

Abstract
Cancer metastasis is a significant challenge in colorectal cancer (CRC) therapy. SET and MYND domain-containing protein 2 (SMYD2) is highly expressed in multiple cancers but is rarely studied in CRC. This study aims to identify whether abnormal expression of SMYD2 is associated with cancer metastasis in CRC. In this study, we demonstrated that SMYD2 not only promoted cell proliferation but also increased the metastatic ability of CRC. The expression of adenomatous polyposis coli 2 (APC2), an inhibitor of the Wnt/β-catenin pathway, was suppressed by SMYD2 overexpression. Overexpression of SMYD2 activated the Wnt/β-catenin pathway and then induced the epithelial-mesenchymal transition (EMT) program in CRC. Mechanistically, low APC2 expression in CRC cells was due to SMYD2-mediated DNA methylation modification. This modification might require synergism with DNMT1. In summary, our study provides new insights into SMYD2-related transcriptional regulation patterns and indicates that SMYD2 could be a potential therapeutic target for CRC patients.
Keywords: Colorectal cancer, SMYD2, APC2, DNA methylation, Wnt/β-catenin pathway, Epithelial-mesenchymal transition
Introduction
Colorectal cancer (CRC) ranks as the third leading cause of cancer-related death worldwide [1] CRC has a high incidence rate but a relatively favorable prognosis. However, patients with advanced CRC often have poor outcomes because of cancer metastasis [2]. Researchers have verified that epithelial-mesenchymal transition (EMT) is a major factor in promoting CRC metastasis. The EMT process endows cancer cells with mesenchymal properties, which results in enhanced invasiveness in CRC [3]. Therefore, it is of considerable significance to explore the underlying mechanism of EMT in CRC metastasis.
Over the past decades, SMYD2 was found to be differentially expressed in a variety of cancers [4]. However, most studies on SMYD2 have emphasized its function in regulating cancer cell proliferation. For instance, SMYD2 overexpression in gastric cancer plays a crucial role in cell proliferation [5]. Overexpression of SMYD2 also results in cervical cancer cell growth and was associated with a poorer survival rate [6]. However, these papers also suggest that high expression of SMYD2 is associated with lymph node and distant metastasis. To date, far too little attention has been paid to the relationship between SMYD2 and cancer metastasis. Whether SMYD2 is associated with metastasis remains unknown and needs further exploration in the field of CRC.
SMYD2, which belongs to a group of SET and MYND domain-containing transcriptional regulators, functions by regulating epigenetic modification. It was first demonstrated to be an H3K36-specific methyltransferase [7]. Nevertheless, in more in-depth studies, SMYD2 was verified to methylate H3K4 and even non-histone proteins [8]. The substrates of SMYD2 are diverse, and SMYD2 itself plays an intricate role in transcriptional regulation. The regulatory mechanism of SMYD2 is still incomplete and needs further explanation.
In our study, we found that SMYD2 was markedly upregulated in CRC and correlated with poor prognosis. In vivo and in vitro experiments confirmed that SMYD2 not only promoted proliferation but also induced EMT in CRC cells. Mechanistically, we first verified that APC2 might be a novel target of SMYD2. SMYD2 might recruit DNMT1 to decrease APC2 expression via DNA modification, thus activating the Wnt/β-catenin pathway and inducing EMT progression.
Materials and methods
Cell lines and cell culture
The human CRC cell lines HCT116, SW480, DLD1, HT29, SW480, and SW620, were purchased from the Chinese Academy of Science (Shanghai, China). HCT116, SW480, FHC, and SW620 cells were cultured in DMEM supplemented with 10% fetal bovine serum, 100 μ/ml penicillin, and 100 μg/ml streptomycin, while DLD1 and HT29 cells were cultured in RPMI 1640 medium. All CRC cells were incubated in a 37°C humidified incubator with 5% CO2.
Patient specimens and immunohistochemistry (IHC)
All CRC patient specimens were obtained from the Third XiangYa Hospital of Central South University. These patients underwent radical colorectal surgery, and colorectal adenocarcinoma was confirmed by postoperative pathology. The clinicopathologic information of the patients was obtained from their hospital medical records, and informed consent was obtained from all patients. The patient studies were conducted according to the Declaration of Helsinki. The use of these specimens and data for research purposes was approved by the Ethics Committee of the Hospital. IHC assays and scoring standards for SMYD2 and APC2 staining intensity were performed as previously described [9].
RNA extraction, RT-PCR, and quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from cells and tissues with TRIzol reagent (Invitrogen, California, USA), and cDNA was generated using HiScript Q RT SuperMix for qPCR (+gDNA wiper) (Vazyme Biotech, Nanjing, China). Real-time PCR was carried out on LightCycler® 480 using SYBR Green Real-Time PCR Master Mix (Vazyme Biotech, Nanjing, China). β-actin was used as a standard internal control. All samples were analyzed in triplicate. The data were calculated with the comparative threshold cycle (Ct) method. All qRT-PCR primer sequences are available in Table S1.
Western blotting (WB) and immunofluorescence (IF) assays
WB was performed as previously described [10]. Primary antibodies against SMYD2 (21290-1-AP), DNMT1 (24206-1-AP), cyclinD1 (60186-1-Ig), β-catenin (51067-2-AP), c-MYC (10828-1-AP), E-cadherin (20874-1-AP), N-cadherin (22018-1-AP), Vimentin (10366-1-AP) and GAPDH (60004-1-Ig) were purchased from Proteintech (Wuhan, China). Anti-APC2 antibody (113370) was purchased from Abcam (Shanghai, China). For IF experiments, cells were seeded in 24-well plates overnight, fixed in 4% paraformaldehyde for 10 minutes, washed twice with phosphate-buffered saline (PBS), and then permeabilized with 0.2% Triton X-100 in PBS for 10 minutes. Fixed cells were pre-incubated with PBS containing 2% BSA for 30 minutes at room temperature. The cells were stained with primary antibody (E-cadherin and Vimentin antibody, 1:100 dilutions) for 1 hour at 37°C, followed by incubation with a secondary antibody conjugated with Alexa Fluor 488. DAPI (4, 6-diamidino-2-phenylindole) emitted blue fluorescence and was used as a nuclear indicator. The Alexa Fluor 488 signal was visualized as green. Fluorescence images were captured and analyzed by confocal microscopy.
Plasmid infection and siRNA transfection
CRC cells were transiently transfected using Lipofectamine 3000 transfection reagent (Life Technologies, Shanghai, China) according to the manufacturer’s protocol. Small interfering RNA (siRNA) and SMYD2/APC2 overexpression plasmids were purchased from GenePharma (Shanghai, China). After 48 hours of transfection, the treated cells were used for various cell assays. The transfection effect was verified by Western blotting. The siRNA sequences used were as follows: Si-SMYD2#1: 5’-GAUUUGAUUCAGAGUGACATT-3’; Si-SMYD2#2: 5’-GGUUAAGAGAUUCUUAUUUTT-3’; Si-Ctrl: 5’-UUCUCCGAACGUGUCACGUTT-3’; Si-APC2: 5’-CCUACAGGGAAAACUGGAGTT-3’; Si-DNMT1: 5’-CCCACUUCACAUUCAAGAATT-3’.
Stable cell line establishment and animal work
A SMYD2 knockdown lentivirus with luciferase was purchased from Shanghai GenePharma Company. SMYD2 knockdown stable cell lines were generated by infection with the lentivirus according to the manufacturer’s protocol and selected with 2 mg/mL puromycin for approximately two weeks. Female BALB/c nude mice (6-8 weeks old) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). All animal experiments were approved by the Animal Care Committee of Xuzhou Medical University. Mice lived in pathogen-free cages with wood shavings at constant room temperature and with a 12 hours light/dark cycle. They had enough water and standard food, which were regularly replaced. All the mice were randomly divided into two groups: sh-Ctrl and sh-SMYD2. Each group contained six mice. In our experiments, 2×106 HCT116-sh-Ctrl and HTC116-sh-SMYD2 cells were suspended in 200 μl PBS and then injected into these mice via the tail vein. From the 21st day after injection, animals were imaged weekly using the In Vivo Imaging System (IVIS) to monitor the pulmonary metastatic situation. After isoflurane induction, mice were injected with luciferin (150 mg/kg) by intraperitoneal injection and then placed onto the imaging platform. IVIS imaging settings, exposures, and images were taken in accordance with the protocol. At eight weeks after injection, mice were sacrificed, and lungs were harvested. The lungs were fixed in 4% paraformaldehyde for hematoxylin/eosin (HE) staining and immunohistochemistry (IHC). The relative number of metastatic lung nodules of lung tissues of mice was counted.
Cell proliferation assay
Cell Counting Kit-8 (CCK-8) assays were carried out according to the manufacturer’s protocol to determine the regulation of cell proliferation by SMYD2. After 48 hours of transfection, cells were seeded in 96-well plates at a density of 4000 cells per well. The absorbance at 450 nm was measured 1 hour after adding the CCK-8 solution. The experiments were repeated four times.
Cell migration and invasion assays
Scratch wound healing and Transwell migration and invasion assays were used to measure CRC cell metastasis ability. For the scratch wound healing assay, cells were seeded in six-well plates and scratched using a 200 μl pipette tip. After scratching, we gently washed the cells twice with medium to remove the detached cells. After growing for an additional 24-48 hours with serum-free medium, photographs were obtained using phase-contrast microscopy. Scratch area and wound healing percentage was evaluated quantitatively using ImageJ. Transwell assays were performed as previously described [11]. Migrated cells were stained with a 0.1% crystal violet solution and counted with ImageJ. Three independent experiments were performed.
Chromatin immunoprecipitation (ChIP) assay
4×106 sh-Ctrl and sh-SMYD2 transfected HCT116 cells were prepared for ChIP assay using the SimpleChIP® Plus Enzymatic Chromatin IP Kit (CST, Shanghai, China). According to the manufacturer’s protocol, 10 μl sample of the diluted chromatin was used as input. Immunoprecipitate (IP) products were incubated with antibodies overnight at 4°C with rotation. 10 μl anti-DNMT1, anti-H3K4me3 and anti-H3K36me2 antibodies were added to the IP sample. A normal rabbit IgG was used as negative control. The purified DNA was obtained and used for qRT-PCR. The amplification efficiency was calculated as enrichment related to the input. The primers targeting the APC2 promoter sequence were listed in Table S1.
DNA methylation analysis and 5-aza-20-deoxycytidine (5-aza-dc) treatment
DNA methylation analysis was carried out as previously described [10]. Bisulfite sequencing PCR (BSP) primers located in the APC2 promoter were generated by MethPrimer. The primers were as shown in Table S1. Bisulfite-treated DNA was amplified with these primers. PCR products were cloned using the pGEM-T Easy Vector system (Promega, Beijing, China). Three individual clones were sequenced. For 5-aza-dc Treatment, CRC cells were treated with 5 μmol/L of 5-aza-dc for 72 h.
Immunoprecipitation (IP)
Immunoprecipitation was carried out according to the manufacturer’s protocols. In brief, HCT116 lysates were incubated overnight at 4°C on a rotating platform with antibodies and protein A/G beads (Cell Signaling Technology). After incubation, the beads were washed, and the immunoprecipitants were eluted off the beads with loading buffer. After boiling for 5 minutes, the samples were analyzed by WB.
RNA sequencing (RNA-seq)
RNA-seq was performed by BGI (Wuhan, China). The samples were 6 in total and divided into two groups, one of which was the control group, and the other was SMYD2 knockdown group. DEseq2 algorithm was used to identify differentially expressed genes (DEGs) between control and SMYD2 siRNA-treated samples. In order to improve the accuracy of DEGs, we defined genes with Fold Change ≥ 1.5 and Adjusted P-value ≤ 0.05 as significantly differentially expressed genes.
Bioinformatics analysis
Gene expression in CRC were analyzed by the Gene Expression Profiling Interactive Analysis (GEPIA) online database [12]. We used the Kaplan-Meier plotter to assess the prognostic value of SMYD2 in CRC patients with GSE12945 microarray data. The genes co-expressed with SMYD2 in CRC patients were downloaded from LinkedOmics, which is a publicly available portal that includes multi-omics data from all 32 cancer types from The Cancer Genome Atlas (TCGA) [13]. DEGs were analyzed by two online analysis tools: DAVID (The Database for Annotation, Visualization and Integrated Discovery) and PANTHER (Protein ANalysis THrough Evolutionary Relationships) [14,15]. The cBioPortal for Cancer Genomics was used to explore multidimensional cancer genomics data.
Statistical analysis
All data were analyzed by SPSS version 25.0 (SPSS, Inc. Chicago, IL, USA) and GraphPad Prism 7. The values are expressed as the mean ± standard deviation (SD). The X2 test was used to evaluate the relationship between SMYD2 expression and clinicopathological characteristics. Student’s t-test and one-way ANOVA were used to determine the significance of differences. P < 0.05 was considered statistically significant.
Results
SMYD2 was upregulated in CRC tissues and positively correlated with its copy number
Recently, it has been confirmed that lysine methyltransferases are frequently deregulated in human cancers [16]. With GEPIA analysis, we found that some lysine methyltransferases were differentially expressed in CRC. SMYD2, SMYD3, and EZH2 were highly expressed in colorectal cancer, while SMYD1 and EZH1 were poorly expressed (Figures 1A, S1A). In these overexpressed molecules, previous researchers have verified the relationship between SMYD3, EZH2 and tumorigenesis, while SMYD2 has rarely been studied in CRC. SMYD2 is widely and highly expressed in different types of human cancers according to GEPIA (Figure S1B). First, we verified the mRNA levels of SMYD2 in 24 pairs of flash-frozen CRC tissues and adjacent normal tissues by real-time PCR. The SMYD2 mRNA level was higher in CRC tissues than in normal adjacent tissues (Figure 1B). Then, we examined the protein level of SMYD2 in 7 pairs of tissues by WB and found that the protein level of SMYD2 was also much higher in CRC tissues (Figure 1C) and that SMYD2 was pervasively highly expressed in CRC cell lines but expressed at relatively low levels in normal colonic epithelial cells (FHC) (Figure 1D). In addition, the expression of SMYD2 was positively correlated with its copy number according to cBioPortal. The mutation rate of SMYD2 was low in CRC compared with that in other cancer types (Figure 1E-G). The differential expression of SMYD2 may be due to copy number variations in CRC patients.
Figure 1.

SMYD2 was upregulated in CRC tissues and positively correlated with its copy number. A. Expression of lysine methyltransferases in CRC tissues (n=275) and adjacent normal tissues (n=349) from GEPIA. B, C. The relative mRNA and protein expression levels of SMYD2 were assessed by qRT-PCR and Western blotting in 24 and 7 paired CRC tissues and adjacent normal tissues, respectively. T = tumor, N = normal tissue. D. SMYD2 expression levels in CRC cell lines and FHC cells were assessed by Western blot and normalized to GAPDH. E. The mutation rate of SMYD2 in CRC was analyzed by cBioPortal. F, G. Relationships between SMYD2 mRNA expression and copy number were assessed by cBioPortal. H. Representative images of SMYD2 protein expression in paired CRC tissue and adjacent normal tissue were captured by IHC. Patients diagnosed with TNM IV had higher protein expression of SMYD2 than patients diagnosed with TNM II. I. Kaplan-Meier survival curves of overall survival and disease-free survival in patients with different mRNA levels of SMYD2 were analyzed by SurvExpress, an online bioinformatics tool. ***P < 0.001.
Clinical and pathologic features were correlated with SMYD2 in CRC patients
To determine the relationship between SMYD2 and the clinicopathologic features of colorectal cancer patients, we selected 80 fresh tissues from patients who had undergone radical colorectal surgery for IHC analysis, and those patients were confirmed to have colorectal adenocarcinoma by postoperative pathology. The IHC results showed that patients diagnosed with TNM stage IV disease had higher SMYD2 protein expression than patients diagnosed with early stage disease (TNM II) (Figure 1H). Among those tissues, 47 (58.75%) samples had high expression of SMYD2, while 33 (41.25%) tissues had relatively low expression. The correlations between clinicopathological features and SMYD2 expression in CRC patients are shown in Table 1. High SMYD2 protein expression was significantly correlated with tumor diameter (P=0.002), TNM stage (P=0.049), lymph node metastasis (P=0.026), and distant metastasis (P=0.015). In contrast, there was no prominent association of SMYD2 protein expression with age, sex, or differentiation. In addition, we used SurvExpress to perform overall survival and disease-free survival analyses for SMYD2 with the GSE12945 microarray data. Patients with higher SMYD2 expression showed significantly worse overall survival and disease-free survival (P < 0.05) (Figure 1I).
Table 1.
Relationships between SMYD2 expression and the clinicopathological characteristics of CRC patients
| Variables | Cases | SMYD2 expression | P value | χ2 | |
|---|---|---|---|---|---|
|
| |||||
| Low (n=33) | High (n=47) | ||||
| Age (years) | 0.063 | 3.449 | |||
| ≤ 4 years | 41 | 21 | 20 | ||
| >62 years | 39 | 12 | 27 | ||
| Gender | 0.474 | 0.512 | |||
| Femal | 45 | 17 | 28 | ||
| Male | 35 | 16 | 19 | ||
| Tumor size | 0.002** | 10.012 | |||
| ≤ 0.0 cm | 49 | 27 | 22 | ||
| >5.0 cm | 31 | 6 | 25 | ||
| Distant metastasis | 0.015* | 5.937 | |||
| Negative | 65 | 31 | 34 | ||
| Positive | 15 | 2 | 13 | ||
| Differentiation | 0.947 | 0.004 | |||
| Poor | 27 | 11 | 16 | ||
| Well to moderate | 53 | 22 | 31 | ||
| Lymph node metastasis | 0.026* | 4.952 | |||
| Negative | 32 | 18 | 14 | ||
| Positive | 48 | 15 | 33 | ||
| TNM stage | 0.049* | 3.869 | |||
| I-II | 38 | 20 | 18 | ||
| III-IV | 42 | 13 | 29 | ||
P < 0.05;
P < 0.01.
SMYD2 promoted EMT in CRC cells
We wondered whether abnormally expressed SMYD2 played a functional role in CRC. Some experiments were performed. Since the expression levels of SMYD2 in HCT116 and SW480 cells were high but relatively lower in DLD1 cells, we chose to knockdown SMYD2 expression in the HCT116 and SW480 cell lines and overexpress it in DLD1 cells. The effects of silencing and overexpression are shown in Figure 2A. Then, we used the treated cells for further experiments. As shown (Figures 2B-G, S2A-C), suppressed SMYD2 expression in HCT116 and SW480 cells reduced cell migration and invasion compared with those in the control group, while overexpression of SMYD2 had opposing effects in DLD1 cells. In a CCK-8 cell proliferation assay, the proliferation ability of HCT116 and SW480 cells decreased significantly after knocking down SMYD2 expression, while the proliferation ability of DLD1 cells overexpressing SMYD2 was substantially enhanced (Figure 2H). We found that SMYD2 regulated the migration and invasion of CRC cells. Therefore, we speculated that SMYD2 might be involved in the regulation of EMT progression in CRC. Then, we validated this conjecture by IF and WB. As shown (Figure 3B), SMYD2 regulated EMT-related proteins such as E-cadherin, N-cadherin and vimentin. Moreover, the E-cadherin fluorescence intensity of the HCT116 and SW480 cell lines increased with the knockdown of SMYD2, while the fluorescence intensity of vimentin decreased (Figure 3A). In vitro, we used a tail vein injection mouse model to detect the role of SMYD2 in cancer metastasis. On the 28th day after infection, we used IVIS to detect lung metastasis. The bioluminescent signals of the lungs in the SMYD2 knockdown group were lower than those in the control group (Figure 3C). Eight weeks after injection, the mice were dissected, and the lungs were sectioned for HE staining and IHC staining. SMYD2 knockdown cells formed fewer metastatic foci in the lungs compared with the control group. SMYD2 knockdown prominently reduced the number of lung metastases per mouse (Figure 3D, 3E). Furthermore, IHC staining for lung tissue sections was performed to evaluate the expression of SMYD2 and APC2. The representative images showed that knockdown of SMYD2 resulted in a weaker staining intensity of SMYD2 in metastatic lung foci but a stronger staining intensity of APC2 compared with that in the control group (Figure 3F).
Figure 2.
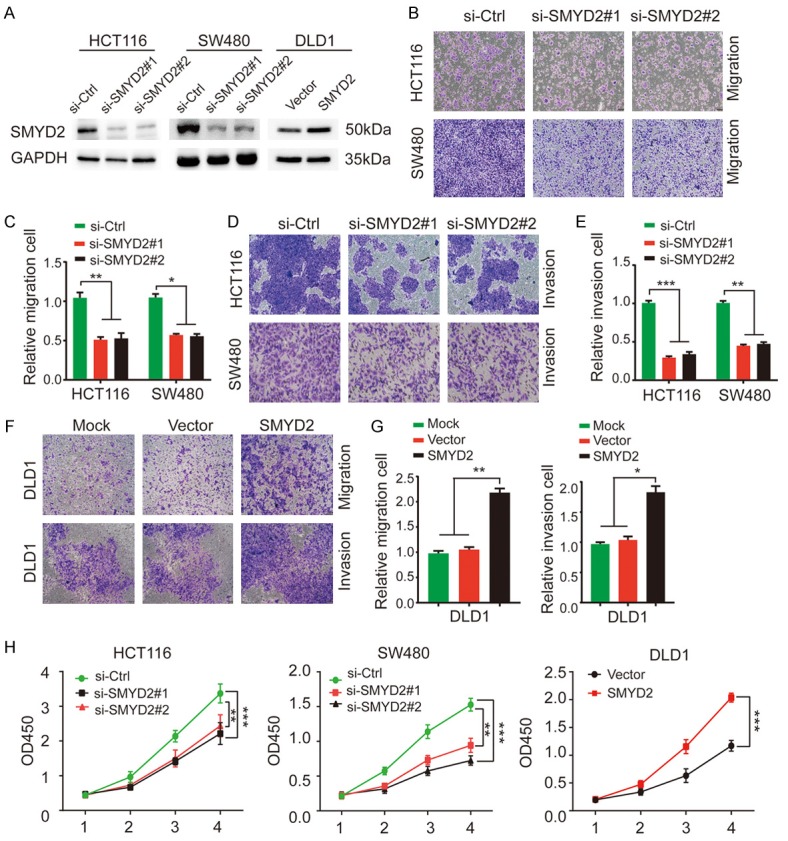
SMYD2 promoted EMT in CRC cells. A. Transfection efficacy in HCT116, SW480 and DLD1 cells was verified by Western blotting. GAPDH was used as an internal control. B-E. SMYD2 knockdown significantly suppressed the migration and invasion ability of HCT116 and SW480 cells. F, G. Overexpression of SMYD2 distinctly enhanced the metastatic ability of DLD1 cells. H. CCK8 assays were used to evaluate the proliferation ability of HCT116, SW480 and DLD1 cells compared to that of negative control cells with SMYD2 knockdown or overexpression. All experiments were performed in triplicate. The results were shown as the mean ± standard deviation. An unpaired t-test was used to analyze significant differences. n=3, *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 3.
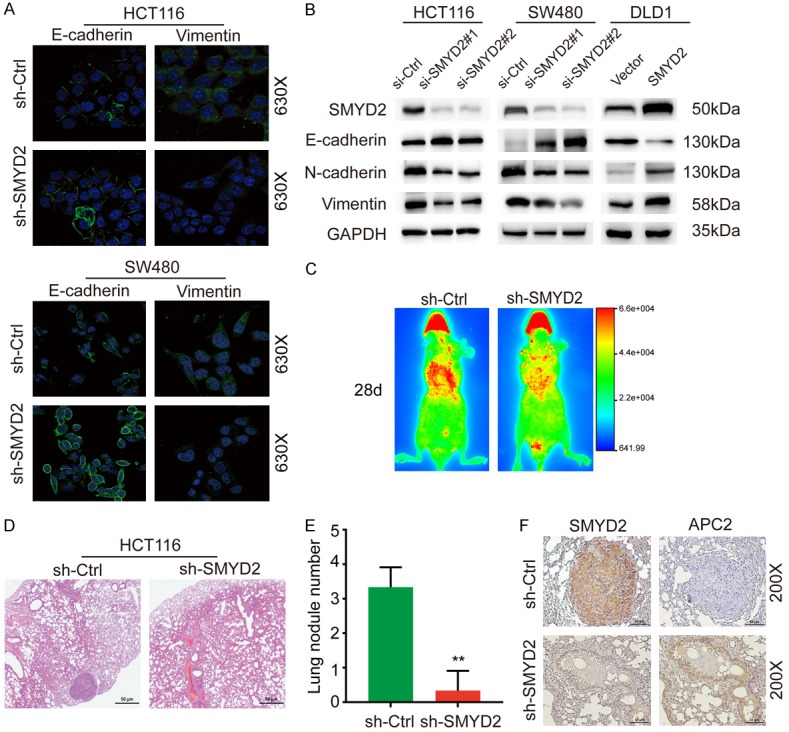
SMYD2 promoted EMT in CRC. A. Immunofluorescence of the epithelial marker E-cadherin and the mesenchymal marker Vimentin in si-SMYD2 transfected HCT116 and SW480 cells. B. Relative protein expression levels of EMT markers in CRC cells with SMYD2 knockdown or overexpression. C. Representative images acquired by IVIS after 28-day’s injection. Bioluminescent signals were detected in the pulmonary region of mouse. D. HE staining of lung sections was shown. E. Lung nodules were analyzed as the numbers of nodules per mouse. n=6, **P<0.01. F. Representative images of IHC staining for SMYD2 and APC2 were shown (×200 magnification).
APC2 was negatively correlated with the expression of SMYD2
To detect the potential targets of SMYD2, we knocked down SMYD2 in HCT116 cells for RNA-seq. Some DEGs between the control group and SMYD2-knockdown group are shown (Figure 4A). All DEGs were subjected to functional enrichment analyses via DAVID. As shown (Figure 4C). GO analysis results indicated that in the biological process (BP) category, DEGs were significantly enriched in five terms: angiogenesis, cell adhesion, cell proliferation, apoptosis, and cell migration. The cellular component (CC) and molecular function (MF) results are shown in the supplementary figure (Figure S3A, S3B). For a deeper understanding of these DEGs, a PANTHER pathway enrichment analysis was performed. These genes were differentially enriched in some cancer-related pathways (Figure 4B). For instance, the Wnt signaling pathway showed a considerable correlation. We verified that SMYD2 was involved in the metastasis and proliferation of CRC cells in previous phenotypic experiments; thus, we took an intersection between the cell proliferation and cell migration categories (Figure 4D). Five genes were enriched in these two categories: CSPG4, MATK, BCAR1, FSCN1, and APC2. We were surprised to find that APC2 was also enriched in the Wnt/β-catenin signaling pathway. Additionally, public data from the TCGA database also confirmed this conclusion. We conducted an analysis using LinkedOmics (Figure S3C). APC2 showed a negative correlation with SMYD2 expression. According to GEPIA, APC2 expression in CRC was relatively low compared with that in normal tissues (Figure 4E).
Figure 4.

APC2 was negatively correlated with the expression of SMYD2. A. The heatmap of the differentially expressed genes after SMYD2 knockdown. B. Kyoto Encyclopedia of Genes and Genomes pathway analysis was performed for screening DEGs using the PANTHER database. C. DAVID was used to identify potential biological process (BP) categories. D. Five Intersecting genes, including APC, were enriched in cell proliferation and cell migration. E. Expression levels of APC2 in CRC tissues (n=275) and adjacent normal tissues (n=349) from GEPIA.
SMYD2 activated the Wnt/β-catenin signaling pathway by suppressing APC2 expression
According to GEPIA, SMYD2 was found to be negatively correlated with APC2 but positively correlated with some Wnt-related markers, such as β-catenin, c-MYC, and cyclin D1 (Figure S4A, S4B). It has been reported that APC2 functions as an essential inhibitor of the Wnt/β-catenin signaling pathway [17]. We verified the effect of APC2 on the Wnt/β-catenin signaling pathway by WB. After silencing APC2, markers of the Wnt/β-catenin signaling pathway, including β-catenin, c-MYC and cyclin D1, were upregulated (Figure S4C). Furthermore, SMYD2 knockdown resulted in decreased protein levels of β-catenin, c-MYC, and cyclin D1 in HCT116 cells. However, APC2 was markedly upregulated with SMYD2 depletion (Figure 5A). To prove that changes in these proteins were due to altered APC2 expression, we silenced APC2 in SMYD2-depleted HCT116 cells and overexpressed APC2 in SMYD2-overexpressing DLD1 cells. The regulation of Wnt-related genes induced by SMYD2 knockdown or overexpression was restored (Figure 5B). Similarly, the expression of EMT-related proteins and the impaired migration, invasion, and proliferation abilities of CRC cells were restored (Figure 5C-F). These data demonstrated that SMYD2 activated the Wnt/β-catenin signaling pathway by repressing APC2 expression and promoting EMT in colorectal cancer.
Figure 5.

SMYD2 activated the Wnt/β-catenin signaling pathway by suppressing APC2 expression. A. WB results showed the effects of SMYD2 knockdown on the Wnt/β-catenin pathway-related markers in HCT116 cells. B. Western blot analysis revealed that the expression of Wnt-related proteins in SMYD2 knockdown HCT116 cells was significantly rescued by APC2 silenceing when compared with the corresponding controls. While in SMYD2 overexpression DLD1 cells, upregulation of these proteins was recovered with APC2 plasmid. C. Regulation of the protein levels of the EMT-related markers caused by SMYD2 knockdown was restored by suppressing APC2 in HCT116 and SW480 cells. D-F. Inhibition of metastasis and proliferation caused by SMYD2 knockdown was suppressed by decreasing APC2 in HCT116 and SW480 cells. n=3, *P < 0.05, **P < 0.01, ***P < 0.001.
SMYD2 suppressed APC2 by controlling DNA methylation of its promoter
Previous studies suggested that SMYD2 specifically induced H3K4me3 and H3K36me2 modifications and predominantly led to the activation of gene expression [8]. We conducted a ChIP assay to verify whether SMYD2 depletion could change the H3K4me3 or H3K36me2 methylation level in the APC2 promoter region. As shown in Figure S5A, SMYD2 expression did not alter histone methylation levels in the APC2 promoter region. When analyzed by MethHC, APC2 methylation levels were higher in CRC tissues than in normal tissues (Figure S5B). We assumed that the suppression of APC2 might be derived from DNA methylation of its promoter. To verify this hypothesis, we examined the methylation status of the APC2 promoter in CRC cell lines by BSP. CpG islands were largely methylated in CRC cells (Figure 6A, 6B). After treating CRC cells with 5-aza-dc for 72 h, APC2 expression was significantly increased (Figure S5C). Moreover, we found that SMYD2 knockdown increased the expression of APC2 by altering the hypermethylation status in HCT116 and SW480 cells. Methylation-specific PCR (MSP) analysis showed that the unmethylated bands increased and the methylated bands decreased after SMYD2 knockdown in both HCT116 and SW480 cells (Figure 6C). However, there have been no previous reports that SMYD2 can directly regulate DNA methylation. DNA methylation maintenance is regulated mainly by DNMT1, a maintenance DNA methyltransferase [18]. SMYD2 might require synergism with DNMT1. We found a positive correlation between SMYD2 and DNMT1 expression in CRC via GEPIA (Figure S5D). To assess whether the DNA occupation of DNMT1 in the promoter of APC2 was directly affected by SMYD2, a ChIP assay was performed in CRC cells. As expected, SMYD2 knockdown led to a decrease in DNA occupation of DNMT1 in the APC2 promoter region. Conversely, SMYD2 overexpression led to an increase in the DNA occupation of DNMT1 (Figure 6D). Then, we conducted an IP experiment, which indicated that SMYD2 and DNMT1 could bind with each other (Figure 6E). After silencing DNMT1, the inhibition of APC2 caused by SMYD2 overexpression was reduced (Figure 6F). These results led us to believe that SMYD2 repressed APC2 expression by recruiting DNMT1 in CRC cells.
Figure 6.

SMYD2 suppressed APC2 by controlling DNA methylation of its promoter. A. CpG islands were predicted using MethPrimer and the percentage of CpG islands in the APC2 promoter region was shown. The regions analyzed by BSP were indicated. The sequence of the APC2 promoter region analyzed by BSP was shown. The CpG dinucleotides within this region were numbered as 1-21. B. The methylation status of the APC2 promoter region in HCT116, SW620, SW480, DLD1, HT29 and FHC cell lines. The white and black circles indicated unmethylated and methylated CpGs, respectively. C. The methylation status of the APC2 promoter was examined by BSP in HCT116 and SW480 cells after the transfection of si-Ctrl or si-SMYD2. M: methylated, U: unmethylated. D. ChIP-qPCR analysis of DNMT1 enrichment upstream of APC2 in sh-Ctrl or sh-SMYD2 transfected HCT116 cells and Vector or SMYD2 transfected DLD1 cells (n=3, *P < 0.05). E. SMYD2 combined with DNMT1 according to IP experiments. F. The inhibition of APC2 caused by SMYD2 overexpression reversed by DNMT1 knockdown in HCT116 cells.
Discussion
Lysine methyltransferases have emerged as attractive regulators in transcriptional regulation, epigenetics, and tumorigenesis [19,20]. The SMYD family is a cluster of protein lysine methyltransferases with SET and MYND domains [4]. This family contains five proteins, SMYD1, SMYD2, SMYD3, SMYD4, and SMYD5, and each member has different distributions and functions [16]. For instance, SMYD1 functions as an epigenetic regulator of cardiomyocyte differentiation and myogenesis [16]. SMYD3 promotes hepatocellular carcinoma metastasis through H3K4me3 modification [21]. According to the analysis of the TCGA database, SMYD2 is broadly and highly expressed in several cancers. A strong relationship between SMYD2 and cancer proliferation has been reported in the literature. SMYD2 promotes cell proliferation in triple-negative breast cancer (TNBC) [11]. It is also reported to be highly expressed in pediatric acute lymphoblastic leukemia and related to poor prognosis [22]. Our results are in line with those of previous studies. SMYD2 was found to be overexpressed in CRC tissues and cells at both the protein and mRNA levels. Interestingly, the high expression of SMYD2 was positively correlated with tumor diameter and was a risk indicator of cancer metastasis. In vivo and in vitro experiments confirmed that SMYD2 could induce EMT progression and promote cancer cell metastasis in CRC. These results provide further support for the hypothesis that there might be a new target of SMYD2 that is involved in regulating CRC metastasis.
According to RNA-seq analysis and bioinformatics analysis, APC2 was the most potential molecule to regulate metastasis and proliferation in CRC. APC2 is identified as a homolog of adenomatous polyposis coli (APC) and has comparable functions in cancer [17]. APC2 regulates the formation of a destruction complex, which results in the activation of the Wnt/β-catenin pathway [23]. Abnormal activation of the Wnt/β-catenin pathway is widely present in cancers and plays a vital role in promoting EMT [24]. The Wnt/β-catenin signaling can stabilize SNAI2 and initiate EMT programs in breast cancer cells [25]. FOXP3 facilitates the Wnt/β-catenin signaling pathway, inducing EMT and metastasis in NSCLC [26]. Our study revealed that SMYD2 expression suppressed APC2 expression, and the downregulation of APC2 was responsible for SMYD2-mediated cell metastasis. Reduced APC2 expression resulted in the activation of the Wnt/β-catenin signaling pathway and then promoted EMT in CRC.
A novel discovery of this study was that SMYD2 suppressed APC2 expression via DNA methylation, and this modification required the participation of DNMT1. SMYD2, as a kind of protein lysine methyltransferase, is mainly involved in regulating histone methylation modification. It was identified as a histone methyltransferase that can deposit methyl groups on histones H3K4 and H3K36, two epigenetic marks of active transcription [27]. However, some reports confirmed that SMYD2 dimethylates H3K36 and represses transcription via interaction with the Sin3A histone deacetylase complex [28]. In addition, it was also reported that SMYD2 only methylates H3K4 in the presence of HSP90 [8]. The exact mechanism by which SMYD2 regulates histone methylation remains unclear, and so far, there is no consensus on this issue. According to our results, APC2 is a new target of SMYD2. At first, we assumed that SMYD2 regulated APC2 expression through histone methylation modification. But SMYD2 knockdown did not affect H3K4me3 and H3K36me2 levels in the APC2 promoter area according to our results. And there was also no conclusive evidence that APC2 was a new nonhistone methylation substrate for SMYD2. With our further research on APC2, aberrantly low expression of APC2 was identified in CRC, but the methylation level of the APC2 promoter region was significantly higher than that in normal tissues. This result suggested that DNA methylation might be responsible for the low expression of APC2 in CRC. After treatment with the DNA methylation inhibitor 5-aza-dc, APC2 expression was upregulated in CRC cells. Methylation PCR confirmed that knockdown of SMYD2 could significantly affect the methylation level of the APC2 promoter region. However, there was no obvious evidence that SMYD2 could directly regulate DNA methylation. DNA methylation is a pivotal epigenetic modification that participates in regulating gene expression. Previous studies have shown that some lysine methyltransferases can promote DNA methylation by binding to DNMT1, which is an active DNA methyltransferase (DNMT) responsible for the maintenance of DNA methylation [29]. EZH2 recruits DNMT1, leading to further modifications such as DNA methylation [30]. DNMT1 and G9a can form a functional complex that enhances DNA methylation [31]. Therefore, we speculated that SMYD2 may work by binding to DNMT1. We performed a ChIP assay in SMYD2 knockdown or overexpressing CRC cells. Our results confirmed that SMYD2 overexpression or knockdown affected the DNA occupation of DNMT1 at the APC2 promoter area. Then, we verified that SMYD2 could bind to DNMT1 by IP experiments, and after inhibiting DNMT1, the downregulation of APC2 by SMYD2 was repaired. These findings indicated that SMYD2 interacts with DNMT1 and that this functional complex methylated CpG islands in the APC2 promoter, thereby suppressing APC2 expression.
In summary, we found that SMYD2 not only promoted the proliferation of CRC cells but also regulated the EMT process and increased the metastasis ability of CRC. Additionally, we demonstrated that SMYD2 might play a role in promoting CRC metastasis by suppressing APC2 and thus activating the Wnt/β-catenin pathway. The downregulation of APC2 may be due to the synergistic effects of SMYD2 and DNMT1. This combination results in the hypermethylation of APC2, thus inhibiting the expression of APC2 in CRC. Our study provides a new insight for the regulation pattern of SMYD2 and implies that SMYD2 could be a potential therapeutic target in CRC.
Acknowledgements
This research was supported in part by the National Natural Science Foundation of China (No. 81702435, No. 81702336), the China Postdoctoral Science Foundation (2018M630606) and the Natural Science Foundation of Jiangsu (BK20170264).
Disclosure of conflict of interest
None.
Abbreviations
- SMYD2
SET and MYND Domain Containing 2
- APC2
Adenomatosis Polyposis Coli 2
- TCGA
The Cancer Genome Atlas
- IHC
immunohistochemistry
- GO
gene ontology
- NSCLC
non-small cell lung cancer
- BRCA
Breast invasive carcinoma
- HCC
hepatocellular carcinoma
- UCS
Uterine Carcinosarcoma
- UVM
Uveal Melanoma
- LUSC
Lung squamous cell carcinoma
- OV
Ovarian serous cystadenocarcinoma
- STES
Stomach and Esophageal carcinoma
- BLCA
Bladder Urothelial Carcinoma
- PRAD
Prostate adenocarcinoma
- COAD
Colon adenocarcinoma
Supporting Information
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Miller KD, Nogueira L, Mariotto AB, Rowland JH, Yabroff KR, Alfano CM, Jemal A, Kramer JL, Siegel RL. Cancer treatment and survivorship statistics, 2019. CA Cancer J Clin. 2019;69:363–385. doi: 10.3322/caac.21565. [DOI] [PubMed] [Google Scholar]
- 3.Vu T, Datta PK. Regulation of EMT in colorectal cancer: a culprit in metastasis. Cancers (Basel) 2017;9 doi: 10.3390/cancers9120171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spellmon N, Holcomb J, Trescott L, Sirinupong N, Yang Z. Structure and function of SET and MYND domain-containing proteins. Int J Mol Sci. 2015;16:1406–1428. doi: 10.3390/ijms16011406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Komatsu S, Ichikawa D, Hirajima S, Nagata H, Nishimura Y, Kawaguchi T, Miyamae M, Okajima W, Ohashi T, Konishi H, Shiozaki A, Fujiwara H, Okamoto K, Tsuda H, Imoto I, Inazawa J, Otsuji E. Overexpression of SMYD2 contributes to malignant outcome in gastric cancer. Br J Cancer. 2015;112:357–364. doi: 10.1038/bjc.2014.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun JJ, Li HL, Ma H, Shi Y, Yin LR, Guo SJ. SMYD2 promotes cervical cancer growth by stimulating cell proliferation. Cell Biosci. 2019;9:75. doi: 10.1186/s13578-019-0340-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh PK. Histone methyl transferases: a class of epigenetic opportunities to counter uncontrolled cell proliferation. Eur J Med Chem. 2019;166:351–368. doi: 10.1016/j.ejmech.2019.01.069. [DOI] [PubMed] [Google Scholar]
- 8.Abu-Farha M, Lambert JP, Al-Madhoun AS, Elisma F, Skerjanc IS, Figeys D. The tale of two domains: proteomics and genomics analysis of SMYD2, a new histone methyltransferase. Mol Cell Proteomics. 2008;7:560–572. doi: 10.1074/mcp.M700271-MCP200. [DOI] [PubMed] [Google Scholar]
- 9.Hou PF, Jiang T, Chen F, Shi PC, Li HQ, Bai J, Song J. KIF4A facilitates cell proliferation via induction of p21-mediated cell cycle progression and promotes metastasis in colorectal cancer. Cell Death Dis. 2018;9:477. doi: 10.1038/s41419-018-0550-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Tang B, Song J, Yu S, Li Y, Su H, He S. Lnc-PDZD7 contributes to stemness properties and chemosensitivity in hepatocellular carcinoma through EZH2-mediated ATOH8 transcriptional repression. J Exp Clin Cancer Res. 2019;38:92. doi: 10.1186/s13046-019-1106-2. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Li LX, Zhou JX, Calvet JP, Godwin AK, Jensen RA, Li X. Lysine methyltransferase SMYD2 promotes triple negative breast cancer progression. Cell Death Dis. 2018;9:326. doi: 10.1038/s41419-018-0347-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98–W102. doi: 10.1093/nar/gkx247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vasaikar SV, Straub P, Wang J, Zhang B. LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 2018;46:D956–D963. doi: 10.1093/nar/gkx1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 15.Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Du SJ, Tan X, Zhang J. SMYD proteins: key regulators in skeletal and cardiac muscle development and function. Anat Rec (Hoboken) 2014;297:1650–1662. doi: 10.1002/ar.22972. [DOI] [PubMed] [Google Scholar]
- 17.Van Es J, Kirkpatrick C, Van de Wetering M, Molenaar M, Miles A, Kuipers J, Destree O, Peifer M, Clevers H. Identification of APC2, a homologue of the adenomatous polyposis coli tumour suppressor. Curr Biol. 1999;9:105–108. doi: 10.1016/s0960-9822(99)80024-4. [DOI] [PubMed] [Google Scholar]
- 18.Ishiyama S, Nishiyama A, Saeki Y, Moritsugu K, Morimoto D, Yamaguchi L, Arai N, Matsumura R, Kawakami T, Mishima Y, Hojo H, Shimamura S, Ishikawa F, Tajima S, Tanaka K, Ariyoshi M, Shirakawa M, Ikeguchi M, Kidera A, Suetake I, Arita K, Nakanishi M. Structure of the Dnmt1 reader module complexed with a unique two-mono-ubiquitin mark on histone h3 reveals the basis for dna methylation maintenance. Mol Cell. 2017;68:350–360. e357. doi: 10.1016/j.molcel.2017.09.037. [DOI] [PubMed] [Google Scholar]
- 19.Chang CJ, Hung MC. The role of EZH2 in tumour progression. Br J Cancer. 2012;106:243–247. doi: 10.1038/bjc.2011.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yi X, Jiang XJ, Fang ZM. Histone methyltransferase SMYD2: ubiquitous regulator of disease. Clin Epigenetics. 2019;11:112. doi: 10.1186/s13148-019-0711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Xie BH, Lin WH, Huang YH, Ni JY, Hu J, Cui W, Zhou J, Shen L, Xu LF. Amplification of SMYD3 promotes tumorigenicity and intrahepatic metastasis of hepatocellular carcinoma via upregulation of CDK2 and MMP2. Oncogene. 2019;38:4948–4961. doi: 10.1038/s41388-019-0766-x. [DOI] [PubMed] [Google Scholar]
- 22.Zipin-Roitman A, Aqaqe N, Yassin M, Biechonski S, Amar M, van Delft MF, Gan OI, McDermott SP, Buzina A, Ketela T, Shlush L, Xie S, Voisin V, Moffat J, Minden MD, Dick JE, Milyavsky M. SMYD2 lysine methyltransferase regulates leukemia cell growth and regeneration after genotoxic stress. Oncotarget. 2017;8:16712–16727. doi: 10.18632/oncotarget.15147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jarrett CR, Blancato J, Cao T, Bressette D, Cepeda M, Young PE, King CR, Byers SW. Human APC2 localization and allelic imbalance. Cancer Res. 2001;61:7978–7984. [PubMed] [Google Scholar]
- 24.Sebio A, Kahn M, Lenz HJ. The potential of targeting Wnt/β-catenin in colon cancer. Expert Opin Ther Targets. 2014;18:611–615. doi: 10.1517/14728222.2014.906580. [DOI] [PubMed] [Google Scholar]
- 25.Wu ZQ, Li XY, Hu CY, Ford M, Kleer CG, Weiss SJ. Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic breast cancer 1, early onset (BRCA1) repression. Proc Natl Acad Sci U S A. 2012;109:16654–16659. doi: 10.1073/pnas.1205822109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang S, Liu Y, Li MY, Ng CSH, Yang SL, Wang S, Zou C, Dong Y, Du J, Long X, Liu LZ, Wan IYP, Mok T, Underwood MJ, Chen GG. FOXP3 promotes tumor growth and metastasis by activating Wnt/β-catenin signaling pathway and EMT in non-small cell lung cancer. Mol Cancer. 2017;16:124. doi: 10.1186/s12943-017-0700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang X, Tanaka K, Yan J, Li J, Peng D, Jiang Y, Yang Z, Barton MC, Wen H, Shi X. Regulation of estrogen receptor α by histone methyltransferase SMYD2-mediated protein methylation. Proc Natl Acad Sci U S A. 2013;110:17284–17289. doi: 10.1073/pnas.1307959110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keogh MC, Kurdistani SK, Morris SA, Ahn SH, Podolny V, Collins SR, Schuldiner M, Chin K, Punna T, Thompson NJ. Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell. 2005;123:593–605. doi: 10.1016/j.cell.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 29.Liao J, Karnik R, Gu H, Ziller MJ, Clement K, Tsankov AM, Akopian V, Gifford CA, Donaghey J, Galonska C, Pop R, Reyon D, Tsai SQ, Mallard W, Joung JK, Rinn JL, Gnirke A, Meissner A. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat Genet. 2015;47:469–478. doi: 10.1038/ng.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu X, Gong Y, Yue J, Qiang B, Yuan J, Peng X. Cooperation between EZH2, NSPc1-mediated histone H2A ubiquitination and Dnmt1 in HOX gene silencing. Nucleic Acids Res. 2008;36:3590–3599. doi: 10.1093/nar/gkn243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fritsch L, Robin P, Mathieu JR, Souidi M, Hinaux H, Rougeulle C, Harel-Bellan A, Ameyar-Zazoua M, Ait-Si-Ali S. A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Mol Cell. 2010;37:46–56. doi: 10.1016/j.molcel.2009.12.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.