

Abstract
Internal N6-methyladenosine (m6A) modification is one of the most common and abundant modifications of RNA. However, the biological role(s) of viral RNA m6A remains elusive. Using human metapneumovirus (hMPV) as a model, we demonstrate that m6A serves as a molecular marker for innate immune discrimination of self from nonself RNAs. We show that hMPV RNAs are m6A methylated and that viral m6A methylation promotes hMPV replication and gene expression. Inactivating m6A addition sites with synonymous mutations or demethylase resulted in m6A deficient recombinant hMPVs and virion RNAs that induced significantly higher expression of type I interferon (IFN) which was dependent on the cytoplasmic RNA sensor RIG-I, not MDA5. Mechanistically, m6A-deficient virion RNA induces higher expression of RIG-I, binds more efficiently to RIG-I, and facilitates the conformational change of RIG-I, leading to enhanced IFN expression. Furthermore, m6A-deficient rhMPVs triggered higher IFN in vivo and were significantly attenuated in cotton rats yet retained high immunogenicity. Collectively, our results highlight that (i) virus acquires m6A in their RNAs as a means of mimicking cellular RNA to avoid detection by innate immunity; and (ii) viral RNA m6A can serve as a target to attenuate hMPV for vaccine purposes.
Introduction
The innate immune system provides the first response to virus infections and must discriminate cellular (self) from viral (nonself) nucleic acids to mount a protective immune response against the latter. Microbial nucleic acids are important activators of innate immune responses by triggering so-called pattern recognition receptors (PRRs) 1, 2. For cytoplasm replicating RNA viruses, this involves detection of viral RNA by RIG-I-like receptors (RLRs), including retinoic acid-induced gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) 3, 4. Upon recognition, the intracellular signaling cascades are activated, triggering the expression of type I interferons and many other proinflammatory cytokines and chemokines to establish an antiviral state. Interferon-α (IFN-α) and IFN-β, the hallmark of innate immune responses, are secreted from infected cells, restricting virus replication and spread and regulating adaptive immunity.
RNA structure and posttranscriptional modifications are important molecular markers for the host to discriminate between self and nonself. The primary ligand for RIG-I is a 5′ triphosphorylated or diphosphorylated single-stranded RNA (ssRNA) 5, 6. Both MDA5 and RIG-I are also capable of recognizing double-stranded RNAs (dsRNAs). However, the optimal ligand for MDA5 is long dsRNAs whereas RIG-I preferentially detects short dsRNAs 7, 8, 9. Eukaryotic “self” mRNA possesses a unique 5′ cap structure typically methylated at the guanine N7 and ribose-2′-O positions 10, 11 and thus is not detected by RIG-I or MDA5 12. However, mRNA lacking ribose-2′-O methylation is recognized as the nonself RNA. Therefore, many RNA viruses have evolved their own mRNA cap methyltransferases to acquire ribose-2′-O methylation 13, 14, 15, 16, 17 to avoid detection by the host innate immune system 18, 19, 20.
Among more than 150 known nucleotide modifications, internal N6-methyladenosine (m6A) modification is the most prevalent 21, 22. Although m6A modification was first discovered in the 1970s, its biological function(s) have been a mystery for four decades. Recent studies have found that m6A modification is involved in RNA metabolism, protein translation, embryonic development, and diseases 22, 23, 24. Since the early 1970s, it has been known that the mRNAs of several DNA and RNA viruses contain m6A modification 25, 26, 27, 28. However, the impact of m6A modification on viral replication was not described until recently. It appears that m6A modification can play either an anti-viral or pro-viral role, depending on the virus species 29, 30, 31, 32, 33, 34, 35, 36, 37. However, whether m6A modification plays a role in innate sensing remains elusive.
Here, we demonstrate that m6A modification serves as a molecular marker for innate sensing by cells to discriminate self from nonself RNA and that m6A regulates viral pathogenesis. Specifically, m6A-deficient HMPV or virion RNA induces significantly higher type I interferon (IFN) in a RIG-I-dependent manner. Mechanistically, m6A-deficient hMPV virion RNA induces higher RIG-I expression, enhances its binding affinity to RIG-I, and facilitates the conformational change of RIG-I. In addition, m6A may serve as a target for rational design of improved live attenuated vaccine candidates.
Results
The hMPV genome, antigenome, and mRNAs contain m6A modifications.
To determine whether hMPV RNA contains m6A, RNA was extracted from purified hMPV virions, sonicated, and subjected to m6A immunoprecipitation followed by high throughput sequencing (m6A-seq). Interestingly, sequencing reads from m6A-seq aligned to both the genome and the antigenome. Indeed, both the N protein-encapsidated genome and antigenome are packaged into hMPV virions (Extended Data Fig.1). In the genomic RNA, a total of 5 m6A peaks were detected spread through the P and G gene regions (Fig. 1a and Supplementary Table 1). A total of 12 m6A peaks were found in the antigenome RNA, including regions complementary to the N, P, M, F, G, and L genes (Fig. 1a and Supplementary Table 1). The strongest m6A peaks are in the G gene in both genome and antigenome. The regions of m6A peaks identified in both strands are largely overlapping.
Fig. 1. The hMPV RNAs are m6A methylated and m6A methylation promotes hMPV replication.

(a) Distribution of m6A peaks in the hMPV antigenome and genome. A schematic diagram of the hMPV antigenome encoding 8 genes is shown. Total RNAs were extracted from purified rhMPV virions grown in A549 cells and were subjected to m6A immunoprecipitation followed by m6A-seq. Top panel: The m6A-seq of hMPV RNA showing the distribution of m6A-IP reads (blue block) mapped to the hMPV antigenome. The baseline signal from input samples is shown as a blue line. Lower panel: The distribution of m6A-IP reads (pink block) from m6A-seq mapped to the hMPV genome. The baseline signal from input samples is shown as a pink line. The red arrows indicate the m6A peaks. (b) Distribution of m6A peaks in the hMPV mRNAs. Polyadenylated mRNAs were isolated from hMPV-infected cells and subjected to m6A-seq. m6A reader (c) and writer (g) proteins enhance hMPV protein expression. A549 cells were transfected with plasmids encoding reader (c) or writer (g) genes. At 24 h, cells were infected with rhMPV at an MOI of 5.0. Total cell extracts were analyzed by Western blot. m6A reader (d) and writer (f) proteins increase hMPV progeny virus production. The release of infectious hMPV particles was monitored by a single-step growth curve. (e) YTHDF1, 2, 3 enhance GFP expression in rghMPV-infected cells. HeLa cells stably overexpressing YTHDF proteins were infected with rghMPV at an MOI of 1.0, and GFP expression was monitored at 48 h post-infection. (h) Strong co-localization of METTL14 with hMPV N protein. A549 cells were infected by rhMPV at an MOI of 5.0. At 24 h, cells were stained with anti-METTL14 antibody (green) and anti-hMPV N antibody (red), and analyzed by confocal microscope. Nuclei were labeled with DAPI (blue). The results of n = 2 (a-b), n = 3 (c, e-h), or n = 4 (d) biologically independent experiments are shown (representative immunoblots (c, g) and images (e, h) are shown). Data (a and b) are the average results from two samples (n = 2). Viral titers are the geometric mean titers (GMT) ± standard deviation. Statistical significance was determined by two-sided student’s t-test. Exact P values are included in Data Source. *P<0.05, **P<0.01, and ***P<0.001.
We next determined whether hMPV mRNAs were m6A modified by performing m6A-seq of polyadenylated mRNAs from hMPV-infected A549 cells. This analysis revealed m6A peaks in 3 of the 8 mRNA species, P, G, and L (Fig.1b and Supplementary Table 1). Notably, several m6A-modified regions in hMPV mRNAs largely overlapped with those found in the antigenome. Again, the G mRNA showed the strongest m6A enrichment. In addition, we found that hMPV infection had a minimal impact on the methylome of cellular mRNAs but significantly upregulated genes that are involved in innate immune signaling (Extended Data Fig.2 and Supplementary Tables 2–4).
m6A methylation promotes hMPV replication and gene expression.
The biological functions of m6A modification could be mediated by m6A-binding YTH domain family proteins (YTHDF1, YTHDF2, YTHDF3, and YTHDC1) 22. We examined the effects of overexpression of m6A-binding proteins on hMPV replication. We found that transient overexpression of m6A-binding proteins increased the levels of viral G and N proteins (Fig.1c), the release of infectious virus (Fig.1d), antigenome, N and G mRNAs (Extended Data Fig.3a–e) in A549 cells. This pro-viral function was also observed in HeLa cells stably overexpressing YTHDF1, YTHDF2, or YTHDF3 using recombinant GFP-expressing HMPV (rghMPV) (Fig.1e, Extended Data Fig.4).
Internal m6A on RNA is installed by m6A writer methyltransfer-like proteins (METTL3 and METTL14) and can be reversibly removed by m6A eraser proteins (ALKBH5 and FTO). Overexpression of writer proteins significantly upregulated hMPV replication (Fig.1f), G and N protein (Fig.1g), and RNA levels (Extended Data Fig.3f–i). Consistently, knockdown of m6A eraser proteins also enhanced hMPV replication and gene expression (Extended Data Fig.5). In addition, knockdown of writer proteins significantly reduced the m6A content of total host RNA and mRNA, and reduced host protein translation (Supplementary Fig.1). Overexpression of eraser proteins had similar effects on host RNA m6A level. Taken together, viral m6A modification plays a pro-viral role in the hMPV replication cycle. The phenotype of overexpression and knockdown m6A proteins is probably due to changes in both viral and host RNA m6A.
Cytoplasmic inclusion bodies are the sites where hMPV replication and ribonucleoprotein (RNP) assembly occurs. We determined whether host m6A machinery is co-localized with viral N protein, a component of the RNP. Interestingly, viral N protein strongly co-localized with METTL14 (Fig.1h) and partially with METTL3 (Supplementary Fig.2). However, N protein did not co-localize with the eraser proteins (ALKBH5) (Supplementary Fig.3). In addition, N protein partially co-localized with m6A binding proteins elsewhere in the cytoplasm (Supplementary Fig.4).
Abrogation of m6A results in attenuation of hMPV in cell culture.
There are 14 potential m6A sites in the G gene region of the antigenome and G mRNA and 6 m6A sites in G gene in the genome (Supplementary Figs.5 and 6). We mutated these m6A sites without changing the amino acids they encode. We generated five rhMPV mutant viruses lacking m6A sites in the G gene region of the antigenome and G mRNA (rhMPV-G1-2, G1-7, G8-9, G8-14, and G1-14) and one recombinant virus [rhMPV-G(-)1-6] carrying 6 m6A site mutations in the G gene region of the genome. All rhMPV mutants were attenuated in cell culture: formed smaller immunospots (Fig.2a), had defects in replication kinetics, and synthesized less viral N and G proteins (Extended Data Fig.6b–c) compared to rhMPV. Interestingly, rhMPV mutants developed cytopathic effects (CPE) earlier in A549 cells (Extended Data Fig.6a, Supplementary Fig.7).
Fig. 2. m6A-deficient rhMPVs and their virion RNAs induce higher type I IFN responses.

(a) Immunostaining spots formed by rhMPVs. (b) Quantification of m6A level. Total m6A level of each virion RNA was quantified by m6A RNA Methylation Assay Kit. (c) m6A-deficient rhMPV RNA has reduced binding efficiency to reader proteins. Cell lysate containing HA-tagged YTHDF1 or YTHDF2 was incubated with virion RNA and anti-HA Magnetic beads. The amount of virion RNA captured by the YTHDF1 or YTHDF2 was quantified by real-time RT-PCR. Percent of bound RNA of hMPV mutants relative to rhMPV was calculated. IFN-β secretion in A549 cells infected by hMPV at MOI of 4.0 (d, h) or an MOI of 1.0 (e). A549 cells were infected with each rhMPV at an MOI of 4.0, and IFN-β in cell supernatants were measured by ELISA. Dynamics of IFN-β secretion in THP-1 cells infected by hMPV at an MOI of 4.0 (f-g). (i) IFN-β response in A549 cells transfected with total RNA. Total RNA was extracted from hMPV-infected A549 cells, and the antigenome was quantified by real-time RT-PCR. A549 cells were transfected with 108 antigenome RNA copies of total RNA with or without treatment CIP. IFN-β was measured by ELISA. (j) IFN-β response in A549 cells transfected with viral G mRNA. A549 cells were transfected with 109 RNA copies of G mRNA either with or without CIP treatment. (k) IFN-β response in A549 cells transfected with virion RNA. A549 cells were transfected with 2×107 antigenome copies of virion RNA either with or without CIP treatment. (l and m) Comparison of IFN response of virion RNA of rhMPV-G1-14, G1-2, G8-9, and rhMPV. A549 cells were transfected with 107 (l) or 106 (m) RNA copies of virion RNA. (n and o) Natural m6A-deficient virion RNA induces IFN response. A549 cells were transfected with 107 (n) or 106 (o) RNA copies of virion RNA of rhMPV-G1-14, G(-)1-6, ALKBH5, and rhMPV. Immunospots (a) shown are the representatives of n = 3 biologically independent experiments. Data shown are means of n = 3 (c-o) or n = 4 (b) biologically independent experiments ± standard deviation. Statistical significance was determined by two-sided student’s t-test. Exact P values are included in Data Source. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001, NS, no significant.
The m6A level was significantly reduced in these hMPV mutants as determined by the quantitative m6A-RNA immunoprecipitation (MeRIP assay) methods (Fig.2b). For those antigenome mutants, the m6A-specific antibody bound the genome well, but bound the antigenome and G mRNA less efficiently (Extended Data Fig.6d). In addition, the binding efficiency of antigenome from rhMPV-G1-14 and rhMPV-G8-14 to YTHDF1 and YTHDF2 only retained 12-34% of the binding to rhMPV RNA. However, the antigenome of rhMPV-G1-2 and G8-9 retained 80-90% of their binding activity (Fig.2c and Supplementary Fig.8). Therefore, m6A-deficient rhMPVs indeed produce antigenome, G mRNA, or genome deficient in m6A methylation.
We also generated another m6A-deficient virus (rhMPV-ALKBH5) in which viral RNA m6A was removed by overexpressing the m6A eraser protein, ALKBH5, in A549 cells. As expected, virion RNA from rhMPV-ALKBH5 was defective in m6A content (Fig.2b). Thus, these RNAs are ‘naturally’ deficient in m6A methylation, as their nucleotide sequences are not changed.
m6A-deficient rhMPVs induce more IFN-I.
The m6A-seq showed that hMPV infection caused broad alterations in the expression of genes involved in innate immunity. Thus, we measured the dynamics of the type I interferon (IFN-I) response to these m6A-deficient rhMPVs in virus-infected cells. At an MOI of 4.0, all rhMPV mutants induced significantly more IFN-α (Extended Data Fig.7a) and IFN-β (Fig.2d) than rhMPV. At a lower MOI (1.0), rhMPV-G1-14 induced significantly more IFN-β than rhMPV-G1-2 and G8-9 (Fig.2e). In THP-1 cells, the increase of IFN-β was much more dramatic than in A549 cells: a 14-33-fold increase of IFN-β was observed for rhMPV-G1-14 (Fig.2f–g, Extended Data Fig.7b). At an MOI of 4.0, rhMPV-G1-14 induced significantly more IFN-β than rhMPV-G1-2 and G8-9 at 16 and 24 h post-infection (Fig.2f). Similarly, rhMPV-G(-)1-6 had significantly higher IFN-β responses in both THP-1 (Fig.2g) and A549 cells (Fig.2h) compared to rhMPV though their IFN-β responses were less than that of rhMPV-G1-14. Therefore, m6A-deficient rhMPV induced significantly higher IFN-I.
m6A-deficient antigenome and genome, but not the G mRNA, trigger higher IFN-I.
To further investigate the mechanism underlying the enhanced IFN-I response associated with m6A-deficient hMPVs, we transfected A549 cells with RNAs isolated from virus-infected cells or virions. Under these conditions, there would be no viral replication. Equal amounts of total RNA isolated from A549 cells infected with rhMPV-G8-14 and rhMPV-G1-14 triggered significantly more IFN than with rhMPV (P<0.01) (Fig.2i). This IFN production was abrogated when the RNAs were dephosphorylated by calf intestinal phosphatase (CIP) treatment (Fig.2i). However, transfection of G mRNA samples into A549 cells only induced a minimal amount of IFN-β (Fig.2j). After removal of any contamination of triphosphorylated RNA by CIP, G mRNA from rhMPV-G8-14, rhMPV-G1-14, and rhMPV failed to induce IFN (P>0.05) (Fig.2j), demonstrating that m6A-deficient G mRNA did not contribute to the enhanced type I IFN response.
Finally, we isolated hMPV genome and antigenome from highly purified virions and assessed their ability to induce IFN expression. Transfection of virion RNA of rhMPV-G8-14 and rhMPV-G1-14 stimulated significantly more IFN induction in A549 cells than that of rhMPV (P<0.0001 or P<0.05) (Fig.2k). IFN induction was completely abrogated when virion RNA was dephosphorylated by CIP. Virion RNA of mutants triggered an earlier cell death (Supplementary Figs.9 and 10).
We next directly compared the IFN responses to virion RNA derived from rhMPV-G1-14, G1-2 and G8-9. RNA from rhMPV-G1-14 triggered significantly more IFN-β than RNA from rhMPV-G1-2 and G8-9 at all three RNA concentrations (Fig.2l and m, Extended Data Fig.7c). RNA from rhMPV-G1-14 and rhMPV-ALKBH5 induced significantly more IFN-β than RNA from rhMPV-G(-)1-6 (Fig.2n and o, Extended Data Fig.7d). Overall, m6A-deficient hMPV antigenome and genome induced more IFN and the level of IFN was correlated with the degree of m6A deficiency.
RIG-I plays a dominant role in the activation of higher IFN-I and NF-κB.
To determine which RNA sensors are involved in the detection of m6A-deficient RNA, we examined the IFN response of m6A-deficient rhMPVs in A549-Dual cells lacking RIG-I, MDA5, or their downstream adaptor protein, MAVS. As expected, both rhMPV-G8-14 and rhMPV-G1-14 triggered a higher and earlier IFN-β response in wild type (WT) A549 cells than rhMPV (P<0.05; two-sided Student’s t-test) (Fig.3a). Notably, IFN-β production in response to rhMPV-G8-14 and rhMPV-G1-14 was completely abrogated in A549 cells knocked out for RIG-I (Fig.3c) and MAVS (Fig.3d). In contrast, a high level of IFN-β was still detectable in MDA5-knockout A549 cells (Fig.3b). Similar to virus infection, IFN-β induction by virion RNA from m6A-deficient rhMPVs was completely abrogated in RIG-I and MAVS but not in MDA5-knockout A549 cells (Fig.3e–h). Since there is a cross-talk between the IFN and NF-κB pathways 3, we measured the activation of the NF-κB pathway. Similarly, m6A-deficient rhMPVs (Fig.3i–l) and their virion RNAs (Fig.3m–p) induced higher NF-κB, which was also dependent on RIG-I and MAVS. Therefore, RIG-I plays a dominant role in the activation of the IFN-I and NF-κB pathways by m6A-deficient rhMPVs and virion RNA.
Fig. 3. IFN response and NF-κB activation in A549 cells infected with m6A deficient hMPVs or transfected with m6A deficient virion RNA.

Confluent WT (a), MDA5 (b), RIG-I (c), or MAVS (d)-knockout A549 cells were infected by rhMPV, rhMPV-G8-14, or rhMPV-G1-14 at an MOI of 1.0, cell culture supernatants were harvested at 24 and 48 h post-inoculation. IFN-β in cell supernatants was measured by ELISA. Confluent wild-type (e), MDA5 (f), RIG-I (g), or MAVs (h)-knockout A549 cells were transfected with 107 antigenomic RNA copies of virion RNA of rhMPV, rhMPV-G8-14, rhMPV-G1-14 or 2 μg poly(I:C), cell culture supernatants were harvested at 24 and 40 h post-inoculation. IFN-β in cell supernatants was measured by ELISA. Confluent WT (i), MDA5 (j), RIG-I (k), or MAVs (l)-knockout A549 cells were infected by rhMPV, rhMPV-G8-14, or rhMPV-G1-14 at an MOI of 1.0, cell culture supernatants were harvested at 24 and 48 h post-inoculation. These A549 cells also express a secreted embryonic alkaline phosphatase (SEAP) reporter gene under the control of the IFN-β minimal promoter fused to five NF-κB binding sites, allowing us to measure the activation of the NF-κB pathway. SEAP secreted in cell supernatants was measured by colorimetric enzyme assay with substrate Quanti-Blue™ and read by microplate reader on OD value at 620nm. Confluent WT (m), MDA5 (n), RIG-I (o), or MAVs (p)-knockout A549 cells were transfected with 107 antigenomic RNA copies of virion RNA of rhMPV, rhMPV-G8-14, rhMPVG1-14 or 2 μg poly(I:C) with or without CIP treatment, cell culture supernatants were harvested at 16, 24, and 40 h post-inoculation. SEAP secretion in cell supernatants was measured. Data shown (a-l) are means of n =3 or n = 4 (m-p) biologically independent experiments ± standard deviation. All data of rhMPV-G8-14 and rhMPV-G1-14 were compared to those of rhMPV. Statistical significance was determined by two-sided student’s t-test. Exact P values are included in Data Source. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
m6A-deficient antigenome and genome enhance expression of RIG-I.
We next asked whether m6A-deficient rhMPVs enhance RIG-I expression in virus-infected cells. rhMPV-G8-14 and rhMPV-G1-14 infection induced significantly higher expression of RIG-I compared to rhMPV (Fig.4a). Similarly, transfection of virion RNA from rhMPV-G8-14 and rhMPV-G1-14 triggered significantly higher expression of RIG-I compared to that of rhMPV (Fig.4b). We next compared the RIG-I expression triggered by transfection of virion RNA from rhMPV-G1-14, G1-2, and G8-9. Virion RNA of rhMPV-G1-14 induced higher expression of RIG-I than virion RNA of rhMPV-G1-2 and G8-9 (Fig.4c). However, RIG-I protein was barely detectable when virion RNA was treated with CIP (Fig.4d, Extended Data Fig.8a). Also, rhMPV-G(-)1-6 and rhMPV-ALKBH5 triggered higher expression of RIG-I compared to rhMPV in virus-infected cells (Fig.4e). Similarly, RNA from rhMPV-G(-)1-6 and rhMPV-ALKBH5 triggered higher expression of RIG-I compared to RNA from rhMPV in RNA-transfected cells (Fig.4f). These results suggest that m6A-deficient RNAs enhance RIG-I expression and that the 5’triphosphate of the virion RNA is required for inducing RIG-I expression.
Fig. 4. m6A-deficient hMPVs and virion RNA induce a higher expression of RIG-I.

(a) m6A-deficient rhMPVs stimulate a higher expression of RIG-I. A549 cells were infected by each hMPV at an MOI of 0.2, 1.0, and 5.0. At indicated times, cell lysates were subjected to Western blot analyses using antibody specific to RIG-I, hMPV N, or β-actin. (b) m6A-deficient virion RNA induces a higher expression of RIG-I. A549 cells were transfected with an increasing amount of poly (I:C) (0.5 and 2.0 µg/well) or virion RNAs (2×105, 2×106, or 2×107 copies/well) of rhMPV, rhMPV-G8-14, or rhMPV-G1-14. At indicated times, cell lysates were subjected to Western blot analysis using antibody against RIG-I or β-actin. (c) Comparison of RIG-I expression triggered by virion RNA. A549 cells were transfected with increasing amounts (105, 106, and 107 RNA copies) of virion RNA of rhMPV, rhMPV-G1-2, rhMPV-G8-9, or rhMPV-G1-14. (d) Removal of 5’ triphosphate abolished RIG-I expression and IRF3 phosphorylation. A549 cells were transfected with virion RNA with or without CIP treatment. At indicated times, cell lysates were subjected to Western blot using antibody specific to IRF3 or phosphorylated IRF3 (pIRF3) on site S386 or S396. (e) Natural m6A-deficient rhMPVs induce higher phosphorylation of IRF3. A549 cells were infected by each hMPV at an MOI of 5.0. At indicated times, RIG-I expression and IRF3 phosphorylation was detected by Western blot. (f) Natural m6A-deficient virion RNA induces higher RIG-I expression. 107 copies of virion RNA were used for transfection. Western blots (a-f) shown are the representatives of n =3 biologically independent experiments. (g) Model for RIG-I mediated IFN signaling pathway. Upon hMPV entry, the RNP complex is delivered into the cytoplasm where RNA synthesis and viral replication occur. During replication, the RdRP initiates at the extreme 3’ end of the genome and synthesizes a full-length complementary antigenome, which subsequently serves as template for synthesis of full-length progeny genomes. The newly synthesized genome and antigenome was methylated by m6A writer proteins and encapsidated by viral N protein. Viral genome and antigenome are recognized by cytoplasmic RNA sensor RIG-I and induces signaling to the downstream adaptor protein MAVS which subsequently activates IRF3 and NF-κB pathways, leading to the production of type-I IFN. The internal m6A methylation on virion RNA inhibits RIG-I mediated IFN signaling pathway.
m6A-deficient antigenome enhances IRF3 phosphorylation.
To demonstrate the activation of the type I IFN downstream signaling cascade, we measured the phosphorylation of IRF3 at S386 and S396 upon hMPV infection or virion RNA transfection. Phosphorylation of IRF3 was significantly higher in rhMPV-G18-14, rhMPV-G1-14, rhMPV-G(-)1-6, and rhMPV-ALKBH5-infected cells than in the rhMPV-infected cells (Fig.4e, Extended Data Fig.8c). Similarly, we observed higher IRF3 phosphorylation in A549 cells transfected with virion RNA derived from rhMPV-G8-14 and rhMPV-G1-14 than those transfected with virion RNA from rhMPV (Fig.4d, Extended Data Fig.8d). In addition, CIP treatment of virion RNA abolished IRF3 phosphorylation (Fig.4d). Thus, m6A deficient hMPVs led to a significantly higher amount of IRF3 phosphorylation, which is consistent with the observation that they induced higher expression of IFN-I (Fig.4g).
Enhanced recognition of m6A-deficient antigenome by RIG-I.
We next directly compared the binding affinity of m6A-containing and -deficient antigenome to RIG-I protein. We first used biotinylated virion RNA to pull down endogenously expressed RIG-I in A549 cell extract. Virion RNA of rhMPV-G8-14 and rhMPV-G1-14 pulled down significantly more RIG-I protein compared to virion RNA of rhMPV (Fig.5a). After removal of triphosphate by CIP, virion RNA from rhMPV and rhMPV mutants failed to pull down RIG-I (Fig.5a).
Figure 5. m6A-deficient virion RNA increases RIG-I binding affinity and facilitates RIG-I:RNA conformation change.

(a) Biotinylated virion RNA pulldown RIG-I. Biotinylated virion RNA was conjugated to Streptavidin beads and incubated with A549 cell lysate containing overexpressed RIG-I. The pull-down RIG-I protein was detected by Western blot. (b and c) RIG-I pulldown hMPV RNA. RIG-I conjugated magnetic beads were incubated with virion RNA, N or G mRNA. One aliquot of beads was subjected for Western blot (b). RNA bound to magnetic beads was quantified by real-time RT-PCR (c). (d) Purified Flag-tagged RIG-I protein. (e) Competitive binding of WT virion RNA and m6A-deficient virion RNA to RIG-I. Streptavidin beads-bound rhMPV-G1-14 and rhMPV RNA were mixed at different ratios and incubated with RIG-I protein in the presence of AMP-PNP. RIG-I pulldown was detected by Western blot. (f) Domain structure of RIG-I protein. CARD, caspase activation and recruitment domains; Helicase, helicase domain; CTD, C-terminal domain. Red flashes indicate trypsin cleavage sites. (g) Model for mechanisms of enhanced RIG-I-mediated IFN signaling by m6A-deficient hMPV RNA. RIG-I is in an autorepressed conformation in the absence of ligand. RIG-I CTD recognizes and binds to 5’triphosphate of RNA. 5’triphosphate RNA without m6A has a higher binding affinity to helicase domain of RIG-I. RIG-I is an RNA translocase, moving from 5’-ppp to RNA chain. Internal m6A may serve as a “brake” to prevent RIG-I translocation (indicated by question mark). The RIG-I helicase domain binds the RNA, triggering RIG-I conformational change and subsequent oligomerization. RNAs without m6A more easily induce RIG-I conformational change. The released CARDs of the activated RIG-I:RNA complex are ubiquitinated for downstream signaling. (h-k) Analysis of RIG-I:RNA conformation by limited trypsin digestion. Limited trypsin digestion of RIG-I protein in the absence of RNA ligand for 0–2 h (h), or in the presence of poly (I:C) (i) or virion RNA (j) for 2h was shown. (k) Competition assay. RIG-I incubated with mixtures containing different ratios of RNA of rhMPV-G1-14 and rhMPV, and digested by trypsin for 2h. RIG-I fragments were detected by Western blot. Black arrows in h–k indicate the 80 kDa trypsin-resistant protein band. Western blots (a, b, e, h-k) and SDS-PAGE (d) shown are the representatives of n = 3 biologically independent experiments. Data shown (c) are means of n =3 independent experiments ± standard deviation. Statistical significance was determined by two-sided student’s t-test. *P<0.05; **P<0.01; ***P<0.001.
As a complementary approach, we used Flag-tagged RIG-I protein to pull down virion RNA (Fig.5b). Antigenome (without CIP) of rhMPV-G1-14 and rhMPV-G8-14 bound 90- and 33-fold better to RIG-I than the rhMPV antigenome (Fig.5c). After CIP treatment, the binding of antigenome to RIG-I was abrogated (Fig.5c). The controls, capped viral mRNAs (N and G), had minimal binding to RIG-I (Fig.5c). In addition, N and G mRNAs from rhMPV-G1-14 and rhMPV-G8-14 had a similar level of binding efficiency compared to those of rhMPV (Fig.5c). This observation is in agreement with the fact that capped mRNA is not recognized by RIG-I. To further confirm that m6A-deficient RNA has an increased binding affinity to RIG-I, we performed an in vitro RNA competitive binding assay. The amount of pulldown RIG-I significantly increased when more m6A-deficient RNA (rhMPV-G1-14) was added (Fig.5e). Therefore, m6A-deficient antigenome RNA increases binding affinity to RIG-I.
m6A-deficient viral RNA facilitates RIG-I conformational changes.
We hypothesized that m6A-deficient hMPV RNA facilitates the RIG-I conformational change needed for downstream IFN signaling (Fig.5f and g). To test this hypothesis, we used limited trypsin proteolysis of RIG-I:RNA complexes (Fig.5f), which has often been used for examining the RIG-I conformational changes 38, 39. In the absence of ligand RNA, trypsin treatment of the full-length RIG-I protein (approximately 107kDa) yielded a 55-kDa RIG-I fragment (helicase domain), representing the trypsin sensitivity of autorepressed RNA-free RIG-I conformation (Fig. 5h). As positive controls, trypsin treatment of polyinosinic:polycytidylic acid (poly(I:C)) bound RIG-I yielded an 80-kDa fragment (CARD-Helicase domain) which represents the trypsin resistance of the RNA-bound RIG-I (Fig.5i). RNA of rhMPV-G1-14 and G8-14 yielded significantly more 80-kDa fragment than RNA of rhMPV-G1-2, G8-9, or rhMPV (Fig.5j). In addition, we performed a competition assay in which different ratios of RNAs from rhMPV-G1-14 and rhMPV were mixed and incubated with RIG-I and β,γ-imidoadenosine 5’-triphosphate (AMP-PNP), followed by trypsin treatment. The RIG:RNA mixture containing more rhMPV-G1-14 RNA yielded more of the trypsin-resistant 80-kDa RIG-I fragment (Fig.5k). Collectively, these data suggest that the 5’ppp- on the genome is essential for RIG-I binding, but the m6A-deficient virion RNA either enhances RIG-I binding or facilitates the conformational change of RIG-I in the RIG-I:RNA complex which is known to enhanced downstream IFN induction (Fig.5g).
Restoration of the replication of m6A-deficient rhMPVs in RIG-I and MAVS knockout A549 cells.
If RIG-I is indeed involved in recognition of nonself rhMPV RNA, the growth of m6A-deficient rhMPVs should be restored when RIG-I and its adaptor MAVS proteins are depleted. Both rhMPV-G1-14 and rhMPV-G8-14 had significant defects in growth in WT A549 cells (Fig.6a). Interestingly, the growth of rhMPV-G1-14 was restored in RIG-I (Fig.6c) and MAVs (Fig.6d) knockout A549 cells although it exhibited a delay in replication kinetics in early time points. Also, the growth of rhMPV-G8-14 was partially restored in RIG-I and MAVS knockout A549 cells. In contrast, the growth of neither rhMPV-G1-14 nor rhMPV-G8-14 was restored in MDA5 knockout cells (Fig.6b), similar to the WT cell line. Both mutants exhibited earlier CPE in WT and MDA5 knockout cells but not RIG-I or MAVS knockout cells (Extended Data Fig.9). These results further support the notion that RIG-I but not MDA5 plays a major role in recognition of m6A-deficient RNA. Since our mutagenesis was also designed to remove m6A sites in G mRNA, we found that abrogation of m6A sites in G mRNA diminished G protein translation (Extended Data Fig.10). Thus, the enhanced innate immune sensing and the reduced viral protein translation may collectively contribute to the attenuation of these m6A-deficient rhMPVs.
Fig. 6. Replication, interferon response, pathogenicity, and immunogenicity of m6A-deficient rhMPVs. Replication Kinetics of m6A deficient rhMPVs in WT.

(a), MDA5 (b), RIG-I (c), or MAVS (d)-knockout A549 cells. Cells in 24-well plates were infected by each hMPV at an MOI of 1.0, and viral growth curve was determined. The arrows indicate the degree of titer difference compared to rhMPV. (e) Interferon response of rhMPV in cotton rats. Six-week-old SPF female cotton rats (n = 5) were inoculated intranasally with 100 µl of PBS or 2.0×105 p.f.u. of rhMPV-G8-14, rhMPV-G1-14 or rhMPV. At 48 h post-inoculation, cotton rats were sacrificed. BAL from the right lung was collected for IFN-β bioactivity assay. (f) hMPV titer in lungs and nasal turbinates. Six-week-old SPF cotton rats (n = 5) were inoculated intranasally with 2.0×105 p.f.u. of each rhMPV. At day 4 post-infection, lungs and nasal turbinates were collected for virus titration. (g) m6A deficient rhMPVs had less lung histopathological changes compared to rhMPV. Representative pathological changes from each group are shown. Micrographs with original magnification, ×20 are shown. The parental hMPV caused moderate interstitial pneumonia, mononuclear cell infiltration. In contrast, fewer histological changes were found in the lungs of cotton rats infected with m6A-deficient rhMPVs. (h) m6A deficient rhMPV provides complete protection against hMPV challenge. Four-week-old SPF cotton rats (n = 5) were inoculated intranasally with 2.0×105 p.f.u. of each rhMPV. At week 4 post-immunization, cotton rats were challenged with 2.0×105 p.f.u. of hMPV. At day 4 post-challenge, the cotton rats were sacrificed, lungs and nasal turbinates were collected for virus titration by an immunostaining plaque assay. (i) m6A deficient rhMPV induced a high level of neutralizing antibody. Blood samples were collected from each rat weekly by retro-orbital bleeding. The hMPV-neutralizing antibody titer was determined using a plaque reduction neutralization assay. Viral titers (a-d) are the geometric mean titers (GMT) of n = 3 biologically independent experiments ± standard deviation. Viral titers (f and h) and antibody titers (i) are the geometric mean titers (GMT) of five cotton rats (n = 5) ± standard deviation. Detection limit is 2.0 log [p.f.u.] per g tissue. Statistical significance was determined by two-sided student’s t-test. Exact P values are included in Data Source. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001.
m6A-deficient rhMPVs induces higher IFN-I in vivo.
We next determined whether m6A-deficient rhMPVs induce higher IFN-I in vivo. Cotton rats were inoculated intranasally with each hMPV, terminated at day 2, and bronchoalveolar lavage (BAL) was collected. The IFN-β bioactivity of BAL from rhMPV and PBS-inoculated cotton rats was below the detection limit (Fig.6e). In contrast, IFN-β in BAL from rhMPV-G8-14 and rhMPV-G1-14 had average bioactivity of 150.2 and 175.3 U/ml, respectively (Fig.6e). Therefore, m6A-deficient rhMPVs induce higher IFN-I responses in vivo.
m6A-deficient rhMPVs are attenuated but immunogenic in cotton rats.
We next determined whether m6A-deficient rhMPVs are attenuated in cotton rats. In the lungs, rhMPV-G1-2, rhMPV-G8-9, and rhMPV-G1-14 displayed 1.80, 2.03, and 2.7 log virus reductions compared to rhMPV, respectively (Fig.6f). These rhMPV mutants also had significant, though not as large, reductions (0.30–0.71 log, P<0.05) in viral replication in the nasal turbinates (Fig.6f). Histologically, rhMPV mutants caused fewer lung lesions than the parental hMPV (Fig.6g). Therefore, m6A-deficient rhMPVs are significantly attenuated and less pathogenic in cotton rats.
We next determined whether m6A-deficient rhMPVs were immunogenic. Cotton rats immunized with rhMPV, rhMPV-G1-2, rhMPV-G8-9, and rhMPV-G1-14 were completely protected from virus replication in both lungs and nasal turbinates (Fig.6h). Also, rhMPV mutants triggered significantly higher neutralizing antibody titers at week 2 than rhMPV (P<0.05) (Fig.6i). Thus, m6A-deficient rhMPV retained WT or even higher levels of immunogenicity and provided complete protection against hMPV infection.
Discussion
One of the most prevalent epigenetic modifications in eukaryotic mRNA is m6A methylation. Viruses are obligatory intracellular parasites; their RNAs are also m6A methylated during replication in host cells. The presence of m6A in viral mRNA enhances translation and mRNA stability 40, 41, 42. However, the biological function of m6A in the viral genome and its replicative intermediate RNA has been mysterious. Using hMPV as a model, we have demonstrated that m6A methylation of the antigenome and genome acts as a molecular signature for discriminating self from nonself RNA through the RNA sensor RIG-I. Several lines of evidence support this finding. First, m6A-site mutated rhMPVs, naturally m6A-deficient hMPV, and their antigenome and/or genome RNA trigger a higher type I IFN response. Second, m6A methylation protects the antigenome and genome from recognition by RIG-I thereby inhibiting RIG-I-dependent production of type I interferon in virus-infected cells and virion RNA-transfected cells. Third, m6A methylation of the viral antigenome and genome contributes to the evasion of the interferon-mediated restriction of viral replication. Finally, the deficiency of m6A methylation in the viral antigenome and genome RNA enhances the activation of the RIG-I signaling pathway including RIG-I expression, RIG-I binding affinity, RIG-I conformational change, and IRF3 phosphorylation. The replication of m6A-deficient rhMPVs was restored when the RIG-I and MAVS were knocked out. Our data demonstrate that the hMPV antigenome and genome may have aquired m6A methylation as a means of mimicking host RNA to avoid detection by the innate immune system.
A model consistent with our findings is depicted in Fig.4g. The RNA-dependent RNA polymerase (RdRp) carries out two distinct RNA syntheses: transcription to yield eight mRNAs which are m6A methylated and translated into 9 proteins; and replication to yield genome and antigenome 43, which are methylated by m6A writer proteins. RNA m6A methylation likely occurs prior to or concomitant with encapsidation by N protein. The antigenome and genome without m6A are recognized as a “nonself RNA” by RIG-I. The deficiency of m6A in virion RNA induces higher RIG-I expression, an enhanced RIG-I binding affinity and an enhanced ability to trigger the conformational change in RIG-I that corresponds to enhanced signaling to the downstream adaptor protein MAVS, activating the IRF3 and NF-κB pathways, leading to higher IFN-I. In addition, RIG-I is a 5’-triphosphate-dependent translocase, traveling from the 5’-ppp into the RNA chain to trigger oligomerization 44, 45, which has been shown to be hindered by internal 2’-O methylation in dsRNA 46. Thus, it is possible that m6A methylation may also serve as a “brake” or “throttle” to prevent RIG-I translocation, oligomerization, and downstream signaling (Fig.5g).
Using short synthetic RNA, several studies have suggested that RNA lacking post-transcriptional modifications can be recognized by host innate immunity as non-self-RNAs 38, 47, 48. Incorporation of modified nucleosides m5C, m6A, m5U, s2U, or Ψ into short RNAs suppressed Toll-like receptor 3 (TLR3), TLR7, and TLR8 signaling 47. Using the in vitro transcribed 106-nucleotide (nt) polyU/UC sequence derived from the 3′ untranslated region (UTR) of hepatitis C virus, it was shown that RNAs containing modified nucleotides (such as Ψ, mΨ, and m6A) impact multiple steps in the RIG-I signaling pathway 38. More recently, it was found that m6A modification on human circular RNAs (circRNAs) inhibits innate immunity 49. Mechanistically, m6A reader YTHDF2 sequesters m6A-circRNA and is essential for suppression of innate immunity 49. Also, it was recently shown that manipulation of host writer and eraser proteins can modulate IFN response during VSV and HCMV infection 50, 51, 52. Our study provides the first evidence that m6A modifications in viral RNA indeed serve as a molecular marker for distinguishing self and nonself RNA during natural virus infection.
One important application of this work is the rational design of live attenuated vaccine candidates for hMPV and other pneumoviruses by reducing m6A methylation in viral RNAs. A distinct advantage is that m6A-deficient hMPVs induce a higher IFN response will likely enhance adaptive immunity. This work highlights that possibility of using m6A as an approach for the development of live attenuated vaccines.
Materials and Methods
Ethics statement.
The cotton rat experiments were conducted in strict accordance with the Guide for the Care and Use of Laboratory Animals of the National Research Council and was approved by The Ohio State University Institutional Animal Care and Use Committee (IACUC; animal protocol no. 2009A0221). For animal experiments, 5 cotton rats were assigned to each group (n = 5). All the groups were randomized and the researchers blinded to the experimental groups. The animal care facilities at The Ohio State University are AAALAC accredited. Every effort was made to minimize potential distress, pain, or discomfort to the animals throughout all experiments.
Cell lines.
Vero E6 cells (ATCC CRL-1586), A549 cells (ATCC CCL-185), and THP-1 (ATCC TIB-202) were purchased from the American Type Culture Collection (Manassas, VA). A549-Dual™, A549-Dual™ KO-RIG-I, A549-Dual™ KO-MDA5, and A549-Dual™ KO-MAVS knockout cells were purchased from InvivoGen (San Diego, CA). BHK-SR19-T7 cells were kindly provided by Apath, LLC, Brooklyn, NY. All cell lines were grown in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies) supplemented with 10% FBS. The medium for the BHK-SR19-T7 cells was supplemented with 10 μg/ml puromycin (Life Technologies) during every other passage to select for T7 polymerase-expressing cells. A549-Dual™ and knockout cell lines were supplemented with Normocin™ (100 μg/ml), blasticidin (10 μg/ml) and Zeocin™ (100 μg/ml). HeLa cells overexpressing the empty vector (pPB-CAG), YTHDF1, YTHDF2, or YTHDF3 were maintained in DMEM supplemented with 10% FBS and 1 µg/ml of puromycin every passage to select for YTHDF1-3 overexpressing cells. All cell lines used in this study were free of mycoplasma, as confirmed by the LookOut Mycoplasma PCR Detection Kit (Sigma). Cell lines were authenticated by the ATCC or InvivoGen.
Plasmids and site-directed mutagenesis.
Plasmids encoding the full-length genomic cDNA of hMPV strain NL/1/00 (phMPV), and support plasmids expressing hMPV N protein (pCITE-N), P protein (pCITE-P), L protein (pCITE-L), and M2-1 protein (pCITE-M2-1) were kindly provided by Ron A. M. Fouchier at the Department of Virology, Erasmus Medical Center, Rotterdam, The Netherlands 53. The F cleavage site in the genome of hMPV NL/1/00 was modified to a trypsin-independent F cleavage site, as described previously 54. A GFP gene was cloned into the gene junction between N and P in plasmid phMPV, resulted in the construction of phMPV-GFP. The G gene of hMPV strain NL/1/00 was cloned into pCAGGS resulted in the construction of pCAGGS-G. Mutations to the potential m6A sites in G gene were introduced into the plasmids pCAGGS-G and phMPV using QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). The pPB-CAG plasmid vector was used to overexpress the readers (YTHDF1-3 and YTHDC1), writers (METTL3, METTL14), and erasers (FTO, ALKBH5) as described previously 40, 41. In general, m6A sites occur at the consensus RRACH motif (where R indicates G or A; H indicates A, C, or U) 21. These m6A sites mutants in G gene region of the antigenome include: site 1,  site 2,
site 2,  site 3,
site 3,  site 4,
site 4,  site 5,
site 5, 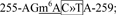 site 6,
site 6, 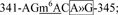 site 7,
site 7,  site 8,
site 8,  site 9,
site 9,  site 10,
site 10,  site 11,
site 11,  site 12,
site 12,  site 13,
site 13,  and site 14,
and site 14,  (where >> indicates nucleotide substitution, Supplementary Fig.5). The m6A sites mutants in the G gene of the genome include: site 1,
(where >> indicates nucleotide substitution, Supplementary Fig.5). The m6A sites mutants in the G gene of the genome include: site 1,  site 2,
site 2,  site 3,
site 3, 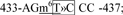 site 4,
site 4,  site 5,
site 5,  and site 6,
and site 6,  (Supplementary Fig.6). The A or C within the m6A consensus motif was mutated to a T or G in these sites without changing the encoded amino acid. Mutants G1-2, G8-9, G1-7, G8-14, and G1-14 were combinations of mutations in sites 1 and 2, sites 8 and 9, sites 1 to 7, sites 8 to 14, and sites 1 to 14, respectively. Mutant G(-)1-6 is the combined mutations of all six m6A sites in the G gene in the genome. These mutations were introduced in pCAGGS-G or an infectious hMPV cDNA clone, lineage A strain NL/1/00. We avoided changes that would alter the RNA secondary structures or codon usage, as predicted by the M-fold and Genscript software. All constructs were sequenced at The Ohio State University Plant Microbe Genetics Facility.
(Supplementary Fig.6). The A or C within the m6A consensus motif was mutated to a T or G in these sites without changing the encoded amino acid. Mutants G1-2, G8-9, G1-7, G8-14, and G1-14 were combinations of mutations in sites 1 and 2, sites 8 and 9, sites 1 to 7, sites 8 to 14, and sites 1 to 14, respectively. Mutant G(-)1-6 is the combined mutations of all six m6A sites in the G gene in the genome. These mutations were introduced in pCAGGS-G or an infectious hMPV cDNA clone, lineage A strain NL/1/00. We avoided changes that would alter the RNA secondary structures or codon usage, as predicted by the M-fold and Genscript software. All constructs were sequenced at The Ohio State University Plant Microbe Genetics Facility.
Virus stocks and purification.
Parental hMPV strain NL/1/00 was propagated and titrated in Vero E6 cells. To prepare highly purified hMPV for m6A sequencing, twenty T150 flasks of A549 cells were infected by hMPV at an MOI of 0.5. Cell culture supernatants harvested at 72 h post-infection were clarified by centrifugation at 5,000 ×g for 30 min. Virus was concentrated by centrifugation at 30,000×g for 2 h at 4°C in a Ty 50.2 rotor (Beckman). The pellet was resuspended in NTE buffer (0.05 M Tris-HCl, 0.15 M NaCl, 15 mM CaCl2 [pH 6.5]) supplemented with 10% trehalose and further purified through a sucrose gradient by centrifugation at 35,000×g for 18 h at 4°C in an SW55 rotor (Beckman). Solution layer containing virus was extracted with syringe, diluted with NTE buffer and centrifuged at 30,000×g for 2 h at 4°C in SW55 rotor. The final pellet was resuspended in 0.5 ml of NTE buffer.
m6A-seq.
High-throughput sequencing of the hMPV and host methylome was carried out using m6A-seq as described previously 55. For m6A-seq of the hMPV genome and antigenome, RNAs were extracted from highly purified hMPV virions and purified with the RiboMinus Eukaryote System v2 kit (Thermo Fisher). For m6A-seq of host transcripts, total RNAs were extracted from mock or hMPV-infected A549 cells and polyadenylated RNAs were isolated using Dynabeads mRNA DIRECT Purification kit (Thermo Fisher). Purified RNAs were sonicated with Bioruptor Pico (Diagenode) with 30 s ON 30 s OFF for 30 cycles, mixed with 1.0 μl of anti-m6A monoclonal antibody (NEB cat.E1610) in IP buffer (150 mM NaCl, 0.1% NP-40, 10 mM Tris-HCl, pH 7.4) and incubated for 2 h at 4°C. Enriched RNA fragments were purified with RNA Clean & Concentrator kit (Zymo) and used for library generation with Kapa RNA HyperPrep kit (Roche). Sequencing was carried out on Illumina HiSeq-4000 at SE50bp mode according to the manufacturer’s instructions. Two replicates of RNA samples from virions, virus-infected cells, and mock-infected cells were subjected to m6A-seq. For data analysis, after removing the adapter sequences, the reads were mapped to the human genome (hg38) and hMPV genome using Hisat2 56. Peak calling for the viral genome RNA was done by first dividing the hMPV genome into 30bp consecutive bins where read counts were quantified. Then we applied Fisher’s exact test to assess the enrichment of coverage by m6A-IP in these bins. The odds ratios were computed by (IP/overall IP)/(Input/overall Input) where overall IP/Input were represented by the median of read counts of bins across the same strand of the whole virus genome. Note, when calling peaks for mRNAs of the hMPV, the overall IP/Input were represented by the median of bins across the gene instead of the whole virus genome. Finally, we merged all neighboring bins that were significant (at FDR < 0.05 cutoff) in all replicates and reported them as consistent peaks.
Differential expression analysis of host cells.
The input of m6A-seq is equivalent to regular RNA-seq, therefore we quantified the gene-level read counts of input samples that aligned to hg38 for differential gene expression analysis. DESeq2 57 was used to make inferential tests where differentially expressed genes were identified at FDR < 0.1 cutoff .
Differential methylation analysis of host cell.
To compare the m6A-methylome of the mock-infected and hMPV-infected A549 cells, we first called peaks using fisher’s exact test on 50bp consecutive bins as described in previous section. We then used QNB package for differential methylation test with default setting 58.
Colorimetric quantification of viral m6A methylation.
Virion RNA was extracted from WT and mutant rHMPVs purified by sucrose gradient ultracentrifugation. Total m6A level on virion RNA was quantified by m6A RNA Methylation Assay Kit (Abcam, ab185912). Briefly, m6A was detected using a specific capture anti-m6A antibody and then quantified colorimetrically by reading the absorbance at 450 nm. A standard curve was generated using known m6A methylated RNA (range from 0.02 to 1 ng of m6A) as a positive control. The m6A level was calculated from each RNA samples based on their OD450 values. The percent change was calculated by dividing m6A levels in viral RNA from the treated group by those from the control group.
Gene Ontogeny (GO) analysis.
GO analysis was performed using the online analysis software metascape www.metascape.org 59.
siRNA and plasmid transfection.
siRNAs against METTL3, METTL14, FTO, ALKBH5, YTHDF1, YTHDF2, YTHDF3 or non-targeting AllStars negative control siRNA were purchased from Qiagen (Valencia, CA, sequences listed in Supplementary Table 5). All siRNA and plasmid transfections were performed using the Lipofectamine 3000 transfection reagent (Thermo-Fisher) according to the manufacturer’s instructions. Briefly, ninety percent confluent A549 cells in 12-well plates were transfected with 1 μg of plasmid or 30 pmol of siRNA and 24 hours later infected with hMPV. At 12, 18, 24 and 48 hours post infection cells were lysed in RIPA buffer (Abcam) on ice and collected for Western blot.
Antibodies and Western blotting.
The antibodies used in this study were anti-YTHDF1 (Proteintech, Rosemont, IL), anti-YTHDF2 (Abcam, Cambridge, MA), anti-YTHDF3 (Abcam), anti-METTL3 (Proteintech), anti-METTL 14 (Abcam), anti-ALKBH5 (Sigma-Aldrich), anti-FTO (Abcam), anti-hMPV serum (prepared in cotton rats), anti-hMPV N antibody (US Biological), anti-RIG-I (Abcam, ab180675), anti-MDA5 (Abcam), anti-IRF3 (Phospho S386) (Abcam, ab76493), anti-IRF3 (Phospho S396) (Abcam, ab138449), anti-FLAG (Sigma-Aldrich), anti-actin (Abcam), and anti-HA antibody(Abcam). Cells were harvested and lysed in RIPA buffer (Abcam) supplemented with protease inhibitor cocktail (Sigma-Aldrich). Western blotting was performed as described. Actin was used as a loading control.
Immunofluorescence analysis and confocal microscopy.
Mock or hMPV-infected cells were fixed in acetone and methanol at the ratio of 1:1 for 30 min, and blocked with goat serum (Sigma-Aldrich, G0923). Slides were stained with all primary antibodies (1:100), washed 3 times with PBS, and stained with conjugated Alexa Fluor secondary antibodies Alexa Fluor 488/594 (Thermo-Fisher; 1:300), and mounted with SlowFade™ Diamond Antifade Mountant with 4,6-diamidino-2-phenylindole (DAPI) (Thermo-Fisher). Imaging was performed on an Olympus FV 1000 confocal microscopy system at The Ohio State University Campus Microscopy & Imaging Facility.
Recovery of rhMPVs from the full-length cDNA clones.
rhMPVs or rghMPV (rhMPV expressing GFP) were rescued using a reverse genetics system as described previously 53, 60. Briefly, T25 flasks of BHK-SR19-T7 cells (kindly provided by Apath LLC), which stably express T7 RNA polymerase, were transfected with 3.75 μg of plasmid phMPV, 3.0 μg of pCITE-N, 1.5 μg of pCITE-P, 1.5 μg of pCITE-L, and 1.5 μg of pCITE-M2-1 using Lipofectamine 2000 (Life Technologies). At day 6 post-transfection, the cells were harvested using cell scrapers and were co-cultured with Vero-E6 cells at 50–60% confluence. When extensive cytopathic effects (CPE) were observed, the cells were subjected to three freeze-thaw cycles in the presence of 10% trehalose, followed by centrifugation at 3,000×g for 10 min. The supernatant was subsequently used to infect new Vero E6 cells. The successful recovery of the rhMPVs was confirmed by methylcellulose overlay plaque assay, immunostaining, and PCR with reverse transcription (RT-PCR). We successfully recovered a total of 5 rhMPVs carrying mutations in m6A sites in the G gene region in antigenome/G mRNA: the first two potential m6A sites (G1 and G2) were combined to produce rhMPV-G1-2; the first 7 mutations of m6A sites (G1 to G7) were combined to produce rhMPV-G1-7; the 8th and 9th m6A sites (G8 and G9) were combined to produce rhMPV-G8-9; the last 7 m6A sites (G8 to G14) were combined to produce rhMPV-G8-14; and all 14 m6A sites were combined to produce rhMPV-G1-14. In addition, we generated one recombinant virus [rhMPV-G(-)1-6] carrying 6 m6A site mutations in the G gene in genome. All hMPV mutants were plaque purified and sequenced.
Generation of naturally m6A-defective rHPMV-ALKBH5.
A549 cells in T150 flask were transfected with 20 µg of a plasmid encoding ALKBH5. After 24 h, cells were infected by hMPV at an MOI of 0.1. At 72 h post-infection, cell culture supernatants were harvested and hMPV virions were purified by sucrose gradient ultracentrifugation. RNA was extracted from rhMPV-ALKBH5, and the m6A level was measured by m6A RNA Methylation Assay Kit (Abcam, ab185912). These virion RNAs are expected to be ‘naturally’ deficient in m6A methylation, as some of the m6A modifications have been removed by ALKBH5 and their nucleotide sequences are not changed.
Immunostaining plaque assay.
Vero E6 cells were seeded in 24-well plates, infected with serial dilutions of rhMPV, and overlayed with methylcellulose. At day 5 post-infection, cells were fixed with 10% neutral buffered formaldehyde at room temperature for 30 min. Then the mixture of overlay and formaldehyde was removed. Cells were permeabilized in phosphate-buffered saline (PBS) containing 0.4% Triton X-100 at room temperature for 10 min and blocked at 37°C for 1 h using 1% bovine serum albumin (BSA) in PBS. The cells were then incubated with anti-hMPV N-protein primary monoclonal antibody (Millipore, Billerica, MA) at a dilution of 1:2,000 overnight at 4°C, followed by incubation with horseradish peroxidase (HRP)-labeled goat anti-mouse secondary antibody (Thermo Scientific, Waltham, MA) at a dilution of 1:5,000. After incubation with 3-amino-9-ethylcarbazole (AEC) chromogen substrate (Sigma, St. Louis, MO), positive cells were visualized under a microscope. The viral titer was calculated as the number of plaque-forming units (p.f.u) per ml.
Viral replication kinetics in A549 cells.
Confluent A549 cells or knockout cells in 24-well plates were infected with parental rhMPV or rhMPV mutant at an MOI of 1.0 or 5.0. After 1 h of adsorption, the inoculum was removed and the cells were washed three times with PBS. Fresh DMEM (supplemented with 1% FBS) was added and the infected cells were incubated at 37°C. At different time points post-infection, the supernatant and cells were harvested by three freeze-thaw cycles, followed by centrifugation at 1,500×g at room temperature for 15 min. The virus titer was determined by an immunostaining assay in Vero E6 cells.
Flow cytometry.
WT HeLa cells or HeLa cells stably overexpressing these YTHDF proteins were infected with rghMPV at an MOI of 1.0, and GFP expression was monitored at the indicated times by fluorescence microscopy. At the indicated time points, cells were trypsinized and fixed in 4 % of paraformaldehyde solution and the number of GFP-positive cells quantified by flow cytometry using Attune NxT Flow Cytometer (ThomoFisher Scientific). The mock-infected cells (GFP negative) were used for gating controls. Then, the number of GFP-positive and negative cells in rghMPV-infected cells were sorted. Attune NxT software was used to collect and analyze the data. The percent of GFP positive cells were calculated. An example of the gating strategy was listed in Supplementary Fig.11.
Quantification of viral genome, antigenome, and mRNA by real-time RT-PCR.
Ninety percent confluent A549 cells were infected with each rhMPV mutant at an MOI of 1.0 or 5.0. At indicated time points, total RNA was isolated from virus-infected cells using the TRizol reagent (Life Technologies). The first strand of DNA was generated from genomic and antigenomic RNA with primers targeting leader and trailer sequence, respectively, and real-time PCR was performed in TB-Green premix Ex Taq™ (TaKaRa, Japan) with the primer pairs located on N and L gene, respectively. A cDNA pool was generated from total RNA with Oligo (dT)23 (Sigma-Aldrich), and hMPV N and G-mRNA copies were quantified with the primer pairs located on N and G gene, respectively. RNA and mRNA copies of each sample were normalized by respective mRNA copies of human GAPDH.
RT-PCR and sequencing.
All plasmids, viral mutants and stocks, and virus isolates from the nasal turbinates and lungs of cotton rats were sequenced. Viral RNA was extracted from 100 µl of each recombinant virus using an RNeasy mini kit (Qiagen, Valencia, CA) and total RNA from infected tissue was extracted with TRizol reagent. A 1-kb DNA fragment spanning the hMPV G gene was amplified by RT-PCR. The PCR products were purified and sequenced using a sequencing primer at The Ohio State University Plant Microbe Genetics Facility to confirm the presence of the designed mutations.
Isolation of total viral RNA, virion RNA, and G mRNA.
Confluent A549 cells in 150-mm dishes were mock infected or infected with WT or mutant rhMPV at an MOI of 0.5. At day 2 postinfection, total RNA was isolated from virus-infected cells using the TRizol reagent (Life Technologies) and dissolved in RNase-free water. Subsequently, poly(A)-containing RNA was isolated from total RNA using a Dynabeads mRNA Direct™ kit (Life Technologies) according to the manufacturer’s recommendations. Finally, hMPV G mRNA was isolated by Dynabeads MyOne™ Streptavidin C1 (ThermoFisher Scientific) conjugated with poly T-tailed G gene specific primer. Virion RNA was extracted from sucrose-gradient purified virions of rhMPV or rhMPV mutant. HMPV genome, antigenome, and G mRNA were quantified by real-time RT-PCR.
[35S]-Methionine metabolic labeling.
A549 cells were transfected with siRNA against METTL3 and METTL14 or control siRNA. After 24 h, cells were incubated in methionine- and cysteine-free media for 1 h, and 50 µCi of [35S]-methionine was added. At indicated time points, cells were washed with PBS and disrupted in lysis buffer. Cell lysates were resolved on by SDS–polyacrylamide gel electrophoresis (SDS–PAGE) and exposed to film. Quantification of [35S]-labeled proteins was performed using ImageJ software. 5 μl of each protein sample was used for measuring total [35S] incorporation by scintillation counting (Beckman).
MeRIP assay.
MeRIP assay was carried out using a procedure provided by Millipore Magna MeRIP m6A kit (Catalog No. 17-10499). Magnetic Beads A/G blend (25µl) was washed and incubated with anti-m6A antibody (5µl) at room temperature for 30 min and washed three times to remove any unbound antibody. Total RNA (15µg) was extracted from rhMPV or m6A deficient rhMPV-infected A549 cells. The RNA samples were treated at 85ºC for 5 min and chilled on ice immediately, and incubated with m6A antibody-associated beads at 4 ºC for 2 h with rotation. The RNA-associated magnetic beads were then washed for 3 times. Total RNA was extracted from beads by TRizol reagent and was quantified by real-time RT-PCR using primers annealing to hMPV antigenome, genome, and G mRNA.
Measurement of interferon in virus-infected or RNA-transfected cells.
For virus-infection, A549 cells or THP-1 cells infected by rhMPV or hMPV mutant at MOI of 1.0 or 4.0, cell supernatants were harvested at 16, 24, and 48 h post-infection and IFN-α and -β concentrations were determined by commercial enzyme-linked immunosorbent assays (ELISA) according to the manufacturer’s instructions (PBL, Piscataway, NJ). A known concentration of human IFN-α and –β was used to generate the standard curve. Prior to RNA transfection, viral RNA was treated with or without calf intestinal alkaline phosphatase (CIP; Promega) at the dose of 107 copies/ 10 U for 30 min at 37°C. After inactivation of CIP at 65°C for 15 min, viral RNA was further purified by TRizol reagent and quantified by real-time RT-PCR. A549 cells or A549-Dual cell lines in 24-well plates were transfected with CIP-treated or untreated viral RNA by Lipofectamine 3000. At 24 and 48 h post-transfection, culture medium was harvested for IFN-β quantification by ELISA.
Immunoprecipitation assay of RIG-I and virion RNA.
Confluent six-well-plates of A549 cells were transfected with 2 μg of plasmid pEF-BOS-RIG-I-Flag (provided by J. Yount, Ohio State University College of Medicine). At 24 h post-transfection, cells were lysed in lysis buffer (Abcam, ab152163). Cell lysates were harvested after centrifugation at 13,000×g for 10 min and incubated with Anti-FLAG® M2 magnetic beads (Sigma-Aldrich, M8823) at room temperature for 80 min. The mixture was then divided into 13 aliquots (150µl/tube). 12 aliquots were incubated with 2×108 copies of virion RNA (with or without CIP treatment) or 2×109 copies of hMPV mRNA respectively at 37°C for 1 h. Beads associated RNA:protein complex were washed in lysis buffer for three times, and total RNA was extracted from beads by TRizol reagent and quantified by real-time RT-PCR. The 13th aliquot was washed and subjected to Western blot.
RIG-I pull-down assay.
109 copies of virion RNA with or without CIP treatment was biotinylated with Pierce™ RNA 3’ End Biotinylation Kit (Thermo Fisher Scientific) according to the product instruction. Purified 3’ end biotinylated RNA was incubated with MyOne™ Streptavidin C1 beads (Thermo Fisher Scientific) in the presence of RNase inhibitor at room temperature for 30 min with rotation. RNA-associated beads were then washed three times and incubated with 50 µl of A549 cell lysate containing overexpressed RIG-I and 20 unit of RNase inhibitor at room temperature for 1 h with rotation. Beads were then washed for 3 times and subjected to SDS-PAGE. The pull-down RIG-I protein on Streptavidin beads were detected by Western blot using anti-RIG-I antibody. For control, mixture of cell lysate and RNA-associated beads were loaded as input.
Limited trypsin digestion of RIG-I.
Recombinant human RIG-I protein was purified from HEK-293T cells transfected with a plasmid encoding Flag-tagged RIG-I (pEF-BOS-RIG-I-Flag). The concentration of RIG-I protein was measured by Bradford assay. Recombinant RIG-I (50 nM) was incubated with 2×107 copies of virion RNA of WT or mutant hMPV in 30 μL MOPS buffered reaction system (10 mM MOPS pH 7.4, 1 mM DTT, 1 mM MgCl2, 0.002% Tween20) in the presence of RNase inhibitor and AMP-PNP (2 mM). The reaction mixtures were incubated at 37°C for 30 min to permit RIG-I:RNA complex formation and mixed with 10μL of tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-trypsin (2.5 ng/μL) and incubated at room temperature. At indicated time points (0 to 120 min), 10μL was removed, mixed with 5×SDS-PAGE loading dye, and boiled for Western blot probed with an anti-RIG-I helicase antibody (Abcam). Poly(I:C) (2–10 μg) was used as a positive control. For the competition assay, WT and rhMPV-G1-14 virion RNA were diluted to 2×106 copies/μL and different ratios of these two RNAs were mixed (10:0, 7.5:2.5, 5:5, 2.5:7.5, and 0:10), and were incubated with purified RIG-I and AMP-PNP and the conformation of RIG-I was examined by limited trypsin digestion, as described above.
Interferon response of rhMPV and mutants in cotton rats.
Six-week-old specific-pathogen-free (SPF) female cotton rats (Envigo, Indianapolis, IN) were inoculated intranasally with 100 µl of PBS or PBS containing 2.0×105 p.f.u. of rhMPV-G8-14, rhMPV-G1-14 or rhMPV. Each group contains 5 cotton rats (n = 5). The experimental groups were blinded to researchers. Forty-eight hours post-inoculation, cotton rats were sacrificed and 1 ml of PBS was injected into the right lung of each cotton rats. Approximately 1 ml of bronchoalveolar lavage (BAL) was collected for IFN-β bioactivity assay on CCRT cells (an osteosarcoma cell line of cotton rats). Briefly, CCRT cells were cultured in 96-well plates with 100 μl of DMEM medium supplemented with 2% FBS. BAL supernatant (250 μl) was mixed with the same volume of DMEM containing 0.1 M HCl, incubated at room temperature for 2 h to destroy type II interferon, and neutralized with 27.8 μl of DMEM containing 7.5% NaHCO3. The treated BAL mixture was 2-fold serially diluted (1:2~1:128) and added to CCRT cell culture medium in duplicate in a volume of 100μl/well. A known concentration of human IFN-β was 2-fold serially diluted (250 U~7.8 U/ml) and used to generate a standard curve. Cells were incubated at 37°C in 5% CO2 for another 24 h and infected by 104 p.f.u. of recombinant vesicular stomatitis virus expressing GFP reporter (rVSV-GFP) per well. GFP positive cells were observed under microscope at 24 h post-infection. The IFN-β concentration of each BAL sample was calculated according to the highest dilution of samples and the lowest concentration of standard human IFN-β which inhibited rVSV-GFP replication therefore GFP expression.
Replication and pathogenesis of rhMPV in cotton rats.
Twenty-five 6-week-old female SPF cotton rats (Envigo, Indianapolis, IN) were randomly divided into 5 groups (5 cotton rats per group, n = 5). The experimental groups were blinded to researchers. Prior to virus inoculation, the cotton rats were anesthetized with isoflurane. The cotton rats in groups 1–4 were intranasally inoculated with 2.0×105 p.f.u. of rhMPV, rhMPV-G1-2, rhMPV-G8-9, and rhMPV-G1-14. The cotton rats in group 5 were mock infected with 100 μl of PBS and served as uninfected controls. Each cotton rat was inoculated intranasally with a volume of 100 μl. After inoculation, the animals were evaluated on a daily basis for any clinical signs. At day 4 postinfection, the cotton rats were sacrificed, and lungs and nasal turbinates were collected for both virus isolation and histological analysis.
Immunogenicity of rhMPV in cotton rats.
For the immunogenicity study, thirty 4-week-old cotton rats (Envigo) were randomly divided into four groups (5 cotton rats per group, n = 5). The experimental groups were blinded to researchers. Cotton rats in groups 1 were mock-infected with PBS and served as uninfected unchallenged control. Cotton rats in groups 2–5 were intranasally inoculated with 2.0×105 p.f.u. of rhMPV, rhMPV-G1-2, rhMPV-G8-9 and rhMPV-G1-14, respectively. Cotton rats in groups 4 were mock-infected with PBS and served as uninfected challenged control. After immunization, the cotton rats were evaluated daily for any possible abnormal reaction and blood samples were collected from each cotton rat weekly by facial vein retro-orbital bleeding, and serum was used for detection of neutralizing antibodies. At 4 weeks post-immunization, the cotton rats in groups 2 to 6 were challenged with 2.0×105 p.f.u. of parental rhMPV via intranasal route, and evaluated twice daily for the presence of any clinical symptoms. At 4 days post-challenge, all cotton rats were euthanized by CO2 inhalation, and their lungs and nasal turbinates were collected for virus titration. The immunogenicity of rhMPV mutant was assessed based on their ability to trigger neutralizing antibody, the ability to prevent hMPV replication in lungs and nasal turbinates, and the ability to protect lung from pathological changes.
Genetic stability of rhMPV mutants in cell culture.
Confluent Vero-E6 cells in T25 flasks were infected with each rhMPV mutant at an MOI of 0.1. At day 3 post-inoculation, the cell culture supernatant was harvested and used for the next passage in Vero-E6 cells. Using this method, each rhMPV mutant was repeatedly passaged 15 times in Vero-E6 cells. At each passage, the G gene was amplified by RT-PCR and sequenced. At passage 15, the entire genome of each recombinant virus was amplified by RT-PCR and sequenced.
Pulmonary histology.
After sacrifice, the right lung of each animal was removed, inflated, and fixed with 4% neutral buffered formaldehyde. Fixed tissues were embedded in paraffin and sectioned at 5 μm. Slides were then stained with hematoxylin-eosin (H&E) for the examination of histological changes by light microscopy. Histopathological changes were evaluated based on the extent of interstitial inflammation, edema, and peribronchiolar inflammation.
Determination of viral titer in lung and nasal turbinate.
The nasal turbinate and the left lung from each cotton rat were removed, weighed, and homogenized in 1 ml of PBS solution using a Precellys 24 tissue homogenizer (Bertin, MD) following the manufacturer’s recommendations. The presence of infectious virus was determined by an immunostaining plaque assay in Vero E6 cells, as described above.
Determination of hMPV-neutralizing antibody.
hMPV-specific neutralizing antibody titers were determined using a plaque reduction neutralization assay61. Briefly, cotton rat sera were collected by retro-orbital bleeding weekly until challenge. The serum samples were heat inactivated at 56°C for 30 min. Twofold dilutions of the serum samples were mixed with an equal volume of DMEM containing approximately 100 p.f.u./well rhMPV in a 96-well plate, and the plate was incubated at 37 °C for 1 h with constant rotation. The mixtures were then transferred to confluent Vero-E6 cells in a 24-well plate. After 1 h of incubation at 37°C, the virus-serum mixtures were removed and the cells were overlaid with 0.75% methylcellulose in DMEM and incubated for another 5 days before immunostaining plaque titration. The plaques were counted, and 50% plaque reduction titers were calculated as the hMPV-specific neutralizing antibody titers.
Statistical analysis.
Quantitative analysis was performed by either densitometric scanning of autoradiographs or by using a phosphorimager (Typhoon; GE Healthcare, Piscataway, NJ) and ImageQuant TL software (GE Healthcare, Piscataway, NJ) or using Image J (NIH, Bethesda, MD). Statistical analysis was performed by one-way multiple comparisons using SPSS (version 8.0) statistical analysis software (SPSS Inc., Chicago, IL) or student’s t test. A P value of <0.05 was considered statistically significant.
Extended Data
Extended Data Fig.1.

Extended Data Fig.2.

Extended Data Fig.3.

Extended Data Fig.4.

Extended Data Fig.5.

Extended Data Fig.6.

Extended Data Fig.7.

Extended Data Fig.8.

Extended Data Fig.9.

Extended Data Fig.10.

Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grants from the National Institutes of Health (no. R01AI090060) to J.L., no. P01 AI112524 to M.E.P., S.N., and J.L., and nos. R01 HG008688 and RM1 HG008935 to C.H. C.H. is an investigator of the Howard Hughes Medical Institute. We thank Ron A. M. Fouchier for the infectious cDNA clone of hMPV and Jacob Yount for the RIG-I plasmid. We are grateful to members of the J. Li laboratory for critical readings of the manuscript. We thank the three anonymous reviewers whose comments have greatly improved this manuscript.
Footnotes
Competing interests
C.H. is a scientific founder of Accent Therapeutics, Ins. J.L., C.H., M.E.P., and S.N. are filing a provision patent application of 62/748,175.
Data availability
The authors declare that the data supporting the findings of this study are available with the article and its Extended Data and Supplementary Information files, or are available from the corresponding author upon request. The accession number for the raw sequencing data obtained from the MeRIP-seq reported in this paper is GEO: GSE136139. The source data underlying Figures, Extended Data Figures, and Supplementary Figures are provided as a Source Data file.
References
- 1.Takeuchi O & Akira S Innate immunity to virus infection. Immunol. Rev 227, 75–86 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loo YM & Gale M Jr. Viral regulation and evasion of the host response. Curr. Top. Microbiol. Immunol 316, 295–313 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Chow KT, Gale M Jr. & Loo YM RIG-I and Other RNA Sensors in Antiviral Immunity. Annu. Rev. Immunol 36, 667–694 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Wu B et al. Molecular imprinting as a signal-activation mechanism of the viral RNA sensor RIG-I. Mol. Cell 55, 511–523 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hornung V et al. 5’-Triphosphate RNA is the ligand for RIG-I. Science 314, 994–997 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Pichlmair A et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science 314, 997–1001 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Schlee M et al. Recognition of 5’ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 31, 25–34 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kato H et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med 205, 1601–1610 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Runge S et al. In vivo ligands of MDA5 and RIG-I in measles virus-infected cells. PLoS Pathog 10, e1004081 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shuman S What messenger RNA capping tells us about eukaryotic evolution. Nat. Rev. Mol. Cell Biol 3, 619–625 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Furuichi Y, LaFiandra A & Shatkin AJ 5’-Terminal structure and mRNA stability. Nature 266, 235–239 (1977). [DOI] [PubMed] [Google Scholar]
- 12.Hyde JL & Diamond MS Innate immune restriction and antagonism of viral RNA lacking 2-O methylation. Virology 479–480, 66–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Wang JT & Whelan SP A unique strategy for mRNA cap methylation used by vesicular stomatitis virus. Proc. Natl Acad. Sci. U S A 103, 8493–8498 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ray D et al. West Nile virus 5’-cap structure is formed by sequential guanine N-7 and ribose 2’-O methylations by nonstructural protein 5. J. Virol 80, 8362–8370 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y et al. Biochemical and structural insights into the mechanisms of SARS coronavirus RNA ribose 2’-O-methylation by nsp16/nsp10 protein complex. PLoS Pathog 7, e1002294 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J, Fontaine-Rodriguez EC & Whelan SP Amino acid residues within conserved domain VI of the vesicular stomatitis virus large polymerase protein essential for mRNA cap methyltransferase activity. J. Virol 79, 13373–13384 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma Y et al. mRNA cap methylation influences pathogenesis of vesicular stomatitis virus in vivo. J. Virol 88, 2913–2926 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zust R et al. Ribose 2’-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol 12, 137–143 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Daffis S et al. 2’-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468, 452–456 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abbas YM et al. Structure of human IFIT1 with capped RNA reveals adaptable mRNA binding and mechanisms for sensing N1 and N2 ribose 2’-O methylations. Proc. Natl Acad. Sci. U S A 114, E2106–E2115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yue Y, Liu J & He C RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes & Development 29, 1343–1355 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roundtree IA, Evans ME, Pan T & He C Dynamic RNA Modifications in Gene Expression Regulation. Cell 169, 1187–1200 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frye M, Harada BT, Behm M & He C RNA modifications modulate gene expression during development. Science 361, 1346–1349 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao BS et al. m(6)A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature 542, 475–478 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lavi S & Shatkin AJ Methylated simian virus 40-specific RNA from nuclei and cytoplasm of infected BSC-1 cells. Proc. Natl Acad. Sci. U S A 72, 2012–2016 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Furuichi Y, Shatkin AJ, Stavnezer E & Bishop JM Blocked, methylated 5’-terminal sequence in avian sarcoma virus RNA. Nature 257, 618–620 (1975). [DOI] [PubMed] [Google Scholar]
- 27.Moss B, Gershowitz A, Stringer JR, Holland LE & Wagner EK 5’-Terminal and internal methylated nucleosides in herpes simplex virus type 1 mRNA. J. Virol 23, 234–239 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sommer S et al. The methylation of adenovirus-specific nuclear and cytoplasmic RNA. Nucleic Acids Res 3, 749–765 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kennedy EM et al. Posttranscriptional m(6)A Editing of HIV-1 mRNAs Enhances Viral Gene Expression. Cell Host Microbe 22, 830 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lichinchi G et al. Dynamics of the human and viral m(6)A RNA methylomes during HIV-1 infection of T cells. Nat. Microbiol 1, 16011 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xue M et al. Viral N(6)-methyladenosine upregulates replication and pathogenesis of human respiratory syncytial virus. Nat. Commun 10, 4595 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gokhale NS et al. N6-Methyladenosine in Flaviviridae Viral RNA Genomes Regulates Infection. Cell Host Microbe 20, 654–665 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tirumuru N et al. N(6)-methyladenosine of HIV-1 RNA regulates viral infection and HIV-1 Gag protein expression. Elife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan B et al. Viral and cellular N(6)-methyladenosine and N(6),2’-O-dimethyladenosine epitranscriptomes in the KSHV life cycle. Nat. Microbiol 3, 108–120 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Courtney DG et al. Epitranscriptomic Enhancement of Influenza A Virus Gene Expression and Replication. Cell Host Microbe 22, 377–386 e375 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hesser CR, Karijolich J, Dominissini D, He C & Glaunsinger BA N6-methyladenosine modification and the YTHDF2 reader protein play cell type specific roles in lytic viral gene expression during Kaposi’s sarcoma-associated herpesvirus infection. PLoS Pathog 14, e1006995 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Imam H et al. N6-methyladenosine modification of hepatitis B virus RNA differentially regulates the viral life cycle. Proc. Natl Acad. Sci. U S A 115, 8829–8834 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Durbin AF, Wang C, Marcotrigiano J & Gehrke L RNAs Containing Modified Nucleotides Fail To Trigger RIG-I Conformational Changes for Innate Immune Signaling. MBio 7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang F et al. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature 479, 423–427 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang X et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X et al. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 161, 1388–1399 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meyer KD et al. 5’ UTR m(6)A Promotes Cap-Independent Translation. Cell 163, 999–1010 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schildgen V et al. Human Metapneumovirus: lessons learned over the first decade. Clin. Microbiol. Rev 24, 734–754 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Myong S et al. Cytosolic viral sensor RIG-I is a 5’-triphosphate-dependent translocase on double-stranded RNA. Science 323, 1070–1074 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng J et al. HDX-MS reveals dysregulated checkpoints that compromise discrimination against self RNA during RIG-I mediated autoimmunity. Nat. Commun 9, 5366 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Devarkar SC, Schweibenz B, Wang C, Marcotrigiano J & Patel SS RIG-I Uses an ATPase-Powered Translocation-Throttling Mechanism for Kinetic Proofreading of RNAs and Oligomerization. Mol. Cell 72, 355–368 e354 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kariko K, Buckstein M, Ni H & Weissman D Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23, 165–175 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Sioud M, Furset G & Cekaite L Suppression of immunostimulatory siRNA-driven innate immune activation by 2’-modified RNAs. Biochem. Biophys, Res. Commun 361, 122–126 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Chen YG et al. N6-Methyladenosine Modification Controls Circular RNA Immunity. Mol. Cell 76, 96–109 e109 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zheng Q, Hou J, Zhou Y, Li Z & Cao X The RNA helicase DDX46 inhibits innate immunity by entrapping m(6)A-demethylated antiviral transcripts in the nucleus. Nat. Immunol 18, 1094–1103 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Rubio RM, Depledge DP, Bianco C, Thompson L & Mohr I RNA m(6) A modification enzymes shape innate responses to DNA by regulating interferon beta. Genes Dev 32, 1472–1484 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Winkler R et al. m(6)A modification controls the innate immune response to infection by targeting type I interferons. Nat. Immunol 20, 173–182 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Herfst S et al. Recovery of human metapneumovirus genetic lineages a and B from cloned cDNA. J. Virol 78, 8264–8270 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y, Wei Y & Li J Development and optimization of a direct plaque assay for human and avian metapneumoviruses. J. Virol. Methods 185, 61–68 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dominissini D, Moshitch-Moshkovitz S, Salmon-Divon M, Amariglio N & Rechavi G Transcriptome-wide mapping of N(6)-methyladenosine by m(6)A-seq based on immunocapturing and massively parallel sequencing. Nature Protoc 8, 176–189 (2013). [DOI] [PubMed] [Google Scholar]
- 56.Kim D et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu L, Zhang SW, Huang Y & Meng J QNB: differential RNA methylation analysis for count-based small-sample sequencing data with a quad-negative binomial model. BMC Bioinformatics 18, 387 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tripathi S et al. Meta- and Orthogonal Integration of Influenza “OMICs” Data Defines a Role for UBR4 in Virus Budding. Cell Host & Microbe 18, 723–735 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cai H et al. Zinc binding activity of human metapneumovirus M2–1 protein is indispensable for viral replication and pathogenesis in vivo. J. Virol 89, 6391–6405 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y et al. Rational design of human metapneumovirus live attenuated vaccine candidates by inhibiting viral mRNA cap methyltransferase. J. Virol 88, 11411–11429 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.