

Abstract
Dinaciclib is a small molecule cyclin-dependent kinase inhibitor with the potential to treat multiple cancers. To better understand its cytotoxic action in pancreatic ductal adenocarcinoma (PDAC), we evaluated dinaciclib therapeutic effects in the transgenic mouse model (LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx-1-Cre mice; KPC mice). Tumor growth and microenvironment were dynamically monitored by magnetic resonance imaging (MRI). Dinaciclib therapy significantly delayed tumor progression (P < 0.001) and prolonged survival (P = 0.007) in KPC mice. In vitro assays showed that dinaciclib exerted antiproliferative effects on PDAC cells by increasing surface calreticulin expression and release of ATP. Dinaciclib treatment inhibited proliferation and induced apoptosis in KPC tumor as assessed by Ki67 and cleaved caspase 3, respectively. Particularly, the tumor infiltrating CD8+ T cells were increased after dinaciclib treatment in KPC mice. Additionally, the mean apparent diffusion coefficient values of KPC tumor calculated from diffusion weighted MR images were significantly lower after dinaciclib treatment (P = 0.033). These finding suggest that dinaciclib as a single agent can inhibit tumor growth and improve the overall survival in KPC mice.
Keywords: Pancreatic ductal adenocarcinoma, dinaciclib, magnetic resonance imaging, apoptosis
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the most common form of pancreatic cancer, with a median survival of 6 months [1]. PDAC is predicted to become the second leading cause of cancer-related deaths in the United States by 2030 [2]. Hence, there is an urgent need to develop new and effective therapeutic approaches to treat this lethal cancer.
Analyses of human PDAC samples have revealed that TP53 mutation occurs in 50-75% of PDACs following a universal initiating activating mutation in the KRAS gene [3,4]. Activating mutations in the Kras proto-oncogene is one of the most frequent genetic events driving development of human pancreatic low grade precursor lesions (pancreatic intraepithelial neoplasia -PanIN-) [4,5]. Mouse models that endogenously express the mutant Kras in pancreatic exocrine lineages can recapitulate human PanIN [6], but progression to PDAC requires additional genetic events. Remarkably, concurrent genetic inactivation of the Trp53 tumor suppressor gene substantially accelerates the PDAC development in the context of Kras mutation [7,8]. Different pancreatic cancer transgenic mouse models (e.g. KrasG12D; Cre model, KrasG12D; Trp53flox; Cre model, and KrasG12D; Trp53flox/flox; Cre or KrasG12D; Trp53mut/mut; Cre model) have been created for studying the progression of PDAC and for identifying therapeutic targets [9]. Here, we focused in particular on the KPC mouse model, one of the most commonly studied preclinical transgenic mouse models of PDAC, that was established by targeting endogenous expression of oncogenic KrasG12D and mutant Trp53R172H to the mouse pancreas using Cre-loxP technology. KPC transgenic mice stochastically form pre-invasive PanIN lesions that eventually evolve into invasive PDAC with 100% penetrance [7,10].
The cyclin-dependent kinases (CDKs), a family of serine/threonine kinases, regulates cell cycle progression. Aberrant CDK activity as well as cell cycle dysregulation is a hallmark of many tumor cells [11]. As such, inhibition of CDK activities represents a potential therapeutic strategy for cancer treatment. CDK inhibitor dinaciclib, also known as SCH 727965 and MK-7965, is a potent CDK2, CDK5, CDK1, and CDK9 inhibitor in vitro with IC50 values of 1, 1, 3, and 4 nmol/L, respectively [12]. It showed promising results in preclinical trials for a range of solid and hematologic malignancies [13-16]. More importantly, a recent study demonstrated that dinaciclib induced a type I IFN gene signature and immunogenic cell death (ICD) in immunocompetent mouse tumor models [17]. ICD response is characterized by apoptotic cell death accompanied by the expression of calreticulin (CRT) on dying tumor cell surfaces, which can re-initiate immune responses suppressed by the tumor microenvironment [18]. Immunologically, the PDAC microenvironment is considered especially suppressive. While several previous studies showed that treatment with dinaciclib effectively inhibited growth and progression of pancreatic cancer xenografts in immunodeficient mice [19-21], the therapeutic role for dinaciclib in PDAC has not yet been fully clarified.
Assessment of response to cancer therapy in pancreatic cancer can often be performed using magnetic resonance imaging (MRI) which allows non-invasive evaluation of tumors over time [22,23]. Moreover, diffusion-weighted MRI (DW-MRI) can qualitatively and quantitatively assess the molecular function and micro-architecture of solid tumors [24]. Several studies have shown that the apparent diffusion coefficient (ADC) values measured from DW-MRI can serve as an early imaging biomarker for predicting tumor response to chemotherapy for pancreatic cancer [25,26].
In this study, the KPC mice were used to evaluate the therapeutic effects (including overall survival (OS)) of dinaciclib in PDAC. MRI was used to dynamically quantify and serially assess the therapeutic responses in pancreatic tumor growth and cellularity during dinaciclib treatment. Overall, our results revealed that dinaciclib therapy significantly improved the OS of treated KPC mice and supported clinical practice in this context.
Material and methods
Mice and regents
All animal experiments were approved by the Institutional Animal Care and Use Committee of the Northwestern University and conducted in compliance with the National Institute of Health guidelines for animal research. KrasLSL-G12D/+, Trp53LSL-R172H/+, and pdx1-Cre mice strains, purchased from Jackson Laboratory (Bar Harbor, ME), were interbred to generate KPC mice, as previously described [7]. KPC mice (3-6 months old) with spontaneous pancreatic cancer were used. Tumor growth and development were outlined by serial MRIs as outlined below.
Dinaciclib was purchased from Selleckchem. Inc. (Houston, TX) and formulated in the vehicle 20% hydroxypropyl β cyclodextrin (Sigma-Aldrich, St. Louis, MO). Doxorubicin (DOX) was purchased from Selleckchem. Inc. (Houston, TX).
Cell lines
KPC cells were derived from spontaneous pancreatic tumors originating in KPC mice (6-month-old) as previously described [27,28]. Single-cell suspensions from tumor fragments were plated on collagen in DMEM/F12 (50:50) medium supplemented 5% Nu-serum IV, 25 μg/mL bovine pituitary extract, 20 ng/mL epidermal growth factor, 0.5% ITS+ Premix, 100 ng/mL cholera toxin, and 1 μM dexamethasone. After 3 passages, the KPC cells were maintained in DMEM supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM L-glutamine. KPC cells did not surpass 12 passages between thawing and use. A murine PDAC cell line Pan02 cell line was obtained from ATCC (Rockville, MD) and was cultured in complete RPMI 1640 medium containing 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM L-glutamine.
Cell viability assay
Cell Counting Kit-8 (CCK-8) (Dojindo Laboratories, Rockville, MA) was used to conduct the cell viability assay. Cells were seeded in 96-well clear-bottom plates starting from 1 × 104 cells per well. After changing the media 24 h later, the cells were treated with different doses of dinaciclib and 1 μM Dox for 24 h. A mixture of 190 μL of OPTI-MEM and 10 μL of CCK-8 was then added into each well. Two hours later, the absorbance at 450 nm was measured.
CRT expression from the cell lines
Pan02 cells and KPC cells were seeded on microscope coverslips (Thermo, Rockford, IL) in 24-well plates. Each well contained 2 × 104 cells in 1 mL of culture medium. After incubation with 1 μM dinaciclib or 1 μM DOX for 4 h, cells were then fixed and washed 3 times followed by permeabilization with 0.1% TX-100. After incubating for 60 min with blocking buffer, cells were stained with Alexa Fluor® 488-conjugated wheat germ agglutinin (WGA) (Thermo, Waltham, MA) to visualize the cell surface membrane. Then the cells were stained with an anti-CRT antibody (ab2907, 1/500, Abcam, Cambridge, MA) at 4°C overnight followed by Alex-fluor®555 conjugated secondary antibodies (Life technology, Carlsbad, CA). After washing 3 times with PBS, slides were mounted with Hoechst 33,342 nuclear dye. Finally, all images were obtained with a Nikon confocal microscope. The MFI of immunofluorescent images were quantified by ImageJ 1.52a software (http://imagej.nih.gov/ij).
Apoptosis assays
To analyze apoptosis, the cells were seeded into 6-well plates. After growing to 70% to 80% confluence, cells were cultured in increasing doses of dinaciclib for 24 h, as indicated. Dinaciclib-induced PDAC cell death was assessed by flow cytometry using the Annexin V-Propidium Iodide Apoptosis Detection Kit (Life technology, Carlsbad, CA) according to the manufacturer’s instructions.
ATP release assays
Supernatants were harvested from Dinaciclib-treated tumor cells. Extracellular ATP levels were measured by ENLITEN ATP Assay System Bioluminescence Detection Kit for ATP (Promega, Madison, WI) using a Multi-Mode Plate Reader (Thermo Fisher Scientific, Waltham, MA).
Monitoring of tumor growth and therapy
MRI was performed on 3-6 month-old KPC mice on a 7.0-Tesla scanner (Clinscan, Bruker BioSpin, Ettlinggen, Germany) as described previously [29]. KPC transgenic mice underwent treatment or control treatments when the longest tumor diameter reached 0.2-0.5 cm on MRI. Dinaciclib (40 mg/kg) was administrated intraperitoneally twice a week for three weeks. OS was defined as the number of days post injection until predefined endpoints requiring euthanasia have been met such as > 20% weight loss, extreme lethargy, decreased mobility, tumor size > 2 cm, or moribund status.
The MRI sequences and parameters were as follows [29]: (a) coronal T2W images: TSE; TR: due to respiratory gating approx. 2100 ms; TE: 40 ms; ST: 0.5 mm; FA: 180; FOV: 40 × 30 mm2; (b) axial T2-weighted imaging (T2WI): TSE; TR: due to respiratory gating approx. 2100 ms; TE: 40 ms; ST: 0.5 mm; FA: 180º; FOV: 21 × 30 mm2; (c) axial diffusion-weighted imaging (DW-MRI): Echo Planar Imaging (EPI); TR: due to respiratory gating approx. 2700 ms; TE: 40 ms; ST: 1 mm; FA: 90º; FOV: 24 × 30 mm2; b value = 0, and 800 s/mm2. DW-MRI was performed in 3 orthogonal directions of the diffusion gradients. ITK-SNAP was used for volume calculations and 3D reconstruction. MATLAB R2018a (Mathworks, Natick, MA) was used for image processing. ImageJ 1.52a was used for ADC calculations.
Histology analysis and immunohistochemistry
Histology analysis including H&E, Masson’s Trichrome, anti-mouse cytokeratin 19 (CK19) (Developmental Studies Hybridoma Bank, Iowa City, IA), anti-mouse cleaved caspase 3, rat monoclonal anti-mouse CD8 (Clone 4SM15, Invitrogen), and rabbit monoclonal anti-Ki67 (clone SP6) (Invitrogen, Carlsbad, CA) was performed as described previously [29]. Briefly, tissue was fixed with 10% formalin embedded in paraffin. 4-μm sections were deparaffinized and rehydrated. Heat-induced antigen retrieval was performed 10 mmol/L citrate buffer (pH 6.0). After quenching endogenous peroxidase activity with H2O2 and blocking with 10% horse serum and 0.1% Triton-X100 in PBS, the sections were then incubated with the primary antibodies overnight at 4°C. The sections were incubated with polymer-HRP-conjugated second antibodies, followed by DAB reagents (Vector, Burlingame, CA). All stained slides were scanned by 20 × magnification and quantified with ImageJ 1.52a software.
Statistical analysis
All statistical analyses and graphing were performed using GraphPad Prism 7.0 (La Jolla, CA). A P value less than 0.05 was considered significant. At least 3 independent experiments were performed for in vitro experiments. Significant between groups of multiple samples from the same source were determined using one-way ANOVA. Difference between two groups was analyzed by Student t-tests. The OS was assessed using the Kaplan-Meier method, and the survival difference between groups was compared using the log-rank test.
Results
Dinaciclib induces immunogenic death of PDAC cells in vitro
To analyze the cytotoxic effect of dinaciclib in PDAC cell lines, we chose KPC and Pan02 cell lines. We observed that dinaciclib induced both KPC and Pan02 cell apoptosis in vitro (Figures 1A, S1). Our results also showed that dinaciclib treatment significantly inhibited cell proliferation in a dose-dependent manner in both KPC and Pan02 cell lines (Figures 1B, S1). One previous study has reported that tumor cells treated with dinaciclib showed increased expression of the hallmarks of ICD including surface CRT and release of ATP [17]. To test this effect in PDAC cell lines, we initiated a screen for CRT expression in KPC and Pan02 cell lines after dinaciclib treatment. Dinaciclib treatment induced a high level of CRT expression on the surface of both KPC cells and Pan02 cells (Figure 1C-F), which is similar to the effect produced by Doxorubicin (DOX). DOX, the classical example of inducing an ICD response, was used as positive control [30]. We also detected a dose-dependent secretion of ATP in both KPC cells and Pan02 cells by dinaciclib (Figures 1G and S1D). These results together suggested that dinaciclib treatment potently induced an ICD effect in PDAC cell line.
Figure 1.

Dinaciclib induced immunogenic cell death in PDAC cells. (A) Representative Annexin V versus PI flow cytometry plots of KPC cells after dinaciclib or PBS treatment for 6 h. (B) KPC cells were treated with increasing concentrations of dinaciclib and 1 μM DOX for 24 h. Cell viability was then measured by the CCK-8 assay. (C) Confocal microscopy showing the induction of the ICD marker, CRT, in Pan02 cells in the presence of PBS, Dinaciclib (1 μM), and DOX (1 μM) for 4 h, and (E) corresponding quantification of the mean immunofluorescent intensity. Scale bar is 20 μm. (D) Confocal microscopy showing the induction of the ICD marker, CRT, in KPC cells in the presence of PBS, Dinaciclib (1 μM), and DOX (1 μM) for 4 h, and (F) corresponding quantification of the mean immunofluorescent intensity. Scale bar is 20 μm. (G) KPC cells were treated with dinaciclib at the indicated concentrations for 24 h. Graphical data show the release of ATP into the culture supernatants. *, P < 0.05, **, P < 0.01, ***, P < 0.001.
Dinaciclib inhibits tumor growth and prolongs survival in KPC mice
To investigate the antitumor effect of dinaciclib in PDAC, we treated the KPC genetic mice with pancreatic tumors that measured 2-5 mm in diameter on MRI (Figure 2A). Interestingly, dinaciclib treatment significantly delayed tumor growth compared with control group (Figure 2B, 2C). In addition, we evaluated the OS of KPC mice in different groups. Treatment with dinaciclib prolonged the OS of KPC mice from 31 to 57 days (P < 0.01 (Median)) (Figure 2D). Taken together, these results confirmed that dinaciclib can potentially elicit tumor specific antitumor immunity in KPC mice.
Figure 2.
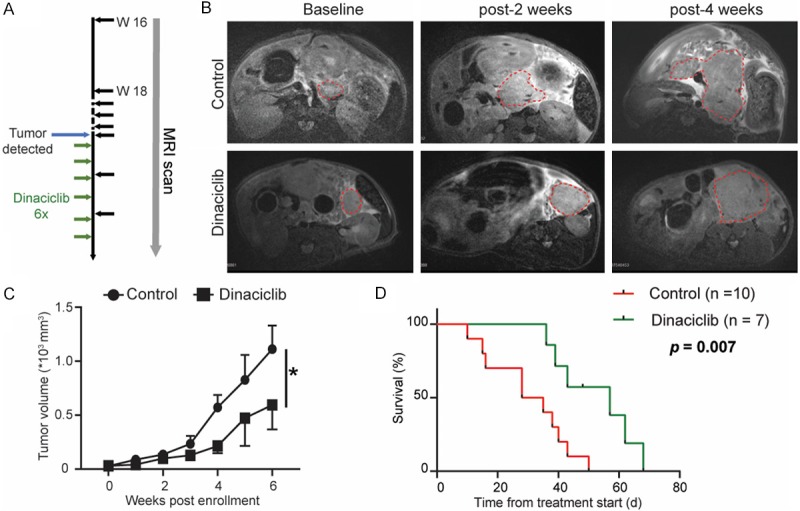
Dinaciclib inhibits tumor growth and prolongs OS in KPC mice. A. Experimental schemes for the MRI scan and treatment of the KPC transgenic mice. B. Representative T2W images at different time points from KPC genetic model of PDAC in control or treated groups. C. Measurement of tumor volumes in KPC transgenic mice (n = 7-8). D. Survival rate of treated KPC transgenic mice. Dinaciclib confers survival advantage to the treated animals (P = 0.007, log-rank test). *, P < 0.001.
Dinaciclib decreases tumor fibrosis in KPC mice
High amounts of fibrosis characterize the tumor microenvironment of human PDAC. We next examined the effect of dinaciclib treatment on fibrosis formation, reflected by collagen deposition as detected by Masson-trichrome staining. We observed that dinaciclib-treated KPC model of PDAC had significantly decreased levels of collagen deposition throughout the pancreas compared with control mice (Figure 3A, 3B).
Figure 3.
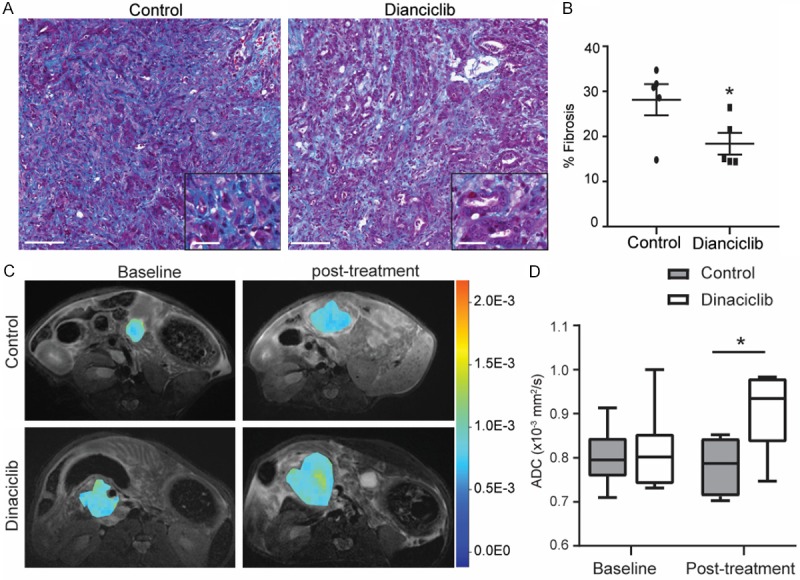
Dinaciclib treatment alters tumor fibrosis and mean ADC values in KPC mice. A. Representative images of trichrome staining for collagen connective tissue deposition in PDAC from treated and control KPC genetic model of PDAC. Scale bars represent 100 μm (inset, 25 μm). B. Relative quantification of collagen stained area for each group. C. Representative ADC pseudocolor maps of the tumor overlaid onto a T2W image from KPC genetic model of PDAC in treated and control cohorts are shown. D. ADC values measured from KPC genetic model of PDAC in treated and control groups at baseline and 3 weeks post treatment. n = 6. *, P < 0.05.
In order to facilitate clinical translation of dinaciclib, we utilized the ADC values calculated from DW-MRI as it is clinically applicable [25,26,31]. Dinaciclib therapy induced a significant increase in tumor mean ADC values, suggesting altered microstructure was induced after the treatment (Figure 3C, 3D). Taken together, these results suggest that dinaciclib treatment reshaped PDAC tumor-induced fibrosis.
Treatment with dinaciclib delays PDAC progression and induces apoptosis
Histological examination of the pancreatic tumor tissues showed the presence of more normal-looking tissue with a desmoplastic reaction in dinaciclib treated KPC mice compared with control group as evaluated by H&E staining (Figure 4A). Importantly, the area of ductal marker CK19 positive structures was larger in the pancreas of dinaciclib treated KPC mice than in control group (Figure 4A, 4B), again corroborating a less extensive desmoplastic transformation after dinaciclib therapy.
Figure 4.
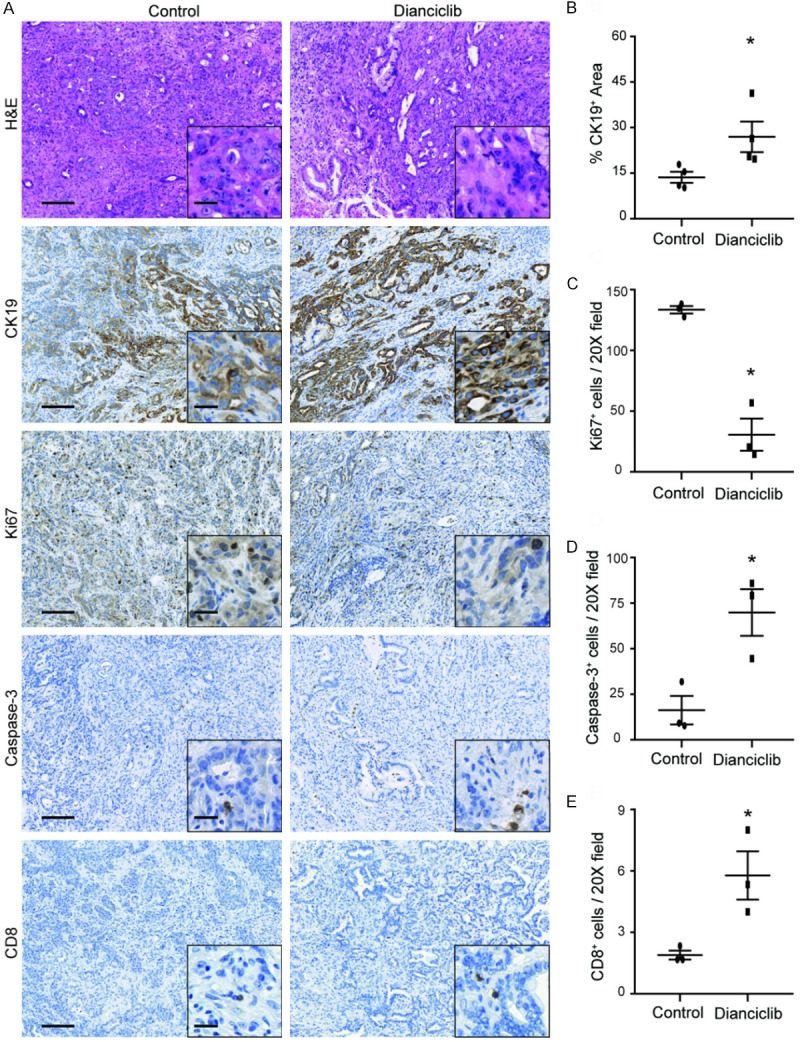
Dinaciclib therapy suppresses PDAC progression and induces apoptosis in KPC mice. A. Representative images of H&E and immunohistochemical analyses in PDAC from KPC mice in treated and control groups are shown. Scale bars represent 100 μm (inset, 20 μm). B. Quantification of CK19 staining from treated and control KPC mice. C. Quantification of cleaved caspase-3 staining from treated and control KPC mice. D. Quantification of stromal and tumor Ki67+ cells from treated and control KPC mice. E. Quantification of stromal and tumor CD8+ T cells from treated and control KPC mice n = 3-4. *, P < 0.05.
To further evaluate whether the induction of apoptosis after dinaciclib treatment occurs in vivo, KPC tumor samples underwent IHC evaluation. Increased PDAC cell apoptosis was observed in dinaciclib treatment KPC mice compared with control mice (Figure 4A, 4C), as assessed by immunohistochemistry for cleaved caspase 3. Cell proliferation assessed by measuring Ki67 levels also revealed a significant decrease in the dinaciclib group compared to control group (Figure 4A, 4D). These data, demonstrating both induction of apoptosis and reduction of cell proliferation, were consistent with the tumor growth delay results shown earlier. Additionally, the tumor infiltrating CD8+ T cells were significantly increased in treated group than in control group (Figure 4A, 4E), further indicating that dinaciclib treatment induced an immune response in KPC mice.
Discussion
In this study, we demonstrated that dinaciclib can exert potent antitumor effect in the KPC genetic model of PDAC. The results showed that dinaciclib can induce an ICD response in PDAC cells, as reflected by enhanced expression of CRT and release of ATP. Furthermore, the dinaciclib treated KPC mice also showed a higher percentage of apoptotic cells, a lower percentage of proliferating cell, and a higher percentage of tumor infiltrating CD8+ T cells than the control group. Moreover, we also observed an inhibition of tumor growth and a significant reduction of average tumor ADC values after dinaciclib therapy in KPC mice. Importantly, a significant improvement of OS was observed in this study.
Dinaciclib is one of the first drugs in its class that exerts antitumor effects as a single agent in PDAC cells [19,32]. However, the mechanism underlying its antitumor activity has not been fully elucidated yet. Hu C et al. evaluated the antitumor effect of dinaciclib in several different patient-derived pancreatic cancer xenograft models in 2015 and showed that dinaciclib can inhibit proliferation and induce apoptosis [20]. Accordingly, we demonstrated that administration of dinaciclib alone has considerable inhibitory effects on tumor growth rate in the highly aggressive KC mouse model. KPC mice are excellent tools for assessing therapeutics and understanding mechanisms of drug action and resistance in PDAC. The KPC model has significant advantages over previously described xenograft and implantation models, including that: i) the KPC model recapitulates many of the clinical (e.g. ascites development, bowel and biliary obstruction, metastatic spread, and cachexia), histopathological (e.g. cellular morphology, poor vascularity, and fibrosis), and genetic (e.g. concomitant expression of oncogenic KrasG12D and of Trp53R172H) features of human disease; ii) the KPC mice are immunocompetent and provide a useful platform for studying changes in the tumor environment; and iii) low passage tumor cell lines can be easily derived from tumor-bearing KPC mice for use in in vitro and in vivo studies. Therefore, the KPC mouse is a better model than the subcutaneous and orthotopic models of PDAC. Although prior studies demonstrated that dinaciclib can inhibit PDAC tumor growth in different subcutaneous and orthotopic xenograft murine models, OS after dinaciclib treatment has never been assessed [19,20,32]. In this study, we showed that treatment with dinaciclib prolonged the OS of KPC mice from 31 to 57 days, which further confirmed the therapeutic effects of dinaciclib in PDAC.
Previous studies have suggested that dinaciclib is a pan-CDK-1/2/5/9 inhibitor that can induce apoptosis in different tumor cells, including PDAC cells [19,32-35]. Dinaciclib has been shown to repress transcription via inhibition of CDK9. One target gene of this repression is the Myc, which is amplified in several different cancer types including PDAC [36,37]. Moreover, a recent study found that tumor cells expressed the hallmarks of ICD including surface CRT expression and release of ATP after dinaciclib treatment [17]. Consistent with this, we showed that in vitro treatment with dinaciclib exerts proapoptotic effects in PDAC cells by increasing expression of CRT and ATP, the key damage-associated molecular patterns of ICD that play a beneficial role in antitumor effects [38-41]. Meanwhile, we found that the tumor infiltrating CD8+ T cells and cleaved caspase 3 positive apoptotic cells are increased after dinaciclib treatment in KPC tumor, which further confirmed the ICD response induced by dinaciclib. Dinaciclib also exerts its cytotoxic effects by inhibiting CDKs 1 and 2 [12]; these CDKs have been shown to be potential therapeutic targets for cancer therapy [42] and thus may also contribute to the antitumor efficacy we have shown. Furthermore, one recent study reported that inhibition of CDK by dinaciclib increases casein kinase 1δ activity in pancreatic cancer cells [43]. In addition, CDK5 signaling is critical for Ras signaling, which served as a pivotal determinant of the malignant phenotype in PDAC cells [44]. Hu C et al. demonstrated that the combination of dinaciclib and AKT inhibitor MK-2206 was quite effective in patient-derived pancreatic cancer xenograft model by blocking RAS/RAL activation and inhibiting PI3K/AKT pathway [20]. We hypothesize that CDK inhibition induced by dinaciclib synergizes with the ICD response to generate antitumor effects in PDAC. Addressing the antitumor mechanisms of dinaciclib monotherapy could translate into novel rational combinatorial approaches for patients. Combination of dinaciclib with other chemotherapeutics like epirubicin and PARP1/2 inhibitor ABT-888 have been shown to be more effective than monotherapy on malignancies [20,45]. Further studies are needed to more carefully and closely evaluate the regulatory mechanisms of dinaciclib on PDAC apoptosis. Understanding tumor mechanisms will also enable the development of rational combinations with cancer immunotherapy, including immune-checkpoint blockade, adoptive T-cell strategies, and dendritic cell vaccines, to overcome suppressive tumor microenvironment to induce effective long-term immunity.
Dinaciclib has been approved to enter clinical trials against multiple types of malignant diseases, including PDAC [13-16,46], but its antitumor efficacy may be limited by the increasing toxicity. Thus, further assessment and optimization of therapeutic strategies are required in preclinical and clinical settings. Herein, we used DW-MRI as an imaging biomarker for early evaluation of the treatment efficacy as it has been found capable of predicting tumor response to chemotherapy for pancreatic cancer [25,26,31,47]. DW-MRI is a highly sensitive imaging technique capable of quantifying and detecting macromolecular and microstructural alterations that impact microscopic water diffusion within tissues [48]. In this study, we showed that DW-MRI is useful for early detection of increases in tumor mean ADC values in KPC mice treated with dinaciclib. The increased mean ADC values indicated that dinaciclib-induced tumor cell apoptosis was achieved in KPC mice. Thus, integration of DW-MRI into a clinical PDAC trial involving evaluation of chemotherapy such as dinaciclib would provide worthful information for assessing therapeutic response.
There are some limitations in this study. One limitation is that we only investigated the ICD responses induced by dinaciclib, and we have not yet evaluated the antitumor effects of CDK inhibition on PDAC, which will need to be elucidated in future studies. Second, in this study, no complete tumor elimination was observed in KPC mice bearing established highly invasive tumors, underlining that dinaciclib combination therapies may be important for achieving durable tumor regression in PDAC. Further studies are needed to clarify whether these combination approaches can induce complete and durable tumor regression in PDAC.
In conclusion, our study demonstrated that dinaciclib as a single agent can inhibit tumor growth and improve the OS in KPC mice.
Acknowledgements
The authors thank the staff of the Pathology Core Facility at the Northwestern University for histology analysis. We also thank Matteo Figini for assistance with the MR imaging studies. This study was supported by the National Cancer Institute (grants R01CA209886, R01CA196967), by 2019 Harold E. Eisenberg Foundation Scholar Award and by the Fishel Fellowship Award at the Robert H. Lurie Comprehensive Cancer Center.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016;388:73–85. doi: 10.1016/S0140-6736(16)00141-0. [DOI] [PubMed] [Google Scholar]
- 2.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–2921. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 3.Scarpa A, Capelli P, Mukai K, Zamboni G, Oda T, Iacono C, Hirohashi S. Pancreatic adenocarcinomas frequently show p53 gene mutations. Am J Pathol. 1993;142:1534–1543. [PMC free article] [PubMed] [Google Scholar]
- 4.Matthias L, Gunter K, Patrick M, Albert BL, Jutta L. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: a meta-analysis. Neoplasia. 2005;7:17–23. doi: 10.1593/neo.04445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murphy SJ, Hart SN, Lima JF, Kipp BR, Klebig M, Winters JL, Szabo C, Zhang L, Eckloff BW, Petersen GM, Scherer SE, Gibbs RA, McWilliams RR, Vasmatzis G, Couch FJ. Genetic alterations associated with progression from pancreatic intraepithelial neoplasia to invasive pancreatic tumor. Gastroenterology. 2013;145:1098–1109. e1091. doi: 10.1053/j.gastro.2013.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, Wright CVE, Hruban RH, Lowy AM, Tuveson DA. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 7.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 8.Guerra C, Barbacid M. Genetically engineered mouse models of pancreatic adenocarcinoma. Mol Oncol. 2013;7:232–247. doi: 10.1016/j.molonc.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gopinathan A, Morton JP, Jodrell DI, Sansom OJ. GEMMs as preclinical models for testing pancreatic cancer therapies. Dis Model Mech. 2015;8:1185–1200. doi: 10.1242/dmm.021055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lo A, Li CP, Buza EL, Blomberg R, Govindaraju P, Avery D, Monslow J, Hsiao M, Pure E. Fibroblast activation protein augments progression and metastasis of pancreatic ductal adenocarcinoma. JCI Insight. 2017;2 doi: 10.1172/jci.insight.92232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 12.Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, Seghezzi W, Paruch K, Dwyer MP, Doll R, Nomeir A, Windsor W, Fischmann T, Wang Y, Oft M, Chen T, Kirschmeier P, Lees EM. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther. 2010;9:2344–2353. doi: 10.1158/1535-7163.MCT-10-0324. [DOI] [PubMed] [Google Scholar]
- 13.Stephenson JJ, Nemunaitis J, Joy AA, Martin JC, Jou YM, Zhang D, Statkevich P, Yao SL, Zhu Y, Zhou H, Small K, Bannerji R, Edelman MJ. Randomized phase 2 study of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus erlotinib in patients with non-small cell lung cancer. Lung Cancer. 2014;83:219–223. doi: 10.1016/j.lungcan.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 14.Mita MM, Joy AA, Mita A, Sankhala K, Jou YM, Zhang D, Statkevich P, Zhu Y, Yao SL, Small K, Bannerji R, Shapiro CL. Randomized phase II trial of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus capecitabine in patients with advanced breast cancer. Clin Breast Cancer. 2014;14:169–176. doi: 10.1016/j.clbc.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 15.Flynn J, Jones J, Johnson AJ, Andritsos L, Maddocks K, Jaglowski S, Hessler J, Grever MR, Im E, Zhou H, Zhu Y, Zhang D, Small K, Bannerji R, Byrd JC. Dinaciclib is a novel cyclin-dependent kinase inhibitor with significant clinical activity in relapsed and refractory chronic lymphocytic leukemia. Leukemia. 2015;29:1524–1529. doi: 10.1038/leu.2015.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar SK, LaPlant B, Chng WJ, Zonder J, Callander N, Fonseca R, Fruth B, Roy V, Erlichman C, Stewart AK, Mayo Phase C. Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma. Blood. 2015;125:443–448. doi: 10.1182/blood-2014-05-573741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hossain DMS, Javaid S, Cai M, Zhang C, Sawant A, Hinton M, Sathe M, Grein J, Blumenschein W, Pinheiro EM, Chackerian A. Dinaciclib induces immunogenic cell death and enhances anti-PD1-mediated tumor suppression. J Clin Invest. 2018;128:644–654. doi: 10.1172/JCI94586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111. doi: 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- 19.Feldmann G, Mishra A, Bisht S, Karikari C, Garrido-Laguna I, Rasheed Z, Ottenhof NA, Dadon T, Alvarez H, Fendrich V, Rajeshkumar NV, Matsui W, Brossart P, Hidalgo M, Bannerji R, Maitra A, Nelkin BD. Cyclin-dependent kinase inhibitor Dinaciclib (SCH727965) inhibits pancreatic cancer growth and progression in murine xenograft models. Cancer Biol Ther. 2011;12:598–609. doi: 10.4161/cbt.12.7.16475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu C, Dadon T, Chenna V, Yabuuchi S, Bannerji R, Booher R, Strack P, Azad N, Nelkin BD, Maitra A. Combined inhibition of cyclin-dependent kinases (Dinaciclib) and AKT (MK-2206) blocks pancreatic tumor growth and metastases in patient-derived xenograft models. Mol Cancer Ther. 2015;14:1532–1539. doi: 10.1158/1535-7163.MCT-15-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayes TK, Neel NF, Hu C, Gautam P, Chenard M, Long B, Aziz M, Kassner M, Bryant KL, Pierobon M, Marayati R, Kher S, George SD, Xu M, Wang-Gillam A, Samatar AA, Maitra A, Wennerberg K, Petricoin EF 3rd, Yin HH, Nelkin B, Cox AD, Yeh JJ, Der CJ. Long-term ERK inhibition in KRAS-mutant pancreatic cancer is associated with MYC degradation and senescence-like growth suppression. Cancer Cell. 2016;29:75–89. doi: 10.1016/j.ccell.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bipat S, Phoa SS, van Delden OM, Bossuyt PM, Gouma DJ, Laméris JS, Stoker J. Ultrasonography, computed tomography and magnetic resonance imaging for diagnosis and determining resectability of pancreatic adenocarcinoma. J Comput Assist Tomogr. 2005;29:438–445. doi: 10.1097/01.rct.0000164513.23407.b3. [DOI] [PubMed] [Google Scholar]
- 23.Lemasson B, Wang H, Galban S, Li Y, Zhu Y, Heist KA, Tsein C, Chenevert TL, Rehemtulla A, Galban CJ, Holland EC, Ross BD. Evaluation of concurrent radiation, temozolomide and ABT-888 treatment followed by maintenance therapy with temozolomide and ABT-888 in a genetically engineered glioblastoma mouse model. Neoplasia. 2016;18:82–89. doi: 10.1016/j.neo.2015.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bihan DL. Molecular diffusion, tissue microdynamics and microstructure. NMR Biomed. 1995;8:375–386. doi: 10.1002/nbm.1940080711. [DOI] [PubMed] [Google Scholar]
- 25.Cuneo KC, Chenevert TL, Ben-Josef E, Feng MU, Greenson JK, Hussain HK, Simeone DM, Schipper MJ, Anderson MA, Zalupski MM, Al-Hawary M, Galban CJ, Rehemtulla A, Feng FY, Lawrence TS, Ross BD. A pilot study of diffusion-weighted MRI in patients undergoing neoadjuvant chemoradiation for pancreatic cancer. Transl Oncol. 2014;7:644–649. doi: 10.1016/j.tranon.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nishiofuku H, Tanaka T, Marugami N, Sho M, Akahori T, Nakajima Y, Kichikawa K. Increased tumour ADC value during chemotherapy predicts improved survival in unresectable pancreatic cancer. Eur Radiol. 2016;26:1835–1842. doi: 10.1007/s00330-015-3999-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reichert M, Rhim AD, Rustgi AK. Culturing primary mouse pancreatic ductal cells. Cold Spring Harb Protoc. 2015;2015:558–561. doi: 10.1101/pdb.prot078279. [DOI] [PubMed] [Google Scholar]
- 28.Reichert M, Takano S, Heeg S, Bakir B, Botta GP, Rustgi AK. Isolation, culture and genetic manipulation of mouse pancreatic ductal cells. Nat Protoc. 2013;8:1354–1365. doi: 10.1038/nprot.2013.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu S, Pan L, Shangguan J, Figini M, Eresen A, Sun C, Wang B, Ma Q, Hu C, Yaghmai V, Velichko Y, Yang J, Zhang Z. Non-invasive dynamic monitoring initiation and growth of pancreatic tumor in the LSL-Kras (G12D/+); LSL-Trp53 (R172H/+); Pdx-1-Cre (KPC) transgenic mouse model. J Immunol Methods. 2019;465:1–6. doi: 10.1016/j.jim.2018.11.009. [DOI] [PubMed] [Google Scholar]
- 30.Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Metivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, Kroemer G. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 31.Niwa T, Ueno M, Ohkawa S, Yoshida T, Doiuchi T, Ito K, Inoue T. Advanced pancreatic cancer: the use of the apparent diffusion coefficient to predict response to chemotherapy. Br J Radiol. 2009;82:28–34. doi: 10.1259/bjr/43911400. [DOI] [PubMed] [Google Scholar]
- 32.Feldmann G, Mishra A, Hong SM, Bisht S, Strock CJ, Ball DW, Goggins M, Maitra A, Nelkin BD. Inhibiting the cyclin-dependent kinase CDK5 blocks pancreatic cancer formation and progression through the suppression of Ras-Ral signaling. Cancer Res. 2010;70:4460–4469. doi: 10.1158/0008-5472.CAN-09-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rajput S, Khera N, Guo Z, Hoog J, Li S, Ma CX. Inhibition of cyclin dependent kinase 9 by dinaciclib suppresses cyclin B1 expression and tumor growth in triple negative breast cancer. Oncotarget. 2016;7:56864–56875. doi: 10.18632/oncotarget.10870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Danilov AV, Hu S, Orr B, Godek K, Mustachio LM, Sekula D, Liu X, Kawakami M, Johnson FM, Compton DA, Freemantle SJ, Dmitrovsky E. Dinaciclib induces anaphase catastrophe in lung cancer cells via inhibition of cyclin-dependent kinases 1 and 2. Mol Cancer Ther. 2016;15:2758–2766. doi: 10.1158/1535-7163.MCT-16-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin SF, Lin JD, Hsueh C, Chou TC, Wong RJ. A cyclin-dependent kinase inhibitor, dinaciclib in preclinical treatment models of thyroid cancer. PLoS One. 2017;12:e0172315. doi: 10.1371/journal.pone.0172315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gregory GP, Hogg SJ, Kats LM, Vidacs E, Baker AJ, Gilan O, Lefebure M, Martin BP, Dawson MA, Johnstone RW, Shortt J. CDK9 inhibition by dinaciclib potently suppresses Mcl-1 to induce durable apoptotic responses in aggressive MYC-driven B-cell lymphoma in vivo. Leukemia. 2015;29:1437–1441. doi: 10.1038/leu.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, metabolism, and cancer. Cancer Discov. 2015;5:1024–1039. doi: 10.1158/2159-8290.CD-15-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martins I, Tesniere A, Kepp O, Michaud M, Schlemmer F, Senovilla L, Seror C, Metivier D, Perfettini JL, Zitvogel L, Kroemer G. Chemotherapy induces ATP release from tumor cells. Cell Cycle. 2009;8:3723–3728. doi: 10.4161/cc.8.22.10026. [DOI] [PubMed] [Google Scholar]
- 39.Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G, Rello-Varona S, Tailler M, Menger L, Vacchelli E, Galluzzi L, Ghiringhelli F, Virgilio Fd, Zitvogel L, Kroemer G. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573–1577. doi: 10.1126/science.1208347. [DOI] [PubMed] [Google Scholar]
- 40.Martins I, Michaud M, Sukkurwala AQ, Adjemian S, Ma Y, Shen S, Kepp O, Menger L, Vacchelli E, Galluzzi L, Zitvogel L, Kroemer G. Premortem autophagy determines the immunogenicity of chemotherapy-induced cancer cell death. Autophagy. 2012;8:413–415. doi: 10.4161/auto.19009. [DOI] [PubMed] [Google Scholar]
- 41.Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, Rubio N, Firczuk M, Mathieu C, Roebroek AJ, Annaert W, Golab J, de Witte P, Vandenabeele P, Agostinis P. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. EMBO J. 2012;31:1062–1079. doi: 10.1038/emboj.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Z, Wang Z, Pang JC, Yu Y, Bieerkehazhi S, Lu J, Hu T, Zhao Y, Xu X, Zhang H, Yi JS, Liu S, Yang J. Multiple CDK inhibitor dinaciclib suppresses neuroblastoma growth via inhibiting CDK2 and CDK9 activity. Sci Rep. 2016;6:29090. doi: 10.1038/srep29090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ianes C, Xu P, Werz N, Meng Z, Henne-Bruns D, Bischof J, Knippschild U. CK1δ activity is modulated by CDK2/E- and CDK5/p35-mediated phosphorylation. Amino Acids. 2016;48:579–592. doi: 10.1007/s00726-015-2114-y. [DOI] [PubMed] [Google Scholar]
- 44.Lim KH, Baines AT, Fiordalisi JJ, Shipitsin M, Feig LA, Cox AD, Der CJ, Counter CM. Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell. 2005;7:533–545. doi: 10.1016/j.ccr.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 45.Alagpulinsa DA, Ayyadevara S, Yaccoby S, Shmookler Reis RJ. A cyclin-dependent kinase inhibitor, dinaciclib, impairs homologous recombination and sensitizes multiple myeloma cells to PARP inhibition. Mol Cancer Ther. 2016;15:241–250. doi: 10.1158/1535-7163.MCT-15-0660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Molenaar JJ, Ebus ME, Geerts D, Koster J, Lamers F, Valentijn LJ, Westerhout EM, Versteeg R, Caron HN. Inactivation of CDK2 is synthetically lethal to MYCN over-expressing cancer cells. Proc Natl Acad Sci U S A. 2009;106:12968–12973. doi: 10.1073/pnas.0901418106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, Chen ZE, Nikolaidis P, McCarthy RJ, Merrick L, Sternick LA, Horowitz JM, Yaghmai V, Miller FH. Diffusion-weighted magnetic resonance imaging of pancreatic adenocarcinomas: association with histopathology and tumor grade. J Magn Reson Imaging. 2011;33:136–142. doi: 10.1002/jmri.22414. [DOI] [PubMed] [Google Scholar]
- 48.Luypaert R, Boujraf S, Sourbron S, Osteaux M. Diffusion and perfusion MRI: basic physics. Eur J Radiol. 2001;38:19–27. doi: 10.1016/s0720-048x(01)00286-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.