

Abstract
Bacterial microcompartments (MCPs) are protein-based organelles that consist of metabolic enzymes encapsulated within a protein shell. The function of MCPs is to optimize metabolic pathways by increasing reaction rates and sequestering toxic pathway intermediates. A substantial amount of effort has been directed toward engineering synthetic MCPs as intracellular nanoreactors for the improved production of renewable chemicals. A key challenge in this area is engineering protein shells that allow the entry of desired substrates. In this study, we used site-directed mutagenesis of the PduT shell protein to remove its central iron–sulfur cluster and create openings (pores) in the shell of the Pdu MCP that have varied chemical properties. Subsequently, in vivo and in vitro studies were used to show that PduT-C38S and PduT-C38A variants increased the diffusion of 1,2-propanediol, propionaldehyde, NAD+ and NADH across the shell of the MCP. In contrast, PduT-C38I and PduT-C38W eliminated the iron–sulfur cluster without altering the permeability of the Pdu MCP, suggesting that the side-chains of C38I and C38W occluded the opening formed by removal of the iron–sulfur cluster. Thus, genetic modification offers an approach to engineering the movement of larger molecules (such as NAD/H) across MCP shells, as well as a method for blocking transport through trimeric bacterial microcompartment (BMC) domain shell proteins.
Keywords: microcompartment, Salmonella, carboxysome, vitamin B12
Introduction
A great deal of effort is being focused on engineering synthetic metabolic pathways for the rapid high-yield production of diverse chemicals and pharmaceuticals. However, efficient product formation will be difficult when pathways have slow reaction rates, volatile or toxic intermediates and/or unfavourable interactions with the cellular milieu [1–4]. In nature, cells mitigate these problems by co-localizing metabolic pathways in multi-protein complexes or by confining them within compartments [4–8]. Hence, there is considerable interest in engineering spatially organized enzyme complexes to improve the efficiency and economy of renewable chemicals production. Within this field, a promising line of research aims to repurpose protein-based organelles known as bacterial microcompartments (MCPs) for the improved production of renewable chemicals by pathway compartmentalization [5, 8–10].
Bacterial MCPs are widespread protein-based organelles whose natural function is to optimize metabolic pathways by compartmentalization [11–14]. MCPs consist of metabolic enzymes encapsulated within a protein shell. They are typically 100–150 nm in diameter and are built from thousands of protein subunits of 10–20 different types [11–13, 15]. MCPs increase reactions rates by creating high local concentrations of enzymes and substrates, confine pathway intermediates that are toxic or rapidly excreted from the cell, and enable the use of private cofactor pools [11–13, 15, 16]. MCPs also have substantial diversity. Based on bioinformatics analyses, MCPs are produced by about 20 % of bacteria distributed across 23 phyla [12–14, 17–21] and are involved in 10 or more metabolic processes, ranging from carbon dioxide fixation to the catabolism of 1,2-propanediol (1,2-PD), ethanolamine, choline, glycerol, rhamnose, fucose and fucoidan [22–32]. Furthermore, because MCPs are made completely of protein subunits, they are amenable to genetic modification [5, 8–10].
Considerable progress has been made in engineering MCPs to contain a desired protein cargo. Empty MCP shells have been produced in Escherichia coli , Corynebacterium and Bacillus [21, 33–36]. Heterologous proteins have been encapsulated within MCP shells using short targeting peptides fused to their N-termini [35–41]. A number of MCP targeting sequences have been identified that may facilitate the encapsulation of multiple enzymes at desired stoichiometries [40, 42]. Targeting systems have also been designed de novo [43–46], and in a few cases the interactions between targeting sequences and shell proteins that are thought to mediate enzyme encapsulation have been investigated [39, 40, 47]. In addition, encapsulation of heterologous proteins within MCPs has been monitored in vivo by protease protection using C-terminal SsrA proteolysis tags [48], and several proof-of-concept synthetic nanobioreactors have been engineered using MCP technology [39, 40, 49].
An important area where more work is needed on MCP-based nanoreactors is the development of methods to control the permeability properties of the MCP shells. The ability of MCPs to enhance reaction rates and sequester problematic metabolites depends on a selectively permeable protein shell that allows the entry of substrates into the MCP while restricting the outward diffusion of pathway intermediates [50–52]. Hence, engineering optimal synthetic MCP-based nanoreactors will likely require the development of methods to control the permeability properties of MCP shells.
The shells of MCPs are built primarily from a family of small proteins known as bacterial microcompartment (BMC) domain proteins, most of which are hexamers or pseudohexameric trimers (Fig. 1) [53–55]. The hexameric BMC domain proteins have small central pores that are thought to act as conduits for MCP substrates and perhaps also MCP products [51–53, 56, 57]. For example, the central pore of the PduA shell protein allows the selective uptake of substrate (1,2-PD) into the Pdu MCP [51, 52]. A widespread type of trimeric BMC domain protein is thought to have a centrally located allosteric gate that opens to form a larger pore that allows the entry of enzymatic cofactors while maintaining the confinement of smaller pathway intermediates [58–61]. MCP shells also typically contain several divergent types of BMC domain proteins presumed to have specialized functions, but their specific roles are currently unknown [19, 20, 54, 62].
Fig. 1.
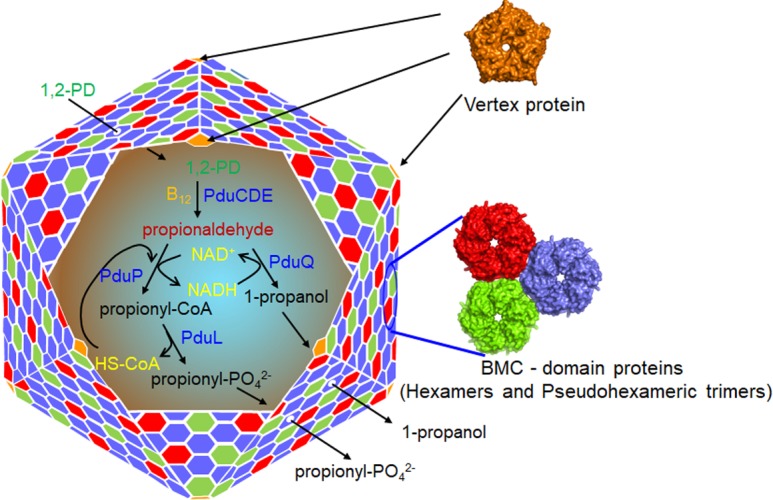
Model for the 1,2-propanediol utilization microcompartment (MCP). The Pdu MCP consists of a protein shell composed of a few thousand proteins that encapsulate a series of enzymes for metabolizing 1,2-propanediol (1,2-PD). A primary function of the Pdu MCP is to sequester the toxic pathway intermediate propionaldehyde. It is also thought to increase reaction rates by concentrating substrates together with enzymes. The function of the Pdu MCP depends on a selectively permeable protein shell that allows the entry of substrates into the MCP while restricting the outward diffusion of pathway intermediates. The central pores of the BMC domain proteins (the major components of the shell) are thought to control shell permeability.
Although some of the basic principles of molecular transport across MCP shells have been determined [20, 51, 53, 57, 63, 64], only a few studies have engineered new properties into MCP shells. Prior work has shown that chimeric shells can be built by using BMC domain hexamers originating from different MCPs [65, 66]. This suggests that the permeability properties of MCPs can be modified by taking advantage of the natural variation in MCP shell proteins that evolved to transport varied substrates. Other studies have used site-directed mutagenesis of the pore region of the PduA hexamer to alter the permeability of the Pdu MCP to 1,2-PD and propionaldehyde [51, 66]. In more recent work, a [4Fe–4S] cluster was engineered into a BMC domain protein that might have application to electron transfer across the MCP shells [67]. However, further work is needed to enable the construction of synthetic MCP shells with desired properties.
In this report, we explore the possibility of engineering the PduT shell protein to modify the permeability properties of the Pdu MCP (Fig. 1). The Pdu MCP is the most advanced MCP system with regard to engineering pathway compartmentalization [8, 10]. The natural function of the Pdu MCP is to enhance the catabolism of 1,2-PD by Salmonella (and other bacteria) while sequestering a toxic metabolic intermediate (propionaldehyde) [23, 68, 69]. PduT is a specialized trimeric BMC domain shell protein that contains a central Fe–S cluster of unknown function [57, 70, 71]. PduT is a minor component of the shell of the Pdu MCP (estimated at 3.2% of the total shell protein), and it is not required for MCP function under standard laboratory conditions [72]. Guided by structural modelling, site-directed mutagenesis was used to remove the central Fe–S cluster of PduT and create pores with different sizes and chemical properties. This approach allowed us to construct pores that were substantially larger (area: ~46 Å2) than those of previously engineered MCP shell proteins such as PduA (area~24 Å2) [51, 73]. We then used in vivo and in vitro studies to evaluate the effects of these larger engineered pores on the permeability of the Pdu MCP.
Methods
Chemicals and reagents
Antibiotics, vitamin B12 (CN-B12), NAD+, NADH and alcohol dehydrogenase (from Saccharomyces cerevisiae) were from Sigma-Aldrich (St Louis, MO, USA). Coenzyme A and dithiothreitol (DTT) were from MP Biomedicals (Santa Ana, CA, USA). KOD DNA polymerase, restriction enzymes and T4 ligase were from Novagen (Cambridge, MA, USA) and New England Biolabs (Beverly, MA, USA), respectively. Isopropyl-β-D-1-thiogalactopyranoside (IPTG) was from Diagnostic Chemicals Ltd (Charlotteville, PEI, Canada). Choice Taq Blue Mastermix was from Denville Scientific (South Plainfield, NJ, USA). The other chemicals were from Fisher Scientific (Pittsburgh, PA, USA).
Bacterial strains, media and growth conditions
The bacterial strains used in this study are listed in Table S1 (available in the online version of this article). All strains are derivatives of Salmonella enterica serovar Typhimurium strain LT2. The rich medium used was lysogeny broth (LB) (Luria–Bertani/Lennox, Difco, Detroit, MI, USA) [74]. Tryptone yeast extract (TYE) medium was used for the selection of transformants. The minimal medium used was no carbon-E (NCE) medium supplemented with 1 mM MgSO4, 0.3 mM each of valine, isoleucine, leucine and threonine, and 50 µM ferric citrate [72, 75]. Growth studies were performed at either limiting (25 nM) or saturating (100 nM) CN-B12 concentrations, as previously described using a Synergy HT Microplate reader (BioTek, Winooski, VT) [72, 76].
Three-dimensional model building and visualization
The three-dimensional structures of PduT mutants were modelled using the Swiss Model server [77] with PDBID 3N79 as a template and all the visualization and graphical presentations were carried out in Pymol [78].
Construction of chromosomal mutations
Chromosomal deletions of pduA, pduT and pduQ had been constructed previously by linear recombination of PCR products [79]. Chromosmal point mutants were constructed by sac-cat recombineering as described elsewhere [51, 79, 80]. All of the mutations were confirmed by DNA sequencing.
P22 transduction
In order to make the required double mutants either a ΔpduT::kanR marker or a sacB-cat cassette (from the PduT pore region) was moved to a pduA-S40L mutant by transduction with phage P22 HT105/1 int-210 [81]. Transductants were tested for phage contamination and sensitivity by streaking on green plates against P22 H5 (81). The kan cassette was removed by expressing flp recombinase from pCP20 [79], whereas the sacB-cat cassette was eliminated by recombination with single-stranded oligonucleotides with the desired mutations at the PduT pore [79, 82]. All mutations were confirmed by PCR followed by DNA sequencing.
MCP purification, Western blotting and diol dehydratase (DDH) assays
Pdu MCPs were purified according to a published protocol [51, 83]. Their protein content was examined by SDS PAGE (4–12 % NuPAGE Bis-Tris gels) (Invitrogen, Carlsbad, CA, USA). For Western blotting, proteins were transferred to nitrocellulose membranes and probed with commercially prepared primary rabbit antisera (GenScript, Piscataway, NJ, USA) against a PduT peptide epitope (CPRPHEAMWRQMVEG) that had been diluted 1 : 1000 in TBST buffer (50 mM Tris-HCl, 150 mM NaCl, 0.05 % Tween 20, pH 7.4). The secondary antibody used was goat anti-rabbit IgG conjugated with horseradish peroxidase at 1 : 3000 dilution in the same buffer (Biorad, Hercules, CA, USA). Colour development was carried out using the Opti-4CN substrate kit (Biorad, Hercules, CA, USA) according to the manufacturer’s instructions. DDH activity was measured using a coupled NADH-dependent alcohol dehydrogenase assay as described elsewhere [51, 84]. For some studies, purified MCPs were broken by overnight dialysis against a buffer containing 50 mM Tris (pH 8.0), 50 mM KCl and 5 mM MgCl2, followed by sonication as described elsewhere [41].
Electron microscopy
Purified MCPs were negatively stained with uranyl acetate (2 %) and visualized using a transmission electron microscope (JEOL 2100, Peabody, MA, USA) as described earlier [51, 83].
Determination of propionaldehyde in culture media
An overnight LB culture was harvested by centrifugation and resuspended in NCE minimal medium. The resuspended cells were used to inoculate 50 ml of NCE minimal medium supplemented with 0.4 % 1,2-PD and 150 nM CN-B12 to a final optical density of 0.1 at 600 nm. Cultures were grown in 250 ml Erlenmeyer flasks at 37 °C with continuous shaking at 275 r.p.m. [72]. Samples were taken at timed intervals. Cells were removed by centrifugation followed by filtration using 0.22 µm Millex-GV syringe filters (Millipore, Darmstadt, Germany). Propionaldehyde was determined by high-performance liquid chromatography (HPLC) using a Bio-Rad Aminex HPX-87H (300 by 7.8 mm) column eluted isocratically with 5 mM H2SO4 as described elsewhere [69].
Determination of cofactor transport
A kanR marker was introduced at the pduQ locus of each of the PduT pore mutants and ΔpduT::frt mutant individually by linear transformation of PCR products as described elsewhere [79]. The kan marker was removed by expressing the flp recombinase from pCP20 plasmid [79]. However, the kan cassette was kept intact in a ΔpduT::frt / ΔpduQ::kan mutant in order to avoid possible deletion of intermediate gene(s) by flippase activity. Growth studies were performed using a Synergy HT Microplate reader (BioTek, Winooski, VT, USA) as described elsewhere [85].
Results
Modelling the PduT pore
The main goal of this study was to determine whether a trimeric BMC domain protein (the PduT shell protein) could be engineered to alter the permeability properties of the Pdu MCP. The rationale for using a trimeric BMC domain protein was that it allowed us to engineer substantially larger pores (~46 Å2) compared to those examined in earlier studies with hexameric shell proteins (pore diameter and area of hexameric PduA: ~5.6 Å and ~24 Å2, respectively) [51, 73]. Prior crystallographic studies showed that a PduT-C38S mutation eliminated the central Fe–S cluster of PduT and created a relatively large pore with an area of ~45.9 Å2 [57]. Structural modelling conducted for this study indicated that various substitutions of residue 38 of PduT could be used to create pores with varied sizes and chemical properties (and remove the Fe–S cluster). Various PduT residue 38 changes were threaded onto the crystal structure of PduT-C38S (PDB 3N79) using the Swiss Model Server [77]. The modelled structures showed that PduT-C38I had the smallest pore area (~3.4 Å2), PduT-C38W had an intermediate pore area (~17.4 Å2) and PduT-C38A had a similar pore area (~45.1 Å2) to that of PduT-C38S (Fig. 2). Additionally, the polarity of the pore region varied with the side-chain of residue 38, since this residue forms the constriction point of the pore. C38S increased the polarity at the pore surface, whereas C38I and C38W had pores with decreased polarity (Fig. S1). This is of interest because prior studies indicated that the electrostatic properties of the smaller pores in hexameric shell proteins influence molecular transport across the MCP shell [51, 52]. Thus, modelling suggested that site-directed mutagenesis could be used to engineer PduT proteins with pores of various sizes and chemical properties.
Fig. 2.
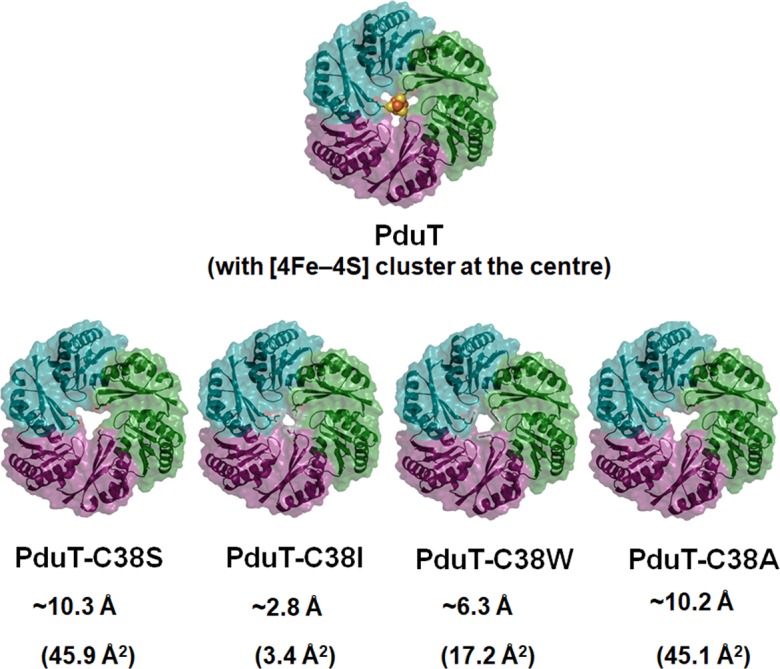
Model structures of PduT pore mutants. For each mutant, the pore diameter is given in Å and the pore area is given in Å2.
Construction and evaluation of pduT mutants
Given the modelling studies described above, site-directed mutagenesis of the Salmonella chromosome was used to construct strains that produce PduT-C38S, -C38A, -C38I and -C38W mutants. To test whether these PduT variants had any effects on MCP assembly, MCPs were purified from each variant and analysed. SDS-PAGE indicated that the MCPs purified from each mutant had a similar protein composition to the wild-type (Fig. S2a). Western blots indicated that each PduT variant was normally incorporated into the Pdu MCP (Fig. S2b), which indicated normal expression and folding. A ΔpduT mutant established that the antibody used was specific for the PduT protein (Fig S2a). Electron microscopy showed that each pduT mutant formed MCPs with a similar size and shape to the wild-type (Fig. S3). We also found that the % yield of purified MCPs from each mutant was similar to that of the wild-type (~97 % compared to wild-type for PduT-C38S, -C38A and -C38I and ~94 % for PduT-C38W). This indicated that the pduT mutants formed MCPs with similar stability to the wild-type during purification [15]. Thus, overall, the results suggested that PduT variants -C38S, -C38A, -C38I and -C38W were efficiently incorporated into the Pdu MCP and did not adversely affect MCP assembly, stability or composition.
Effects of engineered PduT proteins on the diffusion of 1,2-PD across the shell of the Pdu MCP
In Salmonella , the enzymes used for 1,2-PD degradation are encapsulated within the protein shell of the Pdu MCP (Fig. 1). Prior studies indicated that growth of Salmonella on 1,2-PD minimal medium is limited by the diffusion of 1,2-PD across the shell of the Pdu MCP, and that mutations that increase the permeability of the shell to 1,2-PD increase the growth rate [51, 72]. Therefore, growth tests were used to assess the effects of PduT-C38S, -C38A, -C38I and -C38W on the permeability of the Pdu MCP to 1,2-PD. All PduT residue 38 mutants and a pduT deletion grew similarly to the wild-type on 1,2-PD minimal medium (Fig. S4, Table S2). This indicated that a pduT deletion and the pduT variants with engineered pores did not significantly affect the diffusion of 1,2-PD into the Pdu MCP in an otherwise wild-type background.
As a second test of whether the engineered PduT pores altered the diffusion of 1,2-PD across the shell of the Pdu MCP, we measured the coenzyme B12-dependent DDH activity in purified MCPs [51]. DDH is an MCP lumen enzyme that catalyzes the first step of 1,2-PD degradation (the conversion of 1,2-PD to propionaldehyde) and its enzymatic activity is limited by the diffusion of 1,2-PD across the MCP shell [51]. The DDH activities of MCPs purified from of all pduT mutants were similar to those of the wild-type within experimental error (Table S3). Similar results were obtained for a pduT deletion mutant. These results were consistent with the growth studies described above and supported the interpretation that a pduT deletion mutant and the PduT variants tested (-C38S, -C38A, -C38I and -C38W) did not substantially increase the diffusion of 1,2-PD across the shell of the Pdu MCP under the conditions used.
PduT-C38S and PduT-C38A increase the rate of 1,2-PD diffusion in PduA-S40L Mutant
Next, we tested the effects of the PduT variants in a genetic background that included PduA-S40L. Prior studies showed that PduA-S40L impedes 1,2-PD diffusion across the shell of the Pdu MCP by obstructing the central pore of the PduA protein [51]; hence, we reasoned that the effects of the PduT variants on 1,2-PD transport might be more readily observed in a PduA-S40L background.
In contrast to a PduA-S40L strain, which grows slowly on 1,2-PD minimal medium due to restricted 1,2-PD transport, a PduA-S40L/PduT-C38S double mutant grew similarly to the wild-type (Fig. 3, Table 1). Likewise, PduT-C38A also corrected the growth defect of the PduA-S40L mutant (Fig. 3, Table 1). These results indicated that PduT-C38S and PduT-C38A increased the movement of 1,2-PD across the MCP shell when the diffusion of 1,2-PD through the central pore of PduA is restricted by the introduction of S40L. These findings are consistent with the modelling studies that indicated PduT-C38S and -C38A would have relatively large central pores. In contrast, the PduA-S40L/PduT-C38I and PduA-S40L/PduT-C38W double mutants grew similarly to the PduA-S40L mutant (Fig. 3, Table 1). These results suggest that PduT-C38I and -C38W (which were predicted by modelling to form small hydrophobic pores) do not mediate the transport of 1,2-PD to an extent measurable with the growth tests used here.
Fig. 3.
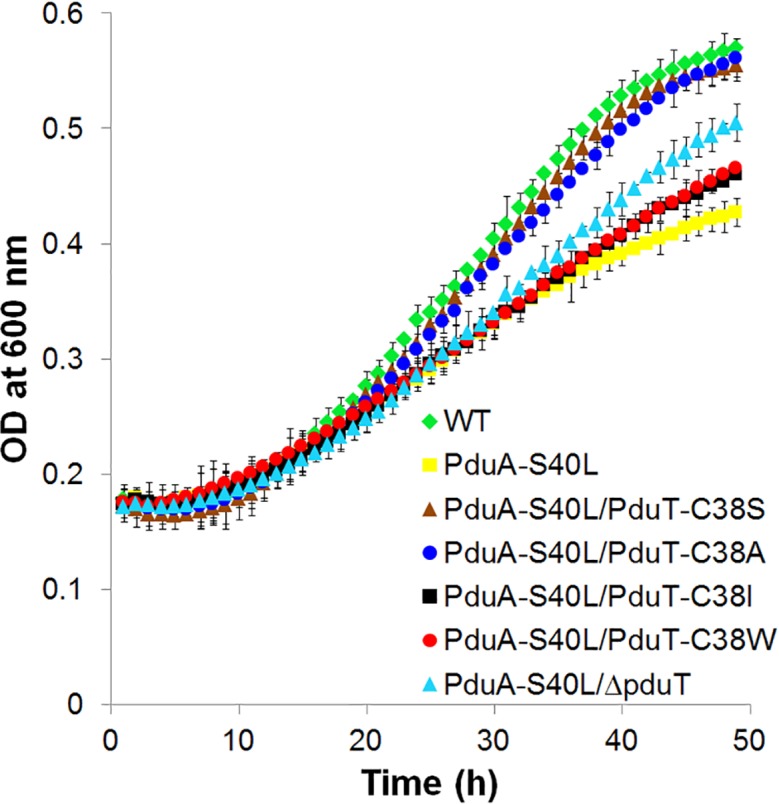
Effects of pduT mutations on the growth of a Salmonella PduA-S40L mutant on 1,2-propanediol minimal medium. All strains were grown on 1,2-PD minimal medium supplemented with limiting B12 (25 nM) as described in the Methods section. Growth assays were performed three or more times with similar results in a microplate reader. The error bars represent one standard deviation and are based on three biological replicates.
Table 1.
Growth of Salmonella PduT variants on 1,2-PD minimal medium
|
Strains |
Doubling time (h) |
|---|---|
|
WT |
16.4±1.1* |
|
PduA-S40L |
23.3±1.2† |
|
PduA-S40L/PduT-C38S |
17.1±0.4 |
|
PduA-S40L/PduT-C38A |
18.5±0.7 |
|
PduA-S40L/PduT-C38I |
22.9±0.8† |
|
PduA-S40L/PduT-C38W |
22.7±0.8† |
|
PduA-S40L/ΔpduT::frt |
20.4±0.8† |
*Growth assays were performed on 1,2-PD minimal medium with limiting B12 (25 nM). Doubling times were calculated from at least three biological replicates measured in triplicate. The error estimate shown is ±one standard deviation.
†P-value<0.01 compared to wild-type (WT) as determined by two-tailed Student’s t-test. The growth rates of PduA-S40L/PduT-C38S and PduA-S40L/PduT-C38A are not significantly different from those of the WT.
To further examine the effects of PduT variants on 1,2-PD transport in a PduA-S40L background, we measured the DDH activity of purified MCPs. Previous studies showed that MCPs purified from a PduA-S40L mutant exhibited lower DDH activity than the wild-type due to impaired diffusion of 1,2-PD across the MCP shell [51]. Therefore, mutations that increase the permeability of the Pdu MCP to 1,2-PD should increase the DDH activity of MCPs purified from the PduA-S40L mutant. Enzyme assays showed that MCPs purified from the PduA-S40L/PduT-C38S and the PduA-S40L/PduT-C38A double mutants had DDH activities that were similar to those of the wild-type and almost twofold higher than those of the PduA-S40L mutant (Table 2). This indicated that the PduT-C38S and -C38A mutants increased the diffusion of 1,2-PD across the shell of the Pdu MCP to an extent that allowed restoration of wild-type levels of DDH activity in the PduA-S40L background. We note that the PduT-C38S and -C38A corrected the PduA S40L phenotype, even though PduT is a minor shell protein (~3.2 % of the total shell protein) and PduA is a major shell protein (~15 % of the total shell protein). This suggests that the larger pore size of these PduT variants has substantial effects on 1,2-PD diffusion across the MCP shell. On the other hand, MCPs purified from the double mutants PduA-S40L/PduT-C38I, PduA-S40L/PduT-C38W and PduA-S40L/ΔpduT had similar DDH activities to the MCPs from the PduA-S40L mutant, indicating that these PduT variants did not increase 1,2-PD diffusion across the MCP shell in a PduA-S40L genetic background (Table 2).
Table 2.
Diol dehydratase activities of MCPs purified from pduT mutants
|
MCPs from Pdu mutants |
Specific activity (μmol/min/mg) |
|---|---|
|
WT |
27.8±1.2* |
|
PduA-S40L |
14.4±0.8† |
|
PduA-S40L/PduT-C38S |
27.4±0.8 |
|
PduA-S40L/PduT-C38A |
25.5±0.7 |
|
PduA-S40L/PduT-C38I |
13.05±0.5† |
|
PduA-S40L/PduT-C38W |
16.9±0.8† |
|
PduA-S40L/ΔpduT::frt |
16.1±0.4† |
|
WT (broken)‡ |
35.1±0.8† |
|
PduA-S40L (broken) |
33.8±1.1† |
|
ΔpduT::frt (broken) |
34.1±0.8† |
|
PduT-C38I (broken) |
34.6±0.6† |
|
PduA-S40L/PduT-C38I (broken) |
36.1±1.2† |
|
PduT-C38W (broken) |
34.3±0.7† |
|
PduA-S40L/PduT-C38W (broken) |
33.5±1.1† |
*Enzyme activities are based on at least three independent replicates. The error estimate shown is ±one standard deviation.
†P-value<0.001 compared to wild-type (WT) as determined by two-tailed Student’s t-test. Diol dehydratase (DDH) activities of PduA-S40L/PduT-C38S and PduA-S40L/PduT-C38A are not significantly different from the WT.
‡MCPs were broken by dialysis and sonication.
As a control, to test for normal recruitment of DDH to the MCP, the purified MCPs were disrupted by dialysis and sonication and reassayed for DDH activity. The broken MCPs from all mutants had similar DDH activities to broken MCPs from the wild-type (Table 2). This indicated normal DDH recruitment to Pdu MCP in the mutants tested. In all cases, the DDH activities were higher in broken MCPs due to increased access of DDH to its substrate (1,2-PD), as shown previously [51, 73].
MCPs with engineered PduT pores still confine toxic propionaldehyde
Next, we tested the effects of PduT pore variants on the efflux of propionaldehyde from the Pdu MCP. HPLC was used to measure the amount of propionaldehyde that diffused out of the MCP, through the bacterial cell membrane, and into the culture medium during the growth of Salmonella on 1,2-PD, as described elsewhere [51, 69, 72]. In this test, the PduT-C38A and PduT-C38S variants excreted slightly more propionaldehyde into the culture medium (about 2.5 mM) than the wild-type (about 2 mM) (Fig. 4). The PduT-C38I excreted similar amounts of propionaldehyde to the wild-type (Fig. 4). None of the mutants liberated propionaldehyde at toxic levels (5–20 mM), as was seen earlier in the case of ΔpduA and ΔpduBB′ mutants [72].
Fig. 4.
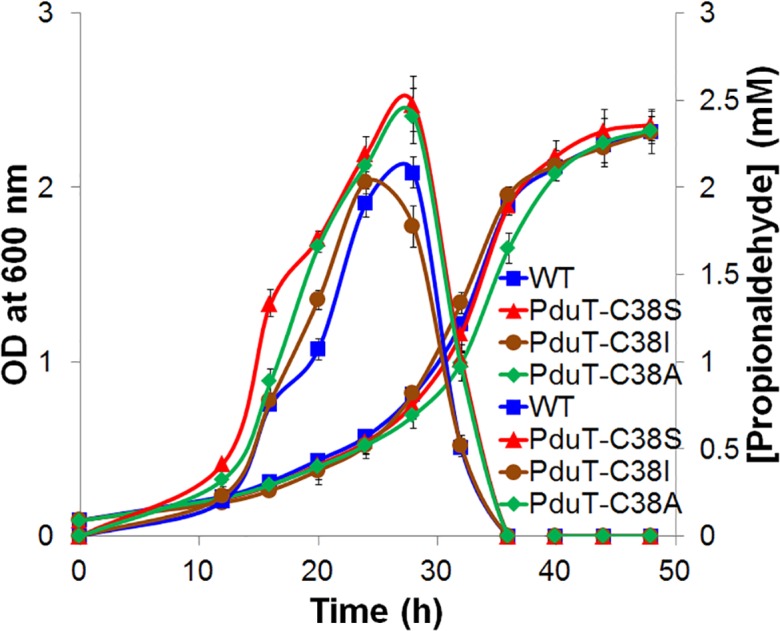
Propionaldehyde release by selected PduT pore mutants. Growth and propionaldehyde production by wild-type (WT) Salmonella and various PduT pore mutants during growth on 1,2-PD minimal medium at a saturating B12 concentration (150 nM). Propionaldehyde released into the growth medium was measured by HPLC. Prior studies indicated that a high level of B12 assists with greater production of propionaldehyde, which can readily be measured by HPLC [51, 69, 72]. The error bars represent one standard deviation and are based on three biological replicates.
PduT-C38S and PduT-C38A increase NAD/H diffusion across the shell of the Pdu MCP
The role of the PduQ enzyme in 1,2-PD degradation is to recycle NADH (produced by the PduP enzyme) back to NAD+ internally within the Pdu MCP (Fig. 1). Prior studies showed that Salmonella pduQ mutants grow slowly on 1,2-PD because (in the absence of internal NAD/H recycling) their growth is limited by the diffusion of NAD/H across the shell of the Pdu MCP [85]. Previous work also showed that the slow growth phenotype of pduQ mutants is corrected if the shell of the Pdu MCP is broken genetically, since this allows the MCP lumen enzymes ready access to cytoplasmic NAD/H [85]. Importantly, correction of the slow growth phenotype of a pduQ mutant provides a facile in vivo test for increased permeability of the Pdu MCP to NAD/H [85]. Therefore, to test the effects of PduT-C38S, -C38A, -C38I, -C38W and ΔpduT on NAD/H permeability, we individually combined these variants with a ΔpduQ mutation and measured growth on 1,2-PD minimal medium as described elsewhere [85]. PduT-C38S and PduT-C38A each increased the growth rate of a ΔpduQ mutant on 1,2-PD, but PduT-C38I, and -C38W had no significant effect (Fig. 5, Table 3). This indicated that the PduT-C38S and -C38A variants increased the permeability of the shell of the Pdu MCP to NAD/H. These results are consistent with modelling studies that predicted PduT-C38S and -C38A would result in the formation of pores of sufficient size to allow NAD/H to cross the MCP shell, while the PduT-C38I would not. The results also showed that a pduT deletion mutant did not have a substantial effect on the permeability of the MCP shell to NAD/H (Fig. S6, Table 3).
Fig. 5.
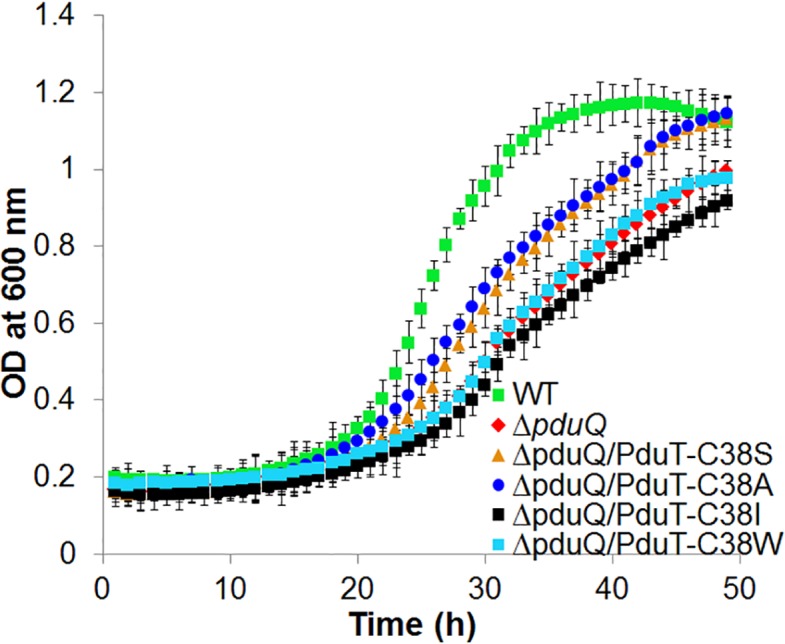
Effects of pduT mutations on the growth of a Salmonella pduQ mutant on 1,2-propanediol minimal medium. Salmonella strains were grown on 1,2-PD minimal medium supplemented with 100 nM B12 in a microplate reader. A prior study indicated that the sensitivity of the phenotype was better at a saturating B12 concentration [85]. The error bars represent one standard deviation and are based on three biological replicates.
Table 3.
Effects of pduT mutations on the growth of a Salmonella pduQ mutant
|
Pdu mutants |
Doubling time (h) |
|---|---|
|
WT |
5.4±0.9* |
|
ΔpduQ::frt |
11.2±0.8† |
|
ΔpduQ::kan |
10.7±0.8† |
|
ΔpduQ::frt /PduT-C38S |
7.4±0.7† |
|
ΔpduQ::frt /PduT-C38A |
7.9±0.7† |
|
ΔpduQ::frt /PduT-C38I |
11.5±0.8† |
|
ΔpduQ::frt /PduT-C38W |
10.1±0.4† |
|
ΔpduQ::kan /ΔpduT::frt |
9.6±0.6† |
|
PduT-C38S |
5.6±0.5 |
|
PduT-C38A |
5.2±0.8 |
|
PduT-C38I |
5.4±0.6 |
|
PduT-C38W |
5.9±0.9 |
|
ΔpduT::frt |
6.2±0.8 |
*Growth assays were performed on 1,2-PD minimal medium containing saturating levels of B12 (100 nM). Doubling times were calculated from at least three biological replicates measured in triplicate. The error estimate shown is one standard deviation.
†P-value <0.01 compared to wild-type (WT) as determined by two-tailed Student’s t-test. PduT-C38S, PduT-C38A, PduT-C38I, PduT-C38W and ΔpduT::frt are not significantly different from the WT.
As a control, we measured growth of PduT-C38S, -C38A, -C38I, -C38W and ΔpduT mutants on 1,2-PD minimal medium (Fig. S5, Table 3). None had a significant effect on the growth rate of Salmonella on 1,2-PD under the same conditions as those used to examine the double mutants. Thus, the PduT-C38S and -C38A mutants described above only increased the growth rate of Salmonella on 1,2-PD minimal medium in the ΔpduQ genetic background, which (in conjunction with prior studies) supports the interpretation that these mutants increase the permeability of the Pdu MCP to NAD/H.
Lastly, as a further control, we purified MCPs from the ΔpduQ/PduT C38S, ΔpduQ/PduT-C38A and ΔpduQ/PduT-C38I double mutants and showed that MCPs were normally formed by these mutants (Fig S7).
Discussion
Bacterial MCPs are a promising basis for engineering compartmentalized pathways to improve the efficiency of renewable chemicals production. MCPs consist of enzymes encapsulated within a protein shell and their native function is to optimize metabolic pathways [11–14, 18, 19, 86]. A challenge moving forward is to engineer MCP shells with the desired permeability properties, since optimal MCP function requires the diffusion of pathway substrates, products and enzymatic cofactors across the shell at the same time pathway intermediates are sequestered within [50–52]. Thus far, two strategies have been used to investigate/modify the permeability properties of bacterial MCPs. Studies of PduA, which is a major shell protein of the Pdu MCP, have used site-directed mutagenesis to change the amino acid that forms the narrowest point of its central pore (residue S40) [51]. These changes altered the permeability of the Pdu MCP to small molecules such as 1,2-PD (the substrate), propionaldehyde (a toxic intermediate) and glycerol (a substrate analogue) [51]. Structural and biophysical analyses of these mutants indicated that the main factors affecting diffusion through the pore of PduA (and presumably other BMC domain hexamers) are its size and electrostatic properties [51]. A second approach to modifying the permeability properties of MCPs was to engineer chimeric shells built from BMC domain hexamers native to two different MCPs [65, 66]. The feasibility of this approach is based on a conserved mechanism of shell assembly among divergent MCP shell proteins [53, 57, 62, 80, 80, 87].
In this report, we modified the permeability of the Pdu MCP by engineering the PduT shell protein. PduT is a pseudohexameric trimer with a central Fe–S cluster and is estimated to comprise about 3.2 % of the total MCP shell protein [57, 70]. Prior crystallography, as well as the modelling studies reported here, indicated that site-directed mutagenesis of PduT-C38 would create pores with varied chemical properties and sizes, including substantially larger pores (up to ~46 Å2) than had been examined previously (up to ~24 Å2). Therefore, we tested the effects of selected C38 mutations on MCP permeability. Studies of the PduT-C38I and -C38W mutants both in vivo (Table 1, Figs 3–5) and in vitro (Table 2) suggested that these variants (which are predicted to have small hydrophobic pores) did not alter the permeability of the Pdu MCP to 1,2-PD (the substrate), propionaldehyde (toxic intermediate) or NAD/H, which are required cofactors for two MCP lumen enzymes. Thus, these mutations suggest an approach to blocking diffusion through certain trimeric BMC domain proteins as well as a means for removing the Fe–S clusters without substantially altering shell permeability. In contrast, the results indicated that the PduT-C38S and PduT-C38A variants were more permeable to 1,2-PD, propionaldehyde and NAD/H. The PduT-C38S and -C38A mutations increased the diffusion of 1,2-PD across the MCP shell in a genetic background where 1,2-PD diffusion was restricted by a PduA-S40L mutation (Fig. 3, Table 1). This suggests that altering the pores of more than one shell protein might be a useful approach for controlling MCP permeability. We also found that the PduT-C38S and -C38A variants were somewhat more permeable to the metabolic intermediate propionaldehyde. Both variants excreted about 25 % more propionaldehyde into the culture medium during growth on 1,2-PD; however, the amount of propionaldehyde did not reach toxic levels, as seen earlier (Fig. 4) [69]. Similarly, the results indicated that the PduT-C38S and -C38A mutants were more permeable to NAD/H. In a genetic background where growth on 1,2-PD is limited by diffusion of NAD/H across the MCP shell, both the PduT-C38S and -C38A mutants increased growth rates by ~40 %, indicating increased NAD/H diffusion across the MCP shell (Fig. 5). This suggests that in at least some cases pore size can be engineered to allow the diffusion of larger molecules across MCP shells.
Lastly, we found that a pduT deletion had relatively little effect on the permeability of the Pdu MCP. If the deletion of PduT left a hole in the shell where PduT would normally be located, the permeability of the MCP should have been altered (PduT is about 72 Å across at its widest point). We speculate that PduT was replaced by another BMC domain protein, such as PduA or PduB, and that this was possible due to the conserved edge-to-edge interactions that mediate the assembly of BMC domain proteins into MCP shells [57, 80, 88].
Supplementary Data
Funding information
This work was supported by grant AI081146 from the National Institutes of Health to T. A. B.
Acknowledgements
We thank the ISU DNA Sequencing and Synthesis Facility for assistance with DNA analyses and the ISU Microscopy and Nanoimaging facility for help with electron microscopy.
Author contributions
C. C. contributed to conceptualization, methodology, investigation, original draft preparation and editing. T. B. contributed to conceptualization, original draft preparations, editing, project administration and funding.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Footnotes
Abbreviations: BMC, bacterial microcompartment; CN-B12, vitamin B12; DDH, diol dehydratase; DTT, dithiothreitol; HPLC, high-performance liquid chromatography; IPTG, isopropyl-β-D-1-thiogalactopyranoside; LB, lysogeny broth; MCP, microcompartment; NAD/H, nicotinamide adenine dinucleotide both reduced and oxidized forms; NCE, no carbon-E; 1,2-PD, 1,2-propanediol; TYE, tryptone yeast extract.
Three supplementary tables and seven supplementary figures are available with the online version of this article.
Edited by: J. Cavet and Y. Li
References
- 1.Lee H, DeLoache WC, Dueber JE. Spatial organization of enzymes for metabolic engineering. Metab Eng. 2012;14:242–251. doi: 10.1016/j.ymben.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 2.Giessen TW, Silver PA. Encapsulation as a strategy for the design of biological compartmentalization. J Mol Biol. 2016;428:916–927. doi: 10.1016/j.jmb.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 3.Conrado RJ, Varner JD, DeLisa MP. Engineering the spatial organization of metabolic enzymes: mimicking nature's synergy. Curr Opin Biotechnol. 2008;19:492–499. doi: 10.1016/j.copbio.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 4.Dueber JE, Wu GC, Malmirchegini GR, Moon TS, Petzold CJ, et al. Synthetic protein scaffolds provide modular control over metabolic flux. Nat Biotechnol. 2009;27:753–759. doi: 10.1038/nbt.1557. [DOI] [PubMed] [Google Scholar]
- 5.Held M, Quin MB, Schmidt-Dannert C. Eut bacterial microcompartments: insights into their function, structure, and bioengineering applications. J Mol Microbiol Biotechnol. 2013;23:308–320. doi: 10.1159/000351343. [DOI] [PubMed] [Google Scholar]
- 6.Hurtley S. Spatial cell biology. Location, location, location. Introduction. Science. 2009;326:1205. doi: 10.1126/science.326.5957.1205. [DOI] [PubMed] [Google Scholar]
- 7.Lopez-Gallego F, Schmidt-Dannert C. Multi-enzymatic synthesis. Curr Opin Chem Biol. 2010;14:174–183. doi: 10.1016/j.cbpa.2009.11.023. [DOI] [PubMed] [Google Scholar]
- 8.Frank S, Lawrence AD, Prentice MB, Warren MJ. Bacterial microcompartments moving into a synthetic biological world. J Biotechnol. 2013;163:273–279. doi: 10.1016/j.jbiotec.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez-Esquer CR, Newnham SE, Kerfeld CA. Bacterial microcompartments as metabolic modules for plant synthetic biology. Plant J. 2016;87:66–75. doi: 10.1111/tpj.13166. [DOI] [PubMed] [Google Scholar]
- 10.Kim EY, Tullman-Ercek D. Engineering nanoscale protein compartments for synthetic organelles. Curr Opin Biotechnol. 2013;24:627–632. doi: 10.1016/j.copbio.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 11.Kerfeld CA, Heinhorst S, Cannon GC. Bacterial microcompartments. Annu Rev Microbiol. 2010;64:391–408. doi: 10.1146/annurev.micro.112408.134211. [DOI] [PubMed] [Google Scholar]
- 12.Chowdhury C, Sinha S, Chun S, Yeates TO, Bobik TA. Diverse bacterial microcompartment organelles. Microbiol Mol Biol Rev. 2014;78:438–468. doi: 10.1128/MMBR.00009-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rae BD, Long BM, Badger MR, Price GD. Functions, compositions, and evolution of the two types of carboxysomes: polyhedral microcompartments that facilitate CO2 fixation in cyanobacteria and some proteobacteria. Microbiol Mol Biol Rev. 2013;77:357–379. doi: 10.1128/MMBR.00061-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bobik TA. Polyhedral organelles compartmenting bacterial metabolic processes. Appl Microbiol Biotechnol. 2006;70:517–525. doi: 10.1007/s00253-005-0295-0. [DOI] [PubMed] [Google Scholar]
- 15.Lehman BP, Chowdhury C, Bobik TA. The N terminus of the PduB protein binds the protein shell of the Pdu microcompartment to its enzymatic core. J Bacteriol. 2017;199 doi: 10.1128/JB.00785-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jakobson CM, Tullman-Ercek D, Slininger MF, Mangan NM. A systems-level model reveals that 1,2-Propanediol utilization microcompartments enhance pathway flux through intermediate sequestration. PLoS Comput Biol. 2017;13:e1005525. doi: 10.1371/journal.pcbi.1005525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bobik TA, Lehman BP, Yeates TO. Bacterial microcompartments: widespread prokaryotic organelles for isolation and optimization of metabolic pathways. Mol Microbiol. 2015;98:193–207. doi: 10.1111/mmi.13117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abdul-Rahman F, Petit E, Blanchard JL. The distribution of polyhedral bacterial microcompartments suggests frequent horizontal transfer and operon reassembly. J Phylogen Evolution Biol. 2013;1:1–7. [Google Scholar]
- 19.Jorda J, Lopez D, Wheatley NM, Yeates TO. Using comparative genomics to uncover new kinds of protein-based metabolic organelles in bacteria. Protein Sci. 2013;22:179–195. doi: 10.1002/pro.2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zarzycki J, Erbilgin O, Kerfeld CA. Bioinformatic characterization of glycyl radical enzyme-associated bacterial microcompartments. Appl Environ Microbiol. 2015;81:8315–8329. doi: 10.1128/AEM.02587-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lassila JK, Bernstein SL, Kinney JN, Axen SD, Kerfeld CA. Assembly of robust bacterial microcompartment shells using building blocks from an organelle of unknown function. J Mol Biol. 2014;426:2217–2228. doi: 10.1016/j.jmb.2014.02.025. [DOI] [PubMed] [Google Scholar]
- 22.Shively JM, Ball F, Brown DH, Saunders RE. Functional organelles in prokaryotes: polyhedral inclusions (carboxysomes) of Thiobacillus neapolitanus . Science. 1973;182:584–586. doi: 10.1126/science.182.4112.584. [DOI] [PubMed] [Google Scholar]
- 23.Bobik TA, Havemann GD, Busch RJ, Williams DS, Aldrich HC. The propanediol utilization (pdu) operon of Salmonella enterica serovar Typhimurium LT2 includes genes necessary for formation of polyhedral organelles involved in coenzyme B12-dependent 1, 2-propanediol degradation. J Bacteriol. 1999;181:5967–5975. doi: 10.1128/jb.181.19.5967-5975.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erbilgin O, McDonald KL, Kerfeld CA. Characterization of a planctomycetal organelle: a novel bacterial microcompartment for the aerobic degradation of plant saccharides. Appl Environ Microbiol. 2014;80:2193–2205. doi: 10.1128/AEM.03887-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Craciun S, Balskus EP. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc Natl Acad Sci USA. 2012;109:21307–21312. doi: 10.1073/pnas.1215689109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petit E, LaTouf WG, Coppi MV, Warnick TA, Currie D, et al. Involvement of a bacterial microcompartment in the metabolism of fucose and rhamnose by Clostridium phytofermentans . PLoS One. 2013;8:e54337. doi: 10.1371/journal.pone.0054337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Talarico TL, Axelsson LT, Novotny J, Fiuzat M, Dobrogosz WJ. Utilization of glycerol as a hydrogen acceptor by Lactobacillus reuteri: Purification of 1,3-propanediol: NAD oxidoreductase. Appl Environ Microbiol. 1990;56:943–948. doi: 10.1128/aem.56.4.943-948.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kofoid E, Rappleye C, Stojiljkovic I, Roth J. The 17-gene ethanolamine (eut) operon of Salmonella typhimurium encodes five homologues of carboxysome shell proteins. J Bacteriol. 1999;181:5317–5329. doi: 10.1128/jb.181.17.5317-5329.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moore TC, Escalante-Semerena JC. The EutQ and EutP proteins are novel acetate kinases involved in ethanolamine catabolism: physiological implications for the function of the ethanolamine metabolosome in Salmonella enterica . Mol Microbiol. 2016;99:497–511. doi: 10.1111/mmi.13243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Price-Carter M, Tingey J, Bobik TA, Roth JR. The alternative electron acceptor tetrathionate supports B12-dependent anaerobic growth of Salmonella enterica serovar Typhimurium on ethanolamine or 1,2-propanediol. J Bacteriol. 2001;183:2463–2475. doi: 10.1128/JB.183.8.2463-2475.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brinsmade SR, Paldon T, Escalante-Semerena JC. Minimal functions and physiological conditions required for growth of Salmonella enterica on ethanolamine in the absence of the metabolosome. J Bacteriol. 2005;187:8039–8046. doi: 10.1128/JB.187.23.8039-8046.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herring TI, Harris TN, Chowdhury C, Mohanty SK, Bobik TA. A bacterial microcompartment is used for choline fermentation by Escherichia coli 536. J Bacteriol. 2018;200 doi: 10.1128/JB.00764-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parsons JB, Frank S, Bhella D, Liang M, Prentice MB, et al. Synthesis of empty bacterial microcompartments, directed organelle protein incorporation, and evidence of filament-associated organelle movement. Mol Cell. 2010;38:305–315. doi: 10.1016/j.molcel.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 34.Mayer MJ, Juodeikis R, Brown IR, Frank S, Palmer DJ, et al. Effect of bio-engineering on size, shape, composition and rigidity of bacterial microcompartments. Sci Rep. 2016;6:36899. doi: 10.1038/srep36899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huber I, Palmer DJ, Ludwig KN, Brown IR, Warren MJ, et al. Construction of recombinant Pdu metabolosome shells for small molecule production in Corynebacterium glutamicum . ACS Synth Biol. 2017;6:2145–2156. doi: 10.1021/acssynbio.7b00167. [DOI] [PubMed] [Google Scholar]
- 36.Wade Y, Daniel RA, Leak DJ. Heterologous microcompartment assembly in Bacillaceae: establishing the components necessary for scaffold formation. ACS Synth Biol. 2019;8:1642–1654. doi: 10.1021/acssynbio.9b00155. [DOI] [PubMed] [Google Scholar]
- 37.Choudhary S, Quin MB, Sanders MA, Johnson ET, Schmidt-Dannert C. Engineered protein nano-compartments for targeted enzyme localization. PLoS One. 2012;7:e33342. doi: 10.1371/journal.pone.0033342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kinney JN, Salmeen A, Cai F, Kerfeld CA. Elucidating essential role of conserved carboxysomal protein CcmN reveals common feature of bacterial microcompartment assembly. J Biol Chem. 2012;287:17729–17736. doi: 10.1074/jbc.M112.355305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lawrence AD, Frank S, Newnham S, Lee MJ, Brown IR, et al. Solution structure of a bacterial microcompartment targeting peptide and its application in the construction of an ethanol bioreactor. ACS Synth Biol. 2014;3:454–465. doi: 10.1021/sb4001118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quin MB, Perdue SA, Hsu S-Y, Schmidt-Dannert C. Encapsulation of multiple cargo proteins within recombinant Eut nanocompartments. Appl Microbiol Biotechnol. 2016;100:9187–9200. doi: 10.1007/s00253-016-7737-8. [DOI] [PubMed] [Google Scholar]
- 41.Fan C, Cheng S, Liu Y, Escobar CM, Crowley CS, et al. Short N-terminal sequences package proteins into bacterial microcompartments. Proc Natl Acad Sci USA. 2010;107:7509–7514. doi: 10.1073/pnas.0913199107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jakobson CM, Kim EY, Slininger MF, Chien A, Tullman-Ercek D. Localization of proteins to the 1,2-propanediol utilization microcompartment by non-native signal sequences is mediated by a common hydrophobic motif. J Biol Chem. 2015;290:24519–24533. doi: 10.1074/jbc.M115.651919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jakobson CM, Slininger Lee MF, Tullman-Ercek D. De novo design of signal sequences to localize cargo to the 1,2-propanediol utilization microcompartment. Protein Sci. 2017;26:1086–1092. doi: 10.1002/pro.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee MJ, Mantell J, Hodgson L, Alibhai D, Fletcher JM, et al. Engineered synthetic scaffolds for organizing proteins within the bacterial cytoplasm. Nat Chem Biol. 2018;14:142–147. doi: 10.1038/nchembio.2535. [DOI] [PubMed] [Google Scholar]
- 45.Ferlez B, Sutter M, Kerfeld CA. A designed bacterial microcompartment shell with tunable composition and precision cargo loading. Metab Eng. 2019;54:286–291. doi: 10.1016/j.ymben.2019.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hagen A, Sutter M, Sloan N, Kerfeld CA. Programmed loading and rapid purification of engineered bacterial microcompartment shells. Nat Commun. 2018;9:2881. doi: 10.1038/s41467-018-05162-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fan C, Cheng S, Sinha S, Bobik TA. Interactions between the termini of lumen enzymes and shell proteins mediate enzyme encapsulation into bacterial microcompartments. Proc Natl Acad Sci USA. 2012;109:14995–15000. doi: 10.1073/pnas.1207516109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim EY, Tullman-Ercek D. A rapid flow cytometry assay for the relative quantification of protein encapsulation into bacterial microcompartments. Biotechnol J. 2014;9:348–354. doi: 10.1002/biot.201300391. [DOI] [PubMed] [Google Scholar]
- 49.Held M, Kolb A, Perdue S, Hsu SY, Bloch SE, et al. Engineering formation of multiple recombinant Eut protein nanocompartments in E. coli . Sci Rep. 2016;6:24359. doi: 10.1038/srep24359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dou Z, Heinhorst S, Williams EB, Murin CD, Shively JM, et al. CO2 fixation kinetics of Halothiobacillus neapolitanus mutant carboxysomes lacking carbonic anhydrase suggest the shell acts as a diffusional barrier for CO2 . J Biol Chem. 2008;283:10377–10384. doi: 10.1074/jbc.M709285200. [DOI] [PubMed] [Google Scholar]
- 51.Chowdhury C, Chun S, Pang A, Sawaya MR, Sinha S, et al. Selective molecular transport through the protein shell of a bacterial microcompartment organelle. Proc Natl Acad Sci USA. 2015;112:2990–2995. doi: 10.1073/pnas.1423672112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park J, Chun S, Bobik TA, Houk KN, Yeates TO. Molecular dynamics simulations of selective metabolite transport across the propanediol bacterial microcompartment shell. J Phys Chem B. 2017;121:8149–8154. doi: 10.1021/acs.jpcb.7b07232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kerfeld CA, Sawaya MR, Tanaka S, Nguyen CV, Phillips M, et al. Protein structures forming the shell of primitive bacterial organelles. Science. 2005;309:936–938. doi: 10.1126/science.1113397. [DOI] [PubMed] [Google Scholar]
- 54.Yeates TO, Jorda J, Bobik TA. The shells of BMC-type microcompartment organelles in bacteria. J Mol Microbiol Biotechnol. 2013;23:290–299. doi: 10.1159/000351347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yeates TO, Thompson MC, Bobik TA. The protein shells of bacterial microcompartment organelles. Curr Opin Struct Biol. 2011;21:223–231. doi: 10.1016/j.sbi.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tanaka S, Sawaya MR, Phillips M, Yeates TO. Insights from multiple structures of the shell proteins from the beta-carboxysome. Protein Sci. 2009;18:108–120. doi: 10.1002/pro.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Crowley CS, Cascio D, Sawaya MR, Kopstein JS, Bobik TA, et al. Structural insight into the mechanisms of transport across the Salmonella enterica Pdu microcompartment shell. J Biol Chem. 2010;285:37838–37846. doi: 10.1074/jbc.M110.160580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cai F, Sutter M, Cameron JC, Stanley DN, Kinney JN, et al. The structure of CcmP, a tandem bacterial microcompartment domain protein from the β-carboxysome, forms a subcompartment within a microcompartment. J Biol Chem. 2013;288:16055–16063. doi: 10.1074/jbc.M113.456897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klein MG, Zwart P, Bagby SC, Cai F, Chisholm SW, et al. Identification and structural analysis of a novel carboxysome shell protein with implications for metabolite transport. J Mol Biol. 2009;392:319–333. doi: 10.1016/j.jmb.2009.03.056. [DOI] [PubMed] [Google Scholar]
- 60.Tanaka S, Sawaya MR, Yeates TO. Structure and mechanisms of a protein-based organelle in Escherichia coli . Science. 2010;327:81–84. doi: 10.1126/science.1179513. [DOI] [PubMed] [Google Scholar]
- 61.Thompson MC, Cascio D, Leibly DJ, Yeates TO. An allosteric model for control of pore opening by substrate binding in the EutL microcompartment shell protein. Protein Sci. 2015;24:956–975. doi: 10.1002/pro.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sutter M, Greber B, Aussignargues C, Kerfeld CA. Assembly principles and structure of a 6.5-MDa bacterial microcompartment shell. Science. 2017;356:1293–1297. doi: 10.1126/science.aan3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takenoya M, Nikolakakis K, Sagermann M. Crystallographic insights into the pore structures and mechanisms of the EutL and EutM shell proteins of the ethanolamine-utilizing microcompartment of Escherichia coli . J Bacteriol. 2010;192:6056–6063. doi: 10.1128/JB.00652-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsai Y, Sawaya MR, Cannon GC, Cai F, Williams EB, et al. Structural analysis of CsoS1A and the protein shell of the Halothiobacillus neapolitanus carboxysome. PLoS Biol. 2007;5:e144. doi: 10.1371/journal.pbio.0050144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cai F, Sutter M, Bernstein SL, Kinney JN, Kerfeld CA. Engineering bacterial microcompartment shells: chimeric shell proteins and chimeric carboxysome shells. ACS Synth Biol. 2015;4:444–453. doi: 10.1021/sb500226j. [DOI] [PubMed] [Google Scholar]
- 66.Slininger Lee MF, Jakobson CM, Tullman-Ercek D. Evidence for improved encapsulated pathway behavior in a bacterial microcompartment through shell protein engineering. ACS Synth Biol. 2017;6:1880–1891. doi: 10.1021/acssynbio.7b00042. [DOI] [PubMed] [Google Scholar]
- 67.Aussignargues C, Pandelia M-E, Sutter M, Plegaria JS, Zarzycki J, et al. Structure and function of a bacterial microcompartment shell protein engineered to bind a [4Fe-4S] cluster. J Am Chem Soc. 2016;138:5262–5270. doi: 10.1021/jacs.5b11734. [DOI] [PubMed] [Google Scholar]
- 68.Havemann GD, Sampson EM, Bobik TA. PduA is a shell protein of polyhedral organelles involved in coenzyme B12-dependent degradation of 1,2-propanediol in Salmonella enterica serovar Typhimurium LT2. J Bacteriol. 2002;184:1253–1261. doi: 10.1128/JB.184.5.1253-1261.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sampson EM, Bobik TA. Microcompartments for B12-dependent 1,2-propanediol degradation provide protection from DNA and cellular damage by a reactive metabolic intermediate. J Bacteriol. 2008;190:2966–2971. doi: 10.1128/JB.01925-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pang A, Warren MJ, Pickersgill RW. Structure of PduT, a trimeric bacterial microcompartment protein with a 4Fe-4S cluster-binding site. Acta Crystallogr D Biol Crystallogr. 2011;67:91–96. doi: 10.1107/S0907444910050201. [DOI] [PubMed] [Google Scholar]
- 71.Parsons JB, Dinesh SD, Deery E, Leech HK, Brindley AA, et al. Biochemical and structural insights into bacterial organelle form and biogenesis. J Biol Chem. 2008;283:14366–14375. doi: 10.1074/jbc.M709214200. [DOI] [PubMed] [Google Scholar]
- 72.Cheng S, Sinha S, Fan C, Liu Y, Bobik TA. Genetic analysis of the protein shell of the microcompartments involved in coenzyme B12-dependent 1,2-propanediol degradation by Salmonella . J Bacteriol. 2011;193:1385–1392. doi: 10.1128/JB.01473-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chowdhury C, Chun S, Sawaya MR, Yeates TO, Bobik TA. The function of the PduJ microcompartment shell protein is determined by the genomic position of its encoding gene. Mol Microbiol. 2016;101:770–783. doi: 10.1111/mmi.13423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bertani G. Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli. J Bacteriol. 1951;62:293–300. doi: 10.1128/jb.62.3.293-300.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Berkowitz D, Hushon JM, Whitfield HJ, Roth J, Ames BN. Procedure for identifying nonsense mutations. J Bacteriol. 1968;96:215–220. doi: 10.1128/jb.96.1.215-220.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu Y, Jorda J, Yeates TO, Bobik TA. The PduL phosphotransacylase is used to recycle coenzyme A within the Pdu microcompartment. J Bacteriol. 2015;197:2392–2399. doi: 10.1128/JB.00056-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 78.DeLano WL, Bromberg S. PyMOL User’s Guide. San Carlos, California, USA: DeLano Scientific LLC; 2004. [Google Scholar]
- 79.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sinha S, Cheng S, Sung YW, McNamara DE, Sawaya MR, et al. Alanine scanning mutagenesis identifies an asparagine-arginine-lysine triad essential to assembly of the shell of the Pdu microcompartment. J Mol Biol. 2014;426:2328–2345. doi: 10.1016/j.jmb.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schmieger H. A method for detection of phage mutants with altered transducing ability. Mol Gen Genet. 1971;110:378–381. doi: 10.1007/BF00438281. [DOI] [PubMed] [Google Scholar]
- 82.Sun W, Wang S, Curtiss R. Highly efficient method for introducing successive multiple scarless gene deletions and markerless gene insertions into the Yersinia pestis chromosome. Appl Environ Microbiol. 2008;74:4241–4245. doi: 10.1128/AEM.00940-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sinha S, Cheng S, Fan C, Bobik TA. The PduM protein is a structural component of the microcompartments involved in coenzyme B12-dependent 1,2-propanediol degradation by Salmonella enterica . J Bacteriol. 2012;194:1912–1918. doi: 10.1128/JB.06529-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fan C, Bobik TA. The N-terminal region of the medium subunit (PduD) packages adenosylcobalamin-dependent diol dehydratase (PduCDE) into the Pdu microcompartment. J Bacteriol. 2011;193:5623–5628. doi: 10.1128/JB.05661-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cheng S, Fan C, Sinha S, Bobik TA. The PduQ enzyme is an alcohol dehydrogenase used to recycle NAD+ internally within the Pdu microcompartment of Salmonella enterica . PLoS One. 2012;7:e47144. doi: 10.1371/journal.pone.0047144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cheng S, Liu Y, Crowley CS, Yeates TO, Bobik TA. Bacterial microcompartments: their properties and paradoxes. Bioessays. 2008;30:1084–1095. doi: 10.1002/bies.20830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tanaka S, Kerfeld CA, Sawaya MR, Cai F, Heinhorst S, et al. Atomic-level models of the bacterial carboxysome shell. Science. 2008;319:1083–1086. doi: 10.1126/science.1151458. [DOI] [PubMed] [Google Scholar]
- 88.Greber BJ, Sutter M, Kerfeld CA. The plasticity of molecular interactions governs bacterial microcompartment shell assembly. Structure. 2019;27:749–763. doi: 10.1016/j.str.2019.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.