

Abstract
Objective: To investigate the effect of the AKT1 gene mutation hotspot E17K on the growth, proliferation, survival, and migration of breast cancer cells, based on the survival and prognosis of breast cancer patients with the AKT1 E17K mutation shown in TCGA database. Methods: The survival and incidence rates of AKT1 E17K mutation hotspots in breast cancer and other cancers were extracted from the Cancer Genome Atlas (TCGA). The recombinant eukaryotic expression plasmid AKT1 E17K-pIRES2-EGFP was constructed and transfected into breast cancer MCF-7, and MDA-MB-231 cell lines. MCF-7 and MDA-MB-231 cell lines were randomly divided into blank control groups, empty plasmid groups, and recombinant plasmid groups. The growth curve was drawn using the cell counting method. The proliferation and division of breast cancer cells were detected by CFSE fluorescent dye tracking. Apoptosis was detected by Annexin V/PI double labeling and cell vitality was detected using MTT assays, and cell migratory ability was detected by cell scratch and transwell chamber tests. Results: In breast cancer, and other cancers, the overall survival rate of patients with an AKT E17K mutation was higher than that of patients with non-point mutation, and this mutation was the most common found in breast cancer. Compared with the wild type, the growth function of mutant MCF-7 cells was inhibited (P < 0.05), as was the proliferation of MCF-7 cells expressing the AKT1 E17K mutation gene (P < 0.001). The late apoptosis rate of mutant breast cancer cells increased (P < 0.05) and the viability was lower than that of wild-type cells (P < 0.05). Mutant MDA-MB-231 cells showed increased migration ability when compared to wild-type MDA-MB-231 cells (P < 0.05). Conclusions: The expression of the AKT1 E17K mutation hotspot can inhibit the growth, proliferation, and survival ability of breast cancer cells, and promote apoptosis, while it also improves their migratory ability. The survival and prognosis of breast cancer patients with this mutation are good, which may be related to the inhibition of the PI3K/AKT/mTOR signaling pathway.
Keywords: Breast cancer, AKT1 E17K, mutation, cell function
Introduction
Breast cancer is one of the most common malignant tumors in women, and its incidence has been on the rise over the past decade [1]. With the development of innovative medical treatments, the survival rate associated with breast cancer of all types has improved, but the recurrence, local invasion, and distant metastasis in the later stages increase the risk of death [2,3].
AKT is an intracellular kinase, also known as protein kinase B, that plays an important role in cell survival and apoptosis. AKT is crucial in the downstream signal pathway of glyceraldehyde 3-kinase [4], regulating the basic processes of cell survival, proliferation, differentiation, angiogenesis, and metabolism. AKT is divided into three subtypes: AKT1, AKT2, and AKT3, each playing different roles [5]. AKT1, for instance, encodes serine/threonine-protein kinase [6]. Of particular interest, however, PI3k/AKT is one of the most frequently activated pathways in cancer. AKT is involved in many steps of tumor formation through the PI3K signaling pathway [7].
AKT1 E17K is a recurrent somatic mutation observed in breast cancer, colorectal cancer, lung cancer, and ovarian cancer, that functions mainly to activate the PI3K/Akt signal pathway [8,9], and the E17K hotspot is the most characteristic mutation of the AKT1 gene. Some studies have shown that this mutation can lead to protein activation, which is activation of the PI3K/AKT/mTOR pathway, leading to cancer [10]. However, it has also been found that E17K has a certain antitumor effect in breast cancer luminal cells, and has a good prognostic effect in the survival analysis of breast cancer patients [11].
Based on the survival analysis of breast cancer patients by the Cancer Genome Atlas (TCGA), the effects of the AKT1 gene mutation hotspot E17K on the tumorigenesis of breast cancer cells were studied, so as to explore the effect of the AKT1 E17K point mutation on the survival and prognosis of breast cancer patients, and to analyze the effect and mechanism of the E17K mutation on breast cancer [12].
Materials and methods
Cell line and reagent
Human breast cancer cell lines MCF-7 and MDA-MB-231 were purchased from Shanghai cell bank of the Chinese Academy of Sciences. AKT1-E17K upstream and downstream primers were synthesized by Qingke Biological Co., Ltd.
LipofectamineTM3000 and Trizol was from Invitrogen Company. Reverse transcription kit, r Taq enzyme, prime Star HS high fidelity enzyme, T4DNA ligase, 19-T vector, receptive Escherichia coli DH5a were purchased from Takara. BamH1, and Sal1 restriction endonuclease were purchased from New England Biologicals. CFSE fluorescent dye, and Annexin V/PI cell apoptosis detection kit were purchased from BD Company. Plasmid extraction kit was purchased from Omega Company. Transwell chamber was purchased from Corning.
Comparison of AKT1 E17K related survival curves
Based on the statistical data of cancer gene mutation hotspots uncovered in TCGA, the overall survival curves for AKT1 E17K mutant and wild type patients were extracted and analyzed by Kaplan-Meier survival analysis. Afterwards, the survival plot was analyzed by a Log-Rank test, where P < 0.05 demonstrated statistical significance.
Discussion of the incidences of AKT1 E17K mutation in different types of cancer utilizing TCGA data
The number of cases with AKTI E17K mutation hotspot was searched using the TCGA database. The average number of breast cancer patients and cancer patients of other types (N=11) was compared using the Pearson Chi-square test, where the number of effective cases n=1626 > 40, and all theoretical frequencies were above 5. By the two-sided test, P < 0.05 was considered significant.
Construction of AKT1 E17K-pIRES2-EGFP recombinant eukaryotic expression plasmid
The extraction of RNA from MCF-7 breast cancer cells, reverse transcription into cDNA, as well as the design and synthesis of upstream and downstream primers for mutant genes. Using the PCR-directed mutagenesis method, the 17th amino acid translated by AKT1 gene was transformed from glutamic acid (E) to lysine (K); that is, the codon changed from GAG to AAG, by transforming base G into A. PCR amplification conditions were as follows: 98°C 10 s, 58°C 5 s, 72°C 90 s, 35 cycles. The high-fidelity enzyme amplification product was identified by 1% agarose gel electrophoresis and the AKT1 gene was cloned into 1443 bp (as detailed in Figure 3A). After poly-A tailing by Taq enzyme was added to the mutant AKT1 gene fragment, it was held at 72°C for 10 min, and it was cleaned with the Purification kit, T4 DNA ligase was linked to a 19-T vector and held at 16°C overnight. The positive clone of receptive Escherichia coli DH5a was screened, and the AKT1 E17K-19T plasmid was extracted. The AKT1 E17K plasmid was ligated to the pIRES2-EGFP plasmid by a specific restriction site (BamH1, Sal1) by double-enzyme digestion and linked overnight at 16°C. Afterwards, it was transformed into receptive Escherichia coli DH5a. After the colony PCR was identified correctly, the target fragment and mutation sequences were verified by sequencing. The recombinant plasmid AKT1 E17K-pIRES2-EGFP was obtained from the correctly sequenced genetically engineered bacteria by removing the endotoxin by plasmid extraction kit. The sequences of the primers were as follows: AKT1-E17K-Forward primer, 5’-ATGAGCGACGTGGCTATTGTGAAGGAGGGTTGGCTGCACAAACGAGGGAAGTACATCAA-3’. AKT1-E17K-Reverse primer, 5’-TCAGGCCGTGCCGCTGGCCGAGTAG-3’.
Figure 3.

Construction of recombinant eukaryotic expression plasmid AKT1 E17K-pIRES2-EGFP. A. PCR amplification of AKT1 E17K point mutation gene electrophoresis. B. Sequencing comparison diagram of cloned AKT1 E17K mutant gene. C. AKT1 E17K-pIRES2-EGFP plasmid sequencing profile.
Transfer efficiency of recombinant plasmid into breast cancer cells
The extracted AKT1 E17K-pIRES2-EGFP recombinant plasmid and pIRES2-EGFP empty plasmid were transfected into MCF-7 cells and MDA-MB-231 cells, respectively, according to the method of liposomes Lipo3000 specification. After 24 hours, the expression of GFP in MCF-7 cells and MDA-MB-231 cells was observed under an inverted fluorescence microscope, with a maximum excitation wavelength at 490 nm (Olympus IX51, Japan). The transfection efficiency was detected by flow cytometry (purchased from Beckman, Gallios). The positive cells expressing GFP fluorescent protein were separated and collected by a flow cell sorter from Beckman, MoFlo XDP, for follow-up experiments.
Drawing the growth curve of MCF-7 cells
The MCF-7 cells were divided into three groups: wild-type MCF-7 cells, MCF-7 cells expressing the AKT1 E17K recombinant plasmid, and MCF-7 cells expressing the empty plasmid. The MCF-7 cells transfected with each group were inoculated into a six-well plate with 2.8×106 cells per well, respectively, and 2 ml/well DMEM medium containing 10% FBS (Gibco). The cultures were incubated in a cell incubator from Thermo, HERA cell 150, at 37°C and 5% CO2. The 3-well cells were extracted and counted under a microscope daily, and the cell growth curve was drawn with the culture time as a transverse coordinate and the number of cells as longitudinal coordinates.
Breast cancer cell CFSE proliferation test
After successful transfer, CFSE dye (final concentration 1 μmol/L) was added to the wild type MCF-7 cells, incubated for 10 min at 37°C, washed twice with PBS (Hyclone) containing 10% FBS, centrifuged, and resuspended. Three groups of cells were inoculated into the six-well plate with 5×106 cells per well, and 2 mL per well DMEM, containing 10% FBS. The cultures were placed in a cell incubator at 37 degrees and 5% CO2. Every day, 1×105 of cells were taken from each of the three groups and assessed using flow cytometry, where cell proliferation was observed continuously for 4 days.
Detection of apoptosis in breast cancer cells
MCF-7 cells and MDA-MB-231 cells, expressing AKT1 E17K recombinant plasmid and empty plasmid, respectively, and wild type cells, were incubated to the 4th day. 1×105 cells were taken out from each group and were detected by the Annexin V/PI double-labeling method. Reagents were added according to instructions, incubated at room temperature for 10 mins, and the apoptosis rate was detected by flow cytometry.
MTT assay to detect the activity of breast cancer cells
MCF-7 cells and MDA-MB-231 cells, expressing AKT1 E17K recombinant plasmid or empty plasmid, and the wild-type cells were inoculated in a 96-well plate with the cell number of cells 1×104/well, and cultured in DMEM, containing 10% FBS (200 ul/well) for 24 hours in an incubator set to 37°C and 5% CO2. Approximately 4 hours before termination, MTT solution (Sigma, 5 mg/mL, 20 ul) was added to each well for 4 hours at 37°C. The culture medium was discarded, 150 μl of DMSO (Sigma) was added, and the crystal was dissolved at room temperature for 10 min. The absorbance values of each well were measured by an automatic microplate reader (Thermo) at a wavelength of 490 nm. Cell viability was calculated accordingly using the standard equation: (%) = Atreated/Acontrol×100%. The reported data represent three independent experiments.
Cell scratch healing test of breast cancer cells
Cultures were seeded into a six-well plate at a density of 1×106/ml on the previous day. After adhering to the wall, the cells were scratched with a small pipette head along the midline of each well, and the surfaces of the cells were washed three times with sterile PBS buffer, then cultured in a cell incubator with 2 mL/well using DMEM containing 2% FBS. The migration of cells was observed and photographed under an inverted fluorescence microscope 24 hours later.
Transwell migration test of breast cancer cells
MDA-MB-231 cells expressing the AKT1 E17K recombinant plasmid, the empty plasmid, and wild type cells (1×105) were cultured in the upper transwell chamber (8 um). 500 μl DMEM medium containing 10% FBS was added to the lower chamber of the 24-well plate, incubated at 37°C and 5% CO2. The upper chamber wells were wiped with a cotton swab after 24 h and washed 3 times with PBS buffer. The transwell chamber was immersed in 4% paraformaldehyde for 15 min, stained with 0.1% crystal violet for 20 min, and washed with distilled water twice. Cell migration was recorded with an inverted microscope, and the cell count was performed at random in 5 visual fields.
Statistical analysis
All data are presented as the mean ± standard deviation. Comparison of multiple groups in this study was completed using variance analysis (ANOVA). Pairwise comparison between groups was completed using Bonferroni posttests, as indicated by GraphPad Prism software 6.0 (GraphPad Software, Inc.). A Student t-test was used to compare the mean between the two groups. Survival analysis was performed using the Kaplan-Meier method with a Log-rank statistical test. Pearson chi-square test was used to compare the average number of cancer patients, with significance set at P < 0.05.
Results
Comparison of the survival curve between AKT1 E17K mutation and non-AKT1 E17K mutation in breast cancer
As can be seen from the survival curve (Figure 1A), the survival rate of breast cancer patients decreases when the 17th amino acid of the AKT1 gene fails to mutate. When the 17th amino acid of the AKT1 gene was changed from E to K, the survival time changed to 6,292 days, and the survival rate to 100%. The correlation of the two overall survival rates was compared using the Log-Rank test, and the P-value was determined to be 0.0481 (P < 0.05), suggesting that the difference between the two survival curves is statistically significant. Thus, the overall survival rate (OS) of breast cancer patients with the AKT1 E17K mutation was higher than that of the non-AKT1 E17K mutated breast cancer patients.
Figure 1.
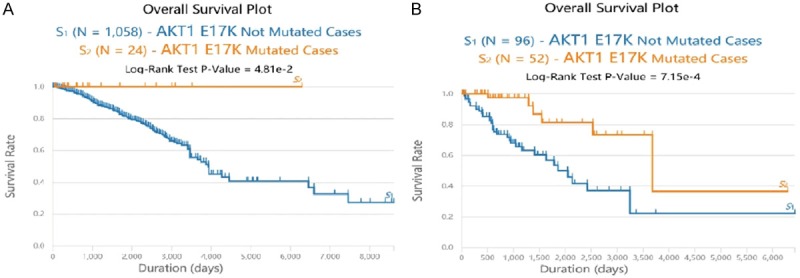
Comparison of AKT1 E17K related survival curves. A. Survival curve of AKT1 E17K mutation and non-AKT1 E17K mutation in breast cancer. Blue survival curve: the number of breast cancer patients with non-AKT1 E17K mutation was 1058. Orange survival curve: the number of breast cancer patients with AKT1 E17K mutation was 24. Compared with the control curve: *P < 0.05. B. Survival plots of AKT1 E17K mutated cancer cases and Non-AKT1 E17K mutated cancer cases. Blue survival curve: the number of cancer patients with non-AKT1 E17K mutation was 96. Orange survival curve: the number of cancer patients with AKT1 E17K mutation was 52. Compared with the control plot: **P < 0.01.
Comparison of survival plots for AKT1 E17K mutated cancer cases and non-mutated cancer cases
Among the 11 types of cancers with AKT1 mutation (Table 1), the survival plot of E17K, at the somatic cell mutation site of AKT1 (orange), was higher than that of other mutation sites (but not E17K, the blue plot). A Log-Rank test verified the P-value of 0.000715 (P < 0.01). Thus, the difference between the two survival plots was statistically significant. It could be concluded that the overall survival for AKT1 E17K mutated cancer cases was higher than that for AKT1 E17K non-mutated cases (Figure 1B).
Table 1.
Number of AKT1 E17K mutated cases in various cancers
| Cancer Types | Number of AKT1 E17K Mutated Cases | Total Cases | Percentage of Mutated Cases (%) |
|---|---|---|---|
| Breast invasive carcinoma (BRCA) | 25 | 987 | 2.53% |
| Uterine Corpus Endometrial Carcinoma (UCEC) | 9 | 530 | 1.70% |
| Colon adenocarcinoma (COAD) | 4 | 400 | 1.00% |
| Cervical and endocervical cancers (CESC) | 3 | 289 | 1.04% |
| Thyroid carcinoma (THCA) | 3 | 492 | 0.61% |
| Lung adenocarcinoma (LUAD) | 2 | 1063 | 0.19% |
| Prostate adenocarcinoma (PRAD) | 2 | 498 | 0.40% |
| Skin Cutaneous Melanoma (SKCM) | 2 | 469 | 0.43% |
| Bladder urothelial carcinoma (BLCA) | 1 | 412 | 0.24% |
| Head and Neck squamous cell carcinoma (HNSC) | 1 | 508 | 0.20% |
| Stomach adenocarcinoma (STAD) | 1 | 440 | 0.23% |
| Total | 53 | 6088 | 0.87% |
| Average | 5 | 609 | 0.82% |
Discussion of the incidences of AKT1 E17K mutation in different types of cancer utilizing TCGA data
Statistics on the TCGA data showed that AKT1 E17K was concentrated in breast cancer, cervical cancer, colon cancer, thyroid cancer, and other cancers (totaling 11). It was at its highest in breast cancer patients (25, 2.53%), with cervical cancer following closely (Figure 2). Among the 11 cancer types, the average number of AKT1 E17K mutated cases was 5, and total average number was 609. As shown in Table 1, Pearson Chi-square test was made according to the average number of breast cancer patients (AKT1 E17K mutated cases 25, totaling 987), and patients with other types of cancers (totaling 11), where P=0.014 (bilateral, P < 0.05). This difference was statistically significant, indicating the incidence of AKTI E17K mutation in breast cancer is higher than other cancer types. Thus, it is of importance to discuss the influence of this mutation hotspot on breast cancer.
Figure 2.
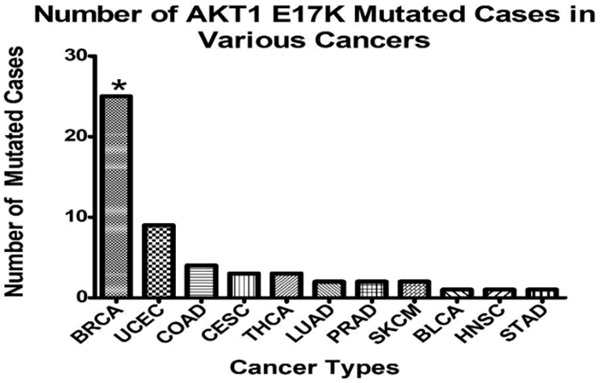
The incidences of AKT1 E17K mutation in different types of cancer. Number of AKT1 E17K mutated cases in various cancers. Compared to the average, by Pearson Chi-square test: *P < 0.05.
Construction of recombinant eukaryotic expression plasmid AKT1 E17K-pIRES2-EGFP
Using the PCR-directed mutagenesis method, the PCR product was identified using 1% agarose gel electrophoresis (Figure 3A). The AKT1 E17K point mutation gene was amplified and the size of the gene fragment was determined to be 1443 bp, the same as the target gene. After sequencing of the mutant gene engineering bacteria, the recombinant plasmid of site-directed mutant AKT1 E17K-pIRES2-EGFP was compared with the wild type AKT1 gene (Figure 3B). The 17th amino acid encoding AKT1 protein changed from glutamic acid (E) to lysine (K), and the codon went from GAG mutation to AAG, base G mutated to A (Figure 3C), while none of the other bases were mutated in the AKT1 gene. The AKT1 E17K mutant gene clone had been successfully constructed in the pIRES2-EGFP plasmid.
Transfer of recombinant plasmid into breast cancer cells and flow cytometry detection of transfer efficiency
After transfection of the pIRES2-EGFP empty plasmid and the AKT1 E17K-pIRES2-EGFP recombinant plasmid into MCF-7 cells and MDA-MB-231 cells, the expression of GFP-green fluorescent protein could be observed post-transfection (Figure 4). The transfection efficiencies of the two plasmids transfected into MCF-7 cells and MDA-MB-231 cells were about 22% to 25%, as determined by flow cytometry (Figure 5). The cells expressing GFP were separated and collected by a flow cell sorter for follow-up experiments.
Figure 4.

Inverted fluorescence microscopy of breast cancer cells transfected with plasmid. A. MCF-7 cells were transfected with empty plasmid (white light). B. MCF-7 cells were transfected with empty plasmid (fluorescence). C. MCF-7 cells were transfected with AKT1 E17K plasmid (white light). D. MCF-7 cells were transfected with AKT1 E17K plasmid (fluorescence). E. MDA-MB-231 cells were transfected with empty plasmid (white light). F. MDA-MB-231 cells were transfected with empty plasmid (fluorescence). G. MDA-MB-231 cells were transfected with AKT1 E17K plasmid (white light). H. MDA-MB-231 cells were transfected with AKT1 E17K plasmid (fluorescence) (Magnification, 10×20).
Figure 5.
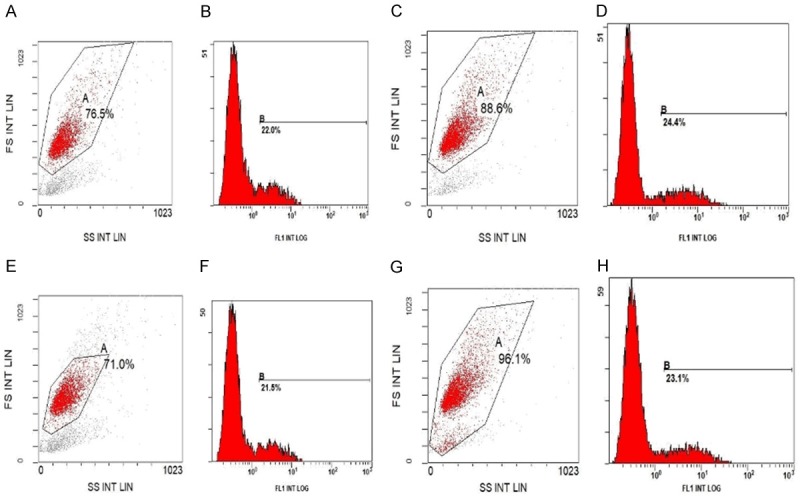
Flow cytometry detection of transfection efficiency. A, B. MCF-7 cells were transfected with empty plasmid. C, D. MCF-7 cells were transfected with AKT1 E17K plasmid. E, F. MDA-MB-231 cells were transfected with empty plasmid. G, H. MDA-MB-231 cells were transfected with AKT1 E17K plasmid.
Breast cancer cell growth curve
As shown in the cell growth curve, it can be seen that the number of cells in the three groups showed an escalating trend as time increased (Figure 6). Compared to the other 2 groups, the number of MCF-7 cells expressing AKT1 E17K was less than that of the wild-type cells or cells expressing empty plasmid. The number of mutant MCF-7 cells was statistically significant, as compared to the MCF-7 cells expressing the empty plasmid at 48 h and 72 h (P < 0.05). Thus, the growth function of mutant-type MCF-7 breast cells is suppressed.
Figure 6.
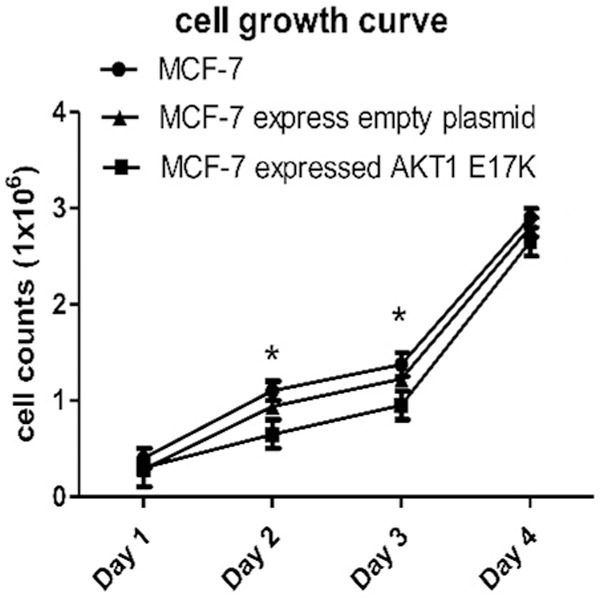
Growth curve of breast cancer cells. At 48 h and 72 h, the number of mutant MCF-7 cells was statistically significant as compared to MCF-7 cells expressing the empty plasmid (*P < 0.05).
Proliferation of breast cancer cells
Through CFSE staining and flow detection of MCF-7 cells, Modfit software was used to analyse the results, so as to determine the proliferation and division of breast cancer cells.
On the 1st, 2nd, 3rd, and 4th day, the proportion of MCF-7 cells that expressed empty plasmid that proliferated and divided into the 7th generation of the cells were 7.64%, 56.06%, 0.39%, and 2.54%, respectively. The proportion of the mutant MCF-7 cells that proliferated and divided into the 7th generation was 6.94%, 37.27%, 0%, 1.62%, respectively. According to Figure 7, on the 1st, 2nd, 3rd and 4th day, the proliferation indexes (PI) of wild type MCF-7 cells were 1.39, 24.56, 13.41 and 11.09, respectively. The PIs of mutant MCF-7 cells were 1.57, 23.45, 11.61 and 7.41, respectively, which was lower than that of wild type cells at 1-4 days. On the 4th day, the PI of mutant and wild-type MCF-7 cells was determined significant by the Bonferroni posts test, t=35.53, P < 0.001 (Figure 8). It is apparent that MCF-7 breast cancer cells expressing the AKT1 E17K mutation gene had significantly inhibited cell proliferation.
Figure 7.

MCF-7 Cell Proliferative Split-Modfit Analysis. A. The proliferation index of wild type MCF-7 cells was 1.39 on the 1st day. B. The proliferation index of mutant MCF-7 cells was 1.57 on the 1st day. C. The proliferation index of wild type MCF-7 cells was 24.56 on the 2nd day. D. The proliferation index of mutant MCF-7 was 23.45 on the 2nd day. E. The proliferation index of wild type MCF-7 cells was 13.41 on the 3rd day. F. The proliferation index of mutant MCF-7 cells was 11.61 on the 3rd day. G. The proliferation index of wild type MCF-7 cells was 11.09 on the 4th day. H. The proliferation index of mutant MCF-7 cells was 7.41 on the 4th day.
Figure 8.
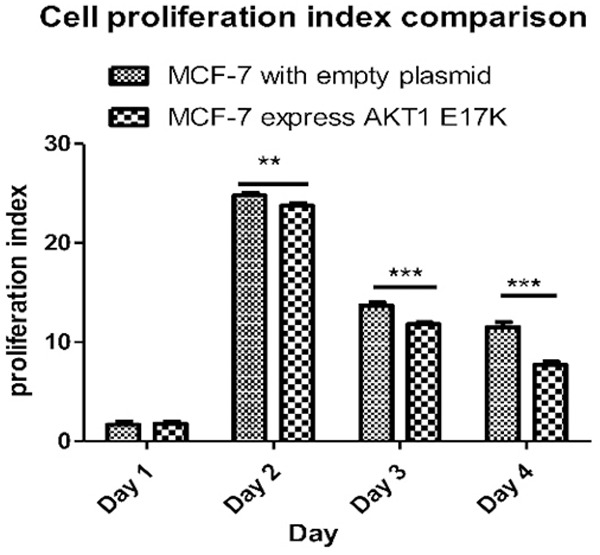
Wild-type and mutant MCF-7 cell proliferation index diagram. On the 2nd, 3rd, and 4th day, the proliferation indexes (PI) of mutant and wild-type MCF-7 cells were statistically significant difference by the Bonferroni posts test, **P < 0.001.
Flow cytometry detection of apoptosis in breast cancer cells
The results of flow cytometry showed that the late apoptosis rates of breast cancer cells expressing the AKT1 E17K gene were all higher than for those expressing empty plasmids (Figure 9). According to the statistical diagram of the late apoptosis rate, as seen in Figure 10, MCF-7 and MDA-MB-231 cells expressing the AKT1 E17K mutant gene increased by 4.4% and 4.2%, respectively, when compared with the control group (cells expressing empty plasmids in each group). The P-values of the two groups were calculated at 0.028, and 0.04, respectively, and the difference was significant (P < 0.05). The results show that the AKT1 E17K mutation gene promoted this increase in late apoptosis rates in breast cancer cells.
Figure 9.
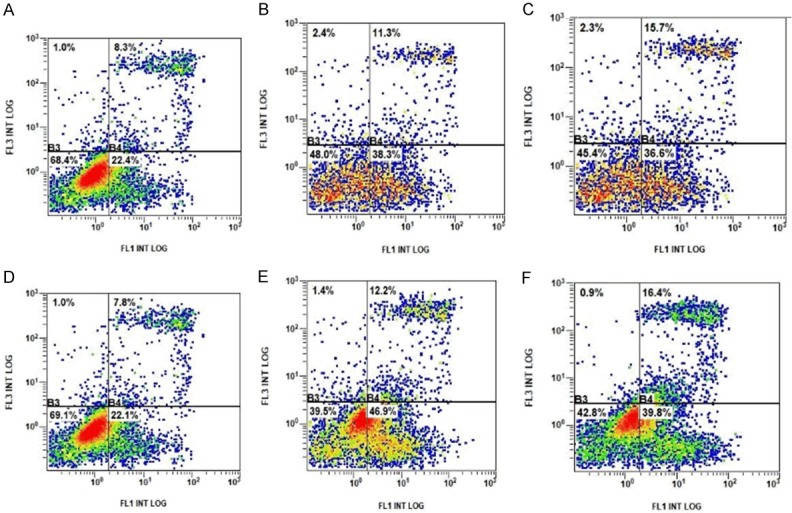
Breast cancer cell apoptosis detection by flow cytometry. A. The late apoptosis rate of MCF-7 cells was 8.3%. B. The late apoptosis rate of MCF-7 cells expressing empty plasmid was 11.3%. C. The late apoptosis rate of MCF-7 cells expressing AKT1 E17K mutated gene was 15.7%. D. The late apoptosis rate of MDA-MB-231 cells was 7.8%. E. The late apoptosis rate of MDA-MB-231 cells expressing empty plasmid was 12.2%. F. The late apoptosis rate of MDA-MB-231 cells expressing AKT1 E17K mutated gene was 16.4%.
Figure 10.
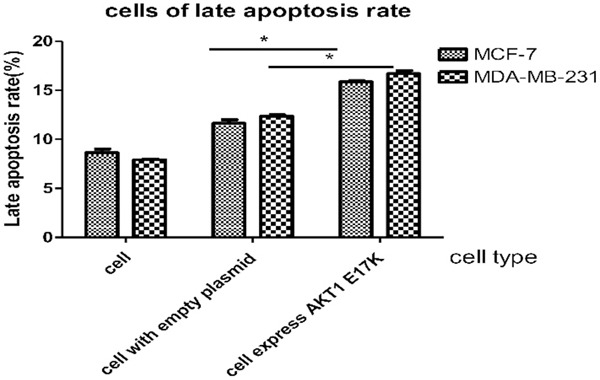
Late apoptosis rate of breast cancer cells diagram. MCF-7 and MDA-MB-231 cells expressing AKT1 E17K mutant gene compared with the control group (cells expressing empty plasmids in each group). P-values of two groups were 0.028, 0.04, respectively (*P < 0.05).
Detection of breast cancer cell viability by MTT assay
According to the cell viability rate 24 hours later (Figure 11), cell survival ability of mutant MCF-7 cells and MDA-MB-231 cells was lower than that of wild type cells. The cell viability rates of mutant MCF-7 cells and MDA-MB-231 cells were 12% and 15% lower than that of wild type cells, respectively. P values of the two groups were 0.0057 and 0.0173 respectively, and the difference was statistically significant (P < 0.05). It is inferred that the expression of AKT1 E17K mutation gene reduces cell viability of breast cancer cells.
Figure 11.
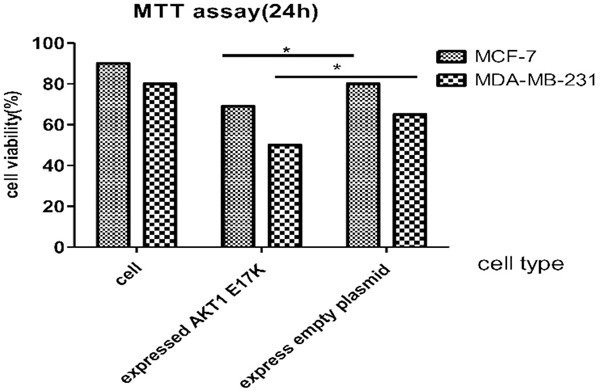
Detection of breast cancer cell viability by MTT assay 24 h later. Cell viability rates of mutant MCF-7 cells and MDA-MB-231 cells were 12% and 15% lower than that of wild type cells. P values of the two groups were 0.0057 and 0.0173, respectively (*P < 0.05).
Breast cancer cell scratch healing test
The number of MDA-MB-231 cells both with wild-type and expression of empty vector did not change significantly after 24 h, but the number of mutant cells was significantly increased (Figure 12). The cell mobility of MDA-MB-231 cells, expressing the AKT1 E17K mutant gene, was determined to be higher than that of wild-type cells by Image J software. The t=4.193 value was obtained by a Bonferroni post hoc test, and the difference was significant (P < 0.05), as shown in Figure 13. These results demonstrate that the expression of the AKT1 E17K mutant gene could increase migration ability of breast cancer cells.
Figure 12.
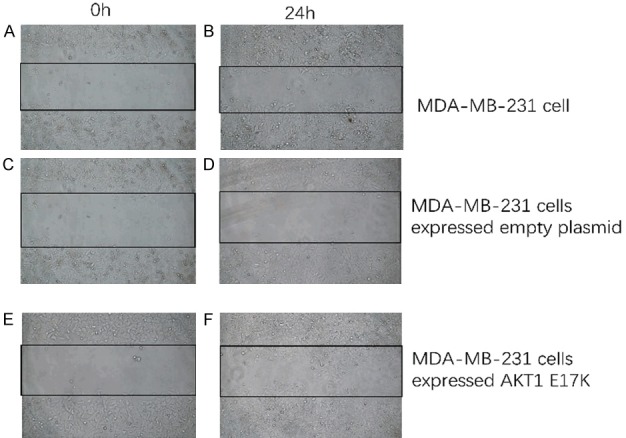
Observation of the migration of wild-type and mutant MDA-MB-231 cells. A. The migration of MDA-MB-231 cells at 0 h. B. The migration of MDA-MB-231 cells after 24 h. C. The migration of MDA-MB-231 expressing empty plasmid at 0 h. D. The migration of MDA-MB-231 expressing empty plasmid after 24 h. E. The migration of MDA-MB-231 expressing AKT1 E17K mutant gene at 0 h. F. The migration of MDA-MB-231 expressing AKT1 E17K mutant gene after 24 h. (Inverted fluorescence microscope, white light, magnification, 10×20).
Figure 13.
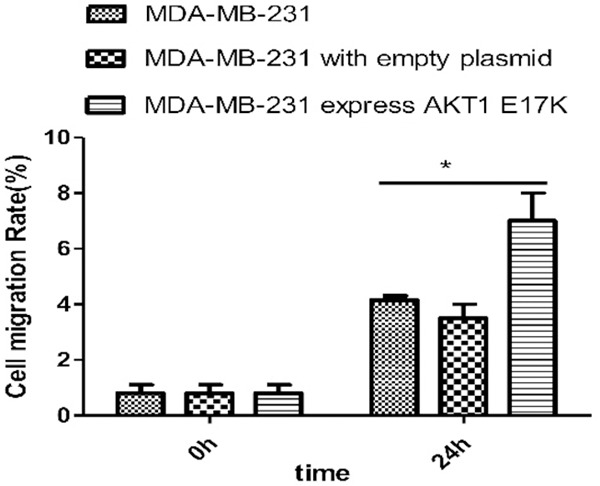
Image J software for statistical cell migration test results. Cell migration rate (%) was calculated by Image J software. The cell mobility of MDA-MB-231 cells expressing AKT1 E17K mutant gene was higher than that of wild-type cells (*P < 0.05).
Transwell migration experiment
Using crystal violet staining and cell counting after 24 hours, it was found that the number of MDA-MB-231 cells expressing the AKT1 E17K mutant gene outnumbered that of wild type cells (Figures 14, 15). The P-value, obtained by unpaired t-test, was significant (P=0.039). It can be concluded that the expression of the E17K point mutation of the AKT1 gene can improve the migratory ability of breast cancer cells.
Figure 14.

Migration of breast cancer cell by transwell experiment 24 h later. A. Migration ability of MDA-MB-231 cells. The number by cell count was 153±9. B. The migration ability of MDA-MB-231 cells expressing empty plasmid. The number of cell count was 160±12. C. The migration ability of MDA-MB-231 cells expressing AKT1 E17K mutant gene. The number of cell count was 200±10. (Cell count was performed at random 5 visual fields. Observed by inverted fluorescence microscope, white light illuminant, magnification, 10×20).
Figure 15.
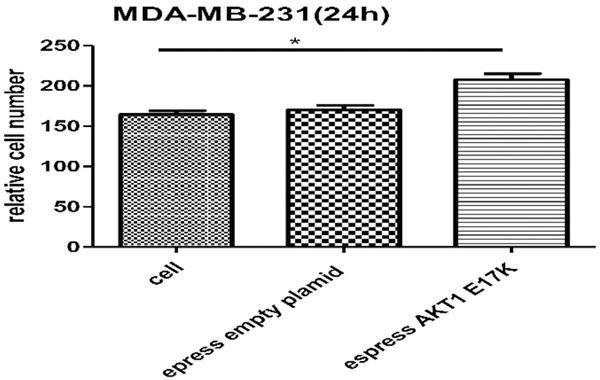
Migration of MDA-MB-231 cells 24 h later. The cell count was performed on random 5 visual fields. The number of MDA-MB-231 cells expressing AKT1 E17K mutant gene was more than that of wild type cells. P=0.039 by unpaired t-test; the difference was significant (*P < 0.05).
Discussion
PI3K/AKT/mTOR is an important regulatory cell signaling pathway, that is usually directly related to cell dormancy, proliferation, and carcinogenesis. In a variety of cancers, the PI3K/AKT/mTOR pathway is overactivated, which inhibits apoptosis and leads to widespread cell proliferation. In previous studies, it has been suggested that mutations in the AKT1 gene can activate the PI3K/AKT/mTOR signaling pathway and lead to cancer [13,14].
However, by extracting survival data of breast cancer patients from the TCGA, it was found that the survival rate of breast cancer patients increases significantly after the 17th AKT1 amino acid sites changed from E to K. The overall survival rate of cancer patients with the AKT1 E17K mutation hot spot in TCGA was also significantly higher than that in patients with other mutation sites at AKT1, such as cervical cancer (UCEC), colon cancer (COAD), and cervical cancer (CESC) [15]. In the cBioPortal database, the median survival time of breast cancer patients with the AKT1 E17K mutation was 13.5 months higher than that of the wild type. In order to explore the causes of the significant effect of this mutation point on survival and prognosis, our basic experimental study on the effect of the AKT1 E17K mutation point on breast cancer cells was carried out.
The recombinant plasmid AKT1-E17K-pIRES2-EGFP was constructed and introduced into breast cancer cells, so as to explore the effects of gene mutation hotspots on the growth, proliferation, survival, migration, and other basic cell functions. After the recombinant plasmid was transferred into MCF-7, and MDA-MB-231 breast cancer cells, it was found that the growth of mutant cells was slower and the proliferation was inhibited, when compared to wild type cells. Apoptosis of breast cancer cells was induced and cell viability decreased. However, the expression of the AKT1 E17K mutation gene promoted the migratory ability of breast cancer cells.
From the results of inhibiting cell growth, inhibiting proliferation, inducing apoptosis, and decreasing cell viability, AKT1 is defined as a carcinogenic gene, but one of its mutation hotspots E17K does not have a definable carcinogenic effect. On the contrary, it appears to have an anti-cancer effect. However, from the phenomenon of increasing the migration ability of breast cancer cells, this mutation hot spot has been implicated in tumorigenesis. Therefore, it can be concluded that this mutation hot spot in the AKT1 gene plays a dual role in cancer.
A study by Bodour Salhi suggests that E17K is largely not pathogenic in non-tumorigenic breast cells. In other words, AKT1 E17K cannot be identified as a carcinogen from all breast cell types. Bodour Salhi demonstrated that E17K was almost entirely present in luminal derived breast cancers and our results of cell survival and immunofluorescence show that, when compared with the cells expressing the wild type AKT1, the growth of mammary cells induced by E17K was reduced by 2 or 3 fold.
However, morphologic changes occurred in breast epithelial cells [11]. DDX21, a nucleolar RNA helicase 2 encoded by the DDX21 gene in the human body, is a DEAD-box protein capable of inducing abnormal expression of an RNA helicase, and a DDX21 immunoblot test confirmed that the expression of DDX21 in mammary cells was downregulated. Bodour Salhi inferred from their experiments that the main mechanism of action is the destruction of the cell transcription mechanism by E17K mutation, which leads to initiation of translation and abnormal function of the ribosome and splice assembly, thus inhibiting cell growth. In the luminal cells of the human mammary gland, the expression of the AKT1 E17K gene can inhibit cell growth, migration, and protein synthesis, and play an anti-tumour role. Due to the decrease of P70S6K (P70 ribosomal protein S6 kinase) protein synthesis and down-regulation of phosphorylation, the AKT signal level connectivity upstream of AKT is blocked, and the downstream mTOR pathway is also naturally suppressed, so it is ultimately related to the overall survival rate and prognosis of patients [11].
Another study (Mancini) found that all genetically engineered mice expressing AKT1 (E17K) developed only breast hyperplasia and did not develop cancer. It is suggested that part of the reason why AKT1 (E17K) can prevent the formation of HER2 positive breast tumors is the negative feedback signal inhibition of RTK tyrosine kinase [16]. Zilberman’s study found an AKT1-E17K mutation in young urothelial carcinoma patients with a favorable prognosis [17]. Therefore, it is assumed that tumors carrying the AKT1 E17 K mutations tend to be less invasive with an improved prognosis. By observing animals and cell lines, Viglietto found that mutations further enhanced AKT2’s motility, invasion, and metastasis, while AKT1 did not [18].
The AKT1 E17K somatic cell mutation was first found in breast cancer, but has also been identified in lung, bladder, endometrium, prostate, and other cancers [19]. At present, it has been found in 4%-8% of breast cancer patients [14]. The study of Bleeker in human solid tumors suggests that the mutation of the AKT1 E17K allele in breast cancer is tissue-specific, existing only in ducts and lobular tissues, but not in mucous and medullary mammary tissues. The mutants of AKT1 E17K were mutually exclusive, compared to PIK3CA E454K or H1047R. Large-scale genome sequencing analysis of human cancer has identified functional acquired mutations of AKT1 in various tumor types. AKT1 E17K is one of the most common kinase domain point mutations [8]. Other studies have also gradually identified point mutations in the kinase domain of AKT1, AKT2, and AKT3. E17K somatic mutations have also been found in AKT2 of breast cancer and AKT3 of melanoma. AKT1 Q79K is the second most common point mutation after E17K [20].
At present, tumor targeting therapy is one of the most promising treatments for cancer [13], which refers to the treatment of drugs that specifically bind to carcinogenic genes or proteins at a molecular level, and preferentially kill tumor cells without damaging normal cells [21,22]. AZD5363, a targeted inhibitor of AKT, has used AKT (AKT1/AKT2/AKT3) as a target for effective treatment in a variety of cancers [20]. The treatment of AZD5363 can lead to a persistent response and tumor regression in a variety of tumor types such as breast cancer (ER-positive and triple-negative type), endometrial cancer, cervical cancer, and lung cancer [23,24]. However, studies have shown that direct or indirect inhibition of intracavitary E17K function in patients with breast cancer is not necessarily an effective strategy. This mutation has dual effects, such as anti-tumor effects (inhibition of cell growth and protein synthesis), and carcinogenic effects, promoting cell migration [11]. Therefore, the effect and mechanism of E17K mutation on tumorigenesis should be further analyzed.
Contemporary cancer care has entered the stage of individualized treatment and precision medicine. The use of molecular detection and individualized targeted therapy [25] is a promising avenue to combat the heterogeneous nature of breast cancer, improving the accuracy of treatment and reducing adverse reactions [26]. The somatic mutation of breast cancer patients was detected by gene sequencing, and the correct targeted treatment is conducive to improving the cure rate, as well as the survival and prognosis rates of patients [27], so as to achieve individualized, accurate treatment.
Acknowledgements
This study was supported by the National Natural Science Foundation of Guangdong Province (grant No. 2018A030313114), the Natural Science Foundation of Guangdong Province (grant No. 2018A030313860), the National Natural Science Foundation of China (grant No. 81703053, No. 31300737), the National Natural Science Foundation of China (grant No. 21771042), the Natural Science Foundation of Guangdong Province (grant No. 2016A030310298), Innovation and university promotion project of Guangdong Pharmaceutical University through No. 2017KCXTD020, 2017KZDXM049. Experiment and Manuscript’s preparation supported from Guangdong Province Key Laboratory for Biotechnology Drug Candidates, School of Life Sciences and Biopharmaceutics, Guangdong Pharmaceutical University. Thanks for the guidance of my tutor (professor Han Shen).
Disclosure of conflict of interest
None.
Abbreviations
- AKT1 E17K
AKT1 gene and the 17th amino acid translated by AKT1 gene was transformed from glutamic acid (E) to lysine (K)
- FBS
Fetal bovine serum
- MTT
trypsin, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- OD
the optical density
- DMSO
dimethylsulfoxide
- DMEM
dulbecco’s modified eagle medium
- TCGA
American Cancer Gene Database
- CFSE
CFDA-SE
- CFDA-SE
Carboxyfluorescein diacetate, succinimidyester
- PI dye
Propidium Iodide
- OS
overall survival rate
- PI
proliferation Index
- DDX21
a nucleolar RNA helicase 2 encoded by the DDX21 gene in the human body
- RTK
tyrosine kinase
- AZD5363
a targeted inhibitor
- DEAD-box
DEAD-box family proteins are a class of ATP-dependent RNA helicase
References
- 1.Ma YF, Wu T, Guo Y, Nan K, Huang X. Based on oncomine and TCGA database, the expression and prognostic significance of p57, CKS1 and SKP2 in hepatocellular carcinoma were explored. Clinical Medical Research and Practice. 2016;1:1–4. [Google Scholar]
- 2.Wang C, Hu H, Lv J, Zhang Y. Breast cancer invasion related molecules research hotspot. Journal of Southeast University (Medical Edition) 2018;37:734–738. [Google Scholar]
- 3.Ye ZL, Qiu MZ, Tang T, Wang F, Zhou YX, Lei MJ, Guan WL, He CY. Gene mutation profiling in Chinese colorectal cancer patients and its association with clinicopathological characteristics and prognosis. Cancer Med. 2020;9:745–756. doi: 10.1002/cam4.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Efremova M, Finotello F, Rieder D, Trajanoski Z. Neoantigens generated by individual mutations and their role in cancer immunity and immunotherapy. Front Immunol. 2017;8:1679. doi: 10.3389/fimmu.2017.01679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang S, Li Z, Zhen C. Expression and clinical significance of AKT3 in gastric cancer were analyzed by TCGA data set. Journal of China Medical University J China Med Univ. 2016;45:398–401. [Google Scholar]
- 7.Jungwirth G, Warta R, Beynon C, Sahm F, von Deimling A, Unterberg A, Herold-Mende C, Jungk C. Intraventricular meningiomas frequently harbor NF2 mutations but lack common genetic alterations in TRAF7, AKT1, SMO, KLF4, PIK3CA, and TERT. Acta Neuropathol Commun. 2019;7:140. doi: 10.1186/s40478-019-0793-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yesiloz U, Kirches E, Hartmann C, Scholz J, Kropf S, Sahm F, Nakamura M, Mawrin C. Frequent AKT1E17K mutations in skull base meningiomas are associated with mTOR and ERK1/2 activation and reduced time to tumor recurrence. Neuro Oncol. 2017;19:1088–1096. doi: 10.1093/neuonc/nox018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang J, Hu S, Wang C, Song J, Chen C, Fan Y, Ben-David Y, Pan W. Fangchinoline derivatives induce cell cycle arrest and apoptosis in human leukemia cell lines via suppression of the PI3K/AKT and MAPK signaling pathway. Eur J Med Chem. 2020;186:111898. doi: 10.1016/j.ejmech.2019.111898. [DOI] [PubMed] [Google Scholar]
- 10.Carpten JD, Faber AL, Horn C, Donoho GP, Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage S, Uhlik M, Lin A, Du J, Qian YW, Zeckner DJ, Tucker-Kellogg G, Touchman J, Patel K, Mousses S, Bittner M, Schevitz R, Lai MH, Blanchard KL, Thomas JE. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 11.Salhia B, Van Cott C, Tegeler T, Polpitiya A, Duquette RA, Gale M, Hostteter G, Petritis K, Carpten J. Differential effects of AKT1(p.E17K) expression on human mammary luminal epithelial and myoepithelial cells. Hum Mutat. 2012;33:1216–1227. doi: 10.1002/humu.22100. [DOI] [PubMed] [Google Scholar]
- 12.Williams SR, Juratli TA, Castro BA, Lazaro TT, Gill CM, Nayyar N, Strickland MR, Babinski M, Johnstone SE, Frosch MP, Silverman IM, Ely HA, Kaplan AB, D’Andrea MR, Bihun IV, Hoang K, Batchelor E, Christiansen J, Cahill DP, Barker FG 2nd, Brastianos PK. Genomic analysis of posterior fossa meningioma demonstrates frequent AKT1 E17K mutations in foramen magnum meningiomas. J Neurol Surg B Skull Base. 2019;80:562–567. doi: 10.1055/s-0038-1676821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rudolph M, Anzeneder T, Schulz A, Beckmann G, Byrne AT, Jeffers M, Pena C, Politz O, Kochert K, Vonk R, Reischl J. AKT1 (E17K) mutation profiling in breast cancer: prevalence, concurrent oncogenic alterations, and blood-based detection. BMC Cancer. 2016;16:622. doi: 10.1186/s12885-016-2626-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Ni J, Song D, Ding M, Huang J, Li W, He G. MAT1 facilitates the lung metastasis of osteosarcoma through upregulation of AKT1 expression. Life Sci. 2019;234:116771. doi: 10.1016/j.lfs.2019.116771. [DOI] [PubMed] [Google Scholar]
- 15.Zeng X, Zhang Y, Kwong JS, Zhang C, Li S, Sun F, Niu Y, Du L. The methodological quality assessment tools for preclinical and clinical studies, systematic review and meta-analysis, and clinical practice guideline: a systematic review. J Evid Based Med. 2015;8:2–10. doi: 10.1111/jebm.12141. [DOI] [PubMed] [Google Scholar]
- 16.Mancini ML, Lien EC, Toker A. Oncogenic AKT1(E17K) mutation induces mammary hyperplasia but prevents HER2-driven tumorigenesis. Oncotarget. 2016;7:17301–17313. doi: 10.18632/oncotarget.8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zilberman DE, Cohen Y, Amariglio N, Fridman E, Ramon J, Rechavi G. AKT1 E17 K pleckstrin homology domain mutation in urothelial carcinoma. Cancer Genet Cytogenet. 2009;191:34–37. doi: 10.1016/j.cancergencyto.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 18.Viglietto G. Activating E17K mutation in the gene encoding the protein kinase AKT1 in a subset of squamous cell carcinoma of the lung. Cell Cycle. 2009;8:2869–2870. [PubMed] [Google Scholar]
- 19.Huo X, Sun H, Liu Q, Ma X, Peng P, Yu M, Zhang Y, Cao D, Shen K. Clinical and expression significance of AKT1 by co-expression network analysis in endometrial cancer. Front Oncol. 2019;9:1147. doi: 10.3389/fonc.2019.01147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brose MS, Cabanillas ME, Cohen EE, Wirth LJ, Riehl T, Yue H, Sherman SI, Sherman EJ. Vemurafenib in patients with BRAF(V600E)-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: a non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17:1272–1282. doi: 10.1016/S1470-2045(16)30166-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang L, Wang l. Common gene mutations in human tumors and the significance of molecular targeted therapy. Theory and Practice of Internal Medicine. 2015;10:221–225. [Google Scholar]
- 22.Zhu Z, Shukla A, Ramezani-Rad P, Apgar JR, Rickert RC. The AKT isoforms 1 and 2 drive B cell fate decisions during the germinal center response. Life Sci Alliance. 2019;2 doi: 10.26508/lsa.201900506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hyman DM, Smyth LM, Donoghue MTA, Westin SN, Bedard PL, Dean EJ, Bando H, El-Khoueiry AB, Perez-Fidalgo JA, Mita A, Schellens JHM, Chang MT, Reichel JB, Bouvier N, Selcuklu SD, Soumerai TE, Torrisi J, Erinjeri JP, Ambrose H, Barrett JC, Dougherty B, Foxley A, Lindemann JPO, McEwen R, Pass M, Schiavon G, Berger MF, Chandarlapaty S, Solit DB, Banerji U, Baselga J, Taylor BS. AKT inhibition in solid tumors with AKT1 mutations. J. Clin. Oncol. 2017;35:2251–2259. doi: 10.1200/JCO.2017.73.0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thirumal Kumar D, Jain N, Evangeline J, Kamaraj B, Siva R, Zayed H, George Priya Doss C. A computational approach for investigating the mutational landscape of RAC-alpha serine/threonine-protein kinase (AKT1) and screening inhibitors against the oncogenic E17K mutation causing breast cancer. Comput Biol Med. 2019;115:103513. doi: 10.1016/j.compbiomed.2019.103513. [DOI] [PubMed] [Google Scholar]
- 25.Beaver JA, Gustin JP, Yi KH, Rajpurohit A, Thomas M, Gilbert SF, Rosen DM, Ho Park B, Lauring J. PIK3CA and AKT1 mutations have distinct effects on sensitivity to targeted pathway inhibitors in an isogenic luminal breast cancer model system. Clin Cancer Res. 2013;19:5413–5422. doi: 10.1158/1078-0432.CCR-13-0884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang J. Application of molecular diagnostic technology and targeted therapy in individualized treatment of breast cancer. Shanghai Medical and Pharmaceutical Journal. 2016;37:10. [Google Scholar]
- 27.Zhou X, Nie Q. Bioinformatics methods: to screen mirnas related to early diagnosis of primary liver cancer. J Gastroenterol Hepatol. 2018;27:609–612. [Google Scholar]