

Abstract
The polyglutamine (polyQ) diseases are a group of nine fatal, adult-onset neurodegenerative disorders characterized by the misfolding and aggregation of mutant proteins containing toxic expansions of CAG/polyQ tracts. The heat shock protein 90 and 70 (Hsp90/Hsp70) chaperone machinery is a key component of cellular protein quality control, playing a role in the regulation of folding, aggregation, and degradation of polyQ proteins. The ability of Hsp70 to facilitate disaggregation and degradation of misfolded proteins makes it an attractive therapeutic target in polyQ diseases. Genetic studies have demonstrated that manipulation of Hsp70 and related co-chaperones can enhance the disaggregation and/or degradation of misfolded proteins in models of polyQ disease. Therefore, the development of small molecules that enhance Hsp70 activity is of great interest. However, it is still unclear if currently available Hsp70 modulators can selectively enhance disaggregation or degradation of misfolded proteins without perturbing other Hsp70 functions essential for cellular homeostasis. This review discusses the multifaceted role of Hsp70 in protein quality control and the opportunities and challenges Hsp70 poses as a potential therapeutic target in polyQ disease.
Keywords: Spinal bulbar muscular atrophy, Huntington disease, Spinocerebellar ataxia, Proteasomal degradation, Autophagy
Introduction
Age-dependent protein aggregation disorders are characterized by the accumulation of damaged or mutant proteins in the central nervous system, most often in neurons. The preferential accumulation of misfolded proteins in neurons underscores the importance of protein quality control mechanisms, such as the heat shock protein 90 and 70 (Hsp90/Hsp70) chaperone machinery, in neuronal homeostasis. The polyglutamine (polyQ) diseases are an example of hereditary protein aggregation disorders and include Huntington disease (HD), spinal bulbar muscular atrophy (SBMA/Kennedy disease), dentatorubral pallidoluysian atrophy (DRPLA), and six autosomal dominant forms of spinocerebellar ataxia (SCAs) [1]. These diseases are caused by the expansion of CAG/polyQ tracts in a collection of unrelated proteins, and penetrance only occurs once polyQ length exceeds disease-specific thresholds. Additional disorders have also been described in which expanded CTG repeats are associated with antisense transcripts containing long CAG repeats [2]. Proteins containing disease length polyQ tracts are prone to misfolding and can self-assemble into soluble oligomers and microaggregates that coalesce into insoluble, amyloid-like fibrils. The polyQ length threshold for disease penetrance is strikingly correlated to the threshold for aggregation of polyQ proteins in vitro [3]. It is well established the misfolding of polyQ containing proteins drives disease pathogenesis. While the exact specie(s) of polyQ proteins that mediate toxicity is unclear, the accumulation and aggregation of misfolded polyQ proteins in neurons implies that protein quality control systems are overwhelmed as an underlying basis of disease.
The role of the Hsp90/Hsp70 chaperone machinery in protein quality control has been well studied in the context of HD, SBMA, and SCA3. Therefore, this review will focus on these three polyQ diseases as representatives of the whole group, with additional examples provided when relevant. HD, the most prevalent of the polyQ diseases, is caused by mutant huntingtin (Htt) harboring an expanded polyQ tract [4]. The normal function of Htt is incompletely understood, however, recent work suggests it acts as a scaffold in retrograde transport, vesicle trafficking, and selective autophagy [5]. PolyQ Htt-dependent neuron dysfunction and death causes progressive motor, cognitive, and psychiatric manifestations in HD patients. SBMA is a neuromuscular degenerative disorder that is characterized by progressive weakness of proximal limb and bulbar muscles [6]. SBMA is caused by a polyQ expansion in the androgen receptor (AR) and pathogenesis is dependent on circulating levels of androgen; therefore, the disease only affects males. Steroid hormone-dependent translocation of polyQ AR to the nucleus leads to ligand-dependent misfolding and formation of nuclear inclusions. PolyQ proteins cause degeneration of the cerebellum in six forms of SCA [7]. The most common polyQ SCA, SCA3, is caused by the expansion of a polyQ tract in ataxin-3 (ATXN3), a deubiquitinating enzyme [8]. All polyQ diseases are ultimately fatal, with disease onset typically occurring in mid-life and disease progression occurring over the next 10–30 years [9]. Even though the genetic cause and progression of these diseases are well understood, there are currently no FDA-approved disease-modifying treatments.
A large body of work has demonstrated that genetic manipulation of the Hsp90/Hsp70 chaperone machinery is therapeutically beneficial in cellular and animal models of polyQ disease [10, 11], making the Hsp90/Hsp70 chaperone machinery an attractive therapeutic target. There are two main pharmacological strategies for targeting this system: inhibition of Hsp90 and activation of Hsp70. While it is well established that cycling into complexes with Hsp90 stabilizes Hsp90 client proteins, such as AR and Htt, and that specific inhibition of Hsp90 enhances client protein degradation, this approach has the potential to cause on-target side effects due a global decrease in the hundreds of proteins reliant on Hsp90 for stabilization [10]. Activation of Hsp70, on the other hand, is focused on selectively enhancing the disaggregation and degradation of already misfolded proteins and should not affect properly folded Hsp90-dependent proteins, thus minimizing side effects. Hsp70 selectively facilitates the degradation of misfolded proteins, thus activation of Hsp70-facilitated degradation may provide a strategy to eliminate misfolded proteins while leaving native proteins untouched. However, targeting Hsp70 is not without problems. To be therapeutically beneficial a small molecule must selectively enhance the anti-aggregation or pro-degradative activity of Hsp70, without disrupting other Hsp70 functions critical for cellular homeostasis. Hsp70 activity is dependent on a conformational cycle determined by nucleotide binding, hydrolysis, and release, and this cycle is adapted to specific functions through regulation by co-chaperones. Indeed, co-chaperone binding sites may provide targets for small molecules that alter Hsp70 function. Designing small molecules that selectively alter one Hsp70 function while leaving others untouched poses a challenge. This review will focus on the role of Hsp70 in protein quality control and the opportunity and obstacles it presents as a therapeutic target in polyQ disease; Hsp90 will be discussed only in context of its cooperation with Hsp70, as comprehensive reviews on Hsp90 are available.
Hsp70 function is driven by nucleotide cycling and regulated by co-chaperones
Heat shock proteins (Hsp) were first discovered due to their increased expression upon heat exposure [12, 13]. These proteins were originally named based on their molecular weight and include Hsp110 (HSPH), Hsp90 (HSPC), Hsp70 (HSPA1), Hsp60 (chaperonins), Hsp40 (J-domain/DNAJ), and small heat shock proteins (sHsp/HspB) [14]. It is now recognized that several environmental and pathological conditions, not just heat, can induce the expression of Hsps, underscoring their importance in the response to cellular stress. The transcription factor HSF1 (heat shock factor 1) is the master regulator of the heat shock response (HSR). Under conditions of stress, localization of HSF1 to the nucleus and binding to the heat shock element in promoters leads to the activation of genes encoding inducible members of the Hsp90/Hsp70 chaperone machinery [15, 16]. Hsp families include constitutively expressed members, such as Hsc70 in the Hsp70 family, and therefore also play a role in protein quality control under basal conditions. To further add to the complexity, Hsp families contain numerous members with distinct subcellular localization and unique functions. The Hsp70 family alone contains 13 members in humans, including the cytoplasmic variants Hsp70 (HSPA1) and Hsc70 (HSPA8), the mitochondrial variant mtHsp70 (mortalin/HSPA9), and the endoplasmic reticulum variant BIP (GRO78/HSPA5) [17]. In this review the term Hsp70 will be used broadly to refer to all family members, and when pertinent, specific family members will be defined.
Interaction of Hsp70 with client proteins is not dependent on a specific sequence or structural motif; instead, Hsp70 interacts with proteins containing exposed hydrophobic stretches [18]. These hydrophobic residues are exposed in proteins during their synthesis, in protein folding (i.e. ligand binding) clefts, in domains regulating protein–protein interactions, or in misfolded proteins. Hsp70 is comprised of two domains, a nucleotide binding domain (NBD) and a substrate binding domain (SBD), connected by a flexible linker. The action of Hsp70 is facilitated by a nucleotide-dependent conformational cycle. In the ATP state, the SBD is docked to the NBD, locking Hsp70 into a conformation that has low affinity for unfolded, hydrophobic substrate (Fig. 1a). Upon hydrolysis of ATP to ADP a large conformational change allows the SBD to move freely, and in the ADP state Hsp70 exhibits high affinity for unfolding proteins, reviewed in [19] (Fig. 1b). However, the intrinsic ATPase activity of Hsp70 is very low, with ATP hydrolysis being the rate limiting step [20].
Fig. 1.
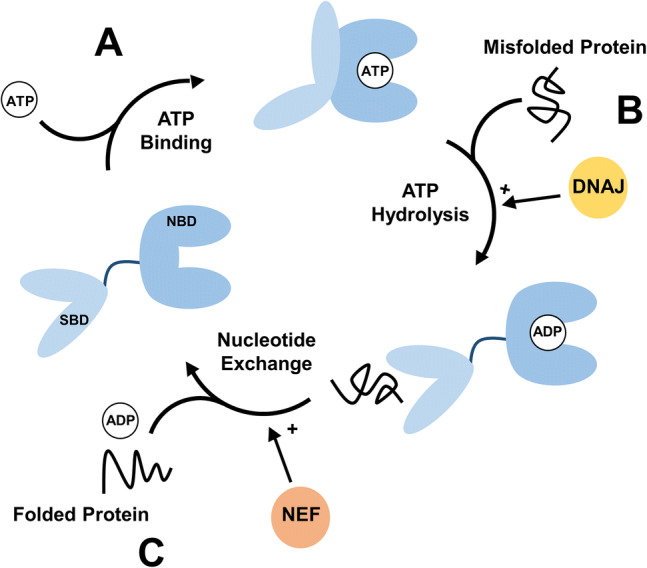
The activity of Hsp70 is driven by a nucleotide-dependent conformational cycle and is regulated by co-chaperones. a Binding of ATP promotes a closed state of Hsp70 where the SBD is docked to the NBD, locking Hsp70 into a conformation with low affinity for misfolded protein. b ATP hydrolysis to ADP induces a large conformational change, increasing the affinity of Hsp70 for misfolded protein. DNAJ binds to and recruits substrate to Hsp70, in addition to stimulating Hsp70 ATP hydrolysis. c Nucleotide exchange promotes the release of protein from Hsp70, and allows the cycle to occur again. NEFs promote nucleotide exchange
ATP hydrolysis is stimulated by the J-domain family of co-chaperones (Fig. 1b) [21]. Nucleotide exchange factors (NEFs), including the Hsp110 and Bcl-2-associated athanogene (BAG) families, are co-chaperones that promote the release of ADP from Hsp70 and allow for the binding of a new ATP molecule and the continuation of the cycle (Fig. 1c) [22]. The nucleotide cycle of Hsp70 is further regulated by the co-chaperone HIP (Hsp70 interacting protein). HIP binds to the NBD of Hsp70 in the ADP-bound state via a tetratricopeptide repeat (TPR) domain, and HIP binding antagonizes NEF activity (Fig. 1c). Therefore, HIP stabilizes the ADP state of Hsp70 and increases the amount of time Hsp70 stays bound to a misfolded protein [23]. Through the regulation of nucleotide cycling these co-chaperones adapt Hsp70 function to different roles in protein quality control.
Multifaceted role of Hsp70 in protein quality control
Hsp70 plays a multifaceted role in protein quality control and is adapted to different pathways through regulation by co-chaperones (Table 1).
Table 1.
Proposed effect of co-chaperones on Hsp70-facilitated protein quality of polyQ proteins
| Co-chaperone | Effect | References |
|---|---|---|
| J-domain containing protein (DNAJ) | ||
| DNAJB1 | Decreases aggregation | [77, 80, 133] |
| Targets Hsp70 substrate to the proteasome | [93, 143] | |
| DNAJB2 | Recruits substrate to Hsp70 | [51] |
| Targets Hsp70 substrate to the proteasome | [51, 52] | |
| DNAJB6 | Decreases aggregationa | [84, 85] |
| Nucleotide exchange factor (NEF) | ||
| BAG1 | Releases Hsp70 substrate to the proteasome | [53, 54] |
| BAG3 | Facilitates the degradation of Hsp70 substrate by autophagy | [72, 111] |
| Hsp110 | Decreases aggregation | [76, 77] |
| Tetratricopeptide repeat (TPR) containing proteins | ||
| CHIP | Ubiquitinates Hsp70 substrate and promotes degradation by the proteasome or autophagy | [50, 53, 150] |
| HIP | Stabilizes ADP state of Hsp70 and promotes degradation by the proteasome | [155] |
| HOP | Coordinates the interaction of Hsp70 and Hsp90 | [40] |
| Small heat shock protein | ||
| HspB8 | Facilitates the degradation of Hsp70 substrate by autophagy | [71, 75] |
aEffect may be independent of cooperation with Hsp70
Folding of newly synthesized proteins
While the Hsp70 family was first recognized for its role in stress response, the constitutive members of the family also play a critical role in protein quality control during basal conditions, including the folding of newly synthesized proteins. In vitro many proteins can fold spontaneously [24]; however, larger proteins and proteins with a high abundance of disordered regions require the assistance of chaperones to properly fold in the crowded cellular environment [25]. The constitutive form of Hsp70, Hsc70, works in concert with specialized J-domain proteins and NEFs to prevent the misfolding of newly synthesized proteins by cycling with the nascent chain as it exits the ribosome, a process known as co-translational folding [26–29]. Binding of Hsc70 stabilizes the unfolded state of the newly synthesized protein and can even reverse misfolded states [30]. Subsequent release of the stabilized protein from Hsc70 allows folding to proceed in the correct order, starting with secondary structure [31]. Under conditions of proteotoxic stress, such as proteasome inhibition, Hsc70 is sequestered from the ribosomes by a build-up of misfolded proteins, resulting in the misfolding of newly synthesized proteins and inhibiting global protein synthesis [32]. The expression of a dominant-negative Hsp70 (K71M), incapable of nucleotide cycling, or treatment with the Hsp70 inhibitors VER-155008 (VER) or Pifithrin-µ (PES) also decreases global protein synthesis [32], demonstrating the importance of Hsc70 for protein quality control under homeostatic conditions.
Maturation of Hsp90 client proteins
Along with Hsp90 itself, Hsp70 is an essential member of the multiprotein chaperone machinery that facilitates the maturation of Hsp90 client proteins. Several hundred Hsp90 client proteins have been identified, including many signaling proteins such as protein kinases and steroid receptors [10, 33]. Hsp90 interacts with client proteins at regions where hydrophobic amino acids are exposed to solvent, such as where ligand binding clefts fuse with the charged protein surface, stabilizing the cleft in an active conformation for ligand binding and stabilizing the protein [34]. For steroid receptors, such as AR, proper folding of the ligand binding cleft, insertion of steroid (Fig. 2a), translocation to the nucleus (Fig. 2b) and therefore transcriptional activity requires the Hsp90 chaperone machinery. This machinery has been studied in detail with relationship to the glucocorticoid receptor (GR), as reviewed in [35]. In the first step of this machinery’s function, Hsp70 in the ATP state [36] interacts with the closed (folded) ligand binding cleft of the steroid receptor and subsequent ATP hydrolysis (promoted by DNAJ proteins) partially unfolds the cleft, priming it for interaction with Hsp90 [37, 38]. The co-chaperone HOP (Hsp Organizing Protein) forms a chaperone machinery by facilitating the interaction of Hsp70 and Hsp90. This interaction occurs through binding of an N-terminal and central TPR domain to EEVD motifs on the C-terminus of Hsp70 and Hsp90 [39], promoting the transfer of the steroid receptor from Hsp70 to Hsp90 [40]. Subsequent ATP dependent cycling of Hsp90 leaves the cleft in an open state and binding of p23 to Hsp90 [41] stabilizes the cleft in an open conformation, allowing for the insertion of steroid [33]. The ATPase dead Hsp70 mutant K71M fails to facilitate the assembly of the multichaperone machinery with the progesterone receptor (PR) [42], and inhibition of Hsp70 by methylene blue (MB) inhibits steroid binding activity of GR, demonstrating Hsp70 is necessary for the proper function of the Hsp90 multichaperone machinery [43]. Steroid insertion leads to rapid retrograde transport of steroid receptors to the nucleus by an Hsp90-immunophilin–dynein complex and passage through nuclear pores in an importin-dependent process, as reviewed in [44]. Notably, Hsp70 also binds to the N-terminal domain of the AR, including the FQNLF motif that interacts with the ligand-binding domain of AR upon steroid binding [45]. For polyQ AR, the conformational change induced by steroid insertion causes misfolding and translocation of the misfolded protein to the nucleus, leading to the formation of nuclear aggregates. Either genetic or pharmacological inhibition of Hsp90 reduces ligand-dependent nuclear translocation of polyQ AR and decreases polyQ AR nuclear aggregates [46].
Fig. 2.

Proposed mechanism for regulation of polyQ AR protein quality control by the Hsp90/Hsp70 chaperone machinery in SBMA. Hsp90 and Hsp70, in cooperation with a multi-protein machinery including HOP, facilitate steroid insertion (a), promoting misfolding and subsequent aggregation of polyQ AR. PolyQ AR is translocated to the nucleus by an Hsp90-immunophilin-dynein complex (b), where it forms nuclear aggregates. Hsp70 facilitates the CHIP-dependent ubiquitination of poly QAR, targeting the protein for degradation by the proteasome (c); the co-chaperones DNAJB2 and BAG3 promote proteasomal degradation of ubiquitinated proteins. PolyQ AR that escapes Hsp70/CHIP-dependent ubiquitination and degradation via (c) forms small oligomers that coalesce into aggregates that are subject to disaggregation by the Hsp70-Hsp110-J-domain complex (d). The CHIP-Hsp70-BAG3-HspB8 complex targets polyQ AR to the aggresome leading to degradation of polyQ AR in the lysosome by chaperone-assisted selective autophagy (e)
Proteasomal degradation
The chaperone machinery plays a critical role in protein quality control by selectively recognizing misfolded proteins and targeting them for degradation by the ubiquitin proteasome system (UPS) [47]. Ubiquitination is a post translational modification by which an 8.6 kDa ubiquitin protein is covalently added to a protein; ubiquitination can target a protein to numerous cellular pathways including degradation by the proteasome. Hsp90 and Hsp70 play opposing roles in the triage of misfolded proteins, as reviewed in [47]. Hsp90 stabilizes proteins in a near-native state, protecting them from degradation. In contrast, if a protein is too misfolded to be stabilized by Hsp90, then Hsp70 facilitates ubiquitination by chaperone-dependent E3 ubiquitin ligases, such as CHIP (C-terminus of Hsp70 interacting protein), a TPR domain containing protein that binds the EEVD domain on the C-terminus of Hsp70 (Fig. 2c) [48, 49]. CHIP interacts with E2 ubiquitin conjugating enzymes, through a C-terminal U box domain, and ubiquitinates proteins bound to Hsp70 [50]. Numerous co-chaperones promote Hsp70-mediated proteasomal degradation of misfolded proteins. For example, the J-domain protein DNAJB2 (HSJ1) recruits misfolded proteins to Hsp70 and promotes ubiquitination by CHIP [51], then DNAJB2 targets the ubiquitinated proteins to the proteasome via ubiquitin interaction motifs (UIMs) [51, 52]. The NEF BAG1 also promotes Hsp70-mediated proteasomal degradation. BAG1 contains a ubiquitin like domain that allows it to associate with the proteasome and promote the release of Hsp70-bound ubiquitinated proteins to the proteasome [53, 54]. Hsp70-mediated ubiquitination and degradation has been shown to regulate the turnover of polyQ disease-related proteins, including AR, Htt, and ATXN3 as well as non-polyQ proteins implicated in age dependent neurodegenerative disorders such as tau and α-synuclein [10, 11].
Autophagy
Hsp70 can also target misfolded proteins to degradation through autophagy, a process by which proteins are degraded in lysosomes. Proteins destined for degradation by autophagy are delivered to the lysosome by different mechanisms. In macroautophagy, cargo is transported to lysosomes inside double membrane vesicles known as autophagosomes [55]. Chaperone-assisted selective autophagy (CASA) is a form of macroautophagy which utilizes Hsp70 to target proteins to the autophagosome. In microautophagy the lysosomal membrane invaginates and pinches off to form vesicles that internalize autophagic cargo [56]. Proteins can also enter the lysosome through a protein translocation system embedded in the lysosomal membrane in a process known as chaperone-mediated autophagy (CMA) [57]. Autophagy has been shown to play an important role in the degradation of polyQ proteins including AR, Htt, and ATXN3 [58]. Autophagy can facilitate the degradation of protein species that are not amenable to degradation by the proteasome, such as insoluble polyQ aggregates and proteolytic fragments of polyQ proteins. For example, N-terminal fragments of polyQ AR are toxic species that lack the ligand binding domain, the site of recognition by the Hsp90/Hsp70 chaperone machinery, and therefore are not readily targeted to the proteasome, being preferentially degraded by autophagy [43]. N-Terminal fragments of polyQ Htt also contribute to toxicity in HD and are preferentially degraded by autophagy [58, 59].
Chaperone-mediated autophagy (CMA)
In CMA, proteins containing a KFERQ-like pentapeptide motif, present in approximately 40% of the mammalian proteome, are targeted to degradation in the lysosome by Hsp70 [60, 61]. Hsp70 binds to the KFERQ domain and targets the protein to the lysosomal membrane, where the protein then binds to LAMP2A (lysosome associated membrane protein type 2A). The target protein is subsequently unfolded and then translocated into the lysosomal lumen [62–64] through the CMA translocon complex, which consists of a trimer of LAMP2A [65]. CMA is observed at low levels under homeostatic conditions in most cells, but upon stress, CMA activity significantly increases, highlighting the importance of this pathway as a compensatory mechanism for proteotoxic stress [62]. Upregulation of CMA activity has been observed in cellular and animal models of polyQ disease [66] and has been shown to promote the clearance of polyQ Htt [67] and polyQ Atxn7, the disease-causing protein in SCA7 [68].
Chaperone-assisted endosomal microautophagy (eMI)
Proteins containing the KFERQ-like pentapeptide motif can also be targeted by Hsc70 to the late endosome for degradation by microautophagy. In eMI proteins do not enter the late endosome through a translocon; instead, the endosomal membrane invaginates to trap cytosolic proteins for degradation [62]. Therefore, unlike CMA, which requires proteins to be unfolded before translocation, chaperone assisted eMI can facilitate the degradation of proteins that are folded, in early stages of misfolding and aggregation, or assembled into higher order aggregation complexes [69]. Hsp70 binds the pentapeptide-containing protein and recruits the protein to the endosomal membrane by binding of a unique site on the lid of Hsp70 to phosphatidylserine moieties [70]. Hsp70 and its cargo are then internalized in ESCRT-mediated microvesicles [69].
Chaperone-assisted selective autophagy (CASA)
Hsp70 also plays a role in assembly of the presentation complex responsible for selectively targeting proteins to the forming autophagosome for degradation by macroautophagy (Fig. 2e) [53]. BAG3 and HspB8 connect Hsp70 to the macroautophagy pathway [71, 72]. First, BAG3 binds Hsp70 in complex with CHIP and ubiquitinated substrate via its BAG domain and then binds the small Hsp, HspB8, through an IPV motif [73]. BAG3 then interacts with dynein to facilitate the transport of the misfolded protein containing CHIP-Hsp70-BAG3-HspB8 complex to the aggresome at the MTOC (microtubule organization center), a region highly enriched in autophagy/lysosomal pathway components. This complex then interacts with the macroautophagy receptor protein p62, which targets the ubiquitinated, misfolded protein to the autophagosome via the autophagosome membrane-associated protein LC3 [74]. The autophagosome then fuses to the lysosome where the misfolded protein is degraded. In a motoneuronal cell model of SBMA it was demonstrated that polyQ AR-induced proteotoxic stress leads to an upregulation of HspB8, promoting the clearance of cytosolic polyQ AR aggregates by autophagy [75].
Disaggregation
Hsp70 in collaboration with specialized J-domain proteins and the Hsp110 family of NEFs can disaggregate and solubilize misfolded proteins (Fig. 2d) [76, 77]. While the structure and composition of the DNAJ-Hsp70-Hsp110 disaggregase complex is still debated, the current ‘nucleation’ model suggests that complex formation is initiated by the recruitment of multiple Hsp70 s to the surface of aggregates by J-domain proteins [78]. J-domain proteins belonging to class A and B, such as DNAJB1 (HDJ1), have been implicated in the targeting of Hsp70 to aggregates [79, 80]. Both class B and A J-domain proteins have high homology to the canonical E. coli J-domain protein, DNAJ, and contain a conserved N-terminal J-domain responsible for promoting ATPase activity [17]. However, their C-terminal domains differ in structure and confer unique substrate specificity [21]. In addition to recruiting Hsp70 to the aggregate surface, J-domain proteins also stimulate ATPase activity. This allows for the binding of Hsp70 to the exposed hydrophobic residues on the ends of aggregated proteins and, through a process known as entropic pulling, liberation of polypeptides from the protein aggregate. Members of the Hsp110 family then reset the disaggregase machinery by promoting dissociation of ADP, allowing for a new round of ATP hydrolysis [81]. It appears that only members of the HSP110 family of NEFs participate in Hsp70-mediated disaggregation [80]. Ubiquilin-2 (UBQLN2) interacts with misfolded proteins liberated from aggregation, through a ubiquitin-associated domain, and targets them to the proteasome via a ubiquitin-like domain [82]. UBQLN2 has been shown to work together with the DNAJ-Hsp70-Hsp110 disaggregase complex to promote the proteasomal degradation of polyQ Htt in cellular and mouse models of HD [83]. Interestingly some J-domain proteins, including DNAJB6 and DNAJB8, have been shown to directly interact with and prevent aggregation of polyQ proteins in a mechanism independent of the DNAJ-Hsp70-Hsp110 disaggregase complex [84, 85].
When it comes to aggregation, most research has focused on Hsp70’s ability to disassemble toxic inclusions of polyQ proteins. However, it is important to keep in mind that controlled sequestration of polyQ proteins can be cytoprotective and Hsp70 also plays a regulatory role in the formation of polyQ aggregates. For example, in a mechanism overlapping with CASA, Hsp70 interacts with dynein motors, in a BAG3-dependent manner [74], to direct retrograde transport of misfolded proteins along microtubules to an aggresome, located at the MTOC [86]. Sequestration of misfolded proteins into the aggresome promotes their clearance by autophagy.
Dysregulation of protein quality control in polyglutamine disease
Dysregulation of the Hsp90/Hsp70 chaperone machinery, the ubiquitin proteasome system, and autophagy leads to diminished protein quality control and accumulation of misfolded proteins in polyQ disease [59, 87]. Disease progression correlates with a decrease in key members of the Hsp90/Hsp70 chaperone machinery in HD, SBMA, and SCA17 [88–90]. One mechanism by which polyQ proteins decrease chaperone levels is through the sequestration of chaperones and related transcription factors into aggregates. For example, levels of soluble DNAJB1 decline over time in the brains of an HD mouse model due to direct sequestration of DNAJB1 into nuclear polyQ Htt aggregates [91]. Consistent with these observations, DNAJB1 also co-localizes with aggregates in a cellular model of SCA3 [92]. In yeast, it has been demonstrated that sequestration of DNAJB1 into aggregates prevents the co-chaperone from targeting misfolded proteins to the proteasome [93]. Hsp70 levels also progressively decline in mouse models of HD. However, this decline is not due to direct sequestration of Hsp70. Instead, polyQ Htt aggregates sequester the transcription factor NF-Y leading to decreased expression of Hsp70 [94]. Sequestration of NF-Y by aggregates was also shown to decrease Hsp70 expression in a cellular model of SCA17 [95]. These findings suggest that sequestration of chaperones and the transcription factors that regulate their expression is a shared mechanism by which polyQ proteins disrupt the function of the chaperone machinery.
Accumulation and aggregation of misfolded proteins activates the HSF1-driven HSR, inducing expression of the Hsp90/Hsp70 chaperone machinery. However, in HD, the HSR is greatly diminished by polyQ Htt-dependent dysregulation of HSF1. For example, cells expressing polyQ Htt have decreased expression of HSF1 and impaired HSR [96]. Consistent with these findings, HSF1 and Hsp70 levels are reduced in the striata of HD knock-in mice compared to wildtype mice [96]. One mechanism by which polyQ Htt decreases HSF1 levels is through the upregulation of CK2α’ kinase and Fbxw7 E3 ligase levels, increasing phosphorylation and proteasomal degradation of HSF1 [14]. In contrast, other studies in both cellular and animal models of HD have not observed changes in HSF1 levels, and instead conclude that altered chromatin structure leads to decreased binding of HSF1 to heat shock response elements and impaired HSR [97, 98]. It is unclear if diminished HSR is a common mechanism amongst all polyQ diseases, but HSR impairment has been observed in other protein folding diseases such as Parkinson disease [99, 100]. Activation of HSR by HSF1 modulators has been shown to alleviate toxicity in both cellular and animal models of polyQ-disease and is therefore being investigated as a potential therapeutic strategy, as reviewed in [11]. Further work is needed to evaluate the long term consequences of pharmacological activation of HSR, as some studies have found that chronic activation of HSR can exacerbate toxicity in cellular models of protein misfolding disease including HD [101] and Alzheimer’s disease [102].
Dysregulation of the ubiquitin proteasome system is also observed in polyQ disease. The Hsp90/Hsp70 chaperone machinery selectively targets misfolded proteins for degradation by the proteasome; therefore, polyQ proteins have an accelerated turnover. This concept is highlighted by cellular experiments where proteasome inhibition causes a greater build-up of ubiquitinated proteins in cells expressing disease length versus wildtype AR and Htt [103, 104]. In polyQ disease it is thought that the increase in proteins targeted for degradation overwhelms the capacity of the proteasome, thus contributing to the accumulation of polyQ proteins [105]. Consistent with this hypothesis ubiquitinated polyQ Htt has been shown to accumulate in the brains of both HD mouse models and patients [106]. This build-up of polyQ proteins has also been shown to compete with other proteasome substrates for a limited degradation capacity. For example, expression of polyQ Htt and Atxn7 in cells has been shown to decrease the degradation of other non-polyQ proteasome substrates [107, 108]. Therefore, the cell has decreased ability to respond to proteotoxic stress [109]. PolyQ proteins can also decrease function of the ubiquitin proteasome system by decreasing the expression of key proteasome components. For example, in a mouse model of SBMA, polyQ AR diminished activity of the proteasome transcription factor NRF1 (nuclear factor erythroid 1) in skeletal muscle in a hormone- and polyQ-tract length-dependent manner, resulting in decreased degradation capacity and a build-up of dysfunctional proteasomes in mutant muscle [110]. Taken together these findings demonstrate that dysregulation of the ubiquitin proteasome system is a compounding factor in the accumulation of misfolded proteins in polyQ disease.
The proteotoxic stress induced by the accumulation of misfolded proteins has been shown to alter autophagic pathways. For example, expression of misfolded proteins or pharmacological inhibition of the proteasome causes a robust increase in HspB8 expression. In contrast, other stressors such as serum starvation and heat shock do not alter HspB8 levels, demonstrating this compensation is specific to proteasome impairment [71, 72]. Both HspB8 and BAG3, co-chaperones that facilitate CASA, are upregulated in polyQ AR knock-in mice [111]. It has been demonstrated that upregulation of HspB8 increases the clearance of cytosolic polyQ AR and reduces toxicity in a motorneuron cellular model of SBMA [75], consistent with the hypothesis that enhancement of autophagy is a beneficial compensatory mechanism to restore proteostasis. However other studies have demonstrated that activation of autophagy is in fact a maladaptive response in SBMA. For example, the activity of the transcriptional factor EB (TFEB) is upregulated in the muscle of polyQ AR knock-in mice leading to an increase in autophagic flux [111, 112]. Activation of autophagy has been shown to increase muscle atrophy in the same mouse model of SBMA [113] whereas genetic inhibition of autophagy decreased muscle atrophy and expanded lifespan [113]. In contrast to findings in SBMA, it has been demonstrated that down regulation of autophagy contributes to diminished protein quality control in HD [11, 114]. For example, transcriptional dysregulation of autophagy-related genes, including TFEB, has been observed in the brains of HD mouse models and patients [115, 116]. PolyQ Htt has also been shown to directly impair cargo recognition and engulfment of cytosoplamic cargo [117, 118].
Not all cell types expressing polyQ proteins experience a decrease in quality control. In HD, polyQ Htt is expressed ubiquitously in the brain but accumulates preferentially in neurons [119]. Interestingly, glial cells such as astrocytes do not experience the same accumulation and are therefore resistant to the toxicity associated with polyQ proteins [120]. This is thought to be related to the variable capacity of each cell type to degrade misfolded proteins, as astrocytes have been shown to degrade polyQ Htt faster than neurons [121]. What differences between neurons and astrocytes determines their capacity to degrade misfolded proteins? One explanation is differential expression of Hsp70-binding protein 1 (HspBP1) a co-chaperone known to antagonize the action of CHIP [122]. HspBP1 decreases CHIP-dependent ubiquitination of polyQ Htt and subsequent proteasomal degradation. Knockdown of HspBP1 enhances degradation of polyQ Htt in cultured neurons and reduces polyQ Htt-dependent neuropathology in a mouse model of HD [122]. These findings highlight the importance of co-chaperone composition in determining the capacity of the protein quality control system in a cell and susceptibility to polyQ protein-mediated toxicity.
Mutations in Hsp70 co-chaperones lead to neurodegenerative disease
Is the disruption of Hsp70-mediated protein quality control observed in polyQ disease a driving mechanism of toxicity? Rare familial neurodegenerative disorders caused by mutations in Hsp70 co-chaperones may shed light on this question. Numerous disease-causing mutations have been identified in genes encoding Hsp70 co-chaperones including J-domain, NEF, and HspB family members, as reviewed in [100–102]. These rare chaperonopathies can manifest with muscular and neurodegenerative phenotypes similar to those observed in polyglutamine disease, suggesting that dysfunction of the Hsp90/Hsp70 chaperone machinery may be a common underlying mechanism. For example, multiple chaperonopathies are linked to mutations in DNAJB2, a J-domain protein shown to work in concert with Hsp70 to shuttle ubiquitinated proteins to the proteasome for degradation [51, 52]. A single amino acid substitution in the J-domain of DNAJB2 causes Charcot Marie Tooth disease type 2 resulting in progressive axonal degeneration involving sensory and spinal cord motoneurons [123]. Splicing mutations in DNAJB2 that result in either decreased DNAJB2 expression or loss of the J-domain lead to distal hereditary motor neuropathies and spinal muscular atrophy, respectively [123, 124]. Mutations in another J-domain protein, DNAJB6 (MRJ), shown to potently inhibit the aggregation of misfolded proteins [125, 126], cause limb-girdle muscular dystrophy type 1 (LGMD1), a disease characterized by the accumulation of abnormal protein aggregates in skeletal muscle fibers and progressive distal greater than proximal muscle atrophy [126, 127]. When expressed in Drosophila, human mutations leading to LGMD1 result in a loss of DNAJB6-mediated anti-aggregation [128]. These studies suggest that disrupting DNAJ protein function can cause muscular and neurodegeneration through dysregulation of pro-degradative and disaggregation functions.
Mutations in co-chaperones essential to Hsp70-mediated autophagy have also been linked to chaperonopathies. Mutations in BAG3 located in the BAG domain responsible for binding Hsp70 or in an isoleucine-proline-valine motif responsible for binding HspB8 cause a dominant form of myopathy that is characterized by protein aggregation in skeletal and cardiac muscle [129]. Characterization of human mutations in vitro demonstrated mutant BAG3 was still capable of binding Hsp70, but surprisingly inhibited Hsp70 ATPase activity. Expression of these mutants in cells caused aggregation of the mutant BAG3 itself, Hsp70, and ubiquitinated Hsp70 clients and impaired CASA [130]. Mutations in HspB8 cause distal hereditary motor neuropathy type II (dHMNII), a disease characterized by neuron degeneration in the peripheral nervous system [131, 132] These HspB8 mutants show decreased interaction with BAG3 [71]. Over-expression of wild type HspB8 in cells promotes association of the autophagosome with lysosomes, whereas expression of dHMNII HspB8 mutants prevents interaction of the autophagosome and lysosome. Consistent with these findings, impaired autophagy was also observed in cells isolated from dHMNII patients [132]. These studies demonstrate disruption of a pro-autophagy Hsp70 co-chaperone leads to muscular and neuronal toxicity.
Genetic manipulation of Hsp70 and co-chaperones in polyQ disease
Numerous studies have shown that genetic manipulation of Hsp70 and associated co-chaperones can ameliorate toxicity in models of polyQ disease by decreasing aggregation or increasing degradation of polyQ proteins (reviewed in detail by Reis et al. [11] and Zarouchlioti et al. [127]). These proof-of-concept studies demonstrate that Hsp70 is an attractive therapeutic target in polyQ disease. For example, over-expression of Hsp70 reduced aggregation of polyQ AR and increased survival in cells [133]. Hsp70 over-expression also reduced aggregation and enhanced degradation of polyQ AR in mice, leading to improved motor function [134]. Additionally, Hsp70 over-expression improved motor function in a mouse model of SCA1 and ameliorated neuropathology in mouse models of both SCA1 and SCA17 [135, 136]. Hsp70 over-expression shows beneficial but less consistent results in models of HD, and most studies focus solely on correlating effects of Hsp70 on polyQ Htt aggregation to toxicity. Over-expression of the ER localized Hsc70 homolog BIP [137] and Hsc70 [138], but not Hsp70, reduced polyQ Htt aggregation in Neuro2a cells. While BIP and Hsc70 both decreased aggregation, only over-expression of BIP increased survival. In contrast, over-expression of Hsp70 in HEK293 cells had no effect on Htt aggregation but did increase cell survival [139]. Each Hsp70 family member plays a unique role in protein quality control, and these experiments highlight the need for further work to investigate if specific Hsp70 isoforms offer better therapeutic targets in different polyQ diseases. Effects of Hsp70 expression have also been mixed in mouse models of HD. In one experiment utilizing R6/2 mice, a transgenic model expressing an N-terminal fragment of the human HD gene containing ~ 150 CAG repeats, Hsp70 over-expression had no effect on polyQ Htt aggregation, but delayed weight loss [140], while in another study Hsp70 over-expression caused a delay in polyQ Htt aggregation but did not affect disease related phenotypes [91]. However, depletion of Hsp70 in R6/2 mice significantly decreased survival and worsened motor dysfunction, re-affirming the importance of Hsp70 in the protein quality control of polyQ Htt [141]. This group of studies also demonstrates that the effect of Hsp70 on polyQ Htt aggregation is not always correlated with survival. While our understanding of the role aggregates play in polyQ protein toxicity is incomplete, these studies suggest that over-expression of Hsp70 may decrease toxicity through a mechanism independent of aggregation.
The role of DNAJ proteins in polyQ diseases has been studied extensively, and family members DNAJB1, DNAJB2, and DNAJB6 have been identified as possible therapeutic targets [127]. Over-expression of DNAJB1 in Neuro2a cells decreased polyQ Htt inclusion formation, and co-expression with Hsp70 reduced toxicity [138, 142]. Similarly, DNAJB1 increased solubility and proteasomal degradation of polyQ AR in cells, and co-expression of Hsp70 further enhanced DNAJB1 action [133, 143]. Over-expression of the Drosophila DNAJB1 homolog, dHDJ1, suppressed polyQ Htt- and ATXN3-dependent toxicity [144, 145], and the effect of dHDJ1 on ATXN3-mediated toxicity was enhanced by over-expression with wildtype Hsp70 but inhibited by co-expression with a dominant negative mutant of Hsp70. DNAJB2 reduced ATXN3 levels by promoting proteasomal degradation or stabilizing ATXN3 in an Hsp70-independent manner in a cellular model of SCA3 [146]. DNAJB2 decreased polyQ AR inclusions in cells by increasing ubiquitination and proteasomal degradation of polyQ AR [52]. DNAJB2 over-expression decreased polyQ Htt aggregation in R6/2 mice and improved neurological performance [147]. These findings suggest that activation of Hsp70-facilitated disaggregation or degradation by upregulation of DNAJB1 and DNAJB2 is beneficial in polyQ disease. Similarly, DNAJB6 over-expression reduced polyQ Htt inclusion formation in the brain of R6/2 mice, and improved both neurological function and lifespan [125]. The extent to which Hsp70 is required for the therapeutic effect of DNAJB6 is uncertain, however, these results suggest decreased aggregation is beneficial in both cellular and animal models of polyQ disease.
Over-expression of co-chaperones that promote degradation of polyQ proteins also has beneficial effects. For example, over-expression of CHIP in models of SBMA decreases polyQ AR aggregation, increases polyQ AR ubiquitination, and enhances proteasomal degradation, leading to improved motor function [148, 149]. CHIP over-expression preferentially enhanced the proteasomal degradation of polyQ AR over wildtype, demonstrating the selectivity of Hsp70-CHIP-facilitated ubiquitination for misfolded proteins. CHIP over-expression also increased polyQ Htt ubiquitination in cells [150] and increased survival in both zebrafish and cellular models of HD [150, 151]. Additionally, over-expression of CHIP has been shown to increase ubiquitination and proteasomal degradation of polyQ proteins in cellular models of SCA3 and SCA1, and to decrease toxicity in a Drosophila model of SCA1 [152]. Surprisingly, CHIP over-expression readily increases the ubiquitination of both wild type and polyQ ATXN1 and ATXN3 [146, 152, 153]. These findings contrast with studies utilizing other polyQ proteins that have demonstrated Hsp70-CHIP-mediated ubiquitination preferentially targets misfolded proteins. With respect to ATXN3, these findings may reflect its role as a deubiquitinating enzyme. Wild type ATXN3 forms a complex with CHIP, regulating its activity by modulating CHIP-autoubiquitination [154]. Over-expression of the co-chaperone HIP, known to stabilize Hsp70 in the ADP state, also appears to enhance proteasomal degradation of polyQ proteins. HIP over-expression increased polyQ AR proteasomal degradation in cells and decreased toxicity in a Drosophila model of SBMA [155]. Over-expression of HspB8 in cellular models of SBMA increased autophagy of polyQ but not wild type AR [75]. Over-expression of HspB8 and BAG3 also enhanced the autophagic degradation of polyQ Htt in cellular models [156]. As a whole, this body of literature demonstrates that the development of pharmacological chaperones that enhance Hsp70 mediated degradation through the UPS or autophagy may be therapeutically beneficial in polyQ disease.
Small molecules targeting Hsp70
Numerous small molecule modulators of Hsp70 have been discovered (reviewed in detail in [157]); however, only a select few have been evaluated in cellular and animal models of polyQ disease (Table 2). Two major strategies have been evaluated thus far: activation of Hsp70-facilitated disaggregation and proteasomal degradation.
Table 2.
Small molecules targeting Hsp70 and their effects in cellular and animal models of polyQ disease
| Compound | Structure | Mechanism of action | PolyQ protein | Model system | Outcome | References |
|---|---|---|---|---|---|---|
| SW02 | 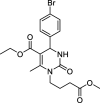 |
ATPase activator | Htt (46/103Q) | Yeast | ↑ Toxicity | [174] |
| ↑ Levels | ||||||
| ↓ Aggregation | ||||||
| PC12 cells | − Toxicity | [174] | ||||
| ↓ Aggregation | ||||||
| CE12 | 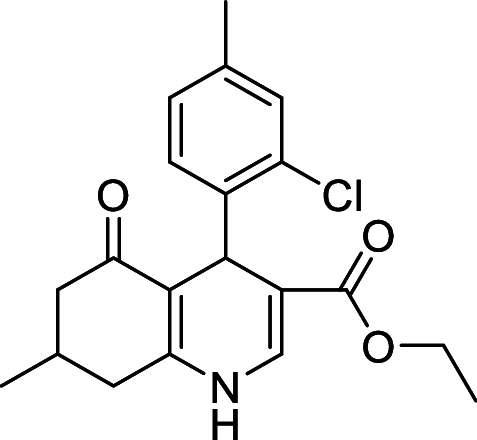 |
ATPase inhibitor | Htt (46/103Q) | Yeast | ↓ Toxicity | [174] |
| ↓ Levels | ||||||
| ↑ Aggregation | ||||||
| PC12 cells | ↓ Toxicity | [174] | ||||
| ↑ Aggregation | ||||||
| Methylene blue (MB) | 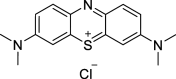 |
ATPase inhibitor | AR (112Q) | HEK293T cells | ↑ Levels | [43] |
| ↓ Degradation | ||||||
| ATXN3 (22/71Q) | HEK293T cells | ↑ Levels | [146] | |||
| Htt (103Q) | Primary mouse neurons | ↓ Toxicity | [181] | |||
| R6/2 mice | ↓ Toxicity | |||||
| Drosophila | ↓ Toxicity | |||||
| Azure C (AC) | 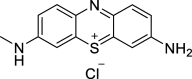 |
ATPase inhibitor | ATXN3 (Q22/71) | HEK293T Cells | ↑ Levels | [146] |
| YM-01 | 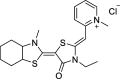 |
Stabilizes ADP state | AR (112Q) | PC12 cells | ↓ Aggregation | [155] |
| ↓ Total levels | ||||||
| ↑ Degradation | ||||||
| AR (52Q) | Drosophila | ↓ Toxicity |
Activation of disaggregase activity
The role of polyQ protein aggregation in disease pathogenesis is a subject of ongoing debate, as some evidence suggests that the formation of large aggregates protects cells from the toxic effects of soluble polyQ proteins [158]. For several polyQ diseases there is good evidence small oligomers are the toxic species [10]. It is also proposed that later stages of aggregation can contribute to toxicity through the sequestration of proteins critical to the maintenance of proteostasis [142]. Nevertheless, as discussed in the previous section, over-expression of co-chaperones that promote Hsp70’s disaggregase activity has been shown to reduce toxicity in models of polyQ disease. Therefore, one therapeutic strategy for the treatment of neurodegenerative disease is to enhance the disaggregase activity of Hsp70 [159, 160], a function that is dependent on nucleotide cycling [161]. Nadler and colleagues discovered that the immunosuppressant 15-deoxyspergualin (15-DSG), a natural product isolated from Bacillus laterosporus, binds to Hsp70 [162, 163] at the EEVD domain on the C-terminus [164]. It was later determined by Brodsky and co-workers that 15-DSG enhanced the basal rate of Hsp70 ATPase activity, but did not affect the stimulation of ATPase activity by J-domain proteins [165]. While the immunosuppressant activity of 15-DSG appeared to be due to Hsp70-dependent dysregulation of NF-κB signaling [166], numerous off target effects including binding to Hsp90 [167] made it difficult to use 15-DSG as a tool to investigate the impact of enhanced Hsp70 ATPase activity. Brodsky and co-workers utilized an in vitro assay for Hsp70 ATPase activity to study the effect of compounds structurally similar to 15-DSG. Groups lead by Brodsky and Gestwicki have discovered that certain dihydropyrimidine compounds activate Hsp70 ATPase activity while others inhibit. Further adding to the complexity, some of the compounds require the presence of co-chaperones for their action [168–171].
The dihydropyrimidine SW02 activates Hsp70 ATPase activity [171] and decreases Aβ aggregation measured by Thioflavin T in an in vitro assay for Aβ aggregation [172]. In contrast, treatment with an inhibitor of ATPase activity, the dihydropyrimidine SW08, enhances Aβ aggregation. 115-7c, a compound structurally similar to SW02, was shown to bind the NBD of Hsp70 in close proximity to the binding site of Hsp40 [173]. Treatment of yeast and PC12 cells expressing polyQ Htt with SW02 decreased aggregation of polyQ Htt, while treatment with the ATPase inhibitor CE12 increased polyQ Htt aggregation [174]. However, in the same model, SW02 enhanced and CE12 decreased polyQ related toxicity [174]. This toxicity correlated with the effect each compound had on the level of proteins, with SW02 increasing and CE12 decreasing soluble polyQ protein. Consistent with these results, treatment of HeLa cells expressing tau with the Hsp70 ATPase activators SW02 and 115-7c increased tau levels, whereas ATPase inhibitors decreased tau levels by enhancing the proteasomal degradation of tau [175]. As a whole these studies suggest that enhancing the disaggregation activity of Hsp70 leads to toxicity by increasing levels of toxic soluble polyQ species.
Enhancing proteasomal degradation
Studies with dihydropyrimidine-containing inhibitors of Hsp70 ATPase demonstrated that pharmacological enhancement of Hsp70-facilitated proteasomal degradation was feasible. Most Hsp70 ATPase inhibitors have been discovered by use of in vitro ATPase activity assays. These assays look at the ability of Hsp70 to hydrolyze nucleotide with or without the presence of J-domain and NEF co-chaperones. The benzothiazine containing compound methylene blue (MB) and its analogue azure C (AC) were discovered to inhibit ATPase activity of the Hsp70 bacterial homolog DnaK [171]. MB inhibits Hsp70 through oxidation of Cys306 (a residue not present on Hsc70), which prevents nucleotide binding [176]. These compounds were first evaluated for their effect on tau. MB and AC were shown to decrease tau levels in HeLa cells, an action that was enhanced by the over-expression of Hsp70 and decreased by inhibition of the proteasome [175], and MB reduced tau aggregation and soluble tau levels in a mouse model of human tauopathy [177]. While it has been demonstrated that MB enhances the proteasomal degradation of tau, this effect has not translated to polyQ-containing Hsp90 client proteins. For example, inhibition of Hsp70 by MB decreases proteasomal degradation of polyQ AR in HeLa cells [43]. Inhibition of Hsp70-mediated proteasomal degradation by MB also induces autophagy in HeLa cells, enhancing the autophagy of N-terminal polyQ AR fragments lacking the ligand binding domain. Additionally, MB and AC increased levels of ATXN3 in a cellular model of SCA3 [146]. MB demonstrated a clinical benefit in an extrapolated phase II clinical trial of patients with moderate and mild Alzheimer’s disease [178]. However, MB has numerous actions unrelated to Hsp70, including inhibition of tau–tau interactions [179] and induction of tau autophagy [180]. Therefore, it is difficult to determine the extent to which inhibition of Hsp70 contributed to clinical benefit. MB has also been studied in models of HD; MB reduced aggregation and prevented neurotoxicity in a Drosophila model and decreased levels of insoluble Htt in R6/2 mice [181]. In both models MB also increased levels of BDNF. BDNF levels have been shown to be protective in HD, thus it is unclear if the positive effects of MB were related to its action on Hsp70.
MKT-077 is a rhodacyanine dye derivative discovered by the Fuji-Chemical Company to have anti-cancer activity [182–185]. A lipophilic cation imparts a positive charge on MKT-077 causing the compound to be retained in cancer cell mitochondria, leading to mitochondrial dysfunction and contributing to its anti-cancer activity [184]. MKT-077 was taken into a phase I clinical trial, however it caused renal toxicity in patients and its clinical development was halted [186]. It was later discovered that MKT-077 binds to numerous species of the Hsp70 family including Hsc70 [187] and mtHsp70 [188]. In search of more clinically applicable compounds, numerous MKT-077 derivatives have been created [183, 189–192]. The MKT-077 derivative YM-01 has decreased retention in the mitochondria and increased localization to the cytosol, factors that are thought to contribute to improved potency in various cancer cells [183, 193]. MKT-077 and its derivatives bind selectively to the ADP state of Hsp70 at an allosteric site on the NBD, located within the interface of subdomains IA and IIA [192, 194, 195]. MKT-077 does not compete with nucleotide binding to Hsp70 [194, 196] and does not appear to alter basal ATPase activity [189, 197]. However, MKT-077 significantly decreases J-protein stimulated ATPase activity [189, 197].
In addition to anti-cancer activity, MKT-077 and YM-01 have also been shown to enhance the degradation of proteins implicated in neurodegenerative disease including polyQ AR and tau (reviewed in [10, 11, 47]). For example, MKT-077 and YM-01 reduced tau levels in primary neurons and brain slices [197]. YM-01 also reduced the levels of polyQ AR in cells and alleviated polyQ AR-mediated toxicity in a Drosophila model of SBMA [155]. YM-01 was shown to substitute for HIP in an in vitro assay of Hsp70-dependent activation of neuronal nitric oxide synthase [198], suggesting that, like HIP, it may stabilize the ADP-bound conformation of Hsp70. Consistent with this hypothesis, HIP competes with YM-01 for binding to Hsp70, and partial trypsin proteolysis experiments have demonstrated YM-01 promotes an ADP-like conformation of Hsp70 [155]. Notably, preliminary studies suggest YM-01 does not inhibit other functions of the Hsp90/Hsp70 chaperone machinery, including GR-Hsp90 heterocomplex assembly and steroid binding [155]. These findings suggest that stabilization of the ADP state of Hsp70 may promote the degradation of damaged client proteins without abolishing other functions of the Hsp90 and Hsp70 chaperone complex.
Conclusion
Hsp70 has been established as an attractive therapeutic target in polyQ disease, and numerous studies in both cellular and animal models demonstrate that the enhancement of Hsp70 activity by over-expression of co-chaperones can decrease polyQ protein aggregation and increase polyQ protein degradation. Therefore, small molecules that target Hsp70 are being investigated in hopes of discovering “pharmacological co-chaperones” that can enhance Hsp70’s protein quality control functions. Compounds that increase Hsp70 ATPase activity have been shown to reduce polyQ aggregation, but are also associated with increased toxicity. Compounds that inhibit Hsp70 ATPase activity have given mixed results, with some compounds increasing and others decreasing proteasomal degradation of polyQ proteins. Additionally, some inhibitors of Hsp70 ATPase activity have been shown to perturb other functions of the Hsp90/Hsp70 chaperone machinery, including the maturation of steroid receptors, and this approach has not been pursued. These findings demonstrate that a major obstacle in development of Hsp70 modulators for the treatment of polyQ disease will be designing compounds that selectively enhance Hsp70’s protein quality control functions while leaving other functions critical to cell survival untouched. Preliminary results suggest that compounds that stabilize the ADP state of Hsp70, such as YM-01, may be able to selectively promote proteasomal degradation of already unfolded proteins, including polyQ proteins, and alleviate toxicity without abolishing other Hsp70 functions.
Acknowledgements
Work in the authors’ laboratories was supported by the National Institutes of Health (NS101030, NS055746 to YO and APL; GM077430 to YO; T32-GM007767 to AKD), and the PhRMA Foundation (Predoctoral Fellowship in Pharmacology/Toxicology to AKD). The authors are all participants in The University of Michigan Medical School’s Protein Folding Diseases Initiative.
Abbreviations
- AC
Azure C
- AR
Androgen receptor
- ATXN
Ataxin
- BAG
Bcl-2 associated athanogene
- CASA
Chaperone-assisted selective autophagy
- CHIP
C-terminus of Hsp70 interacting protein
- CMA
Chaperone-mediated autophagy
- dHMNII
Distal hereditary motor neuropathy type II
- DRPLA
Dentatorubral pallidoluysian atrophy
- eMI
Endosomal microautophagy
- GR
Glucocorticoid receptor
- HD
Huntington’s disease
- HIP
Hsp70 interacting protein
- HOP
Hsp organizing protein
- HSF1
Heat shock factor 1
- Hsp
Heat shock protein
- Hsp70
Heat shock protein 70
- Hsp90
Heat chock protein 90
- HspBP1
Hsp70-binding protein 1
- HSR
Heat shock response
- Htt
Huntingtin
- LAMP2A
Lysosome associated membrane protein type 2A
- LGMD1
Limb-girdle muscular dystrophy type 1
- MB
Methylene blue
- MTOC
Microtubule organization center
- NBD
Nucleotide binding domain
- NEF
Nucleotide exchange factor
- polyQ
Polyglutamine
- PR
Progesterone receptor
- SBD
Substrate binding domain
- SBMA
Spinal bulbar muscular atrophy
- SCA
Spinocerebellar ataxia
- sHsp
Small heat shock protein
- TFEB
Transcriptional factor EB
- TPR
Tetratricopeptide repeat
- UBQLN2
Ubiquilin-2
- UPS
Ubiquitin proteasome system
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lieberman AP, Shakkottai VG, Albin RL. Polyglutamine repeats in neurodegenerative diseases. Annu Rev Pathol. 2019;14:1–27. doi: 10.1146/annurev-pathmechdis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adegbuyiro A, Sedighi F, Pilkington AW, Groover S, Legleiter J, Legleiter J. Proteins containing expanded polyglutamine tracts and neurodegenerative disease. Biochemistry. 2017;56:1199–1217. doi: 10.1021/acs.biochem.6b00936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, Hasenbank R, Bates GP, Lehrach H, Wanker EE. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington’s disease pathology. Proc Natl Acad Sci USA. 1999;96:4604–4609. doi: 10.1073/PNAS.96.8.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, Nance M, Ross CA, Scahill RI, Wetzel R, Wild EJ, Tabrizi SJ. Huntington disease. Nat Rev Dis Prim. 2015;1:15005. doi: 10.1038/nrdp.2015.5. [DOI] [PubMed] [Google Scholar]
- 5.Croce KR, Yamamoto A. A role for autophagy in Huntington’s disease. Neurobiol Dis. 2019;122:16–22. doi: 10.1016/j.nbd.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lieberman AP. Spinal and bulbar muscular atrophy. Handb Clin Neurol. 2018;2018:625–632. doi: 10.1016/B978-0-444-64076-5.00040-5. [DOI] [PubMed] [Google Scholar]
- 7.Buijsen RAM, Toonen LJA, Gardiner SL, van Roon-Mom WMC. Genetics, mechanisms, and therapeutic progress in polyglutamine spinocerebellar ataxias. Neurotherapeutics. 2019 doi: 10.1007/s13311-018-00696-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paulson HL, Shakkottai VG, Clark HB, Orr HT. Polyglutamine spinocerebellar ataxias—from genes to potential treatments. Nat Rev Neurosci. 2017;18:613–626. doi: 10.1038/nrn.2017.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Esteves S, Duarte-Silva S, Maciel P. Discovery of of therapeutic approaches for polyglutamine diseases: a summary of recent efforts. Med Res Rev. 2017;37:860–906. doi: 10.1002/med.21425. [DOI] [PubMed] [Google Scholar]
- 10.Pratt WB, Gestwicki JE, Osawa Y, Lieberman AP. Targeting Hsp90/Hsp70-based protein quality control for treatment of adult onset neurodegenerative diseases. Annu Rev Pharmacol Toxicol. 2015;55:353–371. doi: 10.1146/annurev-pharmtox-010814-124332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reis SD, Pinho BR, Oliveira JMA. Modulation of molecular chaperones in Huntington’s disease and other polyglutamine disorders. Mol Neurobiol. 2017;54:5829–5854. doi: 10.1007/s12035-016-0120-z. [DOI] [PubMed] [Google Scholar]
- 12.Ritossa F. A new puffing pattern induced by temperature shock and DNP in drosophila. Experientia. 1962;18:571–573. doi: 10.1007/BF02172188. [DOI] [Google Scholar]
- 13.Tissiéres A, Mitchell HK, Tracy UM. Protein synthesis in salivary glands of Drosophila melanogaster: relation to chromosome puffs. J Mol Biol. 1974;84:389–398. doi: 10.1016/0022-2836(74)90447-1. [DOI] [PubMed] [Google Scholar]
- 14.Jee H. Size dependent classification of heat shock proteins: a mini-review. J Exerc Rehabil. 2016;12:255–259. doi: 10.12965/jer.1632642.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richter K, Haslbeck M, Buchner J. The heat shock response: life on the verge of death. Mol Cell. 2010;40:253–266. doi: 10.1016/j.molcel.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 16.Gomez-Pastor R, Burchfiel ET, Thiele DJ. Regulation of heat shock transcription factors and their roles in physiology and disease. Nat Rev Mol Cell Biol. 2018;19:4–19. doi: 10.1038/nrm.2017.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kampinga HH, Hageman J, Vos MJ, Kubota H, Tanguay RM, Bruford EA, Cheetham ME, Chen B, Hightower LE. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones. 2009;14:105–111. doi: 10.1007/s12192-008-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rudiger S, Buchberger A, Bukau B. Interaction of Hsp70 chaperones with substrates. Nat Struct Biol. 1997;4:342–349. doi: 10.1038/nsb0597-342. [DOI] [PubMed] [Google Scholar]
- 19.Zuiderweg ER, Hightower LE, Gestwicki JE. The remarkable multivalency of the Hsp70 chaperones. Cell Stress Chaperones. 2017;22:173–189. doi: 10.1007/s12192-017-0776-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brehmer D, Rüdiger S, Gässler CS, Klostermeier D, Packschies L, Reinstein J, Mayer MP, Bukau B. Tuning of chaperone activity of Hsp70 proteins by modulation of nucleotide exchange. Nat Struct Biol. 2001;8:427–432. doi: 10.1038/87588. [DOI] [PubMed] [Google Scholar]
- 21.Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bracher A, Verghese J. The nucleotide exchange factors of Hsp70 molecular chaperones. Front Mol Biosci. 2015;2:10. doi: 10.3389/fmolb.2015.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Z, Hartl FU, Bracher A. Structure and function of Hip, an attenuator of the Hsp70 chaperone cycle. Nat Struct Mol Biol. 2013;20:929–935. doi: 10.1038/nsmb.2608. [DOI] [PubMed] [Google Scholar]
- 24.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 25.Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–579. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- 26.Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16:574–581. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- 27.Willmund F, del Alamo M, Pechmann S, Chen T, Albanèse V, Dammer EB, Peng J, Frydman J. The cotranslational function of ribosome-associated Hsp70 in eukaryotic protein homeostasis. Cell. 2013;152:196–209. doi: 10.1016/J.CELL.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nunes JM, Mayer-Hartl M, Hartl FU, Müller DJ. Action of the Hsp70 chaperone system observed with single proteins. Nat Commun. 2015;6:6307. doi: 10.1038/ncomms7307. [DOI] [PubMed] [Google Scholar]
- 29.Sharma SK, De Los Rios P, Christen P, Lustig A, Goloubinoff P. The kinetic parameters and energy cost of the Hsp70 chaperone as a polypeptide unfoldase. Nat Chem Biol. 2010;6:914–920. doi: 10.1038/nchembio.455. [DOI] [PubMed] [Google Scholar]
- 30.Balchin D, Hayer-Hartl M, Hartl F. In vivo aspects of protein folding and quality control. Science. 2016;353:aac4354. doi: 10.1126/science.aac4354. [DOI] [PubMed] [Google Scholar]
- 31.Sekhar A, Rosenzweig R, Bouvignies G, Kay LE. Hsp70 biases the folding pathways of client proteins. Proc Natl Acad Sci USA. 2016;113:E2794–E2801. doi: 10.1073/PNAS.1601846113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu B, Han Y, Qian S-B. Cotranslational response to proteotoxic stress by elongation pausing of ribosomes. Mol Cell. 2013;49:453–463. doi: 10.1016/j.molcel.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood) 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 34.Pratt WB, Morishima Y, Peng H-M, Osawa Y. Proposal for a role of the Hsp90/Hsp70-based chaperone machinery in making triage decisions when proteins undergo oxidative and toxic damage. Exp Biol Med (Maywood) 2010;235:278–289. doi: 10.1258/ebm.2009.009250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pratt WB, Morishima Y, Murphy M, Harrell M. Chaperoning of glucocorticoid receptors. In: Gaestel M, editor. Molecular chaperones in health and disease. Heidelberg: Springer; 2006. pp. 111–138. [DOI] [PubMed] [Google Scholar]
- 36.Kanelakis KC, Shewach DS, Pratt WB. Nucleotide binding states of hsp70 and hsp90 during sequential steps in the process of glucocorticoid receptor.hsp90 heterocomplex assembly. J Biol Chem. 2002;277:33698–33703. doi: 10.1074/jbc.M204164200. [DOI] [PubMed] [Google Scholar]
- 37.Hutchison KA, Dittmar KD, Czar MJ, Pratt WB. Proof that hsp70 is required for assembly of the glucocorticoid receptor into a heterocomplex with hsp90. J Biol Chem. 1994;269:5043–5049. [PubMed] [Google Scholar]
- 38.Morishima Y, Murphy PJ, Li DP, Sanchez ER, Pratt WB. Stepwise assembly of a glucocorticoid receptor.hsp90 heterocomplex resolves two sequential ATP-dependent events involving first hsp70 and then hsp90 in opening of the steroid binding pocket. J Biol Chem. 2000;275:18054–18060. doi: 10.1074/jbc.M000434200. [DOI] [PubMed] [Google Scholar]
- 39.Brinker A, Scheufler C, Von Der Mulbe F, Fleckenstein B, Herrmann C, Jung G, Moarefi I, Hartl FU. Ligand discrimination by TPR domains. Relevance and selectivity of EEVD-recognition in Hsp70-Hop-Hsp90 complexes. J Biol Chem. 2002;277:19265–19275. doi: 10.1074/jbc.M109002200. [DOI] [PubMed] [Google Scholar]
- 40.Chen S, Prapapanich V, Rimerman RA, Honoré B, Smith DF. Interactions of p60, a mediator of progesterone receptor assembly, with heat shock proteins hsp90 and hsp70. Mol Endocrinol. 1996;10:682–693. doi: 10.1210/mend.10.6.8776728. [DOI] [PubMed] [Google Scholar]
- 41.Dittmar KD, Demady DR, Stancato LF, Krishna P, Pratt WB. Folding of the glucocorticoid receptor by the heat shock protein (hsp) 90-based chaperone machinery. The role of p23 is to stabilize receptor.hsp90 heterocomplexes formed by hsp90-p60-hsp70. J Biol Chem. 1997;272:21213–21220. doi: 10.1074/JBC.272.34.21213. [DOI] [PubMed] [Google Scholar]
- 42.Cintron NS, Toft D. Defining the requirements for Hsp40 and Hsp70 in the Hsp90 chaperone pathway. J Biol Chem. 2006;281:26235–26244. doi: 10.1074/jbc.M605417200. [DOI] [PubMed] [Google Scholar]
- 43.Wang AM, Morishima Y, Clapp KM, Peng H-M, Pratt WB, Gestwicki JE, Osawa Y, Lieberman AP. Inhibition of hsp70 by methylene blue affects signaling protein function and ubiquitination and modulates polyglutamine protein degradation. J Biol Chem. 2010;285:15714–15723. doi: 10.1074/jbc.M109.098806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pratt WB, Galigniana MD, Harrell JM, DeFranco DB. Role of hsp90 and the hsp90-binding immunophilins in signalling protein movement. Cell Signal. 2004;16:857–872. doi: 10.1016/J.CELLSIG.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 45.Eftekharzadeh B, Banduseela VC, Chiesa G, Martínez-Cristóbal P, Rauch JN, Nath SR, Schwarz DMC, Shao H, Marin-Argany M, Di Sanza C, Giorgetti E, Yu Z, Pieratelli R, Felli IC, Brun-Heath I, García J, Nebreda ÁR, Gestwicki JE, Lieberman AP, Salvatella X. Hsp70 and Hsp40 inhibit an inter-domain interaction necessary for transcriptional activity in the androgen receptor. Nat Commun. 2019;10:3562. doi: 10.1038/s41467-019-11594-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thomas M, Harrell JM, Morishima Y, Peng H-M, Pratt WB, Lieberman AP. Pharmacologic and genetic inhibition of hsp90-dependent trafficking reduces aggregation and promotes degradation of the expanded glutamine androgen receptor without stress protein induction. Hum Mol Genet. 2006;15:1876–1883. doi: 10.1093/hmg/ddl110. [DOI] [PubMed] [Google Scholar]
- 47.Pratt WB, Morishima Y, Gestwicki JE, Lieberman AP, Osawa Y. A model in which heat shock protein 90 targets protein-folding clefts: rationale for a new approach to neuroprotective treatment of protein folding diseases. Exp Biol Med (Maywood) 2014;239:1405–1413. doi: 10.1177/1535370214539444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu SJ, Liu FH, Hu SM, Wang C. Different combinations of the heat-shock cognate protein 70 (hsc70) C-terminal functional groups are utilized to interact with distinct tetratricopeptide repeat-containing proteins. Biochem J. 2001;359:419–426. doi: 10.1042/bj3590419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang J, Ballinger CA, Wu Y, Dai Q, Cyr DM, Höhfeld J, Patterson C. CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J Biol Chem. 2001;276:42938–42944. doi: 10.1074/jbc.M101968200. [DOI] [PubMed] [Google Scholar]
- 50.Edkins AL. CHIP: a co-chaperone for degradation by the proteasome. Subcell Biochem. 2015;78:219–242. doi: 10.1007/978-3-319-11731-7_11. [DOI] [PubMed] [Google Scholar]
- 51.Westhoff B, Chapple JP, van der Spuy J, Höhfeld J, Cheetham ME. HSJ1 is a neuronal shuttling factor for the sorting of chaperone clients to the proteasome. Curr Biol. 2005;15:1058–1064. doi: 10.1016/J.CUB.2005.04.058. [DOI] [PubMed] [Google Scholar]
- 52.Howarth JL, Kelly S, Keasey MP, Glover C, Lee Y-B, Mitrophanous K, Chapple JP, Gallo JM, Cheetham ME, Uney JB. Hsp40 molecules that target to the ubiquitin-proteasome system decrease inclusion formation in models of polyglutamine disease. Mol Ther. 2007;15:1100–1105. doi: 10.1038/sj.mt.6300163. [DOI] [PubMed] [Google Scholar]
- 53.Fernández-Fernández MR, Gragera M, Ochoa-Ibarrola L, Quintana-Gallardo L, Valpuesta JM. Hsp70—a master regulator in protein degradation. FEBS Lett. 2017;591:2648–2660. doi: 10.1002/1873-3468.12751. [DOI] [PubMed] [Google Scholar]
- 54.Rujano MAA, Kampinga HHH, Salomons FAA. Modulation of polyglutamine inclusion formation by the Hsp70 chaperone machine. Exp Cell Res. 2007;313:3568–3578. doi: 10.1016/j.yexcr.2007.07.034. [DOI] [PubMed] [Google Scholar]
- 55.Levine B, Klionsky DJ. Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: breakthroughs in baker’s yeast fuel advances in biomedical research. Proc Natl Acad Sci USA. 2017;114:201–205. doi: 10.1073/pnas.1619876114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marzella L, Ahlberg J, Glaumann H. Autophagy, heterophagy, microautophagy and crinophagy as the means for intracellular degradation. Virchows Arch B Cell Pathol. 1981;36:219–234. doi: 10.1007/BF02912068. [DOI] [PubMed] [Google Scholar]
- 57.Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 2018;19:365–381. doi: 10.1038/s41580-018-0001-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ravikumar B, Duden R, Rubinsztein DC. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet. 2002;11:1107–1117. doi: 10.1093/hmg/11.9.1107. [DOI] [PubMed] [Google Scholar]
- 59.Labbadia J, Morimoto RI. Huntington’s disease: underlying molecular mechanisms and emerging concepts. Trends Biochem Sci. 2013;38:378–385. doi: 10.1016/j.tibs.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fred Dice J. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci. 1990;15:305–309. doi: 10.1016/0968-0004(90)90019-8. [DOI] [PubMed] [Google Scholar]
- 61.Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, Hartl FU, Moarefi I. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell. 2000;101:199–210. doi: 10.1016/S0092-8674(00)80830-2. [DOI] [PubMed] [Google Scholar]
- 62.Tekirdag K, Cuervo AM. Chaperone-mediated autophagy and endosomal microautophagy: joint by a chaperone. J Biol Chem. 2018;293:5414–5424. doi: 10.1074/jbc.R117.818237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273:501–503. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- 64.Salvador N, Aguado C, Horst M, Knecht E. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J Biol Chem. 2000;275:27447–27456. doi: 10.1074/jbc.M001394200. [DOI] [PubMed] [Google Scholar]
- 65.Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol. 2008;28:5747–5763. doi: 10.1128/MCB.02070-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koga H, Cuervo AM. Chaperone-mediated autophagy dysfunction in the pathogenesis of neurodegeneration. Neurobiol Dis. 2011;43:29–37. doi: 10.1016/J.NBD.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koga H, Martinez-Vicente M, Arias E, Kaushik S, Sulzer D, Cuervo AM. Constitutive upregulation of chaperone-mediated autophagy in Huntington’s disease. J Neurosci. 2011;31:18492–18505. doi: 10.1523/JNEUROSCI.3219-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Duncan C, Papanikolaou T, Ellerby LM. Autophagy: polyQ toxic fragment turnover. Autophagy. 2010;6:312–314. doi: 10.4161/auto.6.2.11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, Potolicchio I, Nieves E, Cuervo AM, Santambrogio L. Microautophagy of cytosolic proteins by late endosomes. Dev Cell. 2011;20:131–139. doi: 10.1016/J.DEVCEL.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morozova K, Clement CC, Kaushik S, Stiller B, Arias E, Ahmad A, Rauch JN, Chatterjee V, Melis C, Scharf B, Gestwicki JE, Cuervo A-M, Zuiderweg ER, Santambrogio L. Structural and biological interaction of hsc-70 protein with phosphatidylserine in endosomal microautophagy. J Biol Chem. 2016;291:18096–18106. doi: 10.1074/jbc.M116.736744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Crippa V, Sau D, Rusmini P, Boncoraglio A, Onesto E, Bolzoni E, Galbiati M, Fontana E, Marino M, Carra S, Bendotti C, De Biasi S, Poletti A. The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS) Hum Mol Genet. 2010;19:3440–3456. doi: 10.1093/hmg/ddq257. [DOI] [PubMed] [Google Scholar]
- 72.Crippa V, Carra S, Rusmini P, Sau D, Bolzoni E, Bendotti C, De Biasi S, Poletti A. A role of small heat shock protein B8 (HspB8) in the autophagic removal of misfolded proteins responsible for neurodegenerative diseases. Autophagy. 2010;6:958–960. doi: 10.4161/auto.6.7.13042. [DOI] [PubMed] [Google Scholar]
- 73.Rauch JN, Tse E, Freilich R, Mok S-A, Makley LN, Southworth DR, Gestwicki JE. BAG3 is a modular, scaffolding protein that physically links heat shock protein 70 (Hsp70) to the small heat shock proteins. J Mol Biol. 2017;429:128–141. doi: 10.1016/J.JMB.2016.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stürner E, Behl C. The role of the multifunctional BAG3 protein in cellular protein quality control and in disease. Front Mol Neurosci. 2017;10:177. doi: 10.3389/fnmol.2017.00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rusmini P, Crippa V, Giorgetti E, Boncoraglio A, Cristofani R, Carra S, Poletti A. Clearance of the mutant androgen receptor in motoneuronal models of spinal and bulbar muscular atrophy. Neurobiol Aging. 2013;34:2585–2603. doi: 10.1016/j.neurobiolaging.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nillegoda NB, Wentink AS, Bukau B. Protein disaggregation in multicellular organisms. Trends Biochem Sci. 2018;43:285–300. doi: 10.1016/j.tibs.2018.02.003. [DOI] [PubMed] [Google Scholar]
- 77.Kuo Y, Ren S, Lao U, Edgar BA, Wang T. Suppression of polyglutamine protein toxicity by co-expression of a heat-shock protein 40 and a heat-shock protein 110. Cell Death Dis. 2013;4:e833–e833. doi: 10.1038/cddis.2013.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nillegoda NB, Bukau B. Metazoan Hsp70-based protein disaggregases: emergence and mechanisms. Front Mol Biosci. 2015;2:57. doi: 10.3389/fmolb.2015.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nillegoda NB, Stank A, Malinverni D, Alberts N, Szlachcic A, Barducci A, De Los Rios P, Wade RC, Bukau B. Evolution of an intricate J-protein network driving protein disaggregation in eukaryotes. Elife. 2017;6:e24560. doi: 10.7554/eLife.24560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gao X, Carroni M, Nussbaum-Krammer C, Mogk A, Nillegoda N, Szlachcic A, Guilbride D, Saibil H, Mayer M, Bukau B. Human Hsp70 disaggregase reverses Parkinson’s-linked α-synuclein amyloid fibrils. Mol Cell. 2015;59:781–793. doi: 10.1016/J.MOLCEL.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rampelt H, Kirstein-Miles J, Nillegoda NB, Chi K, Scholz SR, Morimoto RI, Bukau B. Metazoan Hsp70 machines use Hsp110 to power protein disaggregation. EMBO J. 2012;31:4221–4235. doi: 10.1038/emboj.2012.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Deng H-X, Chen W, Hong S-T, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H, Jiang H, Hirano M, Rampersaud E, Jansen GH, Donkervoort S, Bigio EH, Brooks BR, Ajroud K, Sufit RL, Haines JL, Mugnaini E, Pericak-Vance MA, Siddique T. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–215. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hjerpe R, Bett JS, Keuss MJ, Solovyova A, McWilliams TG, Johnson C, Sahu I, Varghese J, Wood N, Wightman M, Osborne G, Bates GP, Glickman MH, Trost M, Knebel A, Marchesi F, Kurz T. UBQLN2 mediates autophagy-independent protein aggregate clearance by the proteasome. Cell. 2016;166:935–949. doi: 10.1016/j.cell.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hageman J, Rujano MA, van Waarde MA, Kakkar V, Dirks RP, Govorukhina N, Oosterveld-Hut HM, Lubsen NH, Kampinga HH. A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol Cell. 2010;37:355–369. doi: 10.1016/J.MOLCEL.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 85.Månsson C, Kakkar V, Monsellier E, Sourigues Y, Härmark J, Kampinga HH, Melki R, Emanuelsson C. DNAJB6 is a peptide-binding chaperone which can suppress amyloid fibrillation of polyglutamine peptides at substoichiometric molar ratios. Cell Stress Chaperones. 2014;19:227–239. doi: 10.1007/s12192-013-0448-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jia B, Wu Y, Zhou Y. 14-3-3 and aggresome formation: implications in neurodegenerative diseases. Prion. 2014;8:173–177. doi: 10.4161/PRI.28123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nath SR, Lieberman AP. The ubiquitination, disaggregation and proteasomal degradation machineries in polyglutamine disease. Front Mol Neurosci. 2017;10:78. doi: 10.3389/fnmol.2017.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tsai H-F, Lin SJ, Li C, Hsieh M. Decreased expression of Hsp27 and Hsp70 in transformed lymphoblastoid cells from patients with spinocerebellar ataxia type 7. Biochem Biophys Res Commun. 2005;334:1279–1286. doi: 10.1016/J.BBRC.2005.06.207. [DOI] [PubMed] [Google Scholar]
- 89.Yamanaka T, Miyazaki H, Oyama F, Kurosawa M, Washizu C, Doi H, Nukina N. Mutant Huntingtin reduces HSP70 expression through the sequestration of NF-Y transcription factor. EMBO J. 2008;27:827–839. doi: 10.1038/emboj.2008.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Katsuno M, Sang C, Adachi H, Minamiyama M, Waza M, Tanaka F, Doyu M, Sobue G. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc Natl Acad Sci USA. 2005;102:16801–16806. doi: 10.1073/pnas.0506249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hay DG, Sathasivam K, Tobaben S, Stahl B, Marber M, Mestril R, Mahal A, Smith DL, Woodman B, Bates GP. Progressive decrease in chaperone protein levels in a mouse model of Huntington’s disease and induction of stress proteins as a therapeutic approach. Hum Mol Genet. 2004;13:1389–1405. doi: 10.1093/hmg/ddh144. [DOI] [PubMed] [Google Scholar]
- 92.Seidel K, Meister M, Dugbartey GJ, Zijlstra MP, Vinet J, Brunt ER, van Leeuwen FW, Rüb U, Kampinga HH, den Dunnen WF. Cellular protein quality control and the evolution of aggregates in spinocerebellar ataxia type 3 (SCA3) Neuropathol Appl Neurobiol. 2012;38:548–558. doi: 10.1111/j.1365-2990.2011.01220.x. [DOI] [PubMed] [Google Scholar]
- 93.Park S-H, Kukushkin Y, Gupta R, Chen T, Konagai A, Hipp MS, Hayer-Hartl M, Hartl FU. PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell. 2013;154:134–145. doi: 10.1016/J.CELL.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 94.Merienne K, Helmlinger D, Perkin GR, Devys D, Trottier Y. Polyglutamine expansion induces a protein-damaging stress connecting heat shock protein 70 to the JNK pathway. J Biol Chem. 2003;278:16957–16967. doi: 10.1074/jbc.M212049200. [DOI] [PubMed] [Google Scholar]
- 95.Lee L-C, Chen C-M, Wang H-C, Hsieh H-H, Chiu I-S, Su M-T, Hsieh-Li H-M, Wu C-H, Lee G-C, Lee-Chen G-J, Lin J-Y. Role of the CCAAT-binding protein NFY in SCA17 pathogenesis. PLoS One. 2012;7:e35302. doi: 10.1371/journal.pone.0035302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chafekar SM, Duennwald ML. Impaired heat shock response in cells expressing full-length polyglutamine-expanded huntingtin. PLoS One. 2012;7:e37929. doi: 10.1371/journal.pone.0037929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Labbadia J, Cunliffe H, Weiss A, Katsyuba E, Sathasivam K, Seredenina T, Woodman B, Moussaoui S, Frentzel S, Luthi-Carter R, Paganetti P, Bates GP. Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease. J Clin Invest. 2011;121:3306–3319. doi: 10.1172/JCI57413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Riva L, Koeva M, Yildirim F, Pirhaji L, Dinesh D, Mazor T, Duennwald ML, Fraenkel E. Poly-glutamine expanded huntingtin dramatically alters the genome wide binding of HSF1. J Huntingtons Dis. 2012;1:33–45. doi: 10.3233/JHD-2012-120020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kim E, Wang B, Sastry N, Masliah E, Nelson PT, Cai H, Liao F-F. NEDD4-mediated HSF1 degradation underlies α-synucleinopathy. Hum Mol Genet. 2016;25:211–222. doi: 10.1093/hmg/ddv445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Huang S, Ling JJ, Yang S, Li X-J, Li S. Neuronal expression of TATA box-binding protein containing expanded polyglutamine in knock-in mice reduces chaperone protein response by impairing the function of nuclear factor-Y transcription factor. Brain. 2011;134:1943–1958. doi: 10.1093/brain/awr146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bersuker K, Hipp MS, Calamini B, Morimoto RI, Kopito RR. Heat shock response activation exacerbates inclusion body formation in a cellular model of Huntington disease. J Biol Chem. 2013;288:23633–23638. doi: 10.1074/jbc.C113.481945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Roth DM, Hutt DM, Tong J, Bouchecareilh M, Wang N, Seeley T, Dekkers JF, Beekman JM, Garza D, Drew L, Masliah E, Morimoto RI, Balch WE. Modulation of the maladaptive stress response to manage diseases of protein folding. PLoS Biol. 2014;12:e1001998. doi: 10.1371/journal.pbio.1001998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lieberman AP, Harmison G, Strand AD, Olson JM, Fischbeck KH. Altered transcriptional regulation in cells expressing the expanded polyglutamine androgen receptor. Hum Mol Genet. 2002;11:1967–1976. doi: 10.1093/hmg/11.17.1967. [DOI] [PubMed] [Google Scholar]
- 104.Li X, Wang C-E, Huang S, Xu X, Li X-J, Li H, Li S. Inhibiting the ubiquitin-proteasome system leads to preferential accumulation of toxic N-terminal mutant huntingtin fragments. Hum Mol Genet. 2010;19:2445–2455. doi: 10.1093/hmg/ddq127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hipp MS, Patel CN, Bersuker K, Riley BE, Kaiser SE, Shaler TA, Brandeis M, Kopito RR. Indirect inhibition of 26S proteasome activity in a cellular model of Huntington’s disease. J Cell Biol. 2012;196:573–587. doi: 10.1083/jcb.201110093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bennett EJ, Shaler TA, Woodman B, Ryu K-Y, Zaitseva TS, Becker CH, Bates GP, Schulman H, Kopito RR. Global changes to the ubiquitin system in Huntington’s disease. Nature. 2007;448:704–708. doi: 10.1038/nature06022. [DOI] [PubMed] [Google Scholar]
- 107.Jana NR, Zemskov EA, Wang GH, Nukina N. Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum Mol Genet. 2001;10:1049–1059. doi: 10.1093/hmg/10.10.1049. [DOI] [PubMed] [Google Scholar]
- 108.Wang H-L, He C-Y, Chou A-H, Yeh T-H, Chen Y-L, Li AH. Polyglutamine-expanded ataxin-7 decreases nuclear translocation of NF-κB p65 and impairs NF-κB activity by inhibiting proteasome activity of cerebellar neurons. Cell Signal. 2007;19:573–581. doi: 10.1016/J.CELLSIG.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 109.Ding Q, Lewis JJ, Strum KM, Dimayuga E, Bruce-Keller AJ, Dunn JC, Keller JN. Polyglutamine expansion, protein aggregation, proteasome activity, and neural survival. J Biol Chem. 2002;277:13935–13942. doi: 10.1074/jbc.M107706200. [DOI] [PubMed] [Google Scholar]
- 110.Nath SR, Yu Z, Gipson TA, Marsh GB, Yoshidome E, Robins DM, Todi SV, Housman DE, Lieberman AP. Androgen receptor polyglutamine expansion drives age-dependent quality control defects and muscle dysfunction. J Clin Invest. 2018;128:3630–3641. doi: 10.1172/JCI99042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rusmini P, Polanco MJ, Cristofani R, Cicardi ME, Meroni M, Galbiati M, Piccolella M, Messi E, Giorgetti E, Lieberman AP, Milioto C, Rocchi A, Aggarwal T, Pennuto M, Crippa V, Poletti A. Aberrant autophagic response in the muscle of a knock-in mouse model of spinal and bulbar muscular atrophy. Sci Rep. 2015;5:15174. doi: 10.1038/srep15174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chua JP, Reddy SL, Merry DE, Adachi H, Katsuno M, Sobue G, Robins DM, Lieberman AP. Transcriptional activation of TFEB/ZKSCAN3 target genes underlies enhanced autophagy in spinobulbar muscular atrophy. Hum Mol Genet. 2014;23:1376–1386. doi: 10.1093/hmg/ddt527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yu Z, Wang AM, Adachi H, Katsuno M, Sobue G, Yue Z, Robins DM, Lieberman AP. Macroautophagy is regulated by the UPR–mediator CHOP and accentuates the phenotype of SBMA mice. PLoS Genet. 2011;7:e1002321. doi: 10.1371/journal.pgen.1002321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ochaba J, Lukacsovich T, Csikos G, Zheng S, Margulis J, Salazar L, Mao K, Lau AL, Yeung SY, Humbert S, Saudou F, Klionsky DJ, Finkbeiner S, Zeitlin SO, Marsh JL, Housman DE, Thompson LM, Steffan JS. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc Natl Acad Sci USA. 2014;111:16889–16894. doi: 10.1073/pnas.1420103111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, Hollingsworth ZR, Collin F, Synek B, Holmans PA, Young AB, Wexler NS, Delorenzi M, Kooperberg C, Augood SJ, Faull RL, Olson JM, Jones L, Luthi-Carter R. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum Mol Genet. 2006;15:965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- 116.Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA, Lazarowski ER, Damian VA, Masliah E, La Spada AR. PGC-1α rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med. 2012;4:142ra97. doi: 10.1126/scitranslmed.3003799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, Cuervo AM. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci. 2010;13:567–576. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wong YC, Holzbaur EL. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci. 2014;34:1293–1305. doi: 10.1523/JNEUROSCI.1870-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Saxena S, Caroni P. Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron. 2011;71:35–48. doi: 10.1016/J.NEURON.2011.06.031. [DOI] [PubMed] [Google Scholar]
- 120.Maragakis NJ, Rothstein JD. Mechanisms of disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol. 2006;2:679–689. doi: 10.1038/ncpneuro0355. [DOI] [PubMed] [Google Scholar]
- 121.Zhao T, Hong Y, Li S, Li X-J. Compartment-dependent degradation of mutant huntingtin accounts for its preferential accumulation in neuronal processes. J Neurosci. 2016;36:8317–8328. doi: 10.1523/JNEUROSCI.0806-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhao T, Hong Y, Yin P, Li S, Li X-J. Differential HspBP1 expression accounts for the greater vulnerability of neurons than astrocytes to misfolded proteins. Proc Natl Acad Sci USA. 2017;114:E7803–E7811. doi: 10.1073/pnas.1710549114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gess B, Auer-Grumbach M, Schirmacher A, Strom T, Zitzelsberger M, Rudnik-Schoneborn S, Rohr D, Halfter H, Young P, Senderek J. HSJ1-related hereditary neuropathies: novel mutations and extended clinical spectrum. Neurology. 2014;83:1726–1732. doi: 10.1212/WNL.0000000000000966. [DOI] [PubMed] [Google Scholar]
- 124.Sanchez E, Darvish H, Mesias R, Taghavi S, Firouzabadi SG, Walker RH, Tafakhori A, Paisán-Ruiz C. Identification of a large DNAJB2 deletion in a family with spinal muscular atrophy and parkinsonism. Hum Mutat. 2016;37:1180–1189. doi: 10.1002/humu.23055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kakkar V, Månsson C, de Mattos EP, Bergink S, van der Zwaag M, van Waarde MAWH, Kloosterhuis NJ, Melki R, van Cruchten RTP, Al-Karadaghi S, Arosio P, Dobson CM, Knowles TPJ, Bates GP, van Deursen JM, Linse S, van de Sluis B, Emanuelsson C, Kampinga HH. The S/T-rich motif in the DNAJB6 chaperone delays polyglutamine aggregation and the onset of disease in a mouse model. Mol Cell. 2016;62:272–283. doi: 10.1016/J.MOLCEL.2016.03.017. [DOI] [PubMed] [Google Scholar]
- 126.Ruggieri A, Saredi S, Zanotti S, Pasanisi MB, Maggi L, Mora M. DNAJB6 myopathies: focused review on an emerging and expanding group of myopathies. Front Mol Biosci. 2016;3:63. doi: 10.3389/fmolb.2016.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zarouchlioti C, Parfitt DA, Li W, Gittings LM, Cheetham ME. DNAJ proteins in neurodegeneration: essential and protective factors. Philos Trans R Soc Lond B Biol Sci. 2018;373:20160534. doi: 10.1098/rstb.2016.0534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Li S, Zhang P, Freibaum BD, Kim NC, Kolaitis R-M, Molliex A, Kanagaraj AP, Yabe I, Tanino M, Tanaka S, Sasaki H, Ross ED, Taylor JP, Kim HJ. Genetic interaction of hnRNPA2B1 and DNAJB6 in a Drosophila model of multisystem proteinopathy. Hum Mol Genet. 2016;25:936–950. doi: 10.1093/hmg/ddv627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jaffer F, Murphy SM, Scoto M, Healy E, Rossor AM, Brandner S, Phadke R, Selcen D, Jungbluth H, Muntoni F, Reilly MM. BAG3 mutations: another cause of giant axonal neuropathy. J Peripher Nerv Syst. 2012;17:210–216. doi: 10.1111/j.1529-8027.2012.00409.x. [DOI] [PubMed] [Google Scholar]
- 130.Meister-Broekema M, Freilich R, Jagadeesan C, Rauch JN, Bengoechea R, Motley WW, Kuiper EFE, Minoia M, Furtado GV, van Waarde MAWH, Bird SJ, Rebelo A, Zuchner S, Pytel P, Scherer SS, Morelli FF, Carra S, Weihl CC, Bergink S, Gestwicki JE, Kampinga HH. Myopathy associated BAG3 mutations lead to protein aggregation by stalling Hsp70 networks. Nat Commun. 2018;9:5342. doi: 10.1038/s41467-018-07718-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kakkar V, Meister-Broekema M, Minoia M, Carra S, Kampinga HH. Barcoding heat shock proteins to human diseases: looking beyond the heat shock response. Dis Model Mech. 2014;7:421–434. doi: 10.1242/dmm.014563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kwok AS, Phadwal K, Turner BJ, Oliver PL, Raw A, Simon AK, Talbot K, Agashe VR. HspB8 mutation causing hereditary distal motor neuropathy impairs lysosomal delivery of autophagosomes. J Neurochem. 2011;119:1155–1161. doi: 10.1111/j.1471-4159.2011.07521.x. [DOI] [PubMed] [Google Scholar]
- 133.Kobayashi Y, Kume A, Li M, Doyu M, Hata M, Ohtsuka K, Sobue G. Chaperones Hsp70 and Hsp40 suppress aggregate formation and apoptosis in cultured neuronal cells expressing truncated androgen receptor protein with expanded polyglutamine tract. J Biol Chem. 2000;275:8772–8778. doi: 10.1074/JBC.275.12.8772. [DOI] [PubMed] [Google Scholar]
- 134.Adachi H, Katsuno M, Minamiyama M, Sang C, Pagoulatos G, Angelidis C, Kusakabe M, Yoshiki A, Kobayashi Y, Doyu M, Sobue G. Heat shock protein 70 chaperone overexpression ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model by reducing nuclear-localized mutant androgen receptor protein. J Neurosci. 2003;23:2203–2211. doi: 10.1523/JNEUROSCI.23-06-02203.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yang S, Huang S, Gaertig MA, Li X-J, Li S. Age-dependent decrease in chaperone activity impairs MANF expression, leading to purkinje cell degeneration in inducible SCA17 mice. Neuron. 2014;81:349–365. doi: 10.1016/J.NEURON.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cummings CJ, Sun Y, Opal P, Antalffy B, Mestril R, Orr HT, Dillmann WH, Zoghbi HY. Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum Mol Genet. 2001;10:1511–1518. doi: 10.1093/hmg/10.14.1511. [DOI] [PubMed] [Google Scholar]
- 137.Jiang Y, Lv H, Liao M, Xu X, Huang S, Tan H, Peng T, Zhang Y, Li H. GRP78 counteracts cell death and protein aggregation caused by mutant huntingtin proteins. Neurosci Lett. 2012;516:182–187. doi: 10.1016/J.NEULET.2012.03.074. [DOI] [PubMed] [Google Scholar]
- 138.Jana NR, Tanaka M, Wang GH, Nukina N. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum Mol Genet. 2000;9:2009–2018. doi: 10.1093/hmg/9.13.2009. [DOI] [PubMed] [Google Scholar]
- 139.Zhou H, Li SH, Li XJ. Chaperone suppression of cellular toxicity of huntingtin is independent of polyglutamine aggregation. J Biol Chem. 2001;276:48417–48424. doi: 10.1074/jbc.M104140200. [DOI] [PubMed] [Google Scholar]
- 140.Hansson O, Nylandsted J, Castilho RF, Leist M, Jäättelä M, Brundin P. Overexpression of heat shock protein 70 in R6/2 Huntington’s disease mice has only modest effects on disease progression. Brain Res. 2003;970:47–57. doi: 10.1016/S0006-8993(02)04275-0. [DOI] [PubMed] [Google Scholar]
- 141.Wacker JL, Huang S-Y, Steele AD, Aron R, Lotz GP, Nguyen Q, Giorgini F, Roberson ED, Lindquist S, Masliah E, Muchowski PJ. Loss of Hsp70 exacerbates pathogenesis but not levels of fibrillar aggregates in a mouse model of Huntington’s disease. J Neurosci. 2009;29:9104–9114. doi: 10.1523/JNEUROSCI.2250-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ormsby AR, Ramdzan YM, Mok Y-F, Jovanoski KD, Hatters DM. A platform to view huntingtin exon 1 aggregation flux in the cell reveals divergent influences from chaperones hsp40 and hsp70. J Biol Chem. 2013;288:37192–37203. doi: 10.1074/jbc.M113.486944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bailey CK, Andriola IF, Kampinga HH, Merry DE. Molecular chaperones enhance the degradation of expanded polyglutamine repeat androgen receptor in a cellular model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2002;11:515–523. doi: 10.1093/hmg/11.5.515. [DOI] [PubMed] [Google Scholar]
- 144.Fayazi Z, Ghosh S, Marion S, Bao X, Shero M, Kazemi-Esfarjani P. A Drosophila ortholog of the human MRJ modulates polyglutamine toxicity and aggregation. Neurobiol Dis. 2006;24:226–244. doi: 10.1016/j.nbd.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 145.Kazemi-Esfarjani P, Benzer S. Genetic suppression of polyglutamine toxicity in Drosophila. Science. 2000;287:1837–1840. doi: 10.1126/science.287.5459.1837. [DOI] [PubMed] [Google Scholar]
- 146.Gao XC, Zhou CJ, Zhou ZR, Zhang YH, Zheng XM, Song AX, Hu HY. Co-chaperone HSJ1a dually regulates the proteasomal degradation of ataxin-3. PLoS One. 2011;6:e19763. doi: 10.1371/journal.pone.0019763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Labbadia J, Novoselov SS, Bett JS, Weiss A, Paganetti P, Bates GP, Cheetham ME. Suppression of protein aggregation by chaperone modification of high molecular weight complexes. Brain. 2012;135:1180–1196. doi: 10.1093/brain/aws022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Adachi H, Waza M, Tokui K, Katsuno M, Minamiyama M, Tanaka F, Doyu M, Sobue G. CHIP overexpression reduces mutant androgen receptor protein and ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model. Neurobiol Dis. 2007;27:5115–5126. doi: 10.1523/JNEUROSCI.1242-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Morishima Y, Wang AM, Yu Z, Pratt WB, Osawa Y, Lieberman AP. CHIP deletion reveals functional redundancy of E3 ligases in promoting degradation of both signaling proteins and expanded glutamine proteins. Hum Mol Genet. 2008;17:3942–3952. doi: 10.1093/hmg/ddn296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Jana NR, Dikshit P, Goswami A, Kotliarova S, Murata S, Tanaka K, Nukina N. Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J Biol Chem. 2005;280:11635–11640. doi: 10.1074/jbc.M412042200. [DOI] [PubMed] [Google Scholar]
- 151.Miller VM, Nelson RF, Gouvion CM, Williams A, Rodriguez-Lebron E, Harper SQ, Davidson BL, Rebagliati MR, Paulson HL. CHIP suppresses polyglutamine aggregation and toxicity in vitro and in vivo. J Neurosci. 2005;25:9152–9161. doi: 10.1523/JNEUROSCI.3001-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Al-Ramahi I, Lam YC, Chen H-K, de Gouyon B, Zhang M, Pérez AM, Branco J, de Haro M, Patterson C, Zoghbi HY, Botas J. CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J Biol Chem. 2006;281:26714–26724. doi: 10.1074/jbc.M601603200. [DOI] [PubMed] [Google Scholar]
- 153.Choi JY, Ryu JH, Kim H-S, Park SG, Bae K-H, Kang S, Myung PK, Cho S, Park BC, Lee DH. Co-chaperone CHIP promotes aggregation of ataxin-1. Mol Cell Neurosci. 2007;34:69–79. doi: 10.1016/J.MCN.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 154.Durcan TM, Fon EA. Ataxin-3 and its e3 partners: implications for machado-joseph disease. Front Neurol. 2013;4:46. doi: 10.3389/fneur.2013.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wang AM, Miyata Y, Klinedinst S, Peng H-M, Chua JP, Komiyama T, Li X, Morishima Y, Merry DE, Pratt WB, Osawa Y, Collins CA, Gestwicki JE, Lieberman AP. Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat Chem Biol. 2013;9:112–118. doi: 10.1038/nchembio.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Carra S, Seguin SJ, Lambert H, Landry J. HspB8 chaperone activity toward poly(Q)-containing proteins depends on its association with Bag3, a stimulator of macroautophagy. J Biol Chem. 2008;283:1437–1444. doi: 10.1074/jbc.M706304200. [DOI] [PubMed] [Google Scholar]
- 157.Gestwicki JE, Shao H. Inhibitors and chemical probes for molecular chaperone networks. J Biol Chem. 2019;294:2151–2161. doi: 10.1074/jbc.TM118.002813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 159.Repalli J, Meruelo D. Screening strategies to identify HSP70 modulators to treat Alzheimer’s disease. Drug Des Devel Ther. 2015;9:321–331. doi: 10.2147/DDDT.S72165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Shorter J. Designer protein disaggregases to counter neurodegenerative disease. Curr Opin Genet Dev. 2017;44:1–8. doi: 10.1016/j.gde.2017.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Nadler SG, Tepper MA, Schacter B, Mazzucco CE. Interaction of the immunosuppressant deoxyspergualin with a member of the Hsp70 family of heat shock proteins. Science. 1992;258:484–486. doi: 10.1126/science.1411548. [DOI] [PubMed] [Google Scholar]
- 163.Mazzucco CE, Nadler SG. A member of the Hsp70 family of heat-shock proteins is a putative target for the immunosuppressant 15-deoxyspergualin. Ann N Y Acad Sci. 1993;685:202–204. doi: 10.1111/j.1749-6632.1993.tb35866.x. [DOI] [PubMed] [Google Scholar]
- 164.Nadler SG, Dischino DD, Malacko AR, Cleaveland JS, Fujihara SM, Marquardt H. Identification of a binding site on Hsc70 for the immunosuppressant 15-deoxyspergualin. Biochem Biophys Res Commun. 1998;253:176–180. doi: 10.1006/bbrc.1998.9775. [DOI] [PubMed] [Google Scholar]
- 165.Brodsky JL. Selectivity of the molecular chaperone-specific immunosuppressive agent 15-deoxyspergualin: modulation of HSC70 ATPase activity without compromising DnaJ chaperone interactions. Biochem Pharmacol. 1999;57:877–880. doi: 10.1016/S0006-2952(98)00376-1. [DOI] [PubMed] [Google Scholar]
- 166.Nadler SG, Eversole AC, Tepper MA, Cleaveland JS. Elucidating the mechanism of action of the immunosuppressant 15-deoxyspergualin. Ther Drug Monit. 1995;17:700–703. doi: 10.1097/00007691-199512000-00026. [DOI] [PubMed] [Google Scholar]
- 167.Nadeau K, Nadler SG, Saulnier M, Tepper MA, Walsh CT. Quantitation of the interaction of the immunosuppressant deoxyspergualin and analogs with Hsc70 and Hsp90. Biochemistry. 1994;33:2561–2567. doi: 10.1021/bi00175a027. [DOI] [PubMed] [Google Scholar]
- 168.Fewell SW, Day BW, Brodsky JL. Identification of an inhibitor of hsc70-mediated protein translocation and ATP hydrolysis. J Biol Chem. 2001;276:910–914. doi: 10.1074/jbc.M008535200. [DOI] [PubMed] [Google Scholar]
- 169.Fewell SW, Smith CM, Lyon MA, Dumitrescu TP, Wipf P, Day BW, Brodsky JL. Small molecule modulators of endogenous and co-chaperone-stimulated Hsp70 ATPase activity. J Biol Chem. 2004;279:51131–51140. doi: 10.1074/jbc.M404857200. [DOI] [PubMed] [Google Scholar]
- 170.Evans CG, Chang L, Gestwicki JE. Heat shock protein 70 (hsp70) as an emerging drug target. J Med Chem. 2010;53:4585–4602. doi: 10.1021/jm100054f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Chang L, Bertelsen EB, Wisén S, Larsen EM, Zuiderweg ER, Gestwicki JE. High-throughput screen for small molecules that modulate the ATPase activity of the molecular chaperone DnaK. Anal Biochem. 2008;372:167–176. doi: 10.1016/j.ab.2007.08.020. [DOI] [PubMed] [Google Scholar]
- 172.Evans CG, Wisén S, Gestwicki JE. Heat shock proteins 70 and 90 inhibit early stages of amyloid beta-(1-42) aggregation in vitro. J Biol Chem. 2006;281:33182–33191. doi: 10.1074/jbc.M606192200. [DOI] [PubMed] [Google Scholar]
- 173.Wisén S, Bertelsen EB, Thompson AD, Patury S, Ung P, Chang L, Evans CG, Walter GM, Wipf P, Carlson HA, Brodsky JL, Zuiderweg ER, Gestwicki JE. Binding of a small molecule at a protein-protein interface regulates the chaperone activity of Hsp70-Hsp40. ACS Chem Biol. 2010;5:611–622. doi: 10.1021/cb1000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chafekar SM, Wisén S, Thompson AD, Echeverria A, Walter GM, Evans CG, Makley LN, Gestwicki JE, Duennwald ML. Pharmacological tuning of heat shock protein 70 modulates polyglutamine toxicity and aggregation. ACS Chem Biol. 2012;7:1556–1564. doi: 10.1021/cb300166p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Jinwal UK, Miyata Y, Koren J, 3rd, Jones JR, Trotter JH, Chang L, O’leary J, Morgan D, Lee DC, Shults CL, Rousaki A, Weeber EJ, Zuiderweg ER, Gestwicki JE, Dickey CA. Chemical manipulation of hsp70 ATPase activity regulates tau stability. J Neurosci. 2009;29:12079–12088. doi: 10.1523/JNEUROSCI.3345-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Miyata Y, Rauch JN, Jinwal UK, Thompson AD, Srinivasan S, Dickey CA, Gestwicki JE. Cysteine reactivity distinguishes redox sensing by the heat-inducible and constitutive forms of heat shock protein 70. Chem Biol. 2012;19:1391–1399. doi: 10.1016/j.chembiol.2012.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.O’Leary JC, Li Q, Marinec P, Blair LJ, Congdon EE, Johnson AG, Jinwal UK, Koren J, Jones JR, Kraft C, Peters M, Abisambra JF, Duff KE, Weeber EJ, Gestwicki JE, Dickey CA. Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol Neurodegener. 2010;5:45. doi: 10.1016/j.chembiol.2012.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wischik CM, Staff RT, Wischik DJ, Bentham P, Murray AD, Storey JM, Kook KA, Harrington CR. Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate alzheimer’s disease. J Alzheimer’s Dis. 2015;44:705–720. doi: 10.3233/JAD-142874. [DOI] [PubMed] [Google Scholar]
- 179.Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci USA. 1996;93:11213–11218. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Congdon EE, Wu JW, Myeku N, Figueroa YH, Herman M, Marinec PS, Gestwicki JE, Dickey CA, Yu WH, Duff KE. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy. 2012;8:609–622. doi: 10.4161/auto.19048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sontag EM, Lotz GP, Agrawal N, Tran A, Aron R, Yang G, Necula M, Lau A, Finkbeiner S, Glabe C, Marsh JL, Muchowski PJ, Thompson LM. Methylene blue modulates huntingtin aggregation intermediates and is protective in Huntington’s disease models. J Neurosci. 2012;32:11109–11119. doi: 10.1523/JNEUROSCI.0895-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Koya K, Li Y, Wang H, Ukai T, Tatsuta N, Kawakami M, Shishido T, Chen LB. MKT-077, a novel rhodacyanine dye in clinical trials, exhibits anticarcinoma activity in preclinical studies based on selective mitochondrial accumulation. Cancer Res. 1996;56:538–543. [PubMed] [Google Scholar]
- 183.Koren J, Miyata Y, Kiray J, O’Leary JC, Nguyen L, Guo J, Blair LJ, Li X, Jinwal UK, Cheng JQ, Gestwicki JE, Dickey CA. Rhodacyanine derivative selectively targets cancer cells and overcomes tamoxifen resistance. PLoS One. 2012;7:e35566. doi: 10.1371/journal.pone.0035566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Modica-Napolitano JS, Koya K, Weisberg E, Brunelli BT, Li Y, Chen LB. Selective damage to carcinoma mitochondria by the rhodacyanine MKT-077. Cancer Res. 1996;56:544–550. [PubMed] [Google Scholar]
- 185.Chiba Y, Kubota T, Watanabe M, Otani Y, Teramoto T, Matsumoto Y, Koya K, Kitajima M. Selective antitumor activity of MKT-077, a delocalized lipophilic cation, on normal cells and cancer cells in vitro. J Surg Oncol. 1998;69:105–110. doi: 10.1002/(SICI)1096-9098(199810)69:2<105::AID-JSO11>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 186.Propper DJ, Braybrooke JP, Taylor DJ, Lodi R, Styles P, Cramer JA, Collins WC, Levitt NC, Talbot DC, Ganesan TS, Harris AL. Phase I trial of the selective mitochondrial toxin MKT077 in chemo-resistant solid tumours. Ann Oncol. 1999;10:923–927. doi: 10.1023/a:1008336904585. [DOI] [PubMed] [Google Scholar]
- 187.Tikoo A, Shakri R, Connolly L, Hirokawa Y, Shishido T, Bowers B, Ye LH, Kohama K, Simpson RJ, Maruta H. Treatment of ras-induced cancers by the F-actin-bundling drug MKT-077. Cancer J. 2000;6:162–168. [PubMed] [Google Scholar]
- 188.Wadhwa R, Sugihara T, Yoshida A, Nomura H, Reddel RR, Simpson R, Maruta H, Kaul SC. Selective toxicity of MKT-077 to cancer cells is mediated by its binding to the Hsp70 family protein mot-2 and reactivation of p53 function. Cancer Res. 2000;60:6818–6821. doi: 10.1002/(SICI)1096-9098(199810)69:23.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 189.Miyata Y, Li X, Lee H-F, Jinwal UK, Srinivasan SR, Seguin SP, Young ZT, Brodsky JL, Dickey CA, Sun D, Gestwicki JE. Synthesis and initial evaluation of YM-08, a blood-brain barrier permeable derivative of the heat shock protein 70 (Hsp70) inhibitor MKT-077, which reduces tau levels. ACS Chem Neurosci. 2013;4:930–939. doi: 10.1021/cn300210g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kawakami M, Koya K, Ukai T, Tatsuta N, Ikegawa A, Ogawa K, Shishido T, Chen LB. Synthesis and evaluation of novel rhodacyanine dyes that exhibit antitumor activity. J Med Chem. 1997;40:3151–3160. doi: 10.1021/JM9702692. [DOI] [PubMed] [Google Scholar]
- 191.Kawakami M, Koya K, Ukai T, Tatsuta N, Ikegawa A, Ogawa K, Shishido T, Chen LB. Structure-activity of novel rhodacyanine dyes as antitumor agents. J Med Chem. 1998;41:130–142. doi: 10.1021/jm970590k. [DOI] [PubMed] [Google Scholar]
- 192.Li X, Srinivasan SR, Connarn J, Ahmad A, Young ZT, Kabza AM, Zuiderweg ERP, Sun D, Gestwicki JE. Analogues of the allosteric heat shock protein 70 (Hsp70) inhibitor, MKT-077, as anti-cancer agents. ACS Med Chem Lett. 2013;4:1042–1047. doi: 10.1021/ml400204n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Colvin TA, Gabai VL, Gong J, Calderwood SK, Li H, Gummuluru S, Matchuk ON, Smirnova SG, Orlova NV, Zamulaeva IA, Garcia-Marcos M, Li X, Young ZT, Rauch JN, Gestwicki JE, Takayama S, Sherman MY. Hsp70-Bag3 interactions regulate cancer-related signaling networks. Cancer Res. 2014;74:4731–4740. doi: 10.1158/0008-5472.CAN-14-0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Rousaki A, Miyata Y, Jinwal UK, Dickey CA, Gestwicki JE, Zuiderweg ERP. Allosteric drugs: the interaction of antitumor compound MKT-077 with human Hsp70 chaperones. J Mol Biol. 2011;411:614–632. doi: 10.1016/j.jmb.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Amick J, Schlanger SE, Wachnowsky C, Moseng MA, Emerson CC, Dare M, Luo W-I, Ithychanda SS, Nix JC, Cowan JA, Page RC, Misra S. Crystal structure of the nucleotide-binding domain of mortalin, the mitochondrial Hsp70 chaperone. Protein Sci. 2014;23:833–842. doi: 10.1002/pro.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Li X, Colvin T, Rauch JN, Acosta-Alvear D, Kampmann M, Dunyak B, Hann B, Aftab BT, Murnane M, Cho M, Walter P, Weissman JS, Sherman MY, Gestwicki JE. Validation of the Hsp70-Bag3 protein-protein interaction as a potential therapeutic target in cancer. Mol Cancer Ther. 2015;14:642–648. doi: 10.1158/1535-7163.MCT-14-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Abisambra J, Jinwal UK, Miyata Y, Rogers J, Blair L, Li X, Seguin SP, Wang L, Jin Y, Bacon J, Brady S, Cockman M, Guidi C, Zhang J, Koren J, Young ZT, Atkins CA, Zhang B, Lawson LY, Weeber EJ, Brodsky JL, Gestwicki JE, Dickey CA. Allosteric heat shock protein 70 inhibitors rapidly rescue synaptic plasticity deficits by reducing aberrant tau. Biol Psychiatry. 2013;74:367–374. doi: 10.1016/j.biopsych.2013.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Morishima Y, Lau M, Peng H-M, Miyata Y, Gestwicki JE, Pratt WB, Osawa Y. Heme-dependent activation of neuronal nitric oxide synthase by cytosol is due to Hsp70-dependent, thioredoxin-mediated thiol-disulfide interchange in the heme/substrate binding cleft. Biochemistry. 2011;50:7146–7156. doi: 10.1021/bi200751t. [DOI] [PMC free article] [PubMed] [Google Scholar]