the protein, and in silico functional predictions for missense variants. For our analysis, we considered all reported variants, including variants classified as variants of uncertain significance (VUS). We then removed gene variants that were predicted to have no functional impact on the gene product by in silico algorithms. In order to estimate functional impact of gene variants, we mapped gene variant transcripts to chromosomal coordinates using Mutalyzer [24], then annotated the variants with CADD score of variant deleteriousness [19] and MutPred’s top 5 predicted functional consequences [20] using Ensembl Variant Effect Predictor (VEP) [25]. The 9,499 initial gene variants (excluding CNVs and fusions) extracted from all reports were mapped to 9,436 unique chromosomal coordinates, and 9,279 variants were annotated by VEP. 7,631 variants were classified as potentially deleterious and retained for further analysis based on satisfying at least one of the following criteria: CADD score > 15[26]; mutations classified as stop gained, frameshift, start lost or stop lost, and splice variant changes, as these mutations are expected to result in truncated proteins [27,28]; or mutations with predicted consequences by MutPred. Variants that were not successfully mapped to a chromosomal location, not successfully annotated by VEP, or not deemed to be potentially deleterious were excluded from further analysis.
Out of 9,279 variants recognized by VEP, only 1,242 (13.4%) had a ClinVar significance value [29], meaning that some laboratory had submitted a clinical interpretation of these variants to the ClinVar database. Among mutations without a known ClinVar interpretation, 1,881 mutations are frameshift or premature stop codon variants expected to have high impact.
For patients with multiple reports, we combined mutations and CNVs across all reports. For the purposes of this analysis, we did not address tumor evolution, as would be necessary for use cases such as studying the emergence of drug-resistant phenotypes.
3.2. Extracting and harmonizing clinical data
We extracted clinical data from OHDSI and tumor registry for the relevant set of patients and harmonized data inconsistencies between the two datasets or within each dataset. Number of months elapsed between the first cancer diagnosis and the last observation or death was later used in survival analysis.
3.2.1. Disease classification
We obtained a set of 4,239 SNOMED-CT disease codes for malignant neoplastic diseases by traversing the hierarchical (“Is-A”) relationships in the SNOMED-CT database [21] (US Snapshot database, March 1, 2018 release), starting at node 363346000 (“Malignant neoplastic disease”), and identified 1,165 patients with this diagnosis code. We then attempted to classify patients by the primary cancer site. Both the OHDSI and the Tumor Registry datasets had information on the primary cancer site; however, this information was often incomplete or inconsistent across datasets or even within the dataset. Moreover, patients genuinely present with various primary cancers at different points in their clinical history. For patients with a single entry in the Tumor Registry, we used the primary cancer site recorded in the Tumor Registry. For patients with no record in the Tumor Registry, the ICD-O-3 topology code associated in SNOMED-CT with the first primary neoplastic condition recorded in OHDSI was used as the primary cancer site. For patients with multiple Tumor Registry entries or conflicting primary sites obtained from the Tumor Registry and OHDSI, the primary cancer site was coded as “multiple”. Clinical staging was obtained from the Tumor Registry. We summarize clinical data for the cohort in Table 1.
Table 1.
Demographic and clinical summary of patient cohort (Hema: Hematopoietic, GI: Gastrointestinal Tract; CNS: Central Nervous System; Gyne: Gynecological).
| Cohort summary | N= 1155 | ||
|---|---|---|---|
| Age at diagnosis | Sex | ||
| > 18 | 762 | Male | 476 |
| < = 18 | 146 | Female | 431 |
| Unknown | 247 | Unknown | 248 |
| Primary tumor site | Clinical tumor stage | ||
| Hema. | 207 | 0 | 7 |
| GI | 130 | 1 | 122 |
| CNS | 122 | 2 | 88 |
| Pancreas | 106 | 3 | 85 |
| Gyne. | 71 | 4 | 174 |
| Liver | 58 | Unknown/ Not staged | 689 |
| Lung | 49 | ||
| Breast | 47 | ||
| Skin | 44 | ||
| Multiple | 46 | ||
| Other | 275 |
3.2.2. Dates for survival analysis
In order to handle the right-censored nature of survival data, survival analysis considers the period from diagnosis to death or from diagnosis to the last observation, with the indicator whether the event of death has occurred. We obtained dates of first diagnosis, last observation, and death for all patients from both OHDSI and the Tumor Registry and attempted to reconcile inconsistencies between the two data sources. We obtained the date of the first cancer diagnosis by using the earlier of the first OHDSI diagnosis date with neoplastic disease and the diagnosis date in the Tumor Registry, filtering out invalid dates. For the last observed date, we used the maximum of the last observed date in the Tumor Registry and the date of the last condition or medication record in OHDSI.
Death dates across OHDSI and Tumor Registry also required harmonization: 9 patients had multiple death dates listed in OHDSI death Table 3 patients had multiple dates in the Tumor Registry; 139 patients had a date of death only in OHDSI, and 75 only in the Tumor Registry. Out of 174 patients that had a date in both datasets, both dates did not agree in 42 cases. Moreover, in 16 cases, OHDSI contained records of conditions or medications after that date; and 27 that had a genomic test, which was first administered on March 7, 2011, had recorded dates of death in OHDSI prior to that date. We removed invalid dates of death by filtering out those that occurred before March 7, 2011 and used the Tumor Registry date as the consensus date if available, as our manual review showed it to be more reliable. If no more than one medication or condition record occurred after the reported death date, we considered those records spurious, and retained the death date; however, for cases of OHDSI-reported death dates with more than one condition or medication following that date, we treated the reported death event as unreliable.
Table 3.
Cluster stability evaluation with Clusterboot.
| Cluster stability | |||
|---|---|---|---|
| Based on pathways | Cluster 1 | Cluster 2 | Cluster 3 |
| bootmean | 0.7805 | 0.8963 | 0.9578 |
| # times dissolved | 3 | 0 | 0 |
| cluster size | 213 | 718 | 212 |
| Based on genes | Cluster 1 | Cluster 2 | Cluster 3 |
| bootmean | 0.4468 | 0.4000 | 0.6782 |
| # times dissolved | 71 | 98 | 0 |
| cluster size | 119 | 314 | 722 |
3.3. Subtyping based on pathways harboring mutated genes
Proteins encoded by genes form functional and regulatory networks or pathways; cancer, as well as other diseases, can be viewed as the disease of disrupted processes due to mutations altering healthy pathways. Many gene mutations, including mutations that are rare within the population, can result in the same altered pathway, leading to the same functional consequences [5,6,7,8,11].
For each patient, we mapped gene mutations, CNVs, and fusions to the corresponding affected biological pathways using the Reactome pathway database [8] downloaded on April 13, 2019 and containing 1829 pathways for Homo sapiens. We removed pathways that contained 500 genes or more and those containing fewer than 4 genes, as those pathways would be less informative for analysis; removed duplicate pathways with the same participating genes; and filtered pathways that do not contain any genes in the gene mutation dataset, resulting in 870 Reactome pathways for further analysis. Retaining patients that had mutations in the remaining pathways resulted in a cohort of 1143 patients.
We applied one-hot encoding to the matrix containing patients as rows and pathways as columns, where a value of 1 in the ith row and jth column indicates that the ith patient has a mutation in at least 1 gene in the jth pathway. We then performed unsupervised hierarchical clustering with binary distance on the resulting one-hot encoded matrix of pathways and patients using hclust function in stats R package [30], with method parameter set to “ward.D2”. We determined the optimal number of clusters k = 3 using fviz_nbclust function in factoextra R package [31], which iterates through various k’s and plots within-cluster sum of squares (“elbow plots”) and average silhouette width for each k. We repeated one-hot encoding and clustering on the initial set of genes as well. Hierarchical clustering based on pathways and genes is illustrated in Fig. 3A–B.
Fig. 3.
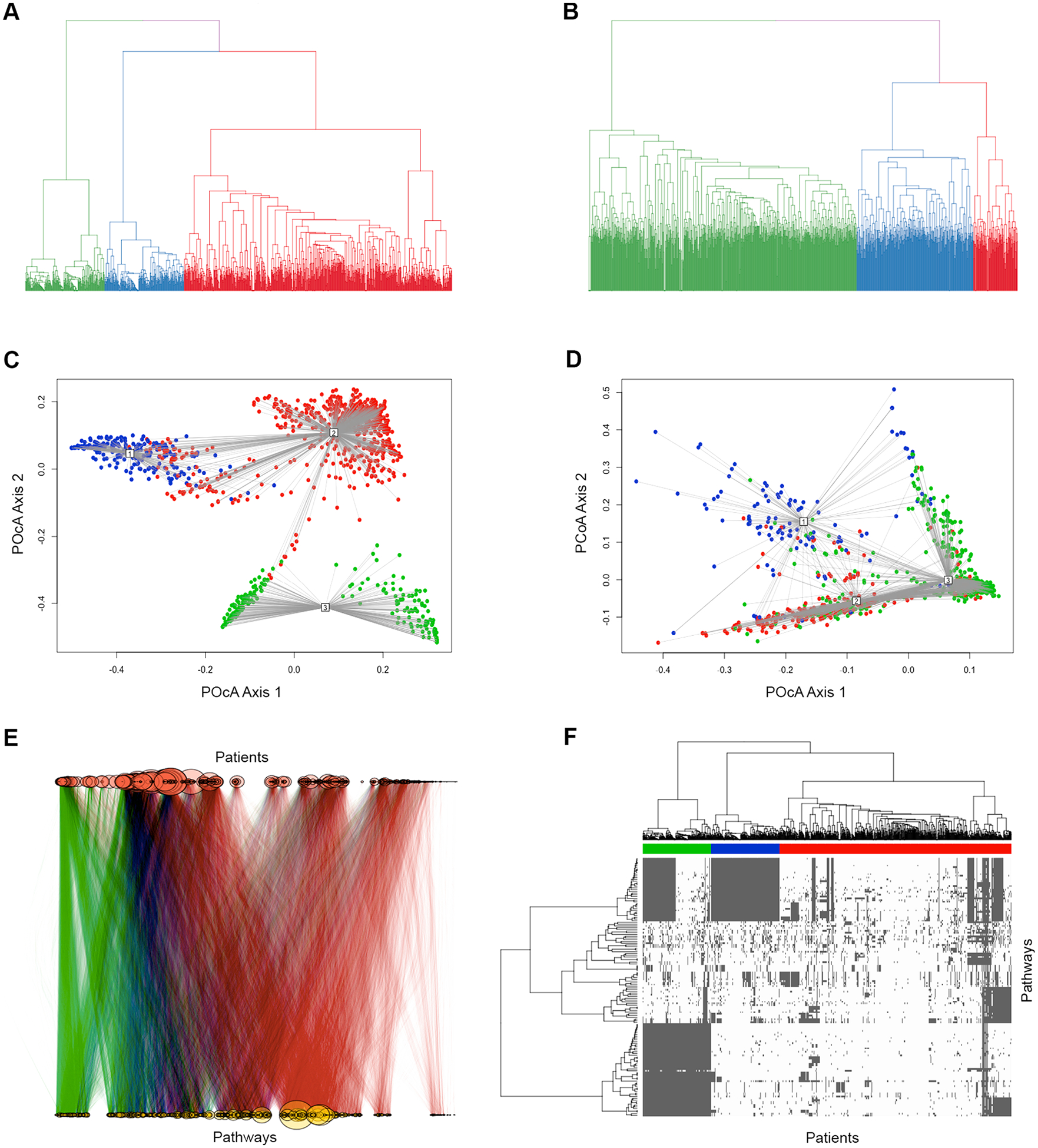
Hierarchical clustering based on the similarity of mutated pathways and genes. A-B: Hierarchical clustering of cancer cases based on altered Reactome pathways (A) and affected genes (B). C-D: PCoA plot of clusters based on altered Reactome pathways (C) and affected genes (D). E: Graph visualization of cancer cases (red circles) connected to altered Reactome pathways (yellow circles), with edges representing presence in a patient’s pathology report of a mutation, CNV, or fusion of a gene participating in the corresponding pathway. F: Heatmap representation of patient clusters based on affected Reactome pathways (150 pathways with most variation among patients are shown). Color bar represents the cluster assignment of those patients. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.4. Predicting survival
Prognosis (survival time, or time from diagnosis to death) is an example of right-censored data where some events are observed, while others have not occurred in the observation time period. We performed Kaplan-Meier survival analysis [32] using SurvFit in survminer R package [33] to determine whether cluster assignment on pathway or gene level was informative of survival, in other words, whether the subtypes identified by hierarchical clustering had prognostic value. We then identified differentially altered pathways between clusters associated with better and worse survival outcomes and evaluated correlation of aberrations in those pathways with survival.
3.5. Reproducibility of clustering methodology and key findings
We applied the proposed methodology to the validation dataset: we annotated the reported variants with VEP and filtered based on CADD and MutPred values using the same criteria as described earlier. We then mapped the genes affected by mutations to Reactome pathways, performed one-hot encoding of pathways and genes separately, and applied hierarchical clustering to identify clusters of similar patients based on altered pathways and affected genes. We then performed survival analysis to determine whether identified clusters are associated with survival outcomes.
We then investigated whether Reactome pathways identified previously as differentially altered between clusters of better and worse survival in the original dataset are in fact predictive of survival in the validation dataset.
4. Results
4.1. Variant and CNV extraction
We evaluated extraction of gene variants from genomic test reports by manually reviewing randomly selected genomic test reports for 5% of patients (77 patients). To evaluate extraction of CNVs and fusions, in addition we manually reviewed reports for 40 patients randomly selected among those who had reports containing one of the following key phrases: “fusion”, “overexpression”, “high copy number”, “increased copy number”, “overexpressed”, “over expressed”, “amplification”, “gain”, “copy number loss”, “deletion”, “decreased copy number”, “copy loss”, “low level of expression”, “low expression level”, “loss”. We measured the accuracy of automatic extraction compared to manual review with F1 metric, Precision (Positive Predictive Value), Recall (True Positive Rate), and Specificity (True Negative Rate). Table 2 lists evaluation measures and demonstrates that overall extraction of all variation was rather accurate, with F1 metric between 0.9969 for gene variants and 0.8485 for CNV. Extraction of gene variants was highly precise, sensitive, and specific; extraction of CNVs was highly specific but less sensitive; and extraction of fusions was highly sensitive and specific but less precise.
Table 2.
Evaluation of data extraction accuracy for gene variants, CNVs, and gene fusions.
| Accuracy measure | Gene variants | CNVs | Fusions |
|---|---|---|---|
| F1 Metric | 0.9969 | 0.8485 | 0.9020 |
| Sensitivity (TPR) | 0.9979 | 0.8092 | 1.0000 |
| Specificity (TNR) | 0.9993 | 0.9969 | 0.9991 |
| Precision (PPV) | 0.9958 | 0.8917 | 0.8214 |
4.2. Patient subtyping with hierarchical clustering
Our dataset consisted of a set of genes smaller than the genome-wide sets of somatic mutations and copy number alterations typically used for bioinformatics analyses to identify biological markers of survival or elucidate disease phenotypes. Analysis of mutations extracted from genomic test reports, when mapped onto pathways using the Reactome pathway database, was able to group patients into three subtypes that had statistically significant prognostic value with a p-value of 0.0207, confirmed by log-rank test. Clusters 2 and 3 had the most marked difference in survival, with a p-value of 0.017. In contrast, clustering patients based on mutated genes themselves did not produce any prognostically significant patient subtypes.
We assessed that cluster assignment was not driven primarily by the overall mutational load (number of mutations in each patient): Pearson correlation between the number of mutations and cluster assignment with Reactome pathways at k = 3 was 0.027, p-value 0.3558. It is notable that the primary cancer site had more prognostic value (Log-rank p-value = 0.0084, Fig. 4B) than the 3 Reactome-based subtypes, suggesting that subtyping could be further improved when performed within each cancer type separately, given sufficient cohort sizes.
Fig. 4.
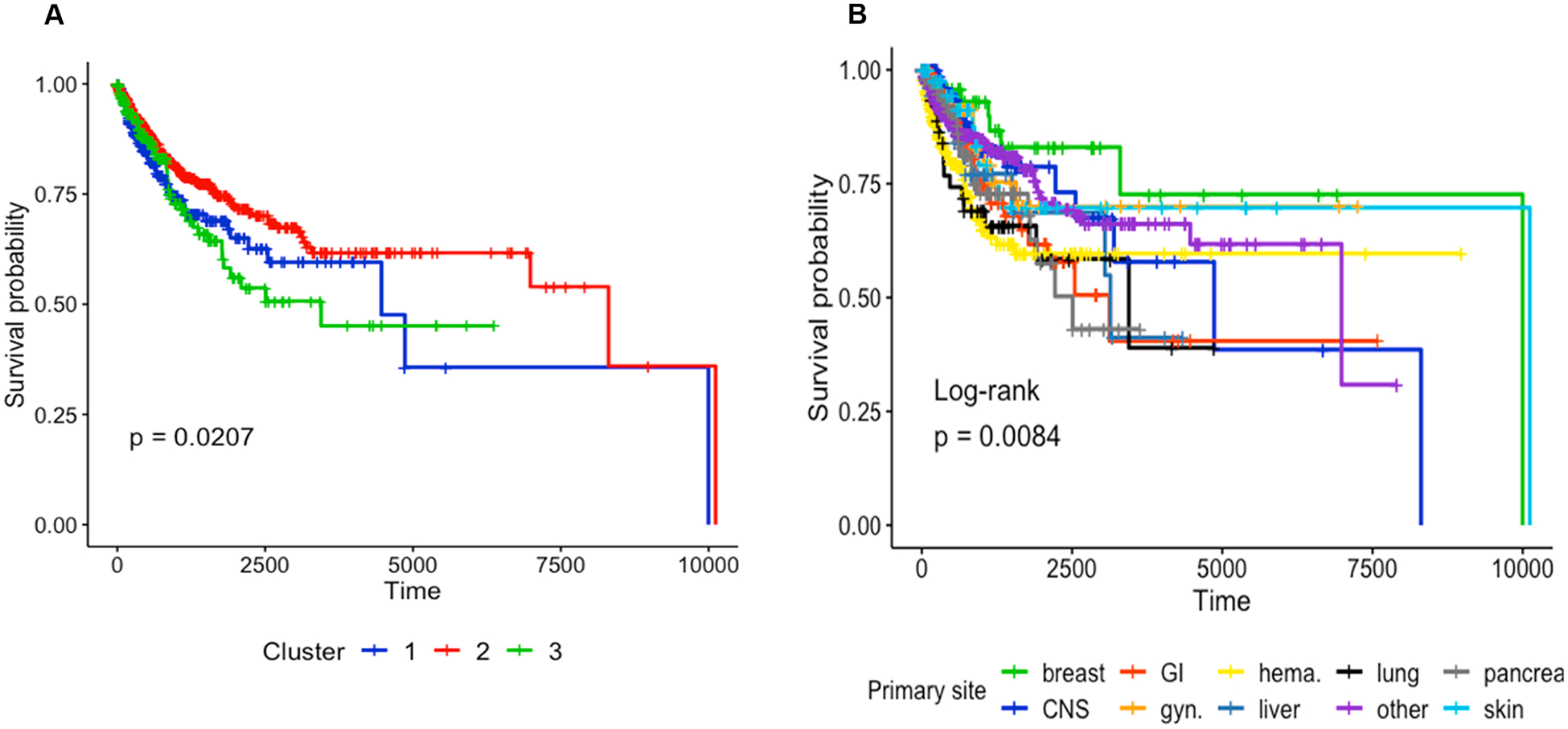
A: Survival analysis with Kaplan-Meier plot based on subtypes derived from altered pathway profiles of 3 patient clusters. B: Kaplan-Meier survival plot stratified by the primary tumor site.
Stability of cluster assignments was evaluated using clusterboot method in fpc R package [34] with the number of bootstrap iterations set to the default value of 100. Bootmean value measures the mean Jaccard similarity between clusters in each bootstrap iteration and the original cluster assignment, with values closer to 1 indicating higher stability. Clusters with Jaccard similarity less than the default value of 0.5 between the bootstrap clusters and the initial cluster assignments are considered dissolved, with the smallest number of dissolutions across all bootstrap iterations indicating higher cluster stability. Hierarchical clustering based on pathways produced more stable cluster assignments than clustering based on altered genes, as seen in Table 3.
Cluster assignments were evaluated for correlation with primary site, sex, age at diagnosis, tumor stage at diagnosis, and the number of different cancer sites recorded in a patient’s clinical history, using Pearson correlation (cor.test function in stats R package [30]). Cluster 1 assignment was positively correlated with age at diagnosis, total number of cancer sites in EHR, gynecological and lung cancer primary sites (p-values of 0.0027, 0.0000, 0.0000, 0.0001 respectively). Cluster 2 was negatively correlated with age at diagnosis (p-value 0.0002) and positively correlated with stage at diagnosis (p-value 0.00000), with a higher proportion of CNS (including brain cancer) and hematopoietic cancer cases (p-values 0.00000 and 0.00004) and a lower representation of pancreatic, GIST, and gynecological cancer. In essence, Cluster 2 represents a younger cohort of patients with cancers more frequent in that age group. Cluster 3 had an over-representation of pancreatic cancer cases and a lower representation of CNS, breast and other primary sites. All statistically significant correlations with p-value < 0.01 are listed in Supplementary Table 2.
4.3. Visualization of cluster separation
Differences in pathway-level mutational signatures among 3 clusters are visualized in Fig. 3E using igraph R package [35]. Pathways are represented with yellow circles, with edges indicating connections to patients. The relative size of each pathway represents its degree of connectedness, or in other words the number of patients with a mutation in that pathway. Cluster 2 and Cluster 3, represented by edges of red and green color respectively, show a visible separation. Pathways that are differentially affected among patients within these two clusters were studied as candidate survival-related biomarkers in further analysis.
Additionally, we visualized the separation of patient clusters based on the similarity of altered pathways and on the similarity of mutated genes, separately, applying Principal Coordinate Analysis (PCoA) with cmdscale function in stats R package [30], with plotting support from vegan R package [36]. PCoA is a technique used to reduce complexity in high-dimensional data while retaining most of the information and its inherent structure or patterns [37]. PCoA plot of patient clusters based on altered pathways in Fig. 3C highlights a visible separation among the three clusters, while the PCoA plot of clusters based on genes (Fig. 3D) displays a much less clear separation.
4.4. Differentially altered pathways between clusters
Results of survival analysis based on Reactome pathway subtyping with 3 clusters showed a significant difference in survival between clusters 2 and 3. If these sub-phenotypes have prognostic value, then identifying differences between them may lead to prognostic markers. We identified Reactome pathways with significant differences between clusters 2 and 3 by calculating fold-change between the number of patients in cluster 2 and 3 with alterations in each pathway, normalized for the size of each cluster, taking into account pathways altered for at least 10 patients. Statistical significance of the difference in altered pathways between clusters was determined by the chi-square test, with p-values adjusted for multiple comparisons for false discovery rate (FDR). A subset of altered pathways with the most significant difference between clusters 2 and 3 is shown in Fig. 5, and the complete list of pathways, their fold-change between clusters, and their association with survival are provided in Supplementary Table 3.
Fig. 5.
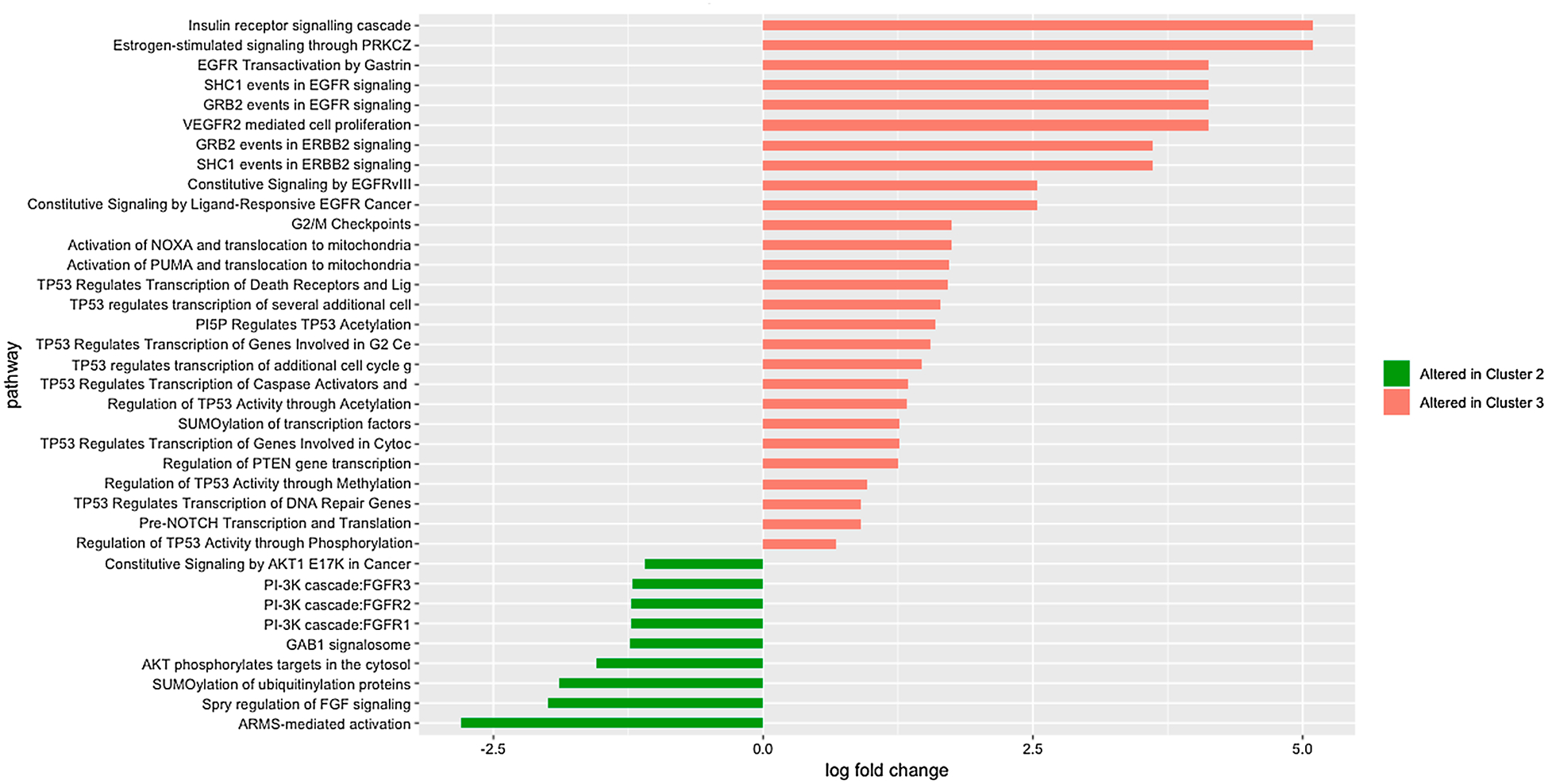
Reactome pathways with the most significant difference in alteration rates between Cluster 2 and Cluster 3, measured by log fold change.
4.5. Identifying prognostic markers
Differentially altered pathways (chi-square test adjusted p-value < 0.05) between clusters 2 and 3 were evaluated for their impact on overall survival with Kaplan Meier estimator [38, R implementation:33]. Kaplan Meier plots in Fig. 6 illustrate the impact of mutations in the top 9 of these pathways to overall survival [33]. Data for all pathways differentially altered between Clusters 2 and 3 is provided in Supplementary Table 3.
Fig. 6.
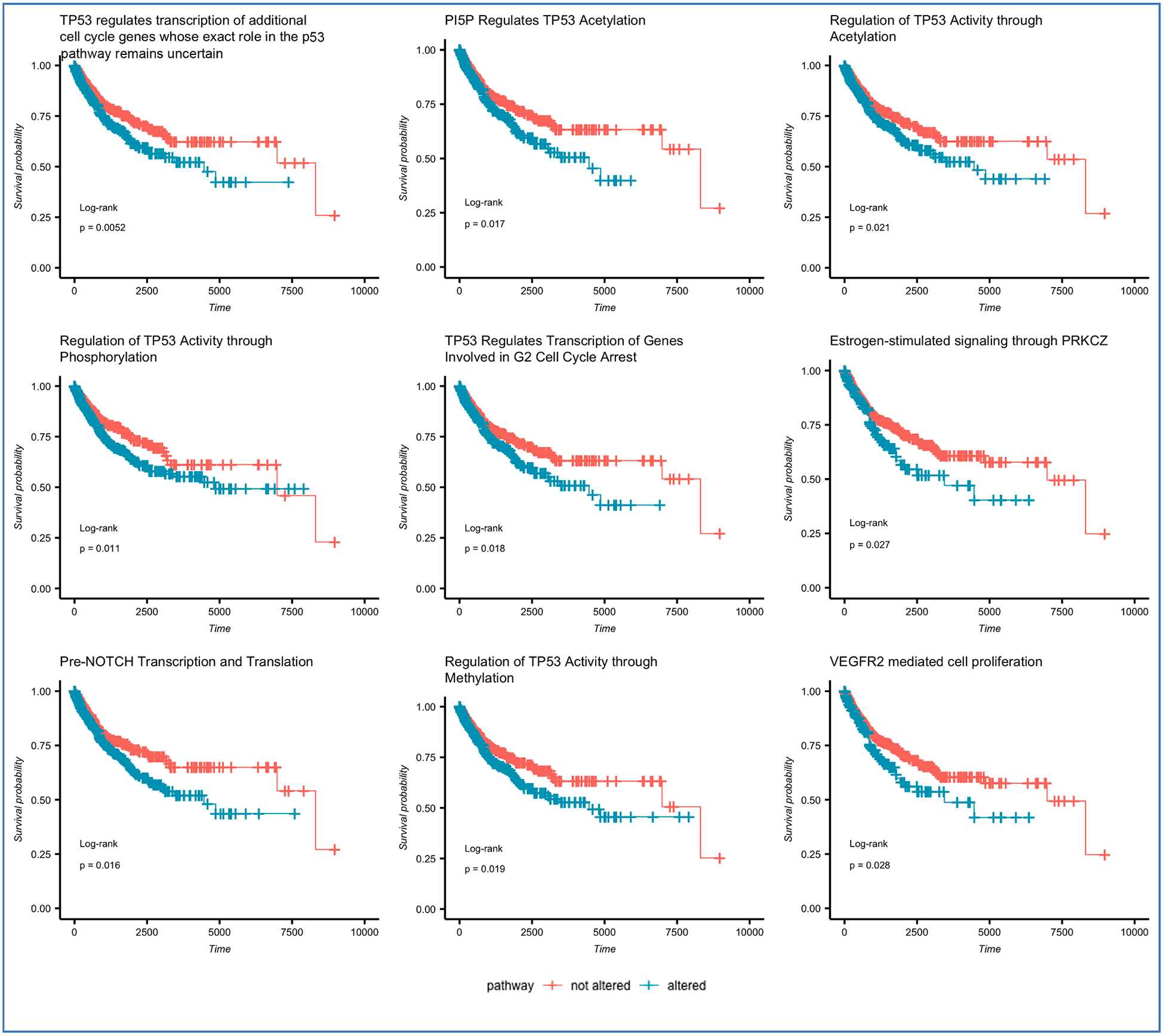
Reactome pathways with the highest difference in alteration frequency between Clusters 2 and 3 and the highest impact on survival outcomes, as evaluated across the full cohort by Log Rank test.
Among pathways found to be associated with survival and differentially altered between the two clusters, many represent processes known to be associated with cancer, such as those involving TP53, VEGFR2, PUMA, EGFRvIII, SHC1, GRB2, ERB2 [39–43]. Several pathways, however, have only recently been implicated in tumorigenesis or response to chemotherapy, and their mechanism of action is not yet fully understood. For example, although the connection of Gastrin to gastric cancers has been known [44], the mechanism of action via EGFR transactivation has only recently been elucidated [45]. Similarly, until recently, few studies implicated PI5P in signaling linked to oncogenesis, largely due to the difficulty of detection of this molecule [46]. The pathway we found to have the strongest association with survival (“TP53 regulates transcription of additional cell cycle genes whose exact role in the p53 pathway remain uncertain”, p = 0.0052 by Kaplan-Meier Log-rank test) suggests that further research into genes participating in p53 pathway may lead to a better understanding of potentially pan-cancer mechanisms of the cell cycle.
4.6. Validating methodology and findings
Although cluster assignments themselves could not be directly compared between the original NYP dataset and the validation dataset, applying the same computational approach to the validation dataset lead to clusters predictive of the clinical variable of interest. In addition, a significant number of pathways identified as associated with survival in the original dataset were also predictive of survival in the validation dataset, despite differences in the cohort composition and the overall set of genes included in the two datasets.
Annotating the validation dataset with VEP and filtering out mutations expected to not have a significant impact based on CADD and MutPred criteria yielded 450,230 variants across 18,775 genes. We mapped these genes to 1,374 Reactome pathways belonging to the Homo Sapiens species and containing between 4 and 499 genes. We used hierarchical clustering to determine similarity between patients based on the common altered biological pathways. Setting the number of clusters at k = 3, 4, 5, 6, and 7 resulted in cluster assignments which, when used in Survival analysis, resulted in statistically significant correlations with survival outcomes. For example, at k = 3, cluster assignment was highly linked to survival, with log-rank test value of 4.11538e-05. This relationship to the survival outcome was retained even when prior to clustering the validation dataset was filtered to only the genes in the original NYP dataset (long-rank test of cluster assignment at k = 3 to survival outcome: 1.139372e-05).
In the original dataset, 27 pathways were significantly more frequently altered in the cluster of poor prognosis compared to the cluster of good prognosis. Of those 27 pathways, we found that 14, or 52%, were also both significantly altered in the poor survival cluster and predictive of poor survival in the validation dataset (log-rank p-value < 0.05, adjusted with Benjamini & Yekutieli correction). In contrast, only 3% of all pathways with mutations reported in the validation dataset, or 54 out of 1,374 pathways, were predictive of survival. When we ranked all pathways in the validation dataset by their relationship to survival, as measured by the adjusted log-rank test, 17 of the 27 pathways identified as significant in the NYP dataset were in the top 10% by significance in the validation dataset.
Our validation demonstrates that both the computational approach and the key findings of our analysis are generalizable to a much larger dataset that is not restricted to genes previously implicated in cancer and composed of a different pan-cancer combination of primary tumors (see Table 4).
Table 4.
Pathways predictive of poor survival and significantly altered in the poor survival cluster in both the original and the validation datasets.
| Pathway | Log rank test (validation data) | |
|---|---|---|
| p-value | p-value adj. | |
| TP53 regulates transcription of additional cell cycle genes whose exact role in the p53 pathway remain uncertain | 1.35E-06 | 0.000142 |
| SHC1 events in EGFR signaling | 6.43E-05 | 0.003378 |
| GRB2 events in EGFR signaling | 0.000209 | 0.007326 |
| Activation of NOXA and translocation to mitochondria | 0.000852 | 0.016340 |
| EGFR Transactivation by Gastrin | 0.000896 | 0.016340 |
| PI5P Regulates TP53 Acetylation | 0.000933 | 0.016340 |
| TP53 regulates transcription of several additional cell death genes whose specific roles in p53-dependent apoptosis remain uncertain | 0.001340 | 0.020119 |
| SUMOylation of transcription factors | 0.001583 | 0.020241 |
| TP53 Regulates Transcription of Death Receptors and Ligands | 0.001734 | 0.020241 |
| Activation of PUMA and translocation to mitochondria | 0.002852 | 0.029962 |
| Regulation of TP53 Activity through Methylation | 0.003273 | 0.031153 |
| TP53 Regulates Transcription of Genes Involved in Cytochrome C Release | 0.003558 | 0.031153 |
| TP53 Regulates Transcription of Genes Involved in G2 Cell Cycle Arrest | 0.004291 | 0.032203 |
| Regulation of PTEN gene transcription | 0.004170 | 0.032203 |
4.7. Comparison to existing approaches
Prior published algorithms have been evaluated in either simulation data or full exome sequencing datasets with mutation data for 5–30 times larger sets of genes and often on cancer-specific cohorts. Thus, direct performance comparison of the proposed approach on a highly restricted gene panel data to existing algorithms is difficult. However, the proposed approach yields a comparable and often superior predictive power of identified clusters on the full exome validation dataset when compared to existing approaches within cancer-specific cohorts. A summary of existing approaches and log-rank test results for Kaplan Meier estimator, wherever available, is provided in Table 5.
Table 5.
Comparison with existing methods.
| Method | Dataset | Number of genes | Type of data | Study cohort | Log-rank test for Kaplan-Meier estimator (p-value) |
|---|---|---|---|---|---|
| This work | Primary: clinical pathology reports | Primary: 467-gene panel (588 genes including cases with whole exome and transcriptome data) | Somatic mutations, transcriptome | Pan-cancer | Primary: P = 0.0207 (pathway-specific: P = 0.0052 to 0.28) |
| Validation: TCGA, 12 cancer types | Validation: full exome | Validation: P = 1.139372e-05 | |||
| Kuijjer et al. [13] | TCGA, 23 cancer types | Full exome, 2,219 cancer-associated genes (COSMIC) | Somatic mutations | Cancer-specific and pan-cancer | Cancer specific: P = 0.0027 to 1.34e-04 No pan-cancer P-value reported |
| Hofree et al. [14] | TCGA, 3 cancer types | Full exome | Somatic mutations | Cancer-specific | Likelihood ratio test with median survival: P = 3.75 × 10–5 to 3.3 × 10–4 (varies by cancer subtype) |
| Altieri et al. [15] | Simulated data, Validation: 3 TCGA datasets (3 cancer types) | Full exome | Somatic mutations | Cancer-specific | P < =0.05 to < − 0.1 on experimental data |
| Fang, Gough [16] | TCGA, 12 cancer types | Full exome (19,171) | Somatic mutations | Pan-cancer | P-value not available; Univariate Cox Hazard Ratio, average across genes: 6.18 |
| Zhao et al. [11] | TCGA, breast cancer | 9,859 | Somatic mutations, transcripttome | Cancer-specific | P = 0.00029 to 0.087 for top 21 routes (not adjusted for multiple hypothesis testing) |
| Yang, Ge, and Zheng [12] | TCGA, 4 cancer types | Full exome | Somatic mutations, DNA methylation, expression | Cancer-specific | P = 2.46E-08 to 3.43E-04 (cancer-specific, combined with additional data) |
5. Discussion
Ongoing systematic analysis of all available clinical data is one of the tenets of the Learning Healthcare System. For diseases with a genomic component, such as cancer, this analysis should include genomic sequencing report data. Implementing truly automated decision support systems based on clinical and genomic data in support of precision medicine requires addressing challenges involving cohort sizes, data incompleteness, biases, and data inconsistency among various sources pointing to potential data inaccuracies.
Much of clinical knowledge resides in unstructured patient notes. While genomic test results represent the inherently structured output of bioinformatics pipelines, in practice the original structured data may be inaccessible, and text extraction from unstructured text format represents another challenge. Moreover, current bioinformatics knowledgebases reflect the incomplete state of knowledge in areas such as assessing the functional impact of genomic variation and mechanisms of action of genes and gene networks. Additionally, although datasets such as the one used in this analysis have the advantage of manual curation by experts, which can reduce germline variants and sequencing or other technical artifacts from contaminating downstream analyses, this curation also restricts the available dataset, reduces the statistical power, and introduces the opportunity for data bias. Finally, clinical data used for such analysis may harbor data inconsistencies and other quality issues. Despite potential data incompleteness, inconsistency and biases, we demonstrate that data contained in genomic pathology test results can be successfully applied to patient classification and pan-cancer research.
We illustrate the application of pathway analysis to clinical genomic data and show that it can reveal structure in the data that is obscured by tumor heterogeneity when looking at individual gene mutations. In future work, pathway-level cancer subtype profiles can be extracted using Principal Component Analysis and combined with clinical features such as primary site, stage, age at diagnosis, and treatment modalities to develop a patient classification model. Such classification models can be trained retrospectively on a cohort of patients within the EHR system and then applied to classify new patients.
This analysis validates known biological pathways and identifies novel ones as predictive of patient survival on a pan-cancer basis, using observational clinical records and clinical pathology report data. In future work, biological pathway information can be obtained from additional datasets such as KEGG [9] and Gene Ontology [10]. Further validation of novel pathways can lead to a better understanding of biological processes implicated in cancer development. Incorporating transcriptome data, the directionality of impact of the gene alteration to a pathway can further improve this analysis. Moreover, patient similarity based on altered biological pathways can be extended to identify an appropriate course of therapy for patients based on the similarity of their tumor mutational profile to best responders of different therapy modalities or targeted therapies.
Limitations of this work include potential data biases introduced by batch effects of test methodologies changing over the course of data collection, as well as different groups of patients and tumor types undergoing testing over time. In addition, this approach yields an ability to distinguish between patient subtypes which may reflect various structural differences among patients’ mutational profiles, not limited to survival and other clinical outcomes. These limitations are partially addressed by validation on an independent dataset and regression of obtained patient subtypes and differences among subtypes on clinical variables of interest. The intention of this work is to study pan-cancer subtypes and mechanisms linked to survival, while further work within cancer types could yield cancer type-specific subtypes and uncover pathways associated with survival or other clinical outcomes.
This analysis was undertaken on mutational data extracted from textual pathology reports. Capturing and reporting in EHR the underlying structured data from the pathology tests would avoid potential information loss and inaccuracies caused by data extraction from text and would enable easier data reuse for downstream research and application of precision healthcare.
Supplementary Material
Acknowledgments
Authors would like to thank David Fasel and Ben May for assistance in the extraction of EHR and Tumor Registry data, Dr. Adler Perotte for insightful guidance on survival analysis, and Dr. Casey Ta for review and valuable feedback.
6.Funding
This study is sponsored by National Library of Medicine Grant R01LM012895-02 (PIs: Weng and Wang) and National Human Genetics Research Institute Grant U01HG008680 (PIs: Weng, Hripcsak, Gharavi).
Footnotes
Human subjects
This study was approved by Columbia University IRB Protocol AAAP7926. IRB approval was not obtained for making the patient pathology report or clinical data publicly available.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jbi.2019.103286
References
- [1].Institute of Medicine (US), Roundtable on Evidence-Based Medicine, in: Olsen LA, Aisner D, McGinnis JM (Eds.), The Learning Healthcare System: Workshop Summary. Washington (DC): National Academies Press; (US: ), 2007. doi: 10.17226/11903. [DOI] [PubMed] [Google Scholar]
- [2].Aronson S et al. , eMERGE Network EHRI Working Group, Empowering genomic medicine by establishing critical sequencing result data flows: the eMERGE example, J. Am. Med. Informatics Assoc, 2018, 05/31/2018. doi: 10.1093/jamia/ocy051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tomczak K, Czerwińska P, Wiznerowicz M, The cancer genome atlas (TCGA): an immeasurable source of knowledge, Contemp. Oncol 19 (1A) (2015) A68–A77, 10.5114/wo.2014.47136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Clough E, Barrett T, The gene expression omnibus database, Methods Mol. Biol 2016 (1418) (2016) 93–110, 10.1007/978-1-4939-3578-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Vogelstein B, et al. , Cancer genome landscapes, Science (New York, N.Y.) 339 (6127) (2013) 1546–1558, 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Creixell P, et al. , Pathway and network analysis of cancer genomes, Nat. Methods 12 (7) (2015) 615–621, 10.1038/nmeth.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tarca AL, et al. , A novel signaling pathway impact analysis, Bioinformatics 25 (1) (2009) 75–82, 10.1093/bioinformatics/btn577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fabregat A, et al. , The reactome pathway knowledgebase, Nucleic Acids Res. 44 (D1) (2015) D481–D487, 10.1093/nar/gkv1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kanehisa M, et al. , KEGG as a reference resource for gene and protein annotation, Nucleic Acids Res. 44 (D1) (2015) D457–D462, 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gene ontology consortium, The gene ontology project in 2008, Nucleic acids research, 36(Database issue), 2007, D440–D444. doi: 10.1093/nar/gkm883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhao Y, et al. , A route-based pathway analysis framework integrating mutation information and gene expression data, Methods 124 (2017) 3, 10.1016/j.ymeth.2017.06.016. [DOI] [PubMed] [Google Scholar]
- [12].Yang C, Ge SG, Zheng CH, ndmaSNF: cancer subtype discovery based on integrative framework assisted by network diffusion model, Oncotarget 8 (51) (2017) 89021, 10.18632/oncotarget.21643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kuijjer ML, et al. , Cancer subtype identification using somatic mutation data, Br. J. Cancer 118 (11) (2018) 1492–1501, 10.1038/s41416-018-0109-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hofree M, et al. , Network-based stratification of tumor mutations, Nat. Methods 10 (11) (2013) 1108–1115, 10.1038/nmeth.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Altieri F, Hansen TV, Vandin F, NoMAS: A computational approach to find mutated subnetworks associated with survival in genome-wide cancer studies, Front Genet. 10 (2019) 265, 10.3389/fgene.2019.00265eCollection2019, 2019 Apr 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fang H, Gough J, The ‘dnet’ approach promotes emerging research on cancer patient survival, Genome Med. 6 (8) (2014) 64, 10.1186/s13073-014-0064-8 eCollection 2014, 2014 Aug 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sireci AN, et al. , Clinical genomic profiling of a diverse array of oncology specimens at a large academic cancer center: identification of targetable variants and experience with reimbursement, J. Mol. Diagn 19 (2) (2017) 277–287, 10.1016/j.jmoldx.2016.10.008. [DOI] [PubMed] [Google Scholar]
- [18].Oberg JA, et al. , Implementation of next generation sequencing into pediatric hematology-oncology practice: moving beyond actionable alterations, Genome Med. 8 (1) (2016) 133, 10.1186/s13073-016-0389-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kircher M, et al. , A general framework for estimating the relative pathogenicity of human genetic variants, Nat. Genet 46 (2014) 310–315, 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mort M, et al. , MutPred Splice: machine learning-based prediction of exonic variants that disrupt splicing, Genome Biol. 15 (1) (2014) R19, 10.1186/gb-2014-15-1-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].SNOMED CT United States Edition, Files available for download. Accessed on April 28, 2018 https://www.nlm.nih.gov/healthit/snomedct/us_edition.html.
- [22].International classification of diseases for oncology, http://codes.iarc.fr/topography. Accessed on Sept 1, 2018.
- [23].Kandoth C, et al. , Mutational landscape and significance across 12 major cancer types, Nature 502 (7471) (2013) 333–339, 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wildeman M, et al. , Improving sequence variant descriptions in mutation databases and literature using the MUTALYZER sequence variation nomenclature checker, Hum. Mutat 29 (2018) 6–13, 10.1002/humu.20654. [DOI] [PubMed] [Google Scholar]
- [25].McLaren W, et al. , The Ensembl variant effect predictor, Genome Biol. 17 (2016) 122, 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dong C, et al. , Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies, Hum. Mol. Genet 24 (8) (2015) 2125–2137, 10.1093/hmg/ddu733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jung S, Lee S, Kim S, Nam H, Identification of genomic features in the classification of loss- and gain-of-function mutation, BMC Med. Inf. Decis. Making 15 (Suppl 1) (2015) S6, 10.1186/1472-6947-15-S1-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pagel KA, et al. , When loss-of-function is loss of function: assessing mutational signatures and impact of loss-of-function genetic variants, ISSN 1367–4803, Bioinformatics 33 (14) (2017) i389, 10.1093/bioinformatics/btx272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Landrum MJ, et al. , ClinVar: public archive of interpretations of clinically relevant variants, Nucleic Acids Res. 44 (Database issue) (2016) D862–D868, 10.1002/0471142905.hg0816s89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].R Core Team R: A language and environment for statistical computing R Foundation for Statistical Computing, Vienna, Austria, 2018. URL https://www.R-project.org/. [Google Scholar]
- [31].Kassambara A, Mundt F, factoextra: Extract and Visualize the Results of Multivariate Data Analyses. R package version 1.0.5, 2017, https://CRAN.R-project.org/package=factoextra. [Google Scholar]
- [32].Bland JM, Altman DG, Survival probabilities (the Kaplan-Meier method), BMJ 317 (7172) (1998) 1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kassambara A, Kosinski M, survminer: Drawing Survival Curves using ‘ggplot2’. R package version 0.4.3, 2018, https://CRAN.R-project.org/package=survminer. [Google Scholar]
- [34].Hennig C, fpc: Flexible Procedures for Clustering. R package version 2.1–11.1, 2018, https://CRAN.R-project.org/package=fpc. [Google Scholar]
- [35].Csardi G, Nepusz T, The igraph software package for complex network research, InterJournal, Complex Syst. 1695 (2006). [Google Scholar]
- [36].Oksanen J, et al. , vegan: Community Ecology Package. R package version 2.5–4, 2019, https://CRAN.R-project.org/package=vegan. [Google Scholar]
- [37].Zuur AF, Leno EN, Smith GM, Statistics for Biology and Health - Analysing Ecological Data, Springer, New York, 2007. ISBN 978-0-387-45967-7 (Print), 978-0-387-45972-1 (Online). [Google Scholar]
- [38].Kaplan EL, Meier P, Nonparametric estimation from incomplete observations, J. Amer. Statist. Assoc 53 (282) (1958) 457–481, 10.2307/2281868. [DOI] [Google Scholar]
- [39].Flørenes VA, et al. , TP53 allele loss, mutations and expression in malignant melanoma, Br. J. Cancer 69 (2) (1994) 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Malaguarnera R, Belfiore A, The insulin receptor: a new target for cancer therapy, Front Endocrinol. (Lausanne) 2 (93) (2011), 10.3389/fendo.2011.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Albert MC, Brinkmann K, Kashkar H, Noxa and cancer therapy: Tuning up the mitochondrial death machinery in response to chemotherapy, Mol. Cell. Oncol 1 (1) (2014) e29906, 10.4161/mco.29906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hikisz P, Kiliańska ZM, PUMA, a critical mediator of cell death–one decade on from its discovery, Cell. Mol. Biol. Lett 17 (4) (2012) 646–669, 10.2478/s11658-012-0032-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].He X, et al. , Probing the roles of SUMOylation in cancer cell biology by using a selective SAE inhibitor, Nat. Chem. Biol 13 (11) (2017) 1164–1171, 10.1038/nchembio.2463. [DOI] [PubMed] [Google Scholar]
- [44].Smith JP, Nadella S, Osborne N, Gastrin and gastric cancer, Cell. Mol. Gastroenterol. Hepatol 4 (1) (2017) 75–83, 10.1016/j.jcmgh.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Moody TW, et al. , Abstract 1793: Gastrin-releasing peptide causes transactivation of the EGFR and HER2 in non-small cell lung cancer cells, in: Proceedings: AACR Annual Meeting 2018; April 14–18, 2018; Chicago, IL, 10.1158/1538-7445. [DOI] [Google Scholar]
- [46].Poli A, et al. , Phosphatidylinositol 5 phosphate (PI5P): From behind the scenes to the front (nuclear) stage, Int. J. Mol. Sci 20 (9) (2019) pii: E2080, 10.3390/ijms20092080. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.