

Abstract
Sox9 is the master transcription factor essential for cartilage development and homeostasis. To investigate the specific role of Sox9 during chondrocyte hypertrophy, we generated a novel Col10a1-Sox9 transgenic mouse model, in which Sox9 is specifically expressed in hypertrophic chondrocytes driven by a well-characterized 10-kb Col10a1 promoter. These mice were viable and fertile, and appeared normal at birth. However, they developed dwarfism by ten weeks of age. The histological analysis of the growth plates from these transgenic mice demonstrated an abnormal growth plate architecture and a significantly reduced amount of trabecular bone and mineral content in the primary spongiosa. Real-time qPCR analysis revealed the reduced expression of Col10a1, and increased expressions of adipogenic differentiation markers in primary hypertrophic chondrocytes isolated from transgenic mice. Concomitantly, the transgenic mouse chondrocyte cultures had increased lipid droplet accumulation. Unexpectedly, we also observed an increased incidence of spontaneous osteoarthritis (OA) development in the transgenic mice by X-ray analysis, micro-computed tomography scanning, and histological examination of knee joints. The manifestation of OA in Col10a1-Sox9 transgenic mice began by six-months of age, and worsened by eleven-months of age. In conclusion, we provide strong evidence that the proper spatiotemporal expression of Sox9 is necessary for normal adult hypertrophic cartilage homeostasis, and that the aberrant expression of Sox9 might lead to spontaneous OA development.
Keywords: Hypertrophic chondrocytes, adipogenesis, osteoarthritis, Sox9, Col10a1
Introduction
Osteoarthritis (OA) is a degenerative disease characterized mainly by articular cartilage degradations and alterations in subchondral bone and synovium [1]. Multiple factors contribute to the OA pathogenesis, including genetics, obesity, and age [2]. One of the leading causes of OA pathogenesis is excessive mechanical loading that results in articular cartilage damages [3,4]. Several strategies, including cell-based approaches [5], biomaterial-guided gene delivery systems [6], and genetic manipulation methods [7] have been proposed to treat OA, but none have matured into standard clinical treatments. At present, the most acceptable surgical treatment for late-stage OA is total knee replacement (TKR). Over 650,000 TKRs were performed in the United States alone in 2008, and the number is projected to increase to over 3.5 million by 2030 as the population ages [8]. OA often begins affecting different joints long before middle age, but evades diagnosis until it becomes symptomatic decades later, at which point the structural degradation is already advanced and irreversible [9]. Thus, it is critical to identify the key factors mediating the early-stage OA pathogenesis to improve its diagnostic and treatment outcomes.
Cartilage homeostasis depends on the controlled degradation of the matrix proteins such as type II collagen and aggrecan by proteolytic enzymes such as matrix metalloproteinases (MMPs), aggrecanase, and other extracellular proteases in the ADAMTS family. These catabolic events are followed by the deposition of newly synthesized extracellular matrix (ECM) proteins [10]. This complex and dynamic process result in a balance between catabolism and anabolism. In OA patients, this delicate balance tips toward degradation due to a variety of factors, including genetics, lifestyle, and differential expression of proteinases, growth factors, cytokines, and other inflammatory factors [9]. Furthermore, the synovium and subchondral bone are also involved in OA pathogenesis [11]. Human and mouse genetics studies have identified multiple factors that may be associated with OA, including, TGF-b/BMP receptors (Bmpr1a and TgfbRII), ECM molecules (Col2a1, Aggrecan), signaling molecules (sFRP3, Gdf5, Opg), transcription factors (Runx2), and microRNAs [12-15].
Long bones are created through a dynamic and well-regulated process called endochondral ossification where cartilage templates are replaced by bone [16]. Chondrocytes in the cartilage templates undergo successive steps of differentiation. These early chondrocytes actively proliferate and produce a highly specific cartilage extracellular matrix (ECM), and later become hypertrophic. These hypertrophic chondrocytes either undergo apoptosis or differentiate into osteoblasts and form bone during the course of chondrocyte maturation [16]. Chondrocytes are ultimately replaced by osteoblasts in the process of endochondral ossification. Sox9, the master transcriptional factor regulating chondrogenesis, is essential for chondrocyte condensation and the successive stages of cartilage formation by transactivation of chondrocyte-specific matrix genes, including Col2a1 and Aggrecan [17,18]. Moreover, SOX9 has been shown to directly regulates connective tissue growth factor (CTGF/CCN2), which is necessary for chondrocyte proliferation and maturation [19]. Additionally, SOX9 regulates multiple genes encoding ECM proteins, ECM modification enzymes, receptors, and transporters in chondrocytes [20].
Sox9 haploinsufficiency in humans causes Campomelic Dysplasia, a severe skeletal malformation syndrome characterized by the generalized hypoplasia of endochondral bones [17,21]. Interestingly, during mouse embryonic development, Sox9 is highly expressed in all chondrogenic precursor cells and in differentiated chondrocytes. However, the Sox9 mRNA and protein expression was extremely low in hypertrophic chondrocytes [22,23]. Notably, Sox9 haploinsufficiency in mice also results in premature skeletal mineralization and an enlarged hypertrophic zone in the growth plates, with increased expression of type X collagen, the major and most specific extracellular component synthesized by hypertrophic chondrocytes [24].
Runx2, the master transcription factor required for osteoblast differentiation, also positively regulates chondrocyte maturation [25]. We have previously shown that Runx2 can directly bind to a consensus cis-element in the Col10a1 promoter and contribute to its hypertrophic chondrocyte-specific expression [26]. Importantly, both Sox9 and Runx2 are expressed in early chondrocytic cells, and we have demonstrated the dominance of Sox9 over Runx2 function during skeletogenesis [17]. Indeed, Sox9 can inhibit Runx2 binding to its target sequences in the Col10a1 promoter and repress the transactivation of a hypertrophic chondrocyte-specific Col10a1 reporter. Furthermore, in Campomelic Dysplasia patients, SOX9 haploinsufficiency results in the up-regulation of the both RUNX2 and COL10A1 [17]. Interestingly, two previous studies have demonstrated that the overexpression of Sox9 in hypertrophic chondrocytes leads to impaired terminal chondrocyte differentiation, with decreased Vegf, Mmp13, Rankl, and Opn expression, likely in part through the inhibition of Runx2 activity in the early postnatal stages [27,28]. Furthermore, Sox9 was also reported to suppress Vegfa expression by directly binding to the cis-element in the Vegfa gene promoter. Thus, Sox9 is proposed to be a major negative regulator of cartilage vascularization and trabecular bone formation in the growth plates [27]. However, the phenotype characterization in all previous Sox9-related studies was limited to embryonic development and early postnatal stages.
Despite many factors contributing to OA, the relationship of OA progression and altered hypertrophic chondrocyte regulation is largely unknown, due to the lack of animal models with targeted and sustained expression of the genes of interest specifically in hypertrophic chondrocytes. Thus, in this study, we aimed to determine the effect of hypertrophic chondrocytes-specific Sox9 overexpression on cartilage homeostasis. To address this question, we generated a novel Col10a1-Sox9 transgenic (TG) mouse and characterized the consequences of Sox9 over-expression in hypertrophic chondrocytes in adult mice. We found that the sustained expression of Sox9 specifically in the hypertrophic chondrocytes of mice results in disorganized chondrocyte columns with reduced mineralization of hypertrophic cartilages. Furthermore, chondrocytes isolated from these mice showed elevated adipogenic markers. Unexpectedly, Col10a1-Sox9 TG mice showed key OA features such as accelerated loss of cartilage matrix in the growth plates of knee joints, narrowed joint spaces, formation of osteophytes, and subchondral sclerosis. Thus, Sox9 overexpression specifically in hypertrophic chondrocytes results in severe osteoarthritis development in adult mice.
Materials and methods
Generation of Col10a1-Sox9 transgenic mice
The Col10a1-Sox9 TG construct, in which flag-tagged Sox9 was specifically expressed in all hypertrophic chondrocytes driven by a 10-kb Col10a1 promoter, was generated in a previously characterized pTYR-WPRE-hGHpA transgenic vector [17]. This 10-kb Col10a1 promoter directed the β-galactosidase reporter expression specifically throughout the whole hypertrophic zone at high levels in a previous report [29]. The transgenic construct (Figure 1C) also includes a tyrosinase (TYR) minigene, which enables rapid genotyping of embryonic TG founders by eye color selection, and a chicken β-globin HS4 insulator to enhance the transgene expression by decreasing chromatin-mediated silencing effect [17]. A woodchuck hepatitis virus posttranscriptional regulatory element (WPRE) was also included in the 3’untranslated sequence to increase the expression level of the transgene [17]. Transgenic founders were generated by pronuclear injection according to standard techniques as reported previously [17]. The transgenic mice were confirmed at birth by eye color change and verified by PCR using the transgene-specific primers as described [17]. Two independent transgenic lines were generated and maintained on a FVB/N background, and they displayed similar phenotypes. All animal protocols have been reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Case Western Reserve University.
Figure 1.
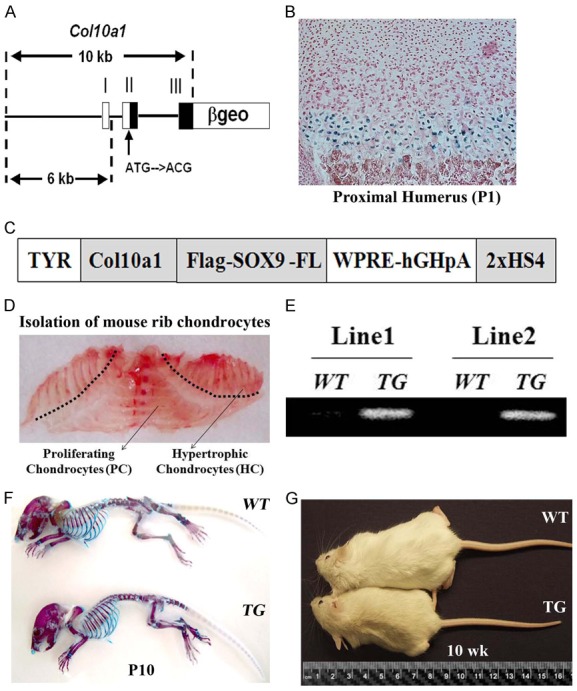
The generation of a novel Col10a1-Sox9 transgenic mouse model. (A) A schematic representation of a Col10a1-β-Gal reporter construct driven by a 10-kb mouse Col10a1 element utilized in this study. (B) The Col10a1-ß-gal transgene drives high-level LacZ reporter activity specifically in the hypertrophic zone of transgenic mice. (C) A schematic representation of the Col10a1-Sox9 transgenic construct that contains the full-length Flag-tagged Sox9 under the control of the same 10-kb Col10a1 element as described in Figure 1 (A). The Col10a1-Sox9 construct was ultimately cloned into a coat color vector as described previously [17]. TYR, tyrosinase minigene; WPRE, woodchuck hepatitis virus posttranscriptional regulatory element; HS4, chicken β-globin insulator. (D) The image of the separation of the hypertrophic and proliferating chondrocytes from a P6 mouse rib cage. The hypertrophic chondrocytes were isolated from the area above the dashed line. (E) The specific transgene expression in the hypertrophic chondrocytes was confirmed by RT-PCR with transgene-specific primers in two independent TG mouse lines. (F) The skeletal preparations of 10-day-old mice by Alizarin red/Alcian blue staining revealed no significant growth differences between WT and TG mice. (G) The gross appearance of 10-week old TG mice was clearly shorter than that of WT littermates.
Isolation of chondrocytes from mouse knee joints
Hind limbs were collected from WT and TG mice upon euthanizing them with isoflurane. Knee joints were cut to separate the ends of the distal femur and proximal tibia under the dissection microscope. Primary chondrocytes were isolated from knee joints following a sequential digestion by pronase (2 mg/ml in PBS) for 1 hour at 37°C, collagenase D (3 mg/ml in DMEM) for 2 hours at 37°C, and overnight at 37°C in collagenase D (1.5 mg/ml in DMEM). Disassociated chondrocytes from WT and TG mice knee joints were collected to perform gene expression analysis.
Skeletal preparations and micro-computed tomography
Skeletons from ten-day-old mice were prepared as described and stained with Alcian blue 8GX for cartilage and Alizarin red S for bone [17]. The dissected hind limbs from six and eleven month old wild-type (WT) and transgenic (TG) mice were analyzed by radiography using a Faxitron radiographic inspection unit. Eleven-month old WT and TG mouse hind limbs were also scanned for micro-computed tomography (micro-CT) analysis on a Scanco µCT40 scanner as described previously [30].
Histological analysis
Hind limbs from ten-week-old and eleven-month-old mice were fixed in 10% buffered formalin overnight and decalcified in 0.5 M EDTA for 3 months. After paraffin embedding, 5-µm sections were prepared and stained with Toluidine Blue and Safranin O/Fast Green (SO/FG). The standard immunostaining was performed on paraffin sections of ten-week and eleven-month-old hind limbs with anti-Sox9 antibody (Cat # AB5535, Millipore Sigma), anti-Type X Collagen antibody (Quartett, Berlin, Germany), anti-Flag antibody (Cat # 14793S, CST), and ABC reagent (Vector Laboratories, Burlingame, CA) was used for detection as previously described [31,32].
Bone histomorphometrical analysis
Undecalcified hind limbs from 10-week-old mice were embedded in methylmethacrylate, and 5-μm sections were generated on a rotation microtome. Sections were then stained with Toluidine Blue and von Kossa reagents. Histomorphometrical analysis was performed on proximal tibiae according to the American Society for Bone and Mineral Research (ASBMR) standards using the OsteoMeasure Analysis System (Osteometrix, Atlanta, GA) [17,33].
Rib hypertrophic chondrocyte isolation and ex vivo chondrocyte culture
Hypertrophic chondrocytes from rib cages of six-day-old Col10a1-Sox9 TG mice and WT littermates were isolated as previously described [31]. After the dissection, soft-tissue was cleaned-up and the hypertrophic potion of rib cartilage was cut at approximately 2-mm distal from the osteochondral junction of each rib, to separate it from the proliferating portion (Figure 1D). The rib cartilages were then digested with pronase (2 mg/ml in PBS) for 30 min at 37°C, collagenase D (3 mg/ml in DMEM) for 1.5 h at 37°C, and collagenase D (1.5 mg/ml in DMEM) overnight at 37°C. After washing with PBS three times, rib hypertrophic chondrocytes were plated at a density of 1×105 cells/ml in 24-well-plate for 9 days in DMEM supplemented with 2 mM GlutaMax, 100 µg/ml Penicillin/100 U Streptomycin, and 10% FBS (Invitrogen, USA). All cells were maintained at 37°C in a humidified atmosphere containing 5% CO2. The medium was changed every other day. At day 9, half of the cells were fixed with formalin and stained with Oil Red O to detect adipogenesis [34], and the remaining cells were utilized for RNA isolation.
RNA isolation, cDNA synthesis, and real-time RT-PCR analysis
Using TRIzol reagent (Invitrogen) and Purelink Micro-To-Midi RNA Kit (Invitrogen), total RNA was extracted from rib hypertrophic chondrocytes after 9 days of culture (n = 3-6/group) as described above. cDNA was then prepared from 1 µg of total RNA with the iScript cDNA Synthesis Kit (Bio-Rad). Power SYBR Green PCR Master Mix (Applied Biosystems) and the 7500 Real Time PCR system (Applied Biosystems) were used for real-time RT-PCR analysis with the gene-specific primers listed in Table 1. The Gapdh gene was used as an internal control of the quantity and quality of the cDNAs. Real time RT-PCR analysis was quantified using the standard ΔΔCT method as described [35].
Table 1.
Primers used in real-time RT-PCR analyses
| Gene | Primers* | Size (bp) | Accession No. |
|---|---|---|---|
| Sox9 | ACTCCCCACATTCCTCCTCCGGCAT | 343 | NM_011448.3 |
| GTGCTGCTGATGCCGTAACTGCCAG | |||
| Col10a1 | GGCAGCAGCATTACGACCCAAGAT | 563 | NM_009925.3 |
| GAATAACAGACACCACCCCCTCAC | |||
| Pparγ2 | ATCATCTACACGATGCTGGCC | 81 | NM_011146.2 |
| CAAGGATTCATGACCAGGGAG | |||
| C/ebpα | CGCAAGAGCCGAGATAAAGC | 81 | NM_007678.3 |
| GCGGTCATTGTCACTGGTCA | |||
| Glut-4 | ATGGCTGTCGCTGGTTTCTC | 81 | NM_009204.2 |
| ACCCATAGTATCCGCAACAT | |||
| Gapdh | ATGGGAAGCTCGTCATCAAC | 152 | XR_030830.1 |
| GTGGTTCACACCCATCACAA |
All primer sequences are written from 5’ to 3’; for each gene, the first sequence is the sense primer, and the second sequence is the anti-sense primer.
Statistical analysis
Results were presented as mean ± standard deviations (SD). Statistical analysis was performed using the Student’s t-test to compare the differences between two groups of age-matched Col10a1-Sox9 TG mice and WT littermates (n = 3-6). P < 0.05 was considered statistically significant.
Results
The sustained Sox9 expression specifically in hypertrophic chondrocytes in a novel Col10a1-Sox9 transgenic mouse model
We previously identified a mouse 10-kb Col10a1 element that was able to direct a reporter activity specifically in hypertrophic zones in transgenic (TG) mice [29]. Indeed, this 10-kb element can direct high-level tissue-specific LacZ reporter activity throughout the hypertrophic zone in Col10a1-βgeo mice (Figure 1A, 1B). For this study, we first confirmed that the ß-galactosidase staining revealed strong LacZ reporter activities in the hypertrophic chondrocytes of the proximal humerus of TG mice (Figure 1A, 1B). Then we generated a Col10a1-Sox9 TG construct that contains the flag-tagged full-length Sox9 under the control of the same 10-kb Col10a1 promoter in a previously described pTYR-WPRE-hGHpA TG vector (Figure 1C) [17]. We generated two independent lines of Col10a1-Sox9 TG mice. To confirm Sox9 overexpression, we isolated hypertrophic chondrocytes from the rib cages of six-day-old Col10a1-Sox-9 TG mice and WT littermate controls (Figure 1D). Our RT-PCR analysis confirmed that both mouse lines exhibited high transgene expression in the hypertrophic chondrocytes (Figure 1E). Interestingly, Alizarin red S and Alcian blue staining of the whole skeleton preparations of ten-day-old mice displayed no gross abnormalities in TG mice compared with WT littermates (Figure 1F). However, by ten-weeks of age, TG mice were clearly shorter compared with the WT littermates (Figure 1G). These results demonstrate that the hypertrophic chondrocyte-specific overexpression of Sox9 in mice results in a postnatal growth delay by 10-weeks of age.
The sustained postnatal hypertrophic chondrocyte-specific Sox9 expression results in disorganized chondrocyte columns and reduced mineralization of hypertrophic cartilages
We performed immunostaining with an antiflag antibody and observed strong transgene expression specifically in the hypertrophic chondrocytes of the proximal tibiae of six-day-old TG mice, but not in the WT littermate controls (Figure 2A). Next, to confirm the sustained Sox9 expression in the cartilage, we performed immunostaining using an anti-Sox9 antibody in the proximal tibiae of 10-week-old WT and TG mice. The immunostaining confirmed Sox9 expression specifically in the hypertrophic zone of the TG mouse growth plates, but not in the WT littermate controls (Figure 2B). Additionally, we observed a severe growth plate disorganization in the TG mice (Figure 2C). Indeed, Toluidine Blue staining in the growth plates of proximal tibiae of 10-week old mice revealed that the chondrocyte columns appeared much more disorganized in TG mice, and that the primary spongiosa was also significantly shorter in TG mice compared with WT controls (Figure 2C). Moreover, von Kossa and toluidine blue staining revealed a slight reduction in the mineralized area of the trabeculae of the primary spongiosa in Col10a1-Sox9 TG mice compared with WT controls (Figure 2D). The reduced mineralization of hypertrophic cartilage in Col10a1-Sox9 TG mice was further confirmed by histomorphometric quantification of von Kossa staining, which revealed over a 10% reduction in von Kossa staining in TG vs. WT mice (Figure 2D and 2E). Together, these results indicate that the sustained overexpression of Sox9 specifically in hypertrophic chondrocytes results in a disrupted growth plate organization, and a reduction in the size and mineralization of trabecular bone in the primary spongiosa postnatally.
Figure 2.
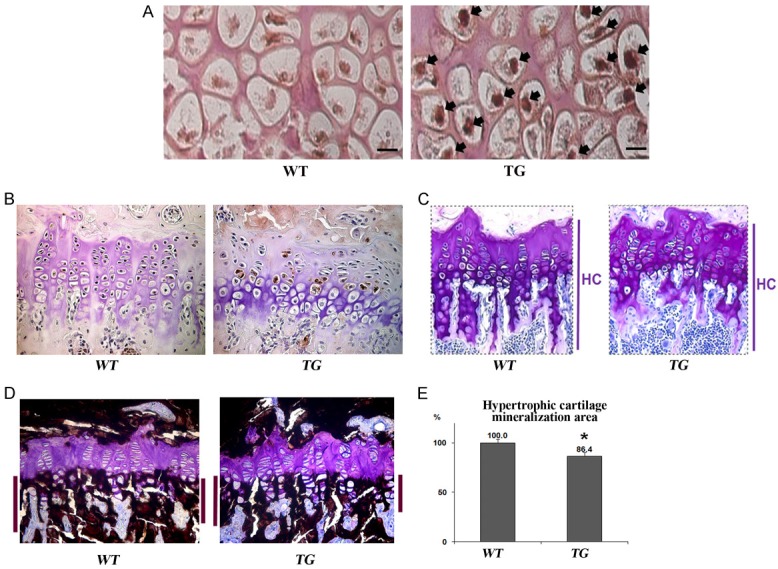
The histological analysis of hypertrophic cartilages in ten-week-old Col10a1-Sox9 transgenic mice. A. Flag-tag immunohistochemistry confirmed the hypertrophic chondrocyte-specific transgene expression in the proximal tibiae of six-day-old TG mice as indicated by black arrows (40×). B. Toluidine blue staining demonstrates disorganized chondrocyte columns and a reduced length of the trabecular bone in the primary spongiosa of TG mice (20×). C. Von Kossa staining demonstrates the slightly reduced mineralization of the primary spongiosa engulfing the hypertrophic zone in TG mice (20×). D, E. The histomorphometric quantification of von Kossa staining confirms a modest but significantly decreased mineralization of the primary spongiosa in TG mice. Error bars represent mean ± SD. (p-value < 0.05).
The increased adipogenesis in Col10a1-Sox9 transgenic mouse hypertrophic chondrocytes
Our histological analysis (Figure 2), which demonstrated altered growth plate morphology and reduced mineralization of primary trabeculae, suggests that chondrocyte differentiation may be perturbed in Col10a1-Sox9 TG mice. Interestingly, a previous study demonstrated a negative correlation between chondrocyte and adipocyte differentiation [36]. Thus, we decided to examine the effect of Sox9 overexpression on adipocyte differentiation ex vivo. To that end, we performed Oil red O staining on hypertrophic chondrocytes after 9 days of ex vivo culture. Interestingly, we found a much stronger Oil Red O staining in TG chondrocytes compared with WT controls, suggesting that sustained Sox9 expression results in increased adipogenesis (Figure 3A). Moreover, our real-time RT-PCR analysis in hypertrophic chondrocytes confirmed that the overexpression of Sox9 was concomitant with the downregulated expression of the most specific marker for hypertrophic chondrocytes, Col10a1 (Figure 3B, 3C). Furthermore, real-time RT-PCR analysis also revealed significantly elevated expressions of adipocyte differentiation markers in TG hypertrophic chondrocyte ex vivo culture, including Pparγ2, C/ebpα, and Glut-4 (Figure 3D-F). Together, these results demonstrate that the sustained Sox9 expression in hypertrophic chondrocytes enhances adipogenesis ex vivo.
Figure 3.
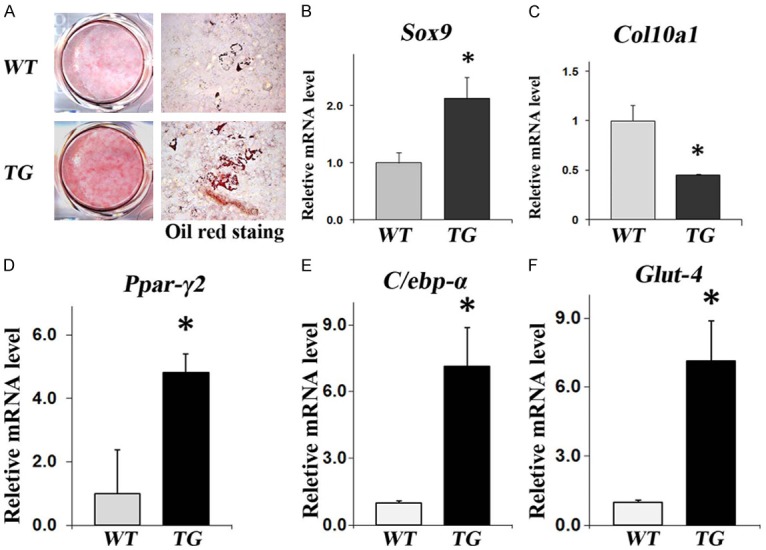
Enhanced adipogenesis in hypertrophic chondrocytes isolated from TG mice in comparison with WT control littermates after nine-days of ex vivo culture. (A) Oil red O staining demonstrates increased adipogenesis in TG mouse hypertrophic chondrocytes after 9-days of ex vivo culture (scale bar = 50 µm). Real-time RT-PCR analysis demonstrated (B) increased Sox9 expression, (C) decreased Col10a1 expression in hypertrophic chondrocytes, (D-F) elevated expressions of key adipogenic markers Pparγ2, C/ebpα and Glut4 in TG verses WT hypertrophic chondrocytes.
The accelerated loss of cartilage matrix in the growth plates of knee joints in Col10a1-Sox9 transgenic mice
Next, we examined the hind limbs of WT and TG mice by X-ray analysis. Surprisingly, radiographs of six-month-old mice revealed some small osteophytes at the knee joints in TG mice, but not in WT mice (Figure 4A, white arrows). We then further analyzed the knee joints of six-month-old WT and TG mice by Safranin O staining. Interestingly, we found a partial loss of Safranin O staining at the articular surface of TG mouse knees, indicating a reduced amount of articular cartilage in TG mice compared with WT littermates (Figure 4B and 4C, red staining at the top). Real-time RT-PCR analysis revealed significantly elevated expressions of Mmp13, Admts5, and Admts4 in the 9 months old TG mice knee chondrocytes. Moreover, pro-inflammatory cytokines including Stat5, Nfat1 and Atf4 were upregulated in TG mice. As expected the expression levels of Col10a1 and Sox9 were increased in the TG mice. Interestingly, Tgfß, which has been shown to boost chondrogenesis, was downregulated in knee chondrocytes of the TG mice (Figure 4D). These results demonstrate that the increased Sox9 expression in hypertrophic chondrocytes impaired cartilage homeostasis and led to the likely early onset of osteoarthritis in TG mice at six month-old.
Figure 4.
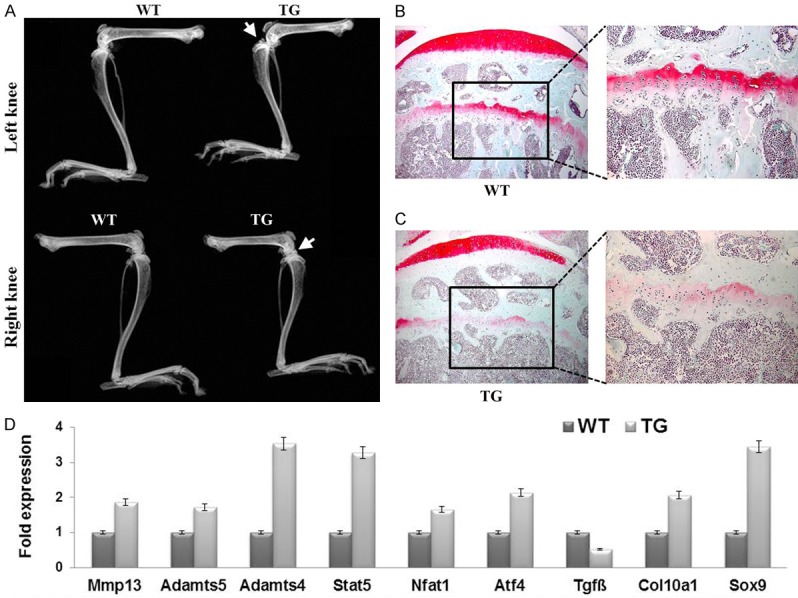
Premature cartilage degradation in the growth plates of knee joints of six-month-old Col10a1-Sox9 transgenic mice vs. wild-type control littermates. A. Radiographs of six-month-old TG and WT mice. White arrows point to osteophytes in the legs of TG mice, which might be associated with the degeneration of cartilage in the TG mice. B, C. Safranin O (SO) staining in the growth plates of proximal tibiae of six-month-old WT mice and TG mice. A reduced SO staining in the TG mice suggests the likely early onset of osteoarthritis. The images on the right represent the enlarged boxed areas of the left images (20×). D. Real-time RT-PCR analysis of the knee chondrocytes isolated from WT and TG mice.
The occurrence of severe osteoarthritis in eleven-month-old Col10a1-Sox9 transgenic mice
At eleven-months of age, 62.5% of Col10a1-Sox9 TG mice developed spontaneous osteoarthritis (OA) (Table 2). In contrast, only 2.5% of WT mice developed spontaneous OA (Table 2), indicating that the TG mice had a much higher incidence of spontaneous OA at this stage. Indeed, X-ray analysis of the knee joints of 11-month-old TG mice showed many typical OA features, including narrowed joint space (Figure 5B, black arrow), osteophyte development (Figure 5B, white arrow), and subchondral sclerosis (Figure 5D, asterisk) (Figure 5A-D). Next, the number of osteophytes were quantified as previously described [37]. WT mice do not show the presence of osteophytes, whereas 3±1 osteophytes were observed in the TG mice (Table 3). Furthermore, micro-CT analysis of 11-month-old TG knee joints revealed rough bone surfaces and numerous osteophytes protruding into the joint cavity in TG mice, but not WT mice (Figure 5E, 5F). Next, we further characterized the OA phenotype of TG mice by performing Safranin-O/Fast Green (SO/FG) staining on knee joints of WT and TG mice (Figure 5G-J). SO/FG staining demonstrated that compared with WT controls, there was significant articular cartilage degeneration in TG mice (Figure 5G-J). OARSI Score analysis for the TG mice is two based on the loss of Safranin-O staining and the percentage of the calcified cartilage erosions extending to articular surface (Table 3). It is well-established that OA pathogenesis is associated with ectopic chondrocyte hypertrophy [15]. Thus, we next examined the expression of the most specific hypertrophic chondrocyte marker, Col10a1, in the articular cartilage by immunostaining. Interestingly, we observed high-levels of Col10a1 expression on the articular cartilage surface in the proximal tibiae of TG compared with WT mice (Figure 5K, 5L). Thus, the sustained Sox9 expression in the hypertrophic zone likely leads to ectopic Type X Collagen expression on articular surfaces, which might contribute in part to the OA occurrence in TG mice. Finally immunostaining confirmed elevated Mmp13 expression in the TG mice (Figure 5M, 5N). These data confirm that the overexpression of Sox9 in hypertrophic chondrocytes results in the accelerated, spontaneous development of OA.
Table 2.
Percentage of OA developed in TG mice compared with WT at eleven-month-old
| Genotype | Joints | OA | Percentage |
|---|---|---|---|
| WT | 40 | 1 | 2.5% |
| TG | 40 | 25 | 62.5% |
Figure 5.
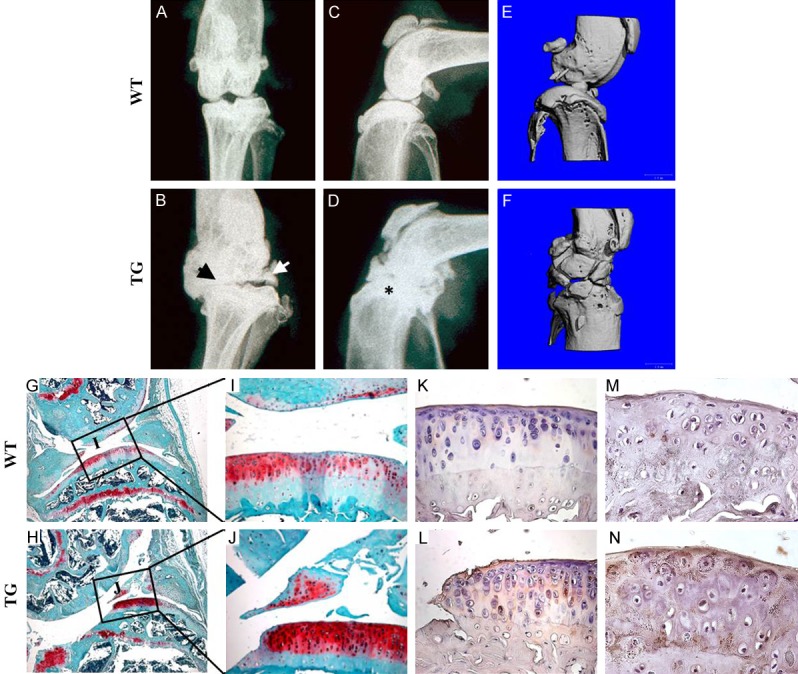
Spontaneous osteoarthritis development in eleven-month-old transgenic mice: (A-D) Radiographs of the knee joints of eleven-month old TG and WT mice. Note the characteristic osteoarthritis features in the TG mice: joint space narrowing (B, black arrow), development of osteophytes (B, white arrow), and sub-chondral sclerosis (D, asterisk). (E, F) The micro-CT analysis of the knee joints of eleven-month-old TG and WT mice revealed rougher bone surfaces and numerous osteophytes protruding into the joint cavity of the TG mice, but not WT mice. (G-J) SO/FG staining of the knee joints of the eleven-month-old TG and WT mice. Note the absence of Safranin-O staining at the articular surfaces in TG mice. (K, L) Type X collagen immunostaining revealed the ectopic Col10a1 expression in the articular cartilages of TG mice, but not of WT mice. (M, N) Immunostaining showed elevated secretion of matrix metalloprotease-13 (Mmp13) in the articular cartilages of 11 months old TG mice.
Table 3.
Osteophyte numbers and the Osteoarthritic Research Society International (OARSI) scores for the eleven-month-old WT and TG mice
| Genotype | # of osteophytes | OARSI Score |
|---|---|---|
| WT | 0 | 0 |
| Normal cartilage | ||
| TG | 3±1 | 2 |
| Loss of Safranin-O | ||
| Erosion of the calcified cartilage extending to < 25% of the articular surface. |
Discussion
As the master transcription factor that is essential for chondrocyte differentiation, the expression of Sox9 was significantly lower in hypertrophic chondrocytes compared with proliferating chondrocytes [23]. Interestingly, it was reported that Sox9 represses Col10a1, the most specific marker of hypertrophic chondrocyte differentiation, in proliferating chondrocytes by binding to its upstream elements [38]. On the other hand, Sox9 downregulation in hypertrophic chondrocytes is proposed to be necessary to initiate the cartilage-to-bone transition in the growth plate [39]. Therefore, the precise expressions of Sox9 and Col10a1 must be tightly controlled in the growth plate to ensure proper chondrocyte differentiation, cartilage formation and endochondral ossification [16]. A previous study has demonstrated that the overexpression of Sox9 in hypertrophic chondrocytes during embryogenesis can lead to impaired terminal chondrocyte differentiation with decreased Vegf, Mmp13, Rankl, and Opn expression [27]. Thus, Sox9 is proposed to act as a major negative regulator of cartilage vascularization and trabecular bone formation in the growth plates by suppressing Vegfa expression [27]. In another mouse model, Kim and his colleagues demonstrated a delayed terminal chondrocyte differentiation as a result of Sox9 overexpression in hypertrophic chondrocytes [28]. Furthermore, a Sox9 conditional knockout mouse model demonstrated that the proper shutdown of Sox9 expression in hypertrophic chondrocytes is necessary for terminal chondrocyte differentiation and cartilage mineralization [23]. Recently a lineage tracing study revealed that the persistent Sox9 expression in hypertrophic chondrocytes hampers terminal chondrocyte differentiation, possibly by suppressing chondrocyte-to-osteoblast transdifferentiation [39]. Together, all these studies highlight the stage-specific and dose-dependent role of Sox9 during endochondral ossification. However, the phenotype characterization in all previous studies was limited to the embryonic and early postnatal stages, and their implication for adult cartilage hemostasis remains to be determined.
In this report, we examined the consequences of sustained Sox9 overexpression in the hypertrophic chondrocytes of adult mice by characterizing a novel Col10a1-Sox9 mouse model. Consistent with the well-established role of Sox9 in repressing Col10a1 expression in vivo [17], we observed that an elevated Sox9 expression was concomitant with the downregulation of Col10a1 expression in the hypertrophic chondrocytes of TG mice (Figure 3B, 3C). Interestingly, in a previous model of Sox9 overexpression in hypertrophic chondrocytes, the mice exhibited dwarfism in the embryonic and early postnatal stages [27]. However, in our TG mouse model, we did not observe any gross size difference at the early postnatal stage. However, we did observe a decrease in TG skeletal size at 10-weeks of age. Furthermore, 10-week-old TG mice had severe growth plate disorganization and reduced mineralization. These differences between ours and the previous report regarding the effect of Sox9 on the early postnatal stage might be due to the different Col10a1 cis-elements used to target Sox9 expression, and/or due to the different levels of Sox9 transgene expression.
The transcriptional regulators C/ebpα and Pparγ2 are considered master transcriptional factors required for both in vivo and in vitro adipogenesis [40]. Interestingly, our study also demonstrated an enhanced expression of C/ebpα along with the other key adipogenic markers, including Pparγ2 and Glut4. Furthermore, we observed an elevated adipogenesis in ex vivo chondrocyte cultures derived from Col10a1-Sox9 TG mice compared with WT littermates (Figure 3). Sox9 activity affects the expression of the key adipogenic transcription factor C/ebpβ in rat bone-marrow-derived mesenchymal stem cells (MSCs). The stable knockdown of Sox9 in rat MSCs resulted in C/ebpβ downregulation, which reduced their adipogenic potential [41]. Consistent with previous studies, we noticed that the increased Sox9 expression in hypertrophic chondrocytes results in elevated adipogenesis, reinforcing the importance of Sox9 in promoting adipogenic differentiation. A recent clinical study showed that the ectopic adipogenesis was a common pathological feature of advanced knee OA [42]. Interestingly, we observed OA pathogenesis in our Col10a1-Sox9 TG mice at six-and eleven-months of age (Table 2; Figures 4 and 5). Because chondrocytes are essential for cartilage homeostasis by producing sufficient extracellular matrix in the knee joints, the increased adipogenic propensity of hypertrophic chondrocytes in our TG mice may render them more prone to OA. Thus, the potential link between increased adipogenesis and increased OA occurrences remains to be further determined.
Moreover, the dietary intervention also needs to be further investigated to reduce the risk of OA. It is important to note that OA occurred spontaneously and developed progressively in our Col10a1-Sox9 TG mice, which recapitulates the natural course of OA progression to some extent. Thus, our novel Col10a1-Sox9 TG mouse model might be suitable for studying the natural course of OA pathogenesis in vivo.
In summary, our work demonstrates that sustained Sox9 expression in hypertrophic chondrocytes results in spontaneous OA. OA is a chronic disease with limited treatment options besides complete knee replacement and it is a huge health and financial burden for the general public [43]. One proposed strategy for improving cartilage tissue engineering and regenerative medicine to treat OA is the transduction of Sox9 into chondrocytes or mesenchymal stem cells to induce cartilage formation to reverse articular cartilage degeneration and to improve fracture healing [44]. Nevertheless, the long-term consequence of the Sox9 overexpression in the cartilage of animal models is still largely unknown. Our study cautions that the Sox9 expression has to be tightly controlled not only during skeletogenesis, but also at the adult stage, to ensure proper cartilage homeostasis. Thus, it is important to carefully design Sox9 overexpression experiments in cartilage tissue engineering and regenerative biology, especially when longer Col10a1 promoter-intron elements are used for hypertrophic chondrocyte-specific expression of target genes as described recently [45]. In conclusion, we provide strong evidence for the crucial roles of Sox9 in adult hypertrophic cartilage homeostasis and OA pathogenesis. Moreover, this study has important implications for the early diagnosis of OA and for skeletal regenerative treatments in patients who might have abnormal and weakened hypertrophic cartilage.
Acknowledgements
We thank Brian C. Dawson for micro-CT analysis and Terasa Pizzuto for expert histology work. We also thank Dongxing Chen for technical support. This work was supported in part by the National Institutes of Health grants NIH R01 AR068361 and K22 DE15139 to Guang Zhou, and T32 AR007505 to William Samsa, the NSFC grants (# 81472047 and # 81672229) and Shenzhen Science and Technology Program (Grant No. KQTD20170810154011370) to Qiping Zheng.
Disclosure of conflict of interest
None.
References
- 1.Loeser RF, Collins JA, Diekman BO. Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2016;12:412–420. doi: 10.1038/nrrheum.2016.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Styrkarsdottir U, Lund SH, Thorleifsson G, Zink F, Stefansson OA, Sigurdsson JK, Juliusson K, Bjarnadottir K, Sigurbjornsdottir S, Jonsson S, Norland K, Stefansdottir L, Sigurdsson A, Sveinbjornsson G, Oddsson A, Bjornsdottir G, Gudmundsson RL, Halldorsson GH, Rafnar T, Jonsdottir I, Steingrimsson E, Norddahl GL, Masson G, Sulem P, Jonsson H, Ingvarsson T, Gudbjartsson DF, Thorsteinsdottir U, Stefansson K. Meta-analysis of Icelandic and UK data sets identifies missense variants in SMO, IL11, COL11A1 and 13 more new loci associated with osteoarthritis. Nat Genet. 2018;50:1681–1687. doi: 10.1038/s41588-018-0247-0. [DOI] [PubMed] [Google Scholar]
- 3.Hunter DJ, Bierma-Zeinstra S. Osteoarthritis. Lancet. 2019;393:1745–1759. doi: 10.1016/S0140-6736(19)30417-9. [DOI] [PubMed] [Google Scholar]
- 4.Chen D, Shen J, Zhao W, Wang T, Han L, Hamilton JL, Im HJ. Osteoarthritis: toward a comprehensive understanding of pathological mechanism. Bone Res. 2017;5:16044. doi: 10.1038/boneres.2016.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mamidi MK, Das AK, Zakaria Z, Bhonde R. Mesenchymal stromal cells for cartilage repair in osteoarthritis. Osteoarthritis Cartilage. 2016;24:1307–1316. doi: 10.1016/j.joca.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 6.Cucchiarini M, Madry H. Biomaterial-guided delivery of gene vectors for targeted articular cartilage repair. Nat Rev Rheumatol. 2019;15:18–29. doi: 10.1038/s41584-018-0125-2. [DOI] [PubMed] [Google Scholar]
- 7.Jin Y, Cong Q, Gvozdenovic-Jeremic J, Hu J, Zhang Y, Terkeltaub R, Yang Y. Enpp1 inhibits ectopic joint calcification and maintains articular chondrocytes by repressing hedgehog signaling. Development. 2018;145 doi: 10.1242/dev.164830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belfer I, Greco CM, Lokshin A, Vulakovich K, Landsittel D, Dai F, Crossett L, Chelly JE. The design and methods of genetic studies on acute and chronic postoperative pain in patients after total knee replacement. Pain Med. 2014;15:1590–1602. doi: 10.1111/pme.12487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martel-Pelletier J, Barr AJ, Cicuttini FM, Conaghan PG, Cooper C, Goldring MB, Goldring SR, Jones G, Teichtahl AJ, Pelletier JP. Osteoarthritis. Nat Rev Dis Primers. 2016;2:16072. doi: 10.1038/nrdp.2016.72. [DOI] [PubMed] [Google Scholar]
- 10.Mokuda S, Nakamichi R, Matsuzaki T, Ito Y, Sato T, Miyata K, Inui M, Olmer M, Sugiyama E, Lotz M, Asahara H. Wwp2 maintains cartilage homeostasis through regulation of Adamts5. Nature Communications. 2019;10:2429. doi: 10.1038/s41467-019-10177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mathiessen A, Conaghan PG. Synovitis in osteoarthritis: current understanding with therapeutic implications. Arthritis Res Ther. 2017;19:18. doi: 10.1186/s13075-017-1229-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li T, Chubinskaya S, Esposito A, Jin X, Tagliafierro L, Loeser R, Hakimiyan AA, Longobardi L, Ozkan H, Spagnoli A. TGF-beta type 2 receptor-mediated modulation of the IL-36 family can be therapeutically targeted in osteoarthritis. Sci Transl Med. 2019;11 doi: 10.1126/scitranslmed.aan2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang F, Liu J, Chen X, Zheng X, Qu N, Zhang B, Xia C. IL-1beta receptor antagonist (IL-1Ra) combined with autophagy inducer (TAT-Beclin1) is an effective alternative for attenuating extracellular matrix degradation in rat and human osteoarthritis chondrocytes. Arthritis Res Ther. 2019;21:171. doi: 10.1186/s13075-019-1952-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang J, Zhao L, Fan Y, Liao L, Ma PX, Xiao G, Chen D. The microRNAs miR-204 and miR-211 maintain joint homeostasis and protect against osteoarthritis progression. Nat Commun. 2019;10:2876. doi: 10.1038/s41467-019-10753-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu Y, Ding M, Li N, Wang Q, Li J, Li X, Gu J, Im HJ, Lei G, Zheng Q. Col10a1-Runx2 transgenic mice with delayed chondrocyte maturation are less susceptible to developing osteoarthritis. Am J Transl Res. 2014;6:736–745. [PMC free article] [PubMed] [Google Scholar]
- 16.Samsa WE, Zhou X, Zhou G. Signaling pathways regulating cartilage growth plate formation and activity. Semin Cell Dev Biol. 2017;62:3–15. doi: 10.1016/j.semcdb.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou G, Zheng Q, Engin F, Munivez E, Chen Y, Sebald E, Krakow D, Lee B. Dominance of SOX9 function over RUNX2 during skeletogenesis. Proc Natl Acad Sci U S A. 2006;103:19004–19009. doi: 10.1073/pnas.0605170103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yasuda H, Oh CD, Chen D, de Crombrugghe B, Kim JH. A novel regulatory mechanism of type II collagen expression via a SOX9-dependent enhancer in intron 6. J Biol Chem. 2017;292:528–538. doi: 10.1074/jbc.M116.758425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oh CD, Yasuda H, Zhao W, Henry SP, Zhang Z, Xue M, de Crombrugghe B, Chen D. SOX9 directly regulates CTGF/CCN2 transcription in growth plate chondrocytes and in nucleus pulposus cells of intervertebral disc. Sci Rep. 2016;6:29916. doi: 10.1038/srep29916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oh CD, Lu Y, Liang S, Mori-Akiyama Y, Chen D, de Crombrugghe B, Yasuda H. SOX9 regulates multiple genes in chondrocytes, including genes encoding ECM proteins, ECM modification enzymes, receptors, and transporters. PLoS One. 2014;9:e107577. doi: 10.1371/journal.pone.0107577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mead TJ, Wang Q, Bhattaram P, Dy P, Afelik S, Jensen J, Lefebvre V. A far-upstream (-70 kb) enhancer mediates Sox9 auto-regulation in somatic tissues during development and adult regeneration. Nucleic Acids Res. 2013;41:4459–4469. doi: 10.1093/nar/gkt140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Sox9 is required for cartilage formation. Nat Genet. 1999;22:85–89. doi: 10.1038/8792. [DOI] [PubMed] [Google Scholar]
- 23.Dy P, Wang W, Bhattaram P, Wang Q, Wang L, Ballock RT, Lefebvre V. Sox9 directs hypertrophic maturation and blocks osteoblast differentiation of growth plate chondrocytes. Dev Cell. 2012;22:597–609. doi: 10.1016/j.devcel.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bi W, Huang W, Whitworth DJ, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Haploinsufficiency of Sox9 results in defective cartilage primordia and premature skeletal mineralization. Proc Natl Acad Sci U S A. 2001;98:6698–6703. doi: 10.1073/pnas.111092198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yano F, Ohba S, Murahashi Y, Tanaka S, Saito T, Chung UI. Runx1 contributes to articular cartilage maintenance by enhancement of cartilage matrix production and suppression of hypertrophic differentiation. Sci Rep. 2019;9:7666. doi: 10.1038/s41598-019-43948-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng Q, Zhou G, Morello R, Chen Y, Garcia-Rojas X, Lee B. Type X collagen gene regulation by Runx2 contributes directly to its hypertrophic chondrocyte-specific expression in vivo. J Cell Biol. 2003;162:833–842. doi: 10.1083/jcb.200211089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hattori T, Muller C, Gebhard S, Bauer E, Pausch F, Schlund B, Bosl MR, Hess A, Surmann-Schmitt C, von der Mark H, de Crombrugghe B, von der Mark K. SOX9 is a major negative regulator of cartilage vascularization, bone marrow formation and endochondral ossification. Development. 2010;137:901–911. doi: 10.1242/dev.045203. [DOI] [PubMed] [Google Scholar]
- 28.Kim Y, Murao H, Yamamoto K, Deng JM, Behringer RR, Nakamura T, Akiyama H. Generation of transgenic mice for conditional overexpression of Sox9. J Bone Miner Metab. 2011;29:123–129. doi: 10.1007/s00774-010-0206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng Q, Keller B, Zhou G, Napierala D, Chen Y, Zabel B, Parker AE, Lee B. Localization of the cis-enhancer element for mouse type X collagen expression in hypertrophic chondrocytes in vivo. J Bone Miner Res. 2009;24:1022–1032. doi: 10.1359/JBMR.081249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tao J, Jiang MM, Jiang L, Salvo JS, Zeng HC, Dawson B, Bertin TK, Rao PH, Chen R, Donehower LA, Gannon F, Lee BH. Notch activation as a driver of osteogenic sarcoma. Cancer Cell. 2014;26:390–401. doi: 10.1016/j.ccr.2014.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen D, Bashur LA, Liang B, Panattoni M, Tamai K, Pardi R, Zhou G. The transcriptional co-regulator Jab1 is crucial for chondrocyte differentiation in vivo. J Cell Sci. 2013;126:234–243. doi: 10.1242/jcs.113795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bashur LA, Chen DX, Chen ZJ, Liang BJ, Pardi R, Murakami S, Zhou G. Loss of Jab1 in osteochondral progenitor cells severely impairs embryonic limb development in mice. J Cell Physiol. 2014;229:1607–1617. doi: 10.1002/jcp.24602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- 34.Kobayashi H, Gao Y, Ueta C, Yamaguchi A, Komori T. Multilineage differentiation of Cbfa1-deficient calvarial cells in vitro. Biochem Biophys Res Commun. 2000;273:630–636. doi: 10.1006/bbrc.2000.2981. [DOI] [PubMed] [Google Scholar]
- 35.Liang B, Cotter MM, Chen D, Hernandez CJ, Zhou G. Ectopic expression of SOX9 in osteoblasts alters bone mechanical properties. Calcif Tissue Int. 2012;90:76–89. doi: 10.1007/s00223-011-9550-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neumann K, Endres M, Ringe J, Flath B, Manz R, Haupl T, Sittinger M, Kaps C. BMP7 promotes adipogenic but not osteo-/chondrogenic differentiation of adult human bone marrow-derived stem cells in high-density micro-mass culture. J Cell Biochem. 2007;102:626–637. doi: 10.1002/jcb.21319. [DOI] [PubMed] [Google Scholar]
- 37.Zhao L, Huang J, Fan Y, Li J, You T, He S, Xiao G, Chen D. Exploration of CRISPR/Cas9-based gene editing as therapy for osteoarthritis. Ann Rheum Dis. 2019;78:676–682. doi: 10.1136/annrheumdis-2018-214724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leung VY, Gao B, Leung KK, Melhado IG, Wynn SL, Au TY, Dung NW, Lau JY, Mak AC, Chan D, Cheah KS. SOX9 governs differentiation stage-specific gene expression in growth plate chondrocytes via direct concomitant transactivation and repression. PLoS Genet. 2011;7:e1002356. doi: 10.1371/journal.pgen.1002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lui JC, Yue S, Lee A, Kikani B, Temnycky A, Barnes KM, Baron J. Persistent Sox9 expression in hypertrophic chondrocytes suppresses transdifferentiation into osteoblasts. Bone. 2019;125:169–177. doi: 10.1016/j.bone.2019.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee JE, Schmidt H, Lai B, Ge K. Transcriptional and epigenomic regulation of adipogenesis. Mol Cell Biol. 2019;39 doi: 10.1128/MCB.00601-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stockl S, Bauer RJ, Bosserhoff AK, Gottl C, Grifka J, Grassel S. Sox9 modulates cell survival and adipogenic differentiation of multipotent adult rat mesenchymal stem cells. J Cell Sci. 2013;126:2890–2902. doi: 10.1242/jcs.124305. [DOI] [PubMed] [Google Scholar]
- 42.Ikemoto-Uezumi M, Matsui Y, Hasegawa M, Fujita R, Kanayama Y, Uezumi A, Watanabe T, Harada A, Poole AR, Hashimoto N. Disuse atrophy accompanied by intramuscular ectopic adipogenesis in vastus medialis muscle of advanced osteoarthritis patients. Am J Pathol. 2017;187:2674–2685. doi: 10.1016/j.ajpath.2017.08.009. [DOI] [PubMed] [Google Scholar]
- 43.Davidson RK, Green J, Gardner S, Bao Y, Cassidy A, Clark IM. Identifying chondroprotective diet-derived bioactives and investigating their synergism. Sci Rep. 2018;8:17173. doi: 10.1038/s41598-018-35455-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cucchiarini M, Terwilliger EF, Kohn D, Madry H. Remodeling of human osteoarthritic cartilage by FGF-2, alone or combined with Sox9 via rAAV gene transfer. J Cell Mol Med. 2009;13:2476–2488. doi: 10.1111/j.1582-4934.2008.00474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen J, Chen F, Bian H, Wang Q, Zhang X, Sun L, Gu J, Lu Y, Zheng Q. Hypertrophic chondrocyte-specific Col10a1 controlling elements in Cre recombinase transgenic studies. Am J Transl Res. 2019;11:6672–6679. [PMC free article] [PubMed] [Google Scholar]