

Abstract
Epithelial ovarian cancer is characterized by universal TP53 mutations, which result in G1/S checkpoint deficiencies. Therefore, it is hypothesized that the abrogation of the G2/M checkpoint with Wee1 inhibitor might preferentially sensitize TP53-defective ovarian cancer cells. Given the extremely high molecular diversity in ovarian cancer, one approach to improving the clinical efficacy is to identify drug combinations that either broaden the applicable spectrum or circumvent resistance. Here, through a high-throughput unbiased proteomic profiling (RPPA), we found the complementary activated mTOR pathway contributes greatly to Wee1 inhibitor resistance. A combination of Wee1 and mTOR inhibits synergistically inhibiting tumor growth in ovarian cancer cell lines and patient-derived xenograft that closely mimic the heterogeneity of patient tumors. Mechanistically, dual Wee1/mTOR inhibition induced massive DNA replication stress, leading to fork stalling and DNA damage. Moreover, we found that the addition of nucleotide metabolic substrate dNTPs alleviated replication stress, restored the cell cycle and reduced apoptosis to some extent, supporting dNTPs depletion is necessary for the synergy between Wee1 and mTOR inhibits. These results suggest that our study opening up a wider therapeutic window of Wee1 inhibitor for the treatment in epithelial ovarian cancers.
Keywords: Wee1 inhibitor, mTOR inhibitor, epithelial ovarian cancer, patient-derived xenograft, replication stress
Introduction
Globally, there are 239,000 new cases and 152,000 deaths annually, making ovarian cancer the most common cause of gynecologic cancer death [1]. Cytoreductive surgery and combination platinum-taxane chemotherapy have remained the mainstay of therapy for decades. Most patients with epithelial ovarian cancers (EOC) have relapsed at some point despite response to initial cytoreductive surgery and platinum-based chemotherapy, and ultimately develop platinum resistance [2]. Despite tremendous research commitment and engagement, 5-year overall survival in ovarian cancer has improved only slightly since 1995 [2]. Recently, PARP inhibitors (PARPi) are recommended as maintenance therapy in Platinum-sensitive recurrent (PSR) ovarian cancer [1], and the latest clinical trials indicate that PARP inhibitors are expected to be used in patients with newly diagnosed advanced ovarian cancer [3]. However, like most other targeted therapies, responses to PARPi are too frequently transient and are limited in patients with a germline BRCA1/2 mutation. More importantly, the efficacy of PARPi in ovarian cancer who is refractory or resistant to first-line platinum-based therapy is still largely debating, the treatment options for recurrence, platinum-resistant epithelial ovarian cancer are exceedingly limited.
Wee1 kinase is a vital regulator of the G2/M transition in the cell cycle [4]. Wee1 checkpoint via phosphorylation of CDK1 (also known as Cdc2) at Tyr15, inhibits CDK1/cyclin B kinase activity [5,6]. AZD1775 (also known as MK1775) was the first-in-class, potent, and selective inhibitor of small molecule Wee1 inhibitor (Wee1i) [7], which is underling investigation in several clinical trials for the treatment in diverse solid tumors [8-12]. High-grade serous ovarian carcinoma (HGSOC) is characterized by universal TP53 mutations [1], which result in G1/S checkpoint deficiencies, leading to the G2/M checkpoint uniquely critical for the survival of such tumor cells. So, it is hypothesized that the abrogation of the G2/M checkpoint with AZD1775 might preferentially sensitize TP53-defective ovarian cancer cells. However, although several studies have shown that Wee1 inhibitors augment the effects of chemotherapy for the treatment of ovarian cancers [9,10], overall response rates are still limited, and resistance eventually occurred. Given the extremely high molecular diversity in ovarian cancer, one approach to improving the clinical efficacy is to identify drug combinations that either broaden the applicable spectrum or circumvent resistance.
Here, through a high-throughput unbiased proteomic profiling (RPPA), we found the complementary activated mTOR pathway contributes greatly to Wee1 inhibitor resistance. Furthermore, our results support combined Wee1/mTOR inhibition induced massive DNA replication stress, leading to fork stalling and DNA damage. Moreover, this synergy is independent of TP53 mutation, and it shows remarked efficacy in multiple cancer cells from various lineage. So, our study opens up a wider therapeutic window of Wee1 inhibitor for the treatment in epithelial ovarian cancers.
Materials and methods
Cell culture
Human ovarian cancer cell lines (HOC7, OVCAR8) were obtained from MDACC characterized Cell line Core, Human ovarian cancer cell lines (A2780, ES2, SKOV3, OVCAR3, Caov3, OV90, TOV-112D, TOV-21G) were obtained from the American Type Culture Collection (ATCC). ID8 is a mouse ovarian cancer derived from C57BL/6, was a gift by Professor K. Roby (Department of Anatomy and Cell Biology, University of Kansas, U.S.A). CT26 is a mouse colon carcinoma, derived from BALB/c mice; MC38 is a mouse colon adenocarcinoma, derived from C57BL/6. B16 is a mouse melanoma, derived from C57BL/6. B16, MC38, and CT26 cells were obtained from the American Type Culture Collection (ATCC).
HOC7, OVCAR8, A2780, OVCAR3 were cultured in RPMI-1640 media supplemented with 10% Fetal Bovine Serum. ES2, SKOV3 cells were cultured in McCoy 5A medium containing 10% fetal bovine. OV90, TOV-112D and TOV-21G were cultured on a medium that used MCDB 105 (sodium bicarbonate containing 1.5 g/L) and M199 (Sodium bicarbonate containing 2.2 g/L) mixed at 1:1, which containing 15% fetal bovine serum. Caov3 cells were cultured in DMEM medium containing 15% fetal bovine serum. ID8, MC38, CT26, B16 were cultured in DMEM medium containing 10% fetal bovine serum. All cell cultures plus with penicillin-streptomycin and cultivated at 37°C and 5% CO2.
Chemical compounds
Chemical compounds (AZD1775, AZD2014). were purchased from Selleck Nucleotide precursor supply was performed by adding a mix of the four dNTPs (20 μM each) for 48 h. Deoxycytidine (Sigma D0776) was solubilized in 1 M NaOH (100 mM). Deoxyadenosine (Sigma D8668) was solubilized in 0.1 M NaOH (20 mM). Thymidine (Sigma T1895) was solubilized in H2O (50 mM). Deoxyguanosine (Sigma D7145) was solubilized in 1 M NH4OH (100 mM).
RPPA
Ovarian cancer cell lines OAW42 were treated for 48 hr, respectively, with DMSO or WEE1i (AZD1775). Protein lysates were analyzed by RPPA in MDACC CCSG (The Cancer Center Support Grant) supported RPPA Core. Antibodies and approaches are described at the RPPA website (https://www.mdanderson.org/research/research-resources/core-facilities/functional-proteomics-rppa-core.html). Heat map represents “rank-ordered” changes induced by WEE1i treatment, calculated by summing median-centered protein amount normalized to DMSO.
Generation of Wee1 inhibitor-resistant clones
To generate Wee1 inhibitor-resistant clones, ID8 cells were subjected to gradual increases in AZD1775 concentrations until cells grew in the presence of 5 μM of the drug (3 months from initial exposure). Cells were cultured in the absence of AZD1775 for a minimum of 1 month before they were used for experiments.
Colony formation assay
Cells were seeded into six-well plates (5,000 per well) and allowed to adhere overnight. Cells were then cultured in the absence or presence of drugs as indicated for 8 days. Remaining cells were fixed with Polyformaldehyde (4%), stained with 0.5% crystal violet, photographed using a digital scanner after drying.
Cell proliferation
Cell proliferation was estimated using the cell counting kit 8 (CCK8, Dojindo Laboratories, Japan) according to manufacturer instructions. The half-maximal inhibitory concentration value was calculated, as the mean drug concentration required to inhibit cell proliferation by 50% compared with vehicle-treated controls. The extent and direction of AZD1775 and AZD2014 anti-proliferation interactions were determined by combination index values that were calculated using CompuSyn software (download from http://www.combosyn.com/). All experiments were performed in triplicate.
Annexin V-FICT/PI apoptosis assay by flow cytometry
Cells were seeded in 6 cm dishes at 50% confluence. After 24 hours, the medium was refreshed and cells were treated with DMSO, Wee1 inhibitor AZD1775, mTOR inhibitor AZD2014, or combinations for 48 hours. After treatment, cells were harvested by trypsinization and washed with ice-cold PBS. We used FITC Annexin V Apoptosis Detection Kit I (Catalog No.556547 BD Biosciences) to stain cells for Annexin V and propidium iodide (PI). After 20 minutes of incubation in the dark, samples were analyzed on a Flow Cytometer (Beckman Coulter). We used FlowJo-V10 software to quantify populations. Apoptosis was calculated as the fractional difference of Annexin V-FITC positive and Annexin V-FITC/PI double-positive population between treated and untreated samples. At least 100,000 events were assessed per measurement.
Western blot analysis
Total cellular proteins were extracted by solubilizing the cells in RIPA buffer, sonicated for 30 seconds. Extracts were clarified by centrifugation and amounts of proteins were normalized with the Coomassie brilliant blue staining. Samples which added loading buffer were boiled for 15 min and subjected to western blotting. Signals were visualized using enhanced chemiluminescence (Bio-Rad). The following antibodies were used for western blotting: Phospho-Akt1 Ser473 (AP0098, ABclonal), Pan-AKT Polyclonal Antibody (A3145, ABclonal), phospho-mTOR-S2448 p-Ab (Ap0094, ABclonal), mTOR Polyclonal Antibody (A2445, ABclonal), phospho-RPS6 (Ser235/36) (AP0538, ABclonal), phospho-RPS6 (Ser240/244) (AP0537, ABclonal), Phospho-cdc2 (Tyr15) (AP0016, ABclonal), Anti-γH2AX (phosphor S140) (ab22551, Abcam), Anti-Tubulin antibody (AC008, ABclonal). HRP Goat Anti-Rabbit IgG (H+L) (AS014, ABclonal). HRP Goat Anti-Mouse IgG (H+L) (AS003, ABclonal).
Immunofluorescence staining
Cells were seeded into 20 mm glass-bottom cell culture dishes at 50% confluence. After 24 hours and treated with AZD1775 and AZD2014 for 72 hours. Cells were fixed with 4% polyoxymethylene, permeabilized with 0.25% Triton X-100, and blocked with 5% goat serum in phosphate-buffered saline (PBS) and then stained overnight at 4°C with Anti-γH2AX (phosphor S140) (ab22551, Abcam). Cells were washed with PBS and stained with secondary antibody (Alexa Fluor 488 goat anti-mouse immunoglobulin G; Invitrogen) for 1 hour. The cells were stained with 4’,6-diamidino-2-phenylindole (DAPI). At last, dishes were observed and photographed. The machine: NIKON Eclipse Ti. DAPI (EX361-389, BA430-490); FITC (EX488, BA500-550). Software: Eclipse C2, Nikon, Tokyo, Japan; Pictures analysis with Image-Pro Plus 6.0.
Flow cytometry
To measure the S-phase population, cells were treated with compounds for 48 hours, then incubated cells with 10 μM EdU. After 2 hours, cells were harvested by trypsinization and washed with ice-cold PBS. Cells were fixed in Polyformaldehyde (3.7%) for 15 minutes and then permeabilized with 0.5% Triton X-100 in PBS for 20 min in room temperature, blocked using 3% BSA in PBS. Use the Click-iT® reaction cocktail within 15 minutes of preparation as the Click-iT® EdU Imaging Kits (C1034, Invitrogen) described. Subsequently, cells were incubated with specific antibodies against γH2AX (Ser139) (1:200) for 2 hours at room temperature. Next, cells were washed three times with PBS and incubated with secondary antibody conjugated to AlexaFluor 488 for 1 hour. Finally, cells were stained with 20 mg/ml propidium iodide (PI) which contained 100 mg/ml RNase A. All samples were detected on a Flow Cytometer (Beckman Coulter). At least 10,000 events were assessed per test. FlowJo-V10 software was used to quantify populations.
DNA fiber assay
Cells were treated with indicated drugs for 24 hours before fiber analysis [13]. First, cells were labeled with 25 mM CIdU (25 μM, last concentration) for 30 min, washed twice with equilibrated medium, and labeled with 250 μM IdU for 30 min. Cells were collected by trypsinization and resuspended in ice-cold PBS at 5 * 105 cells/ml. Spreading buffer (200 mM Tri-HCl pH 7.4, 50 mM EDTA, 0.5% SDS) was prepared. 2 μl cell suspension and 7 spreading buffer were pipetted onto a microscope slide. DNA was allowed to run down the slide slowly, air-dried and fixed in methanol/acetic acid (3:1) for 10 min. Slides were washed with H2O, allowed to dry, and then denatured in 2N HCl for 30 min. Slides were washed with PBS and incubated in blocking solution (PBS containing 5% BSA) for 30 minutes and incubated with Rat anti-BrdU [clone BU1/75 (ICR1), Abcam] antibody (1:300 dilution) and Mouse anti-BrdU antibody [clone B44, BD Biosciences] (1:50 dilution) 3 hours at room temperature. Slides were washed with PBS. After rinsing, the slides were incubated in the anti-Rat AlexaFluor 488 antibody (1:150 dilution) and anti-Mouse AlexaFluor 568 antibody (1:150 dilution) for 1 hr at room temperature. After washing, the slides were mounted in Vectashield and analyzed using a ZEISS LSM 880.
Immunohistochemical staining (IHC)
Tissues were fixed in 4% Polyformaldehyde overnight and embedded in paraffin. Embedded sections were first deparaffinized in xylene. Endogenous peroxidase was blocked by incubation with 0.3% hydrogen peroxide for 15 min. Antigen retrieval was performed by boiling the slides in citrate buffer (10 mM, pH 6.0) in a water bath for 20 min. Slides were rinsed in PBS Tween 0.05% and blocked for 30 min with 5% goat serum albumin (BSA). Slides were incubated overnight at 4°C with primary antibodies (Ki67, p-S6 (S235/236), pRPA32 (S4/8) 1:200, from Cell Signaling Technology; γ-H2AX, 1:200 from Abcam), followed by 1 hour with Labelled Polymer-HRP at room temperature. Negative controls were treated identically but without the primary antibody. Subsequently, slides were incubated with DAB+ Chromogen and then counterstained with hematoxylin. After mounting, slides were observed under a microscope and photographed. The IHC score was using a semi-quantitative five-category grading system, which performed as previously described [14,15].
Human xenograft models
6-week-old female BALB/c-nu mice were obtained from Beijing Vital River Laboratory Animal Technology Co. Ltd. Patient-derived xenograft (PDX), which was established the patient of ovarian cancer. All mice were housed under sterile conditions at the Laboratory Animal Care Center of Tongji Hospital. All animal experiments with these models were conducted in compliance with the National Institute of Health guidelines for animal research. Minced fresh tumor tissue (8-27 mm3 per mouse) was transplanted subcutaneously into flanks of mice approximately 8-10 weeks of age. For treatment, mice were randomized into groups (n=8-10 per group) with similar mean tumor volumes of 100 to 150 mm3. Treatment began on Day 26 after implantation for tumors. AZD1775 was dissolved in 2% DMSO+30% PEG300+5% Tween 80+ddH2O, and administered by oral gavage once a day at 60 mg/kg. AZD2014 was dissolved in 5% DMSO+30% PEG300+ddH2O, and administered by oral gavage once a day at 20 mg/kg. Mice were examined every 2-3 days, and tumor length and width were measured using calipers. Tumor volume was calculated using the following formula: (length * width2)/2. Mice with ovarian cancer tumors received one of the following treatments: 1) vehicle; 2) AZD1775 60 mg/day/Kg, 5 consecutive days per week. 3) AZD2014 (20 mg/kg 5 consecutive days per week), 2 consecutive days per week; or 4) combination of AZD1775 and AZD2014 5 consecutive days per week. At sacrifice, portions of tumors were stored in liquid nitrogen or were fixed in 4% Polyformaldehyde for routine histopathologic processing.
Statistical analysis
Data were recorded as the means ± standard deviation (SD). The differences between the groups were undertaken using the Student two-tailed t-test. P < 0.05 was considered statistically significant. One-way ANOVA was used to compare differences among multiple groups. The statistical analysis and Fig. generation was performed using GraphPad Prism 8 software.
Results
mTOR pathway is complementary activated after Wee1 inhibition in ovarian cancer cells
Wee1 inhibitor has emerged as a promising target for ovarian cancer treatment. Preliminary clinical trial results suggested that AZD1775 has encouraging antitumor activity in patients with TP53-mutated ovarian cancer, but resistance is almost inevasible [10]. Adaptive response to targeted therapies is one of the key mechanisms of drug resistance, blocking adaptive responses to targeted therapies represents an attractive way leading to tumor cell death and improving patient outcomes [16,17]. To explore adaptive response after Wee1 inhibition which would attribute to Wee1i resistance, we applied reverse-phase protein array (RPPA)-based proteomic profiling to assess signaling pathway perturbations after AZD1775 in OAW42, an ovarian cancer cell. As expected, the differential analysis revealed that among all 300 proteins measured, Wee1i markedly decreased its downstream target p-Cdc2-Y15 and increased γH2AX. Notably, the p-mTOR_pS2448, and its downstream target p-70-S6K, and p-S6_pS240_S244 were three of the top increased proteins after AZD1775 (Figure 1A). To confirm the direct activation of the mTOR pathway after Wee1i, exponentially proliferating HOC7 and A2780 cells were treated with AZD1775 for 48 hours, 72 hours, and 5 days. Western blot showed a time-dependent elevation of p-mTOR, and it’s downstream of p-S6 (Ser235/236) and p-S6 (Ser240/244) (Figure 1B and 1C).
Figure 1.

mTOR pathway is complementary activated after Wee1 inhibition in ovarian cancer cells. A. Heatmap of RPPA data representing “rank-ordered” changes of OAW42 induced by AZD1775. Proteins with consistent decreases (Blue) are on the above and increases (red) are on the bottom of the heatmap. Statistically significant changes (Z scores) indicated boxes. B. Western blot of indicated proteins in HOC7 cells treated with AZD1775 300 nM for the indicated length. C. Western blot of indicated proteins in A2780 cells treated with AZD1775 300 nM for the indicated length. D. Dose-response curves for cell viability effects of ID8 and ID8R (ID8 cells resistance to AZD1775) cells treated with graded concentrations of the AZD1775 for 72 hr. IC50 values for inhibition of viability. E. Western blot of indicated proteins in ID8, ID8R cells.
The emergence of acquired resistance limits the efficacy of small molecular inhibitor targeted therapies. To explore molecular mechanism mediated acquired resistant, we established an acquired resistance cells to AZD1775 (ID8-R) by culturing the parental highly Wee1i-sensitive murine ovarian cancer cells (ID8) in the continued presence of AZD1775 for 3 to 4 months. Compared to parental cells, ID8-R cells were highly resistant to AZD1775 with more than 10 folds increased 50% inhibitory concentration (IC50) (Figure 1D). Notably, p-mTOR, p-S6 (Ser235/236), and p-S6 (Ser240/244) were all significantly up-regulated compared to the parental ID8 cells, indicating the mTOR pathway was also highly activated in the acquired Wee1i resistant cells (Figure 1E). AKT was decreased probably because of the feedback suppression (Figure 1E).
Overall, the mTOR signaling was complementary activated not only in adaptive responses after short-term Wee1i treatment but also in acquired resistant cells after long-term treatment.
mTOR and Wee1 inhibition demonstrates synergy in multiple cancer cell lines
The complemental mTOR/S6 activation after Wee1 inhibition raised the possibility that mTOR inhibition (mTORi) would sensitize cells to Wee1i by blocking adaptive responses. So, we evaluated the ability of dual inhibition of Wee1 (AZD1775) and mTOR (AZD2014, a novel ATP-competitive dual inhibitor of both mTORC1 and mTORC2 kinase that is currently in phase II clinical trials [18]) in a panel of ovarian cancer cells. In 8 of 10 (except TOV-21G and OVCAR3) ovarian cancer cell lines, the Wee1i/mTORi combination was synergistic, with a combination index (CI) of less than 0.8 (Figure 2A). Similar results were observed in ID8 murine ovarian cancer cells (Figure 2B). Furthermore, three mouse-derived cancer cell lines (MC38 is a colon cancer cell-derived from C57BL/6J, B16 is a melanoma cell-derived from C57BL/6J, CT26 is a colon cancer cell-derived from BALB/C) exhibited marked sensitivity to combination of AZD1775 and AZD2014 (Figure 2B), indicating the synergy is not ovarian cancer lineage-specific. To further explore genomic features that would responsible for synergy of the Wee1i and mTORi combination. We analyzed the mutation information in human ovarian cancer cell lines. Interestingly, we found the synergistic activity of the combination was independent of ARID1A, ATM, ATR, BRCA1/2, PIK3CA, PTEN, and TP53 status, consistent with its generalizability (Figure 2C).
Figure 2.
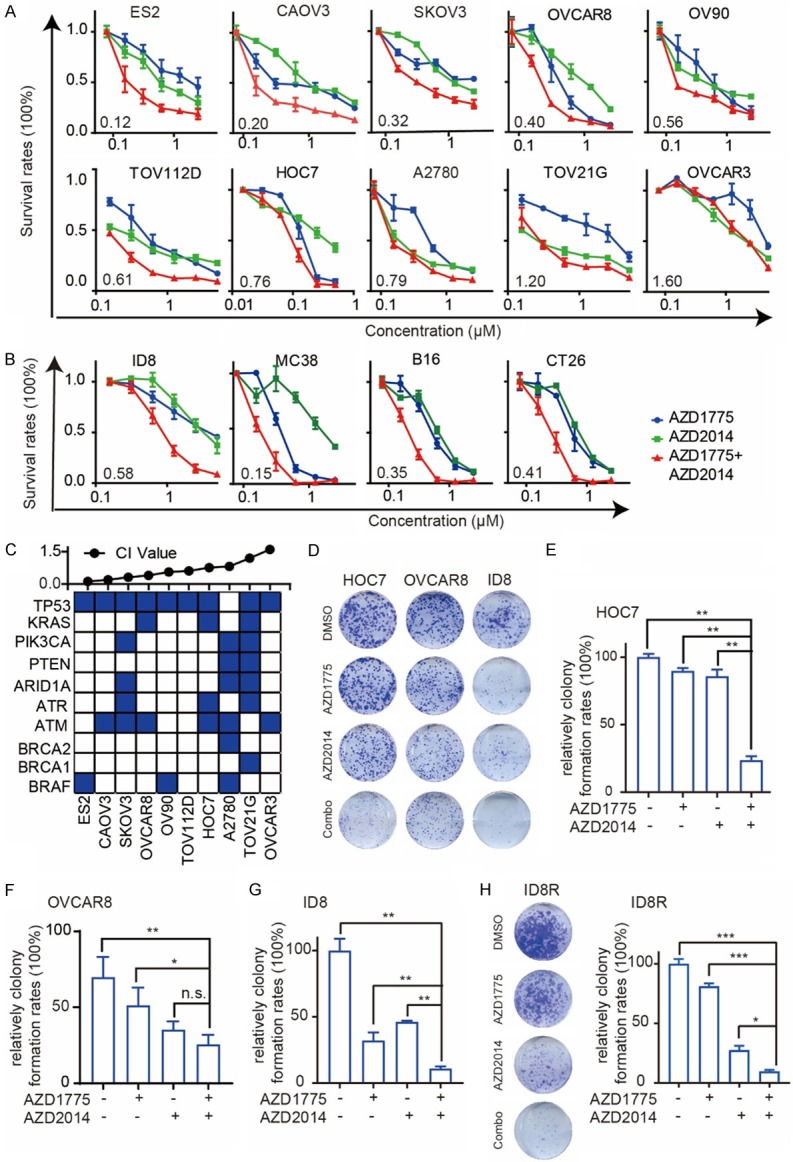
mTOR and Wee1 inhibition demonstrates synergy in multiple cancer cell lines. A. Dose-response curves of AZD1775 or AZD2014 alone or combined in 10 cancer cell lines treated with graded concentrations of the AZD1775 and AZD2014 for 72 hr. The combination index (CI) was calculated using CalcuSyn software with the Chou-Talalay equation. B. Dose-response curves of AZD1775 or AZD2014 alone or combined in 4 mouse-derived cancer cell lines treated with graded concentrations of the AZD1775 and AZD2014 for 72 hr. The combination index (CI) was calculated using CalcuSyn software with the Chou-Talalay equation. C. Top: CI values; cells were arranged by CI values based on A. Bottom: Selected mutations in cell lines (from CCLE). D. Representative pictures of clonogenic assay in HOC7 (treated with AZD1775 50 nM or AZD2014 50 nM alone or combined), OVCAR8 (treated with AZD1775 50 nM or AZD2014 50 nM alone or combined) and ID8 cells (treated with AZD1775 100 nM or AZD2014 100 nM alone or combined) for 8 days. E. Relative colony formation rates of HOC7 cells are presented as percent relative to Vehicle. Data across studies represent mean ± SD of three independent experiments, Student’s t-test: **P < 0.01. F. Relative colony formation rates of OVCAR8 cells are presented as percent relative to Vehicle. Data across studies represent mean ± SD of three independent experiments, Student’s t-test: n.s as nonsense, *P < 0.05, **P < 0.01. G. Relative colony formation rates of ID8 cells are presented as percent relative to Vehicle. Data across studies represent mean ± SD of three independent experiments, Student’s t-test: **P < 0.01. H. Representative pictures of the clonogenic assay in ID8R cells treated with AZD1775 600 nM or AZD2014 200 nM alone or combined for 8 days (left). Relative colony formation rates of ID8R are presented as percent relative to Vehicle (right). Data across studies represent mean ± SD of three independent experiments, Student’s t-test: *P < 0.05, ***P < 0.001.
Colony formation assay showed significantly reduced colony formation after long-term dual inhibition, confirmed the synergistic activity of the combination of Wee1i and mTORi in HOC7, OVCAR8 and ID8 cells (Figure 2D-G). Notably, mTOR inhibitors also reverse the acquired resistance of AZD1775 in ID8R cells, supporting its critical role in overcoming acquired resistance (Figures 2H and S1A).
To further confirm the off-target effects of mTOR inhibition on sensitizing Wee1i, we tested the effect of combination AZD1775 with another mTORC1 inhibitor (AZD8055) [19]. As expected, the combination of AZD1775 with AZD8055 decreased cell viability compared with either drug alone in ID8 and ID8R cell lines (Figure S1B). PI3K/AKT pathway remained unchanged after Wee1 inhibition. So, PI3K inhibitor (AZD8186), or AKT inhibitor (AZD5363) failed to sensitized cells to Wee1 inhibitor AZD1775 in ID8 and ID8R cells (Figure S1C and S1D).
Altogether, these results further support that the mTOR pathway plays a critical role in driving acquired AZD1775 resistance in multiple cancer cells and mTORi sensitized cells to AZD1775 even in cells with acquired resistance.
mTOR and Wee1 inhibition synergistically induces apoptosis and DNA damage in ovarian cancer cells
Consistent with cell viability assays, Wee1i/mTORi combination treatment significantly elevated DNA damage detected by immunofluorescence (IF) analysis of γH2AX foci (a DNA damage marker), as compared with single-drug treatment or vehicle groups in HOC7 cells (Figure 3A and 3B). Furthermore, Western blotting showed that combination of AZD1775 and AZD2014 led to DNA damage accumulation indicated by γH2AX in ID8 and OVCAR8 cells after 72 hours treatments, while treatment with AZD2014 alone was sufficient to abrogate the phosphorylation of S6_S235/236 and S6_S240/244 which are downstream of mTOR, and treatment with AZD1775 decreased CDC2 at tyrosine 15, respectively (Figure 3C and 3D). AZD1775 did not affect the inhibition of p-S6_S235/236 and p-S6_S240/244 by AZD2014 and the addition of AZD2014 did not affect the inhibition of CDC2 by AZD1775 (Figure 3C and 3D). Consistent with the massively induced DNA damage, the dual inhibition of Wee1 and mTOR led to a pronounced increase in cell death via apoptosis detected by Annexin V-FICT/PI apoptosis assay in HOC7, OVCAR8 and A2780 cells, when compared to either agent alone, confirming the observed synergistic anti-tumor effects (Figure 3E-G).
Figure 3.
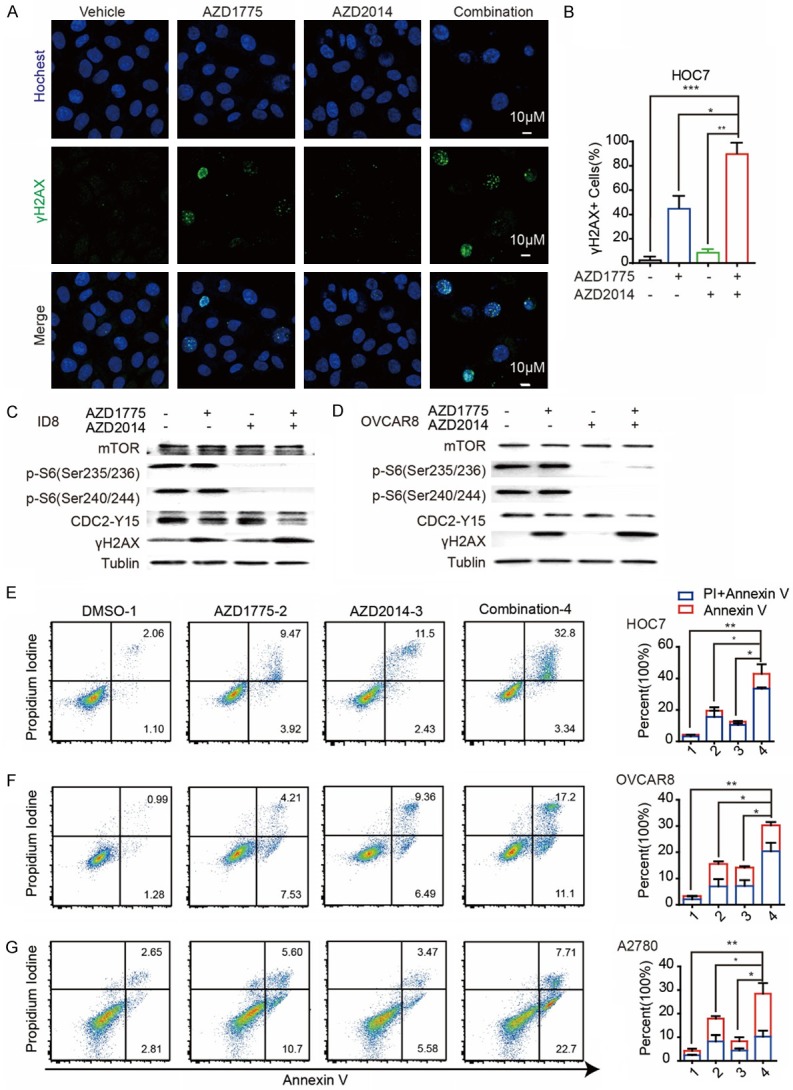
mTOR and Wee1 inhibition synergistically induces apoptosis and DNA damage in ovarian cancer cells. (A) Representative images of positive cells of Hochest and γH2AX foci in HOC7 cells treated with DMSO, AZD1775 300 nM, AZD2014 300 nM and Combination for 72 hr. Scale bar: 10 μM. (B) Quantification of positive cells of γH2AX in HOC7 cells in (A). (C) Data across studies represent mean ± SD of three independent experiments, Student’s t-test: *P < 0.05, **P < 0.01, ***P < 0.001. (C) Western blotting of indicated proteins in ID8 cells treated with single AZD1775 (800 nM), single AZD2014 (800 nM), or combination for 72 hr. (D) Western blotting of indicated proteins in OVCAR8 cells treated with single AZD1775 (500 nM), single AZD2014 (500 nM), or combination for 72 hr. (E) Flow cytometry results for propidium iodide and annexin V staining in HOC7 cells after exposure to DMSO, single AZD1775 300 nM, AZD2014 300 nM or combination for 48 hr (left). Quantification of propidium iodide and annexin V positive cells in HOC7 cells (right). Data across studies represent mean ± SD of three independent experiments, Student’s t-test: *P < 0.05, **P < 0.01. (F) Flow cytometry results for propidium iodide and annexin V staining in OVCAR8 cells after exposure to DMSO, single AZD1775 400 nM, AZD2014 400 nM or combination for 48 hr (left). Quantification of propidium iodide and annexin V positive cells in OVCAR8 cells (right). Data across studies represent mean ± SD of three independent experiments, Student’s t-test: *P < 0.05, **P < 0.01. (G) Flow cytometry results for propidium iodide and annexin V staining in A2780 cells after exposure to DMSO, single AZD1775 500 nM, AZD2014 500 nM or combination for 48 hr (left). Quantification of propidium iodide and annexin V positive cells in OVCAR8 cells (right). Data across studies represent mean ± SD of three independent experiments, Student’s t-test: *P < 0.05, **P < 0.01.
Collectively, these results show that mTOR and Wee1 inhibition interacts synergistically to decrease viability, increase DNA damage and augment apoptosis in ovarian cancer cells.
Dual inhibition of Wee1 and mTOR induces S phase replication stress and triggers DNA damage
To further explore the mechanism underlying, cell-cycle profiles were examined after treatment with AZD1775, AZD2014, and combination of AZD1775 and AZD2014. Consistent with its role as a key G2/M phase checkpoint, AZD1775 (400 nM for 48 hours) resulted in increased G2-M cell population in OVCAR8 (Figure 4A and 4B), and HOC7 cells (Figure S2A and S2B). However, combined with mTORi didn’t further an increased the G2-M cell population (Figures 4A, 4B, S2A, S2B). Next, we applied EDU (5-ethynyl-2’-deoxyuridine), Propidium Iodine (PI) and γH2AX co-labeling to sub-divided cells into G0/G1 phase, early, middle, late S-phase, and G2/M phase. After treatment with single AZD1775, AZD2014, and combination for 48 hours, OVCAR8 and HOC7 cells were labeled with EDU, PI, and γH2AX (see methods). Interestingly, although total S phase population didn’t change (Figures 4A, 4B, S2A, S2B), AZD1775 remarkably increased non-replicating S phase cells (exhibiting a DNA content between 2N and 4N, but not incorporating the synthetic nucleoside EDU) in both OVCAR cells (Figure 4C) and HOC7 cells (Figure S2C). More importantly, the non-replicating S phase cells were further augmented by combination with mTORi, indicating massive DNA replication stress occurring (Figures 4C and S2C).
Figure 4.
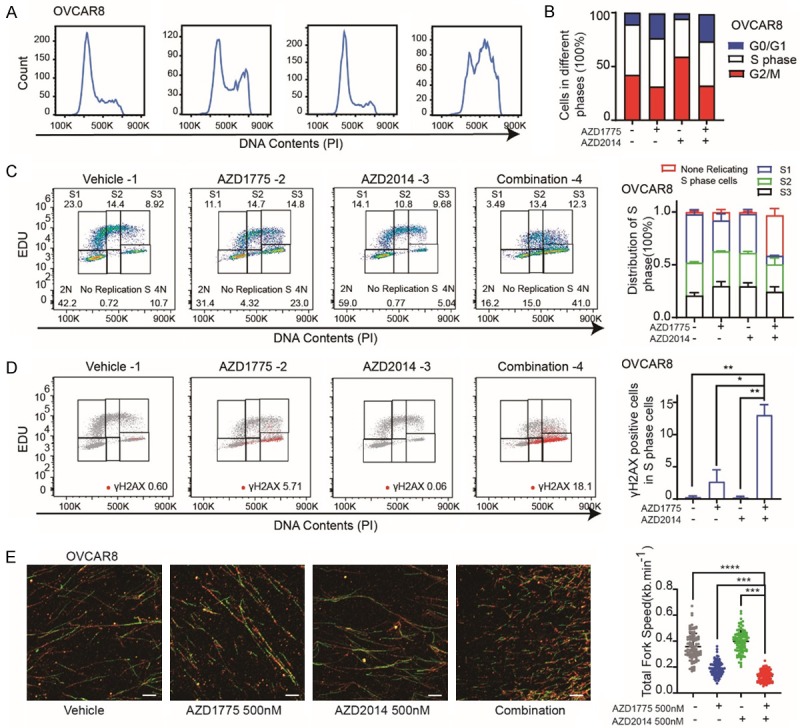
Dual inhibition of Wee1 and mTOR induces S phase replication stress and triggers DNA damage. A. Representative OVCAR8 cell lines were treated with DMSO, AZD1775 400 nM, AZD2014 400 nM or Combination for 48 hr and analyzed for cell-cycle progression. B. The distribution of cell cycle of OVCAR8 cell lines was treated with DMSO, single AZD1775 400 nM, single AZD2014 400 nM or Combination for 48 hr. C. EDU positive cells FACS analysis of the cell cycle distribution of OVCAR8 cells after exposure to DMSO, single AZD1775 (400 nM), single AZD2014 (400 nM) or Combination for 48 hr (left). Quantification and distribution of S1, S2, S3 and no replicated S phase cells in the OVCAR8 cells (right). D. γH2AX positive cells FACS analysis of the cell cycle distribution of OVCAR8 cells after exposure to DMSO, single AZD1775 (400 nM), single AZD2014 (400 nM) or Combination for 48 hr (left). Quantification of γH2AX positive cells in the S phase of OVCAR8 cells (right). Student’s t-test: *P < 0.05, **P < 0.01. E. OVCAR8 cells were treated with DMSO, single AZD1775 (400 nM), single AZD2014 (400 nM) or Combination for 24 hr subjected to DNA fiber analysis (left), scale bars, 10 μM. Mean fork speed (kb/min) is indicated (right), quantification of 100 fibers in each group, Student’s t-test: ***P < 0.001, ****P < 0.0001.
Unresolved replication stress is assumed to trigger DNA damage [20,21]. Indeed, AZD1775 selectively induced phosphorylation of histone H2AX (S139) in G2/M phase cells, and non-replication S phase cells, but not EDU positive replicating S phase cells (Figure 4D). Remarkedly, the γH2AX positive cells in the non-replication S phase were further potentiated in combination groups (Figure 4D). Similar results were observed in the HOC7 cell as well (Figure S2D).
Next, we applied DNA fiber assay, which is a powerful technique for directly qualitative and quantitative analysis of DNA replication programming [13], to quantify the DNA replication stress after treatment. As expected, AZD1775 markedly reduced DNA replication fork speed, which is much slower after a combination of mTORi (Figure 4E).
Together, these data suggest that the dual inhibition of Wee1 and mTOR not only leads to G2-M arrest but also interference of S phase and the increase of non-replicating S phase cells, reflecting the increase of replication stress, leading the increase of DNA damage.
dNTP depletion partially mediates potentiation of Wee1 inhibition by AZD2014
In proliferating cells transiting through S phase, ribonucleotides (NTPs) are converted to deoxyribonucleotides (dNTPs) by ribonucleotide reductase (RNR) to allow DNA replication. Nucleotide depletion is a major reason for replication stress [22,23]. Pfister et al. show that Wee1 inhibition degraded ribonucleotide reductase M2 (RRM2), a critical subunit of ribonucleotide reductase (RNR), enhanced DNA replication stress, increased DNA damage and apoptosis [24]. And, mTORC1 plays an important role in driving nucleotide synthesis and nucleotides pool, while mTORC1 inhibition decreased nucleotides pool, led to replication stress and apoptosis [23,25].
Based on the aforementioned results, we speculated that the mechanism of synergistic killing of cancer cells by dual inhibiting Wee1 and mTOR at least partially (if not totally) rely on nucleotides pool depletion. To verify this hypothesis, HOC7 cells were treated with single AZD1775, AZD2014, and combination for 48 hours with or without exogenous dNTPs mix added in the culture medium. Obviously, ectopic dNTPs supply significantly decreased Wee1i and combination therapy-induced apoptosis and cell death in multiple cancer cells (Figures 5A, 5B, S3A, S3B). Next, OVCAR8 cells were treated with single AZD1775, AZD2014, and combination for 48 hours with or without exogenous dNTPs mix added in the culture medium. Notably, exogenous dNTPs were sufficient to decrease no replicating S phase cell population induced by Wee1i monotherapy and combination with mTORi (Figure 5C and 5D). Subsequently, adding of dNTPs, at least partially, decrease γH2AX after AZD1775 monotherapy and combination therapy with mTORi (Figure 5E and 5F).
Figure 5.

dNTP depletion partially mediates potentiation of Wee1 inhibition by AZD2014. (A) Flow cytometry results for Propidium Iodide and Annexin V staining in the HOC7 cell lines after exposure to DMSO, single AZD1775 (300 nM), AZD2014 (300 nM) or the combination with or without dNTPs mix (30 μM) for 48 hr. (B) Quantification of HOC7 cell lines in (A). p-value from t-test. Error bars represent SD obtained from three independent experiments. n.s as nonsense, *P < 0.05. (C) EDU positive cells FACS analysis of the cell cycle distribution of OVCAR8 cells after exposure to DMSO, AZD1775 (400 nM), AZD2014 (400 nM) or Combination with or without dNTPs mix (30 μM) for 48 hr. (D) Quantification and distribution of S1, S2, S3 and no replicated S phase cells in the OVCAR8 cells treated as (C). (E) γH2AX positive cells FACS analysis of the cell cycle distribution of OVCAR8 cells after exposure to DMSO, AZD1775 (400 nM), AZD2014 (400 nM) or combination with or without dNTPs mix (30 μM) for 48 hr. (F) Quantification of γH2AX positive cells in the S phase of OVCAR8 cells treated as (E). Student’s t-test: n.s as nonsense, *P < 0.05.
These data suggest that dNTPs depletion contributes to the synthetic lethality of Wee1 inhibition and combination therapy with mTORi. Adding of dNTPs partially reverse S phase replication stress, reduce DNA damage and save the cells from synergistic killing by Wee1i and mTORi.
Wee1 and mTOR inhibition significant delay ovarian cancer growth in PDX models
Given the biological implications of our in vitro data, we next tested the anti-tumor activity of AZD1775 and AZD2014 in vivo. Because PDX closely mimics the molecular characteristics and heterogeneity of the original patient tumors [26-28], we established a PDX model using the tumor tissue of the patient. Daily oral administration of single or combined AZD2014 (20 mg/kg/day) and AZD1775 (60 mg/kg/day) treatment at clinically relevant doses [15,29] led to significant delay in tumor growth which was superior to either single agent alone (Figure 6A). At the time of sacrifice, the volume of tumors treated with the combination was significantly less than the single agent alone and vehicle-treated group (Figure 6B).
Figure 6.

Wee1 and mTOR inhibition significant delay ovarian cancer growth in PDX models. A. Tumor volume curves tumor burden changes of patient-derived xenografts. Mice treated with vehicle (5% DMSO+30% PEG300+5% Tween 80+ddH2O), AZD1775 (60 mg/kg, oral gavage, per day), AZD2014 (20 mg/kg, oral gavage, per day), or a combination of AZD1775 and AZD2014. Data across studies represent mean ± SD of three independent experiments, p-value from one-way ANOVA: ****P < 0.0001. B. Photographs of tumors in each group of patient-derived xenografts after sacrifice. C. Representative histologic sections of xenografts from tumors of PDX were immunohistochemical staining with p-S6 (Ser235/236) (left). Percent of p-S6 (Ser235/236) positive cells (right). Student’s t-test: *P < 0.05, **P < 0.01. D. Representative histologic sections of xenografts from tumors of PDX were immunohistochemical staining with p-RPA32 (S4/S8) (left). Percent of p-RPA32 (S4/S8) positive cells (right). Student’s t-test: *P < 0.05, ***P < 0.001. E. Representative histologic sections of xenografts from tumors of PDX were immunohistochemical staining with γH2AX (S139) (left). Percent of γH2AX (S139) positive cells (right). Student’s t-test: n.s as nonsense, **P < 0.01. F. Representative histologic sections of xenografts from tumors of PDX were immunohistochemical staining with Ki67 (left). Percent of Ki67 positive cells (right). Student’s t-test: *P < 0.05, ***P < 0.001.
Consistent with the mTOR activation after Wee1 inhibition in vitro, we confirmed the activation of mTORC1 signaling in vivo after AZD1775 monotherapy by assessing the p-S6 (Ser235/236) in tumors with IHC. And, AZD2014 treatment led to dephosphorylation of the pS6-S235/6 signal even combined with AZD1775, an indication of effective mTORC1 signaling inhibition (Figure 6C). Using pRPA32 (S4/8) as a DNA replication stress marker, we found AZD2014 significantly enhanced Wee1i induced DNA replication stress (Figure 6D). Moreover, tumors were also stained with γH2AX to evaluate the DNA damages induced by treatment. As expected, AZD1775 and combination therapy with AZD2014 induced DNA damage in vivo (Figure 6E). To evaluate the ability of combined therapy to suppress proliferation, Ki67 positive cells were quantified in PDX tumors after combined treatment and compared to vehicle controls. Obviously, we detected significantly decreased Ki67 positive tumor cells in AZD1775 or AZD2014 monotherapy, and the combination-treated tumors compared to vehicle-treated tumors (Figure 6F).
Discussion
With the approval of PARP inhibitors in the treatment of ovarian cancer, targeted therapy provides a new treatment strategy for ovarian cancer which is a fatal disease. But PARP inhibitors are not effective for all patients and disease cannot be cured. Exploiting synthetic lethal interactions to target epithelial ovarian cancer is warranted. Here, we demonstrated that Wee1 inhibition drove activation of the mTOR pathway and combination of Wee1i and mTORi synergistically killing cancer cells and inhibiting tumor growth in ovarian cancer cell lines and patient-derived xenograft that closely mimics the heterogeneity of patient tumors. Further, we provide mechanistic evidence that simultaneous inhibition of Wee1 and mTOR signaling induced massive DNA replication stress, leading to fork stalling and DNA damage. Moreover, this synergy is independent of TP53 mutation, and it shows remarked efficacy in multiple cancer cells from various lineage. The synergy observed with combining AZD1775 with AZD2014 in vitro and in vivo has the potential for rapid translation into the clinical setting.
Initial preclinical studies on AZD1775 focused on its induction of DNA damage by abrogating the G2/M checkpoint and drive cells into mitosis with unrepaired DNA lesions, resulting in cell death especially in cells with G1/S checkpoint defection, such as TP53 mutant cells [30]. However, its role in replication programming in S phase is less known. Our study revealed that Wee1 inhibitor AZD1775 generated replication stress, induced a significant disturbance in the S-phase cells. Consistent with our observations, more recent studies also reveal that the mechanism of AZD1775 cytotoxicity is primarily through DNA damage rather than premature entry into mitosis [9,10,12]. More importantly, we observed the synergistic effects of dual therapy are not only showed in cells containing mutant TP53, but also in TP53 wild-type cells (A2780, ID8, MC38, B16, and CT26 cells), suggesting that the cytotoxic synergy is independent of p53 status.
In mammals, ribonucleotide reductase (RNR) is a unique enzyme that catalyzes the rate-limiting step of de novo synthesis of deoxyribonucleoside triphosphates (dNTPs). RNR consists of two homodimer subunits: the large catalytic dimer RRM1 and the small regulatory dimer RRM2. Recent insights into the mTORC1 demonstrated that mTORC1 increases nucleotide synthesis [23,31], drives tumor anabolic metabolism through ribosome biogenesis and nucleotide synthesis [23]. And, mTORC1 inhibition with rapamycin or mTOR kinase with AZD8055 decrease RRM1 and RRM2 both in vitro and in mouse tumor xenografts [32,33]. Also, recent studies have revealed a novel role of Wee1 in the maintenance of nucleotide (dNTP) pools [34,35]. Wee1 inhibition increased dNTP demand and DNA replication stress through CDK-induced firing of dormant replication origins [35,36]. Pfister et al. identified that loss of methyltransferase SETD2 is synthetically lethal with loss of Wee1 in cancer cells due to dNTP starvation via RRM2 deregulation [34]. These results provided a rationale to explore combined therapy with these two inhibitors. We found that dNTP pool depletion appears to be a major contributor to the synergy of Wee1 and mTOR inhibition. Addition of nucleotide metabolic substrate dNTPs alleviated replication stress, restored the cell cycle and reduced apoptosis to some extent, supporting dNTPs depletion is necessary for the synergy between Wee1i and mTORi.
In conclusion with our study, we propose that dual inhibition of Wee1 and mTOR may be a promising treatment option for epithelial ovarian cancer patients. In particular, the combined treatment of Wee1i and mTORi could be a strategy to convert resistance to Wee1 inhibitor tumors. More evidence is needed to explore the feasibility of this combination therapy before this strategy could be translated into clinical practice. These results provided a new option to patients who suffered epithelial ovarian cancer.
Acknowledgements
This study is supported by grants of the Nature and Science Foundation of China (81572569 to Gang Chen, 81874106 to Gang Chen). Nature and Science Foundation of China (81974408 to Chaoyang Sun).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Lheureux S, Braunstein M, Oza AM. Epithelial ovarian cancer: evolution of management in the era of precision medicine. CA Cancer J Clin. 2019;69:280–304. doi: 10.3322/caac.21559. [DOI] [PubMed] [Google Scholar]
- 2.Cortez AJ, Tudrej P, Kujawa KA, Lisowska KM. Advances in ovarian cancer therapy. Cancer Chemother Pharmacol. 2018;81:17–38. doi: 10.1007/s00280-017-3501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gonzalez-Martin A, Pothuri B, Vergote I, DePont Christensen R, Graybill W, Mirza MR, McCormick C, Lorusso D, Hoskins P, Freyer G, Baumann K, Jardon K, Redondo A, Moore RG, Vulsteke C, O’Cearbhaill RE, Lund B, Backes F, Barretina-Ginesta P, Haggerty AF, Rubio-Perez MJ, Shahin MS, Mangili G, Bradley WH, Bruchim I, Sun K, Malinowska IA, Li Y, Gupta D, Monk BJ PRIMA/ENGOT-OV26/GOG-3012 Investigators. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2019;381:2391–2402. doi: 10.1056/NEJMoa1910962. [DOI] [PubMed] [Google Scholar]
- 4.Igarashi M, Nagata A, Jinno S, Suto K, Okayama H. Wee1(+)-like gene in human cells. Nature. 1991;353:80–83. doi: 10.1038/353080a0. [DOI] [PubMed] [Google Scholar]
- 5.Lundgren K, Walworth N, Booher R, Dembski M, Kirschner M, Beach D. mik1 and wee1 cooperate in the inhibitory tyrosine phosphorylation of cdc2. Cell. 1991;64:1111–1122. doi: 10.1016/0092-8674(91)90266-2. [DOI] [PubMed] [Google Scholar]
- 6.Russell P, Nurse P. The mitotic inducer nim1+ functions in a regulatory network of protein kinase homologs controlling the initiation of mitosis. Cell. 1987;49:569–576. doi: 10.1016/0092-8674(87)90459-4. [DOI] [PubMed] [Google Scholar]
- 7.Mizuarai S, Yamanaka K, Itadani H, Arai T, Nishibata T, Hirai H, Kotani H. Discovery of gene expression-based pharmacodynamic biomarker for a p53 context-specific anti-tumor drug Wee1 inhibitor. Mol Cancer. 2009;8:34. doi: 10.1186/1476-4598-8-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu JY, Cuellar RA, Berndt N, Lee HE, Olesen SH, Martin MP, Jensen JT, Georg GI, Schonbrunn E. Structural basis of Wee kinases functionality and inactivation by diverse small molecule inhibitors. J Med Chem. 2017;60:7863–7875. doi: 10.1021/acs.jmedchem.7b00996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leijen S, van Geel RM, Pavlick AC, Tibes R, Rosen L, Razak AR, Lam R, Demuth T, Rose S, Lee MA, Freshwater T, Shumway S, Liang LW, Oza AM, Schellens JH, Shapiro GI. Phase I study evaluating WEE1 inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J. Clin. Oncol. 2016;34:4371–4380. doi: 10.1200/JCO.2016.67.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leijen S, van Geel RM, Sonke GS, de Jong D, Rosenberg EH, Marchetti S, Pluim D, van Werkhoven E, Rose S, Lee MA, Freshwater T, Beijnen JH, Schellens JH. Phase II study of WEE1 inhibitor AZD1775 plus carboplatin in patients with TP53-mutated ovarian cancer refractory or resistant to first-line therapy within 3 months. J. Clin. Oncol. 2016;34:4354–4361. doi: 10.1200/JCO.2016.67.5942. [DOI] [PubMed] [Google Scholar]
- 11.Cuneo KC, Morgan MA, Sahai V, Schipper MJ, Parsels LA, Parsels JD, Devasia T, Al-Hawaray M, Cho CS, Nathan H, Maybaum J, Zalupski MM, Lawrence TS. Dose escalation trial of the Wee1 inhibitor adavosertib (AZD1775) in combination with gemcitabine and radiation for patients with locally advanced pancreatic cancer. J. Clin. Oncol. 2019;37:2643–2650. doi: 10.1200/JCO.19.00730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, Collins J, Chen AP, Doroshow JH, Kummar S. Phase I study of single-agent AZD1775 (MK-1775), a Wee1 kinase inhibitor, in patients with refractory solid tumors. J. Clin. Oncol. 2015;33:3409–3415. doi: 10.1200/JCO.2014.60.4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nieminuszczy J, Schwab RA, Niedzwiedz W. The DNA fibre technique - tracking helicases at work. Methods. 2016;108:92–98. doi: 10.1016/j.ymeth.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 14.Sun C, Yin J, Fang Y, Chen J, Jeong KJ, Chen X, Vellano CP, Ju Z, Zhao W, Zhang D, Lu Y, Meric-Bernstam F, Yap TA, Hattersley M, O’Connor MJ, Chen H, Fawell S, Lin SY, Peng G, Mills GB. BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell. 2018;33:401–416. e408. doi: 10.1016/j.ccell.2018.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fang Y, McGrail DJ, Sun C, Labrie M, Chen X, Zhang D, Ju Z, Vellano CP, Lu Y, Li Y, Jeong KJ, Ding Z, Liang J, Wang SW, Dai H, Lee S, Sahni N, Mercado-Uribe I, Kim TB, Chen K, Lin SY, Peng G, Westin SN, Liu J, O’Connor MJ, Yap TA, Mills GB. Sequential therapy with PARP and WEE1 inhibitors minimizes toxicity while maintaining efficacy. Cancer Cell. 2019;35:851–867. e857. doi: 10.1016/j.ccell.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Russo M, Crisafulli G, Sogari A, Reilly NM, Arena S, Lamba S, Bartolini A, Amodio V, Magri A, Novara L, Sarotto I, Nagel ZD, Piett CG, Amatu A, Sartore-Bianchi A, Siena S, Bertotti A, Trusolino L, Corigliano M, Gherardi M, Lagomarsino MC, Di Nicolantonio F, Bardelli A. Adaptive mutability of colorectal cancers in response to targeted therapies. Science. 2019;366:1473–1480. doi: 10.1126/science.aav4474. [DOI] [PubMed] [Google Scholar]
- 17.Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature. 2019;575:299–309. doi: 10.1038/s41586-019-1730-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Powles T, Wheater M, Din O, Geldart T, Boleti E, Stockdale A, Sundar S, Robinson A, Ahmed I, Wimalasingham A, Burke W, Sarker SJ, Hussain S, Ralph C. A randomised phase 2 study of AZD2014 versus everolimus in patients with VEGF-refractory metastatic clear cell renal cancer. Eur Urol. 2016;69:450–456. doi: 10.1016/j.eururo.2015.08.035. [DOI] [PubMed] [Google Scholar]
- 19.Faber AC, Coffee EM, Costa C, Dastur A, Ebi H, Hata AN, Yeo AT, Edelman EJ, Song Y, Tam AT, Boisvert JL, Milano RJ, Roper J, Kodack DP, Jain RK, Corcoran RB, Rivera MN, Ramaswamy S, Hung KE, Benes CH, Engelman JA. mTOR inhibition specifically sensitizes colorectal cancers with KRAS or BRAF mutations to BCL-2/BCL-XL inhibition by suppressing MCL-1. Cancer Discov. 2014;4:42–52. doi: 10.1158/2159-8290.CD-13-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dobbelstein M, Sorensen CS. Exploiting replicative stress to treat cancer. Nat Rev Drug Discov. 2015;14:405–423. doi: 10.1038/nrd4553. [DOI] [PubMed] [Google Scholar]
- 21.Macheret M, Halazonetis TD. DNA replication stress as a hallmark of cancer. Annu Rev Pathol. 2015;10:425–448. doi: 10.1146/annurev-pathol-012414-040424. [DOI] [PubMed] [Google Scholar]
- 22.Kotsantis P, Petermann E, Boulton SJ. Mechanisms of oncogene-induced replication stress: jigsaw falling into place. Cancer Discov. 2018;8:537–555. doi: 10.1158/2159-8290.CD-17-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valvezan AJ, Turner M, Belaid A, Lam HC, Miller SK, McNamara MC, Baglini C, Housden BE, Perrimon N, Kwiatkowski DJ, Asara JM, Henske EP, Manning BD. mTORC1 couples nucleotide synthesis to nucleotide demand resulting in a targetable metabolic vulnerability. Cancer Cell. 2017;32:624–638. e625. doi: 10.1016/j.ccell.2017.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gordon DJ, Koppenhafer SL, Goss KL, Terry WW. Inhibition of the ATR-CHK1 pathway in Ewing sarcoma cells causes DNA damage and apoptosis via the CDK2-mediated degradation of RRM2. Mol Cancer Res. 2020;18:91–104. doi: 10.1158/1541-7786.MCR-19-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339:1323–1328. doi: 10.1126/science.1228792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Byrne AT, Alferez DG, Amant F, Annibali D, Arribas J, Biankin AV, Bruna A, Budinska E, Caldas C, Chang DK, Clarke RB, Clevers H, Coukos G, Dangles-Marie V, Eckhardt SG, Gonzalez-Suarez E, Hermans E, Hidalgo M, Jarzabek MA, de Jong S, Jonkers J, Kemper K, Lanfrancone L, Maelandsmo GM, Marangoni E, Marine JC, Medico E, Norum JH, Palmer HG, Peeper DS, Pelicci PG, Piris-Gimenez A, Roman-Roman S, Rueda OM, Seoane J, Serra V, Soucek L, Vanhecke D, Villanueva A, Vinolo E, Bertotti A, Trusolino L. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat Rev Cancer. 2017;17:254–268. doi: 10.1038/nrc.2016.140. [DOI] [PubMed] [Google Scholar]
- 27.Stewart EL, Mascaux C, Pham NA, Sakashita S, Sykes J, Kim L, Yanagawa N, Allo G, Ishizawa K, Wang D, Zhu CQ, Li M, Ng C, Liu N, Pintilie M, Martin P, John T, Jurisica I, Leighl NB, Neel BG, Waddell TK, Shepherd FA, Liu G, Tsao MS. Clinical utility of patient-derived xenografts to determine biomarkers of prognosis and map resistance pathways in EGFR-mutant lung adenocarcinoma. J. Clin. Oncol. 2015;33:2472–2480. doi: 10.1200/JCO.2014.60.1492. [DOI] [PubMed] [Google Scholar]
- 28.Hai J, Liu S, Bufe L, Do K, Chen T, Wang X, Ng C, Li S, Tsao MS, Shapiro GI, Wong KK. Synergy of WEE1 and mTOR inhibition in mutant KRAS-driven lung cancers. Clin Cancer Res. 2017;23:6993–7005. doi: 10.1158/1078-0432.CCR-17-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cosimo E, Tarafdar A, Moles MW, Holroyd AK, Malik N, Catherwood MA, Hay J, Dunn KM, Macdonald AM, Guichard SM, O’Rourke D, Leach MT, Sansom OJ, Cosulich SC, McCaig AM, Michie AM. AKT/mTORC2 inhibition activates FOXO1 function in CLL cells reducing B-cell receptor-mediated survival. Clin Cancer Res. 2019;25:1574–1587. doi: 10.1158/1078-0432.CCR-18-2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, Toniatti C, Ashworth A, Turner NC. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2012;2:524–539. doi: 10.1158/2159-8290.CD-11-0320. [DOI] [PubMed] [Google Scholar]
- 31.Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, Manning BD. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science. 2016;351:728–733. doi: 10.1126/science.aad0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He Z, Hu X, Liu W, Dorrance A, Garzon R, Houghton PJ, Shen C. P53 suppresses ribonucleotide reductase via inhibiting mTORC1. Oncotarget. 2017;8:41422–41431. doi: 10.18632/oncotarget.17440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koppenhafer SL, Goss KL, Terry WW, Gordon DJ. mTORC1/2 and protein translation regulate levels of CHK1 and the sensitivity to CHK1 inhibitors in Ewing sarcoma cells. Mol Cancer Ther. 2018;17:2676–2688. doi: 10.1158/1535-7163.MCT-18-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pfister SX, Markkanen E, Jiang Y, Sarkar S, Woodcock M, Orlando G, Mavrommati I, Pai CC, Zalmas LP, Drobnitzky N, Dianov GL, Verrill C, Macaulay VM, Ying S, La Thangue NB, D’Angiolella V, Ryan AJ, Humphrey TC. Inhibiting WEE1 selectively kills histone H3K36me3-deficient cancers by dNTP starvation. Cancer Cell. 2015;28:557–568. doi: 10.1016/j.ccell.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pai CC, Hsu KF, Durley SC, Keszthelyi A, Kearsey SE, Rallis C, Folkes LK, Deegan R, Wilkins SE, Pfister SX, De Leon N, Schofield CJ, Bahler J, Carr AM, Humphrey TC. An essential role for dNTP homeostasis following CDK-induced replication stress. J Cell Sci. 2019;132 doi: 10.1242/jcs.226969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Puigvert JC, Sanjiv K, Helleday T. Targeting DNA repair, DNA metabolism and replication stress as anti-cancer strategies. FEBS J. 2016;283:232–245. doi: 10.1111/febs.13574. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.