

Abstract
Currently, the acquired resistance of the hepatocellular carcinoma (HCC) first-line therapeutic agent-sorafenib (SOR) remains a major challenge for HCC management. Recent evidence suggested the association between CXCL/CXCRs chemokines and chemotherapy resistant in cancer cells. Hence, exploring the internal mechanism of CXCRs involved in SOR resistance will help to improve the efficacy of HCC. SOR-resistant HCC cells (Huh7-SOR) were established through escalating concentration of SOR. Glucose consumption, lactate production, intracellular ATP levels and oxygen consumption of HCC cells were determined by using the associated detected kits. Effects of CXCR3 on metabolic phenotype of HCC cells, AMPK pathway activity and adipocytokines were demonstrated by knocking down CXCR3 expression with the CXCR3 siRNA technique combined with qPCR and western blot. During the indicated procedure, SOR-resistant HCC cells-Huh7-SOR presented EMT-like morphologic change and underwent glycolysis to OXPHOS switch, representing reduced glucose consumption and lactate production, but increased oxygen consumption level and intercellular ATP levels. Moreover, metabolic alteration in SOR-resistance HCC cells was mediated by CXCR3. Mechanistically, CXCR3 induced metabolic alteration in SOR-resistance HCC cells through downregulating AMPK pathway activity and lipid peroxidation as well as upregulating levels of adipocytokines. The activation of A MPK pathway with metformin achieved the sensitization of HCC to SOR treatment in vivo. These findings unravel the association between metabolic alteration and SOR-resistance in HCC cells and demonstrate an important role of CXCR3 in the development of HCC cells resistance to SOR treatment and a novel mechanism of CXCR3 regulating AMPK pathway activity and adipocytokine signaling, lipid peroxidation resulted in metabolic alteration during the chemoresistance.
Keywords: Hepatocellular carcinoma, chemoresistance, metabolism reprograming, CXCR3, AMPK, adipocytokines, lipid peroxidation
Introduction
Hepatocellular carcinoma (HCC) is one of the most common cancers worldwide, especially in China, and causes most of the global mortality. Currently, effective treatment of HCC is still a rising dilemma. Although surgical resection, ablation or transplantation provide the only chance of cure, there are substantial patients with advanced disease who are struggling with limited effective treatment approaches [1]. Several decades ago, a molecular targeted agent-sorafenib has been approved for advanced HCC as an option of first-line treatment; however, as treatment-time prolongs, many patients experience drug resistance, which represents an unmet clinical need and the acquired resistance remains a major obstacle for its further application [2]. Hence, exploring the internal mechanism of sorafenib resistance will help to discovering more effective therapeutic strategies for HCC patients with previous sorafenib treatment.
Notably, chemotherapies induced upregulation of a variety of chemokines in the C-X-C motif (CXC) family by various mechanisms. It has been found that, compared with parental cells, drug-resistant cells presented higher levels of C-X-C motif chemokine ligands (CXCLs) and C-X-C motif chemokine receptors (CXCRs), as a signature of drug resistance [3]; meanwhile significantly upregulated expression of CXCL/CXCRs chemokines was observed in some chemotherapy resistant cancer cells [4]. Furthermore, the roles of CXCL/CXCRs in the interactions between cancer cells and its microenvironment are recently highlighted [5]. Chemokines have been observed to have functions rather than simply being attractants and are able to alter the biological functions of cancer cells with its expression [6]. As a member of CXCR family and an interferon-inducible chemokine receptor, CXCR3 is not only expressed preferentially on leukocytes and lymphocyte, but also on cancer cells, assisting cancer cells to progress [7] and could induce chemotherapy resistance in HCC [8], suggesting a potential adjuvant therapeutic target for sensitizing therapeutic treatment for HCC [9].
It has been reported that CXC system could affect glucose metabolism in cancer [10]; furthermore, dysregulated metabolism could be associated with increased drug resistance [11,12], suggesting improper cellular metabolism may be linked to drug resistance. Given these associations above, it strongly suggests the involvement of CXCR3 in chemotherapy resistance and metabolic alteration of HCC; hence, we aimed to demonstrate an important role of CXCR3 in metabolic alteration of HCC cells and resistance to sorafenib in the study.
Materials and methods
Reagents and antibodies
Sorafenib (#284461-73-0; Bay 43-9006) was purchased from MCE Inc. (shanghai, China) and solubilized in DMSO. Anti-CXCR3 (ab64714 and ab71864) antibody were respectively mouse monoclonal and rabbit polyclonal antibodies and obtained from Abcam Corp. AMPKα (#2603) and phospho-AMPKα (Thr172) (#2535) were purchased from Cell Signaling Technology. ADIPOQ (CSB-PA618019YA01HU) and ACSL4 (CSB-PA060488EA01HU) were from CUSABIO, ACSBG1 (GTX16561) was from GeneTex. HRP-conjugated monoclonal mouse anti-beta actin was from Kang Chen Biotech.
Cell culture and construction of HCC-SOR cells
Sorafenib-resistant HCC cell line-Huh7-SOR was constructed as follow: parental Huh7 cells were exposed to serial increasing concentrations ranging from 1 to 10 µM of sorafenib (increasing 0.25 µM per cycle) for about 3 months (Figure 1A). To make the cells to acquire steady resistance to sorafenib, the culture medium for Huh7-SOR cells contained 10 µM sorafenib. Resistance index to sorafenib in drug-resistant strains and their parental cells was determined by using CCK-8 assay. Before the experiments, Huh7-SOR cells were cultured without sorafenib for 1 week.
Figure 1.
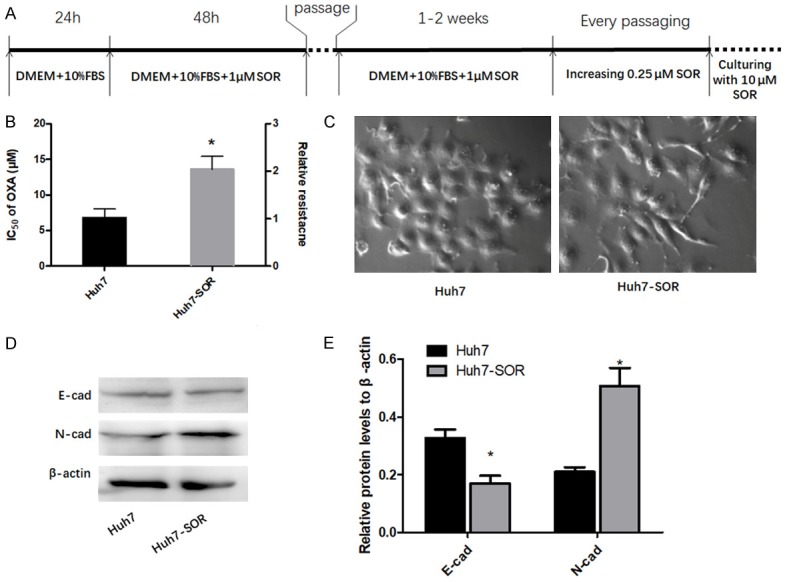
HCC cells underwent glycolysis to OXPHOS switch during resistance to SOR. A. The procedure of establishing SOR-resistant Huh7 cells (Huh7-SOR). The detail description can be found in the METHODS section. B. The IC50 for SOR in Huh7 and Huh7-SOR cells. The left longitudinal axis means the IC50 for SOR; the right longitudinal axis indicates relative resistance based on the definition of Huh7 as 1. C. Huh7-SOR cells presented different morphology from parental Huh7 cells, displaying a spindle shape and pseudopodia formation, consistent with EMT characteristics. All values represent the mean of three independent experiments. D, E. Protein levels of E-cadherin and N-cadherin were detected by western blotting, showing the decrease of E-cadherin and the increase of N-cadherin in Huh7-SOR cells. The results represent means of triplicates. *P<0.05 vs Huh7.
Cytotoxicity analysis by CCK-8 assay
Cells (1,000 cells/well) were plated in 96-well plates and incubated for 24 h in complete medium after treatment as mentioned above and cell viability was measured by using the Cell Counting Kit-8 (CCK-8, Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s instructions. Briefly, 10 μl CCK-8 solution was added to each well with 100 μl medium. After incubation at 37°C for 2 h, a microplate reader (Perkin Elmer Victor 3, USA) was used to detect the optical density at 450 nm. The IC50 values were calculated from the concentration-response curve.
Glucose consumption
Culture medium was collected and glucose contents were measured immediately by using Glucose Uptake Colorimetric Assay Kit (# K676-100; 100 assays; BioVision) following the manufacturer’s instructions. The glucose concentrations were calculated and normalized to control cells.
Lactate production
Lactate production assay was determined by using the Lactate Kit (Nangjing Jiancheng Bioengineering, Nanjing, China) according to manufacturer’s protocol. In brief, culture medium was collected and lactate concentration was measured. Meanwhile, cells were then harvested, stained with trypan blue, and the number of viable cells was counted using a cytometer (Bio-rad, USA). Lactate production rate was defined as lactate concentration/cells/time and normalized to the rate detected in the control.
ATP production
The ATP production assay was performed through using the ATP Colorimetric/Fluorometric Assay Kit (ab83355, Abcam). Harvested cells were washed with cold PBS and centrifuged 2 minutes at 4°C at top speed (13,000 rpm) in a cold microcentrifuge. Supernatant were collected and kept on ice. Measurements were performed with the colorimetric assay, following the manufacturer’s instructions. ATP contents were measured in duplicate and expressed as % control.
Oxygen consumption
The oxygen consumption rate (OCR) was measured using an Oxygen Consumption Rate Assay Kit (MitoXpress-Xtra HS Method, Cayman Chemical, Ann Arbor, MI, USA). Cells were seeded in a 96-well plate and cultured overnight. Spent culture medium were removed from all wells and 150 ul of fresh medium were replaced. Then, 10 ul MitoXpress-Xtra solution was added and 100 ul of HS Mineral Oil was overlaid in each well. Finally, the plate was read immediately and the fluorescence was measured by using the Perkin Elmer Victor 3 reader.
Intracellular MDA content
Intracellular content was measured by OxiSelect™ TBARS Assay Kit (# STA-330; 200 assays; BioVision) according to manufacturer’s specifications. The plate was read at 532 nm. The MDA contents were calculated and normalized to control cells.
Knockdown of CXCR3 by shRNA transfection
In brief, lentiviral vectors pLKO.1 TRC and pWPI.1 were used for constructing recombinant lentiviruses of short interference RNA (shRNA) constructs and CXCR3 shRNA (target sequences: 5’-CCUUCCUGCUGGCUUGUAUTT-3’ (shRNA-1); 5’-GGUCAUGGCCUACUGCUAUTT-3’ (shRNA-2); 5’-GCUAGAUGCCUCG GACUUUTT-3’ (shRNA-3), respectively), non-targeting shRNA (shNT). Recombinant lentivirus was amplified in HEK293T cells.
Immunoblotting analysis
Cells were plated at 6×105 cells per well in six-well plates. After treatment, cells were washed with lysed using RIPA lysis buffer containing 1 mM phenylmethylsulfonyl fluoride (PMSF). Twenty μg proteins underwent electrophoresis on a 10% or 8% SDS- polyacrylamide gel; proteins separated in the gel were transferred to a 0.2 mm polyvinylidene fluoride membrane (PVDF, Millipore, Billerica, USA) using a Bio-Rad semi-dry instrument. After blocking with 5% BSA in TBS containing 0.1% Tween-20 for 1 h at room temperature, the membranes were incubated with a primary antibody overnight at 4°C. After washing, the membranes were incubated with an anti-rabbit secondary antibody and performed using the ECL western blot system (Super Signal West Pico Chemiluminescent Substrate, Pierce, Rockford, IL, USA) according to the manufacturer’s instruction. Quantification of western blots was analyzed densitometrically using QuantityOne software (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Tumorigenicity assay in xenograft mice model
Eighteen male BALB/c nude mice (5-6 weeks, 20-25 g) were purchased from the Shanghai Lab Animal Research Center. For assessment of tumor formation abilities, a total of 5×106 Huh7-SOR cells were resuspended in 100 μL of PBS buffer, and then subcutaneously injected into the right flank of nude mice and tumor volume was measured according to the indicated times. One week after injection, mice were orally administered with 30 mg/kg sorafenib (sorafenib), 200 mg/kg metformin (metformin), or combination every day. After six weeks, mice were sacrificed and tumors were excised and weighted. Tumor volume was calculated as follows: volume = (length × width2 ×0.5).
Statistical analysis
Statistical analysis was performed with SPSS 19.0 software (SPSS, Chicago, IL, USA). Quantitative variables were performed using a one-way analysis of variance (ANOVA). Student’s t-test was used in two-group comparisons. The association between the various factors was determined using Pearson’s correlation. P<0.05 was considered as statistical significance.
Results
HCC cells underwent metabolic alteration during the development of resistance to SOR
At the end of treatment procedure, the IC50 of SOR in these Huh7 cells was significantly higher than that of SOR in parental cells (13.6±1.8 μM vs 6.8±1.2 μM) (Figure 1B), suggesting the resistance of these HCC cells to SOR after continuous enhanced exposure was increased over their parental cells, hence termed as Huh7-SOR. During the development of resistance to SOR, Huh7 cells experienced morphologic change and displayed a spindle shape and pseudopodia formation, which was consistence with epithelial to mesenchymal transition (EMT) (Figure 1C); moreover, compared with parental Huh7 cells, Huh7-SOR cells showed the decrease of E-cadherin as well as the increase of N-cadherin (Figure 1D, 1E).
Recently, the metabolic behavior of cancer cells has been implicated in resistance to chemotherapy [13]. To investigate the metabolism mode of HCC cells during the resistance to SOR, we compared the glucose consumption, lactate production and intracellular ATP levels, oxygen consumption of Huh7-SOR cells with those of parental Huh7 cells. Remarkably, Huh7-SOR cells showed reduced glucose consumption and lactate production, but increased oxygen consumption level and intercellular ATP levels, compared with parental Huh7 cells (Figure 2A-D).
Figure 2.
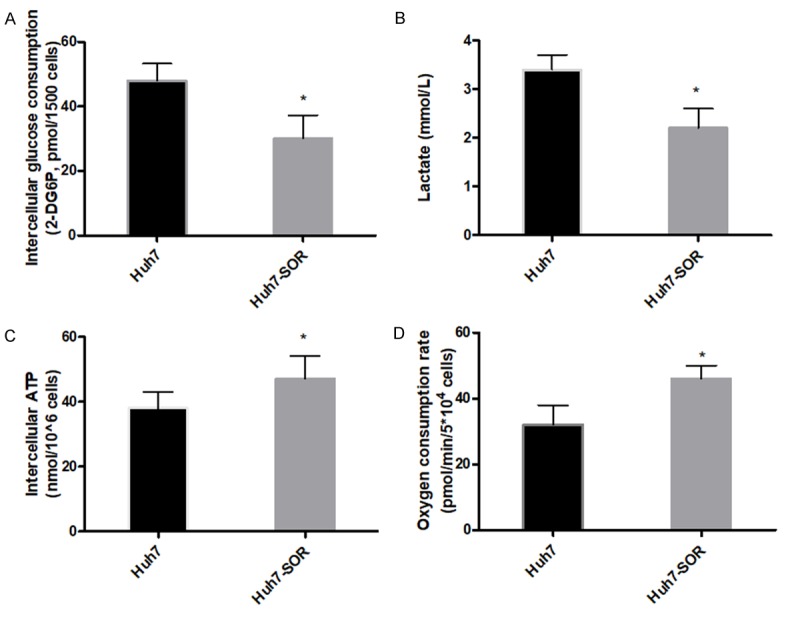
Metabolic parameters alteration in parental Huh7 cells and Huh7-SOR cells. Glucose consumption (A), lactate production (B) and intracellular ATP levels (C), oxygen consumption (D) of Huh7 cells and Huh7-SOR cells were determined by using the associated methods described in METHODS section. All values represent the mean of three independent experiments. *P<0.05.
Metabolic alteration in SOR-resistance HCC cells is mediated by CXCR3
Our previous research has reported CXCR3 was upregulated in HCC tissues compared to para-cancerous tissues [14]. In the study, it was furthermore found that CXCR3 was also significantly upregulated in Huh7-SOR cells compared to parental cells, suggesting CXCR3 may be an important effector in the development of resistance to SOR for HCC cells (Figure 3A-C). To further determine the role of CXCR3 in the development of resistance to SOR, we knockdown the cxcr3 gene in Huh7 cells with three candidate lentivirus harboring shRNA (shRNA-1, shRNA-2 and shRNA-3) and found the highest knockdown effectiveness in cells with lentivirus harboring shRNA-1 (Figure 3D, 3E); hence the lentivirus harboring shRNA-1 was chosen for subsequent experiments. Furthermore, it was strikingly found, in everyday glucose condition, the IC50 of SOR in CXCR3-knockdown Huh7 (Huh7-shCXCR3) cells didn’t reach up to IC50 value as in Huh7-SOR cells, after indicated treatment procedure as in Huh7 cells (8.3±1.3 μM vs 13.6±1.8 μM) (Figure 3F). In addition, we observed that Huh7-shCXCR3 cells didn’t display significant decrease in glucose consumption, lactate production and increase in oxygen consumption level and intercellular ATP levels, compared with parental controls, during about 3 months treatment with series concentrations of SOR (Figure 4A-D).
Figure 3.

Expression of CXCR3 in parental Huh7 cells and Huh7-SOR cells and effect of CXCR3 knockdown on the forming of resistance to SOR. mRNA level (A) and protein level (B, C) of CXCR3 were increased in Huh7-SOR cells compared with parental Huh7 cells. cxcr3 gene in HCC cells was effectively knocked down with three candidate lentivirus harboring shRNA (shRNA-1, shRNA-2 and shRNA-3) and found the highest knockdown effectiveness in cells with lentivirus harboring shRNA-1, the lentivirus harboring shRNA-1 was chosen for subsequent experiments (D, E). After CXCR3 effective knockdown, the relative resistance to SOR was decreased (F). All values represent the mean of three independent experiments. *P<0.05.
Figure 4.
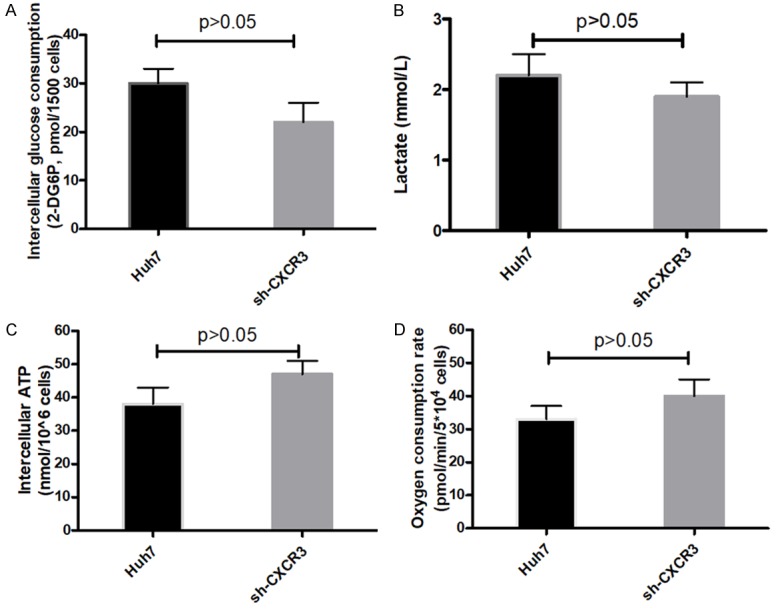
Metabolic parameters alteration in parental Huh7 cells and Huh7-shCXCR3 cells during indicated period with SOR treatment. Glucose consumption (A), lactate production (B) and intracellular ATP levels (C), oxygen consumption (D) of Huh7 cells and Huh7-shCXCR3 cells were determined by using the associated methods described in METHODS section. All values represent the mean of three independent experiments.
AMPK pathway is involved in CXCR3-mediated development of SOR-resistance in HCC cells
The observation that CXCR3 mediated a glycolysis to OXPHOS switch in HCC cells drove us to explore the underlying molecular mechanism. Firstly, RNA-Seq data was analysis and there were total of 291 genes detected with an absolute fold-change ≥2 and P≤0.01 as the cut-off criteria to analyze and compare the RNA expression values between parental Huh7 and Huh7-SOR cells. To further understand potential underlying mechanisms in the development of Huh7 resistance to SOR, we performed KEGG pathway analyses using 291 genes and found these genes were predominantly enriched in networks including adipocytokine signaling pathway, AMPK signaling pathway and FoxO signaling pathway, Insulin signaling pathway, Purine metabolism (Figure 5A). Among These pathways, AMPK signaling was demonstrated to function as a critical factor in the reprogramming metabolism by many studies [15]. In recent years, studies have shown that AMPK is also involved in the physiological role of adipocytokines represented by leptin, adiponectin and resistin, through regulating the secretion of adipocytokines, so elucidating the relationship between AMPK and adipocytokines will provide a new insight for the treatment of cancer [16,17]. Hence, we investigated the activation status of AMPK pathway and levels of adipocytokines including ACSBG1, ADIPOQ, ACSL4 in HCC-SOR cells. Compared with parental Huh7 cells, Huh7-SOR cells presented the lower phosphor-AMPKα/AMPKα ratio and upregulated levels of ACSBG1, ADIPOQ, ACSL4, suggesting the downregulation of AMPK pathway activity and upregulation of adipocytokine signaling pathway in HCC cells resistant to sorafenib. Moreover, we compared the status of lipid peroxidation in HCC-SOR cells with that of parental HCC cells and found the decrease of malondialdehyde amount in HCC-SOR cells, suggesting the inhibition of lipid peroxidation in sorafenib-resistant HCC cells. To investigate the effect of CXCR3 on the indicators mentioned above, we observed the alteration of AMPK pathway activity, levels of adipocytokine and malondialdehyde amount in CXCR3-knockdown HCC cells. The results showed, compared with CXCR3-knockdown parental Huh7 (Huh7-shCXCR3), CXCR3-knockdown Huh7-SOR cells (Huh7-SOR-shCXCR3) didn’t show significant decrease of phosphor-AMPKα/AMPKα ratio and malondialdehyde amount as well as the upregulation of ACSBG1, ADIPOQ, ACSL4 (Figure 5B-D). It indicated that the maintenance of AMPK activity and lipid peroxidation in Huh7-SOR cells might be dependent in the expression of CXCR3. To elucidate the proposal, we treated CXCR3-knockdown Huh7-SOR cells with metformin (10 mM), a well-defined agonist of AMPK pathway in many cancer studies, and found no significant upregulation of AMPK activity and altered status of lipid peroxidation but different change of three adipocytokines in CXCR3-knockdown Huh7-SOR cells compared with that in Huh7-SOR cells (Figure 5E-G).
Figure 5.

AMPK pathway is involved in CXCR3-mediated metabolic phenotype switch in SOR-resistance HCC cells. (A) Corresponding network by sequencing analysis based on the different expression genes of Huh7 cells and Huh7-SOR cells indicated AMPK pathway is a critical node during the forming of resistance to SOR. (B, C) Compared to Huh7 cells, p-AMPKα/AMPKα ratio was significantly decreased, levels of adipocytokines including ACSBG1, ADIPOQ, ACSL4 were increased as well as intracellular level of lipid peroxidation product MDA was decreased in Huh7-SOR cells, as shown in above values (D). After CXCR3 knockdown, compared with CXCR3-knockdown parental Huh7 (Huh7-shCXCR3), CXCR3-knockdown Huh7-SOR cells (Huh7-SOR-shCXCR3) didn’t show significant decrease of phosphor-AMPKα/AMPKα ratio and malondialdehyde amount as well as the upregulation of ACSBG1, ADIPOQ, ACSL4 (B-D). All values represent the mean of three independent experiments. *P<0.05 vs Huh7, #P<0.05 vs Huh7-SOR, &P>0.05 vs Huh7-shCXCR3. Moreover, CXCR3-knockdown Huh7-SOR cells were treated with metformin (10 mM) for 24 h, and no significant upregulation of AMPK activity (E, F) and levels of adipocytokines and intracellular MDA (G) were found, compared with those in control (Huh7-SOR-shCXCR3) cells. &P>0.05 vs Huh7-SOR-shCXCR3.
The sensitization of HCC cells to SOR treatment is induced by activation of AMPK pathway
Based on the findings above, we treated Huh7-SOR cells with metformin (10 mM) or SOR (5 μM) monotherapy or combination for serial times to investigate the effect of AMPK pathway activation by metformin on the response of SOR-resistance HCC cells to SOR. As shown in Figure 6A, compared with control, SOR monotherapy didn’t significantly inhibit the viability of Huh7-SOR cells; moreover, metformin monotherapy suppressed the proliferation of Huh7-SOR cells from 72 h, but compared with metformin monotherapy, combination treatment of metformin and SOR further significantly led to the inhibitory effect.
Figure 6.

Metformin and sorafenib in combination potentiate the antiproliferative effect of the single drug treatments in vitro and in vivo. A: Cell viability of Huh7-SOR cells with metformin (10 mM) or SOR (5 μM) monotherapy or combination for serial times. B: Huh7-SOR cells were implanted subcutaneously into the flank regions of nude mice. when the tumor volume reached ~50 mm3, metformin (200 mg/kg/day, intraperitoneal injection), sorafenib (30 µg/kg/day, intragastric administration), or their combination were administered for 4 weeks, 5 times/week. At 6th weeks, the mice were euthanized and the tumors are shown. C: The growth curve of xenografts was shown. The data were quantified and are represented as the means ± SD from 3 independent experiments. *P<0.05, Combination vs. SOR.
To investigate the in vivo effect of metformin and on SOR-resistance HCC cells, we treated xenograft nude mice bearing SOR-resistance Huh7 cells and observed the growth-curve of xenografts during the indicated procedure. It was found that, compared with SOR monotherapy, although metformin monotherapy led to a slightly slower growth 5 weeks after commencement of treatment, the difference was not significant. In contrast, the combination of metformin and SOR significantly suppressed xenograft growth from the 4th week, the mean size of xenografts was smaller than the tumors treated with SOR or metformin monotherapies (Figure 6B, 6C).
Discussion
As the first molecular targeted-therapeutic drug, SOR has once displayed obvious anti-cancer activity against HCC, the third leading cause of cancer deaths worldwide and the highest incidence in Asia [18]; however, in recent years more and more clinical and preclinical studies observed there is a high-level heterogeneity of individual response to sorafenib treatment, suggesting that sorafenib treatment may have limited efficacy due to tumor progression from the rapid development of acquired resistance [19]. The existence of HCC acquired resistance to SOR has been recognized to be significantly or potentially associated with tumor microenvironment, angiogenesis, inflammation, fibrosis, hypoxia, oxidative stress, autophagy and viral reactivation [20,21]. Nevertheless, the resistance mechanism underlying the impaired sensitivity to sorafenib is still elusive, leading to evasive resistance to SOR is a major challenge in HCC research. More recently, it is observed SOR-resistant HCC cells exhibit higher reductive glutamine metabolism [22], suggesting metabolic alteration maybe associated with therapeutic resistance in cancer cells [23]. Hence, we firstly investigated the metabolic phenotype alteration during the development of HCC cells resistance to SOR and observed decreased glucose consumption, lactate production, and increased oxygen consumption level, intercellular ATP levels in SOR-resistant HCC cells. It suggested that SOR-resistant HCC cells achieved glycolysis to OXPHOS switch in the development period of resistance to SOR.
Of note, we further observed that HCC cells underwent epithelial-to-mesenchymal transition (EMT)-like alteration during the resistance to SOR in the study. In recent years, the role of EMT in the development of drug resistance in HCC has gained increasing attention; EMT has been implicated in the involvement of drug resistance through regulating the generation of cancer stem cells (CSCs) [24] or activating FAK/PI3K/AKT signaling [25]. Although the molecular mechanism of EMT development in HCC resistance to SOR was not elucidated in the current study, it is a fact that EMT is indeed involved in SOR-resistance.
As mentioned above, during the development of resistance to SOR, HCC cells experienced glycolysis to OXPHOS switch; hence, further exploring the internal molecular mechanism will help to elucidate and intervene the forming of resistance to SOR in HCC cells. Accumulated evidence demonstrated that chemokine pathway activation is a critical mechanism of cancer resistance to therapeutic agents through directly promoting cancer stem/tumor-initiating cell phenotype, recruiting stroma cells and promoting angiogenesis [26]. Recently, it was demonstrated the cross-talk of mesenchymal stem cells (MSC) with tumor-initiating cells (TIC) promoted chemotherapy-resistance through the activation of the CXCL10-CXCR3 axis [27]; meanwhile our previous findings indicated that alteration of CXCR3 expression could affect malignant proliferation of HCC cells [14]. These findings encouraged us to elucidate the role of CXCR3 in the development of HCC cells resistance to SOR. In the study, significant upregulation of CXCR3 expression in SOR-resistance HCC cells was identified, which may be related to CXCR3 partner-CXCL10 overexpression, because the overexpression of CXCL10 has been found in the development of HCC cells resistance to cisplatin, which attributed to cisplatin resistance through mediating ATF6/Grp78 ER stress signaling pathway [8]. More-over, as a downstream mediator of the intratumoral interferon response, CXCL10/CXCR3 signaling suppressed antitumor immunity and induced gemcitabine resistance iattention to obesity-associated cancersound CXCR3-knockdown HCC cells could not achieve resistance to SOR during indicated period and meanwhile didn’t present significant alterations in glucose consumption, lactate production, oxygen consumption level and intercellular ATP levels as wildtype cells. These results indicated metabolic phenotype switch in SOR-resistance HCC cells was mediated by CXCR3.
Mechanistically, it was further demonstrated that CXCR3 induced metabolic phenotype switch in SOR-resistance HCC cells through downregulating AMPK pathway activity. Recently, it has been noted that chemokine-receptor network can regulate the homeostasis of bioenergetic signaling [29]. As two critical regulators of bioenergetic signaling in cancers, CXCR3 and AMPK could be therapeutic targets for HCC treatment. In addition to biological importance of CXCR3 in HCC, our work further explored its relevant in clinical management of HCC through treating xenograft nude mice bearing SOR-resistance HCC cells with AMPK agonist-metformin or SOR monotherapy or combination. Compared to monotherapy, combination therapy significantly suppressed xenograft growth, suggesting the activation of AMPK pathway can induce sensitization of HCC cells to SOR treatment, which may provide novel insight for developing new therapeutic strategies for SOR-resistant HCC. Furthermore, in the study, effects of CXCR3 on adipocytokines and lipid peroxidation were at the first time observed in SOR-resistant HCC cells. In these years, the field of cancer research has drawn increased attention to obesity-associated cancers, because adipose tissue provides a unique microenvironment, which confers both cancer initiation and progression [30]. The precise mechanisms elucidating the association of abnormal lipid metabolism with cancer remain elusive. Recently, the inhibition of lipid peroxidation has been recognized as a feature of therapy-resistant cancer cells [31], consistent with our observation in the study. These findings in our study warrants further investigation of the key mechanisms that link CXCR3 with the activity of AMPK pathway, secretion of adipocytokines and the regulation of lipid peroxidation in the SOR-resistant setting.
In summary, our findings unravel the association between metabolic alteration and SOR-resistance in HCC cells and further demonstrate a key role of CXCR3 in the forming of HCC cells resistance to SOR treatment, and a novel mechanism of CXCR3 regulating AMPK pathway activity, adipocytokines and lipid peroxidation, possibly involved in metabolic phenotype alteration during the chemoresistance. In this respect, elucidating metabolic reprogramming in SOR-resistant HCC cells might be a promising strategy for improving responses to sorafenib and overcoming drug resistance. Nevertheless, there is a limitation that extract molecular mechanism of AMPK pathway activation suppressed by CXCR3 was not further elucidated, but this will be included in our next researches.
Acknowledgements
This work was supported by Foundation of Henan Educational Committee (No. 132102310064). We wish to thank Ms. Dongmei Gao, a research assistant at The Laboratory Animal Center, Affiliated to Zhengzhou University, for her assistance in animal experiment.
Disclosure of conflict of interest
None.
References
- 1.Fonseca AL, Cha CH. Hepatocellular carcinoma: a comprehensive overview of surgical therapy. J Surg Oncol. 2014;110:712–719. doi: 10.1002/jso.23673. [DOI] [PubMed] [Google Scholar]
- 2.Klungboonkrong V, Das D, McLennan G. Molecular mechanisms and targets of therapy for hepatocellular carcinoma. J Vasc Interv Radiol. 2017;28:949–955. doi: 10.1016/j.jvir.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Wu S, Saxena S, Varney ML, Singh RK. CXCR1/2 chemokine network regulates melanoma resistance to chemotherapies mediated by NF-kappaB. Curr Mol Med. 2017;17:436–449. doi: 10.2174/1566524018666171219100158. [DOI] [PubMed] [Google Scholar]
- 4.Zhang HH, Zhang ZY, Che CL, Mei YF, Shi YZ. Array analysis for potential biomarker of gemcitabine identification in non-small cell lung cancer cell lines. Int J Clin Exp Pathol. 2013;6:1734–1746. [PMC free article] [PubMed] [Google Scholar]
- 5.Lee NH, Nikfarjam M, He H. Functions of the CXC ligand family in the pancreatic tumor microenvironment. Pancreatology. 2018;18:705–716. doi: 10.1016/j.pan.2018.07.011. [DOI] [PubMed] [Google Scholar]
- 6.Van Raemdonck K, Van den Steen PE, Liekens S, Van Damme J, Struyf S. CXCR3 ligands in disease and therapy. Cytokine Growth Factor Rev. 2015;26:311–327. doi: 10.1016/j.cytogfr.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 7.Karin N. Chemokines and cancer: new immune checkpoints for cancer therapy. Curr Opin Immunol. 2018;51:140–145. doi: 10.1016/j.coi.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 8.Elia G, Fallahi P. Hepatocellular carcinoma and CXCR3 chemokines: a narrative review. Clin Ter. 2017;168:e37–e41. doi: 10.7417/CT.2017.1980. [DOI] [PubMed] [Google Scholar]
- 9.Tokunaga R, Zhang W, Naseem M, Puccini A, Berger MD, Soni S, McSkane M, Baba H, Lenz HJ. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - A target for novel cancer therapy. Cancer Treat Rev. 2018;63:40–47. doi: 10.1016/j.ctrv.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma B, Khazali A, Wells A. CXCR3 in carcinoma progression. Histol Histopathol. 2015;30:781–792. doi: 10.14670/HH-11-594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mikawa T, ME LL, Takaori-Kondo A, Inagaki N, Yokode M, Kondoh H. Dysregulated glycolysis as an oncogenic event. Cell Mol Life Sci. 2015;72:1881–1892. doi: 10.1007/s00018-015-1840-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhattacharya B, Low SH, Soh C, Kamal Mustapa N, Beloueche-Babari M, Koh KX, Loh J, Soong R. Increased drug resistance is associated with reduced glucose levels and an enhanced glycolysis phenotype. Br J Pharmacol. 2014;171:3255–3267. doi: 10.1111/bph.12668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steinbichler TB, Dudas J, Skvortsov S, Ganswindt U, Riechelmann H, Skvortsova II. Therapy resistance mediated by cancer stem cells. Semin Cancer Biol. 2018;53:156–167. doi: 10.1016/j.semcancer.2018.11.006. [DOI] [PubMed] [Google Scholar]
- 14.Ren Y, Kan YZ, Kong LF. Study on the effects of target-silencing CXCR3 expression on malignant proliferation of hepatocellular carcinoma. Zhonghua Gan Zang Bing Za Zhi. 2018;26:508–512. doi: 10.3760/cma.j.issn.1007-3418.2018.07.006. [DOI] [PubMed] [Google Scholar]
- 15.Wang Z, Liu P, Chen Q, Deng S, Liu X, Situ H, Zhong S, Hann S, Lin Y. Targeting AMPK signaling pathway to overcome drug resistance for cancer therapy. Curr Drug Targets. 2016;17:853–864. doi: 10.2174/1389450116666150316223655. [DOI] [PubMed] [Google Scholar]
- 16.Zhao H, Orhan YC, Zha X, Esencan E, Chatterton RT, Bulun SE. AMP-activated protein kinase and energy balance in breast cancer. Am J Transl Res. 2017;9:197–213. [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Z, Shi A, Song D, Han B, Zhang Z, Ma L, Liu D, Fan Z. Resistin confers resistance to doxorubicin-induced apoptosis in human breast cancer cells through autophagy induction. Am J Cancer Res. 2017;7:574–583. [PMC free article] [PubMed] [Google Scholar]
- 18.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 19.Chen J, Jin R, Zhao J, Liu J, Ying H, Yan H, Zhou S, Liang Y, Huang D, Liang X. Potential molecular, cellular and microenvironmental mechanism of sorafenib resistance in hepatocellular carcinoma. Cancer Lett. 2015;367:1–11. doi: 10.1016/j.canlet.2015.06.019. [DOI] [PubMed] [Google Scholar]
- 20.Moris D, Chakedis J, Sun SH, Spolverato G, Tsilimigras DI, Ntanasis-Stathopoulos I, Spartalis E, Pawlik TM. Management, outcomes, and prognostic factors of ruptured hepatocellular carcinoma: a systematic review. J Surg Oncol. 2018;117:341–353. doi: 10.1002/jso.24869. [DOI] [PubMed] [Google Scholar]
- 21.Sun T, Liu H, Ming L. Multiple roles of autophagy in the sorafenib resistance of hepatocellular carcinoma. Cell Physiol Biochem. 2017;44:716–727. doi: 10.1159/000485285. [DOI] [PubMed] [Google Scholar]
- 22.Kim MJ, Choi YK, Park SY, Jang SY, Lee JY, Ham HJ, Kim BG, Jeon HJ, Kim JH, Kim JG. PPARdelta reprograms glutamine metabolism in sorafenib-resistant HCC. Mol Cancer Res. 2017;15:1230–1242. doi: 10.1158/1541-7786.MCR-17-0061. [DOI] [PubMed] [Google Scholar]
- 23.Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013;4:e532. doi: 10.1038/cddis.2013.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu CX, Wang XQ, Chok SH, Man K, Tsang SHY, Chan ACY, Ma KW, Xia W, Cheung TT. Blocking CDK1/PDK1/Beta-Catenin signaling by CDK1 inhibitor RO3306 increased the efficacy of sorafenib treatment by targeting cancer stem cells in a preclinical model of hepatocellular carcinoma. Theranostics. 2018;8:3737–3750. doi: 10.7150/thno.25487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang PF, Li KS, Shen YH, Gao PT, Dong ZR, Cai JB, Zhang C, Huang XY, Tian MX, Hu ZQ. Galectin-1 induces hepatocellular carcinoma EMT and sorafenib resistance by activating FAK/PI3K/AKT signaling. Cell Death Dis. 2016;7:e2201. doi: 10.1038/cddis.2015.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duda DG, Kozin SV, Kirkpatrick ND, Xu L, Fukumura D, Jain RK. CXCL12 (SDF1alpha)-CXCR4/CXCR7 pathway inhibition: an emerging sensitizer for anticancer therapies? Clin Cancer Res. 2011;17:2074–2080. doi: 10.1158/1078-0432.CCR-10-2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Timaner M, Letko-Khait N, Kotsofruk R, Benguigui M, Beyar-Katz O, Rachman-Tzemah C, Raviv Z, Bronshtein T, Machluf M, Shaked Y. Therapy-educated mesenchymal stem cells enrich for tumor-initiating cells. Cancer Res. 2018;78:1253–1265. doi: 10.1158/0008-5472.CAN-17-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delitto D, Perez C, Han S, Gonzalo DH, Pham K, Knowlton AE, Graves CL, Behrns KE, Moldawer LL, Thomas RM. Downstream mediators of the intratumoral interferon response suppress antitumor immunity, induce gemcitabine resistance and associate with poor survival in human pancreatic cancer. Cancer Immunol Immunother. 2015;64:1553–1563. doi: 10.1007/s00262-015-1760-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stone MJ, Hayward JA, Huang C, E Huma Z, Sanchez J. Mechanisms of regulation of the chemokine-receptor network. Int J Mol Sci. 2017;18 doi: 10.3390/ijms18020342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park J, Morley TS, Kim M, Clegg DJ, Scherer PE. Obesity and cancer-mechanisms underlying tumour progression and recurrence. Nat Rev Endocrinol. 2014;10:455–465. doi: 10.1038/nrendo.2014.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, Viswanathan SR, Chattopadhyay S, Tamayo P, Yang WS, Rees MG, Chen S, Boskovic ZV, Javaid S, Huang C, Wu X, Tseng YY, Roider EM, Gao D, Cleary JM, Wolpin BM, Mesirov JP, Haber DA, Engelman JA, Boehm JS, Kotz JD, Hon CS, Chen Y, Hahn WC, Levesque MP, Doench JG, Berens ME, Shamji AF, Clemons PA, Stockwell BR, Schreiber SL. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453–457. doi: 10.1038/nature23007. [DOI] [PMC free article] [PubMed] [Google Scholar]