

Abstract
The current outbreak of viral pneumonia in the city of Wuhan, China, was caused by a novel coronavirus designated 2019‐nCoV by the World Health Organization, as determined by sequencing the viral RNA genome. Many initial patients were exposed to wildlife animals at the Huanan seafood wholesale market, where poultry, snake, bats, and other farm animals were also sold. To investigate possible virus reservoir, we have carried out comprehensive sequence analysis and comparison in conjunction with relative synonymous codon usage (RSCU) bias among different animal species based on the 2019‐nCoV sequence. Results obtained from our analyses suggest that the 2019‐nCoV may appear to be a recombinant virus between the bat coronavirus and an origin‐unknown coronavirus. The recombination may occurred within the viral spike glycoprotein, which recognizes a cell surface receptor. Additionally, our findings suggest that 2019‐nCoV has most similar genetic information with bat coronovirus and most similar codon usage bias with snake. Taken together, our results suggest that homologous recombination may occur and contribute to the 2019‐nCoV cross‐species transmission.
Keywords: 2019‐nCoV, codon usage bias, cross‐species transmission, phylogenetic analysis, recombination
Research Highlights
Taken together, our results suggest that homologous recombination may occur and contribute to the 2019‐nCoV cross‐species transmission.
1. INTRODUCTION
China has been the epicenter of emerging and re‐emerging viral infections that continue to stir a global concern. In the last 20 years, China has witnessed several emerging viral diseases, including an avian influenza in 1997, 1 the severe acute respiratory syndrome (SARS) in 2003, 2 and a severe fever with thrombocytopenia syndrome (SFTS) in 2010. 3 The most recent crisis was the outbreak of an ongoing viral pneumonia with unknown etiology in the city of Wuhan, China. On 12 December 2019, Wuhan Municipal Health Commission (WMHC) reported 27 cases of viral pneumonia with 7 of them being critically ill. Most of them had a history of exposure to the virus at the Huanan Seafood Wholesale Market where poultry, bats, snakes; and other wildlife animals were also sold. 4 On 3 January 2020, WMHC updated the number of cases to a total of 44 with 11 of them in critical condition. On 5 January, the number of cases increased to 59 with 7 critically ill patients. The viral pneumonia outbreak was not caused by severe acute respiratory syndrome coronavirus (SARS‐CoV), Middle East Respiratory Syndrome coronavirus (MERS‐CoV), influenza virus, or adenovirus as determined by laboratory tests. 4 On 10 January, it was reported that a novel coronavirus designated 2019‐nCoV by the World Health Organization (WHO) 5 was identified by high‐throughput sequencing of the viral RNA genome, which was released through http://virological.org. More significantly, the newly identified 2019‐CoV has also been isolated from one patient. The availability of viral RNA sequence has made it possible to develop reverse‐transcription polymerase chain reaction (RT‐PCR) methods for the detection of viral RNA in samples from patients and potential hosts. 6 As a result, 217 patients were confirmed to be infected with the 2019‐nCoV, and 9 patients died as of 20 January 2020. Several patients from Wuhan were also reported in Thailand, Singapore, Hong Kong, South Korea, and Japan. High‐throughput sequencing of viral RNA from patients’ samples has identified a novel coronavirus designated 2019‐nCoV by the World Health Organization. Currently, a total of 14 full‐length sequences of the 2019‐nCoV were released to GISAID and GeneBank.
The coronavirinae family consists of four genera based on their genetic properties, including genus Alphacoronavirus, genus Betacoronavirus, genus Gammacoronavirus, and genus Deltacoronavirus. 7 The coronavirus RNA genome (ranging from 26 to 32 kb) is the largest among all RNA viruses. 8 Coronavirus can infect humans and many different animal species, including swine, cattle, horses, camels, cats, dogs, rodents, birds, bats, rabbits, ferrets, mink, snake, and other wildlife animals. 7 , 9 Many coronavirus infections are subclinical. 7 , 9 SARS‐CoV and MERS‐CoV belong to the Betacoronavirus genus and are zoonotic pathogens that can cause severe respiratory diseases in humans. 7
The outbreak of viral pneumonia in Wuhan is associated with history of exposure to virus reservoir at the Huanan seafood wholesale market, suggesting a possible zoonosis. The seafood market also sold live animals such as snakes, marmots, birds, frogs, and hedgehogs. Currently, there is no evidence suggesting a specific wildlife host as a virus reservoir. Studies of relative synonymous codon usage (RSCU) bias between viruses and their hosts suggested that viruses tends to evolve codon usage bias that is comparable to their hosts. 10 , 11 Results from our analysis suggest that 2019‐nCoV has most similar genetic information with bat coronovirus and has most similar codon usage bias with snake. More interestingly, an origin‐unknown homologous recombination may occured within the spike glycoprotein of the 2019‐nCoV, 5 which may explain its cross‐species transmission, and limited person‐person spread.
2. MATERIALS AND METHODS
2.1. Sequence data collection
The newly sequenced Beta‐coronavirus (MN908947) genome was downloaded from the GenBank database. Five hundred closely related sequences were also downloaded from GenBank. Out of them, 271 genome sequences (>19 000 bp in length) were used in this study together with the above‐described Beta‐coronavirus (2019‐nCoV, MN908947) genome sequence (Table S1). The geographic origins of the sequences were from Bulgaria (n = 1), Canada (n = 2), China (n = 67), Germany (n = 1), Hong Kong (n = 5), Italy (n = 1), Kenya (n = 1), Russia (n = 1), Singapore (n = 24), South Korea (n = 1), Taiwan (n = 11), United Kingdom (n = 2), United States of America (n = 67), and unknown (n = 88). Sequences were aligned using MAFFT v7.222, 12 followed by manual adjustment using BioEdit v7.2.5. 13
2.2. Phylogenetic and simplot analysis
Phylogenetic trees were constructed using maximum‐likelihood methods and general time‐reversible model of nucleotide substitution with gamma‐distributed rates among sites (GTR+G substitution model) in RAxML v8.0.9. 14 Support for the inferred relationships was evaluated by a bootstrap analysis with 1000 replicates and trees were midpoint‐rooted.
To investigate the putative parents of the 2019‐nCoV, we performed Similarity and Bootscanning plot analyses based on the Kimura two‐parameter model with a window size of 500 bp, step size of 30 bp using SimPlot v.3.5.1. 15 We divided our data set into four clades, the newly discovered 2019‐nCoV sequence was grouped as the query sequence. The closest relative coronaviruses (bat‐SL‐CoVZC45 and bat‐SL‐CoVZXC21) obtained from the city of Nanjing, China were grouped as “Clade A.” The other two coronaviruses (BtCoV/BM48‐31/BGR/2008 and BtKY72) from Bulgaria and Kenya were grouped as “Clade B.” The rest sequences were grouped as “Clade C” (Figure 1).
Figure 1.

Maximum likelihood phylogenetic tree of the 2019‐nCoV. Phylogenetic tree inferred from 272 near‐complete genome sequences of coronavirus was midpoint rooted and grouped into 4 clades (2019‐nCoV, Clades A, B, and C). Coronaviruses originating from different countries/regions are highlighted in colors
2.3. Synonymous codon usage analysis
To estimate the RSCU bias of the 2019‐nCoV and its potential host(s), All avaliable coding sequences (retaining coding sequences with ATG primer and mutiple of 3 nucleotides, excluding incorrect coding sequences) of the 2019‐nCoV genome (1CDS's, 9672 codons), bat‐SL‐CoVZC45 genome (1CDS's, 9680 codons), Bungarus multicinctus genes (38 CDS's, 5381 codons), Naja atra genes (64 CDS's, 9587 codons), Erinaceus europaeus genome CDS (28947 CDS's, 16717458 codons), Marmota genes (36055CDS's, 21090600 codons), Manis javanica genome CDS (39192 CDS's, 22980491 codons), Rhinolophus sinicus genes (10 CDS's, 8081 codons) and Gallus gallus genome CDS (49453 CDS's, 36086657 codons) from GenBank were calculated with Codon W1.4.2. 16 , 17 The RSCU of human genes (40662582 codons) was retrieved from the Codon Usage Database (http://www.kazusa.or.jp/codon/). The relationship among these sequences was calculated using a squared Euclidean distance , as we previously reported. 18 A heat map of RSCU was drawn with MeV 4.9.0 software. 19 The coronavirus and their potential hosts were clustered using a Euclidean distance method.
3. RESULTS
3.1. Phylogenetic classification
Phylogenetic analysis of 276 coronavirus genomes revealed that the newly identified coronavirus 2019‐nCoV sequence was monophyletic with 100% bootstrap support. The Clade A (bat‐SL‐CoVZC45 and bat‐SL‐CoVZXC21) derived from bats in the city of Nanjing, China between 2015 and 2017 represents the sister lineage to 2019‐nCoV. The Clade B (BtCoV/BM48‐31/BGR/2008 and BtKY72) obtained from bats in Bulgaria and Kenya between 2005 and 2007 formed a distinct monophyletic cluster with 100% bootstrap support. The Clade C including 267 coronavirus strains was clustered together with 63% bootstrap support (Figure 1). This suggest that 2019‐nCoV has most similar genetic information with bat coronovirus.
3.2. Homologous recombination may occured within the viral spike glycoprotein
Homologous recombination is an important evolutionary force and previous studies have found that homologous recombination occurred in many viruses, including Dengue virus, 20 human immunodeficiency virus, 21 hepatitis B virus, 22 hepatitis C virus, 23 and classical swine fever virus. 18 Similarity plot analysis of the 2019‐nCoV revealed that homologous recombination may occurred between Clade A strains (bat‐coronaviruses) and the origin‐unknown isolates, located within the spike glycoprotein that recognizes cell surface receptor (Figure 2). These characteristics indicate that cross‐species transmission may be caused by homologous recombination.
Figure 2.
Sequence comparison among different coronaviruses. Similarity plot analysis was performed among coronaviruses in Clades A, B, and C. Recombination analysis was conducted with a sliding window of 500 bp and a step size of 30 bp. Recombination sites were located within the viral spike glycoprotein genes, as indicated by an orange box on the top
3.3. Relative synonymous codon usage analysis
As parasitic microorganism, virus codon usage pattern resembles its host to some extent. The RSCU bias shows that the 2019‐nCoV, bat‐SL‐CoVZC45, and snakes from China have similar synonymous codon usage bias (Figure 3A, Table 1). The squared euclidean distance indicates that the 2019‐nCoV and snakes from China have the highest similarity in synonymous codon usage bias compared to those of bat, bird, Marmota, human, Manis, and hedgehog and (Figure 3B). Two types of snakes, containing B. multicinctus (many‐banded krait) and N. atra (Chinese cobra) were used for RSCU analysis. Squared Euclidean distance between the 2019‐nCoV and B. multicinctus is 13.54. The distance between the 2019‐nCoV and another snake N. atra is 16.69. The distance between the 2019‐nCoV and Rhinolophus sinicus is 23.46. However, the distance between the 2019‐nCoV and other animals is greater than 26, specifically 26.93 for bird, 34.79 for Marmota, 35.36 for human, 36.71 for Manis, and 37.96 for hedgehog. These data suggest that the 2019‐nCoV might more effectively use snake's translation machinery than that of other animals.
Figure 3.
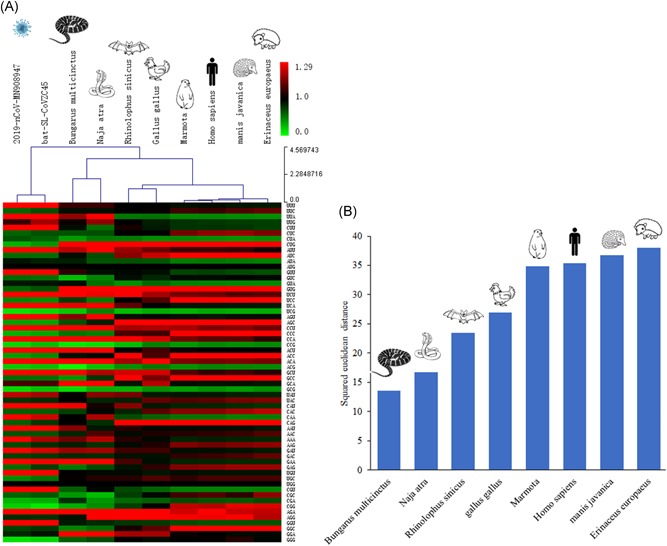
Comparison of relative synonymous codon usage (RSCU) between 2019‐nCoV and its putative wildlife animal reservoir(s). A, Heat map resulting from cluster analysis of the RSCU among the 2019‐nCoV, bat‐SL‐CoVZC45, Bungarus multicinctus, Naja atra, Rhinolophus sinicus, Gallus gallus, Marmota, Homo sapiens, Manis javanica, and Erinaceus europaeus. B, Comparison of squared euclidean distance between 2019‐nCoV and different animal species. Squared Euclidean distance was calculated based on the RSCU
Table 1.
The RSCU analysis of the preferred codons (codons with RSCU >1), the optimal codons and the rare codons for coronaviruses, snakes, hedgehog, bat Marmota, Manis, Gallus, and human genome
| bat‐SL‐CoVZC45 | 2019‐nCoV‐MN908947 | Bungarus multicinctus | Naja atra | Rhinolophus sinicus | Gallus gallus | Marmota | Homo sapiens | manis javanica | Erinaceus europaeus | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Phe | UUU | 1.33 | 1.41 | 1.07 | 1.07 | 1 | 0.99 | 0.92 | 0.93 | 0.9 | 0.88 |
| UUC | 0.67 | 0.59 | 0.93 | 0.93 | 1 | 1.01 | 1.08 | 1.07 | 1.1 | 1.12 | |
| Leu | UUA | 1.37 | 1.64 | 1.17 | 1.32 | 0.52 | 0.54 | 0.45 | 0.46 | 0.44 | 0.43 |
| UUG | 1.19 | 1.07 | 1.02 | 1.17 | 1 | 0.87 | 0.79 | 0.77 | 0.75 | 0.74 | |
| CUU | 1.77 | 1.75 | 0.94 | 0.55 | 1.03 | 0.89 | 0.79 | 0.79 | 0.78 | 0.74 | |
| CUC | 0.66 | 0.59 | 0.66 | 0.67 | 0.99 | 1 | 1.14 | 1.17 | 1.18 | 1.15 | |
| CUA | 0.6 | 0.66 | 0.38 | 0.51 | 0.5 | 0.44 | 0.45 | 0.43 | 0.41 | 0.43 | |
| CUG | 0.4 | 0.3 | 1.83 | 1.77 | 1.95 | 2.26 | 2.37 | 2.37 | 2.44 | 2.5 | |
| Ile | AUU | 1.57 | 1.53 | 1.21 | 1.67 | 1.18 | 1.13 | 1.08 | 1.08 | 1.07 | 1.03 |
| AUC | 0.56 | 0.56 | 0.93 | 0.68 | 1.16 | 1.23 | 1.42 | 1.41 | 1.43 | 1.5 | |
| AUA | 0.86 | 0.91 | 0.87 | 0.65 | 0.66 | 0.64 | 0.5 | 0.51 | 0.5 | 0.48 | |
| Met | AUG | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1.00 | 1 | 1 |
| Val | GUU | 1.89 | 1.95 | 1.06 | 0.94 | 0.97 | 0.93 | 0.71 | 0.73 | 0.69 | 0.67 |
| GUC | 0.55 | 0.57 | 0.26 | 0.47 | 0.94 | 0.83 | 0.95 | 0.95 | 0.98 | 0.96 | |
| GUA | 0.91 | 0.9 | 0.93 | 0.54 | 0.42 | 0.57 | 0.48 | 0.47 | 0.46 | 0.45 | |
| GUG | 0.66 | 0.58 | 1.75 | 2.05 | 1.68 | 1.67 | 1.86 | 1.85 | 1.87 | 1.91 | |
| Ser | UCU | 2.04 | 1.96 | 1.96 | 1.27 | 1.29 | 1.19 | 1.14 | 1.13 | 1.1 | 1.11 |
| UCC | 0.44 | 0.47 | 0.74 | 0.51 | 1 | 1.1 | 1.31 | 1.31 | 1.3 | 1.3 | |
| UCA | 1.66 | 1.66 | 1.26 | 1.43 | 1.11 | 0.99 | 0.9 | 0.90 | 0.89 | 0.88 | |
| UCG | 0.15 | 0.11 | 0.21 | 0.47 | 0.26 | 0.32 | 0.29 | 0.33 | 0.31 | 0.32 | |
| AGU | 1.36 | 1.43 | 1.16 | 1.46 | 0.93 | 0.96 | 0.94 | 0.90 | 0.93 | 0.94 | |
| AGC | 0.36 | 0.37 | 0.66 | 0.86 | 1.4 | 1.45 | 1.42 | 1.44 | 1.46 | 1.45 | |
| Pro | CCU | 1.82 | 1.94 | 1.91 | 1.81 | 1.19 | 1.2 | 1.19 | 1.15 | 1.16 | 1.13 |
| CCC | 0.34 | 0.3 | 0.52 | 0.49 | 1.09 | 1.08 | 1.28 | 1.29 | 1.31 | 1.37 | |
| CCA | 1.59 | 1.6 | 1.47 | 1.57 | 1.46 | 1.25 | 1.14 | 1.11 | 1.11 | 1.06 | |
| CCG | 0.26 | 0.16 | 0.1 | 0.13 | 0.26 | 0.48 | 0.39 | 0.45 | 0.42 | 0.44 | |
| Thr | ACU | 1.75 | 1.78 | 1.27 | 1.28 | 1.01 | 1.08 | 1.02 | 0.99 | 1 | 1 |
| ACC | 0.44 | 0.38 | 0.91 | 0.96 | 1.38 | 1.09 | 1.42 | 1.42 | 1.41 | 1.42 | |
| ACA | 1.58 | 1.64 | 1.79 | 1.52 | 1.19 | 1.32 | 1.15 | 1.14 | 1.15 | 1.14 | |
| ACG | 0.24 | 0.2 | 0.02 | 0.23 | 0.42 | 0.51 | 0.41 | 0.46 | 0.45 | 0.44 | |
| Ala | GCU | 2.13 | 2.19 | 1.95 | 1.21 | 1.24 | 1.24 | 1.1 | 1.06 | 1.08 | 1.05 |
| GCC | 0.55 | 0.57 | 0.41 | 0.78 | 1.57 | 1.14 | 1.59 | 1.60 | 1.62 | 1.65 | |
| GCA | 1.09 | 1.09 | 1.5 | 1.7 | 0.9 | 1.21 | 0.94 | 0.91 | 0.92 | 0.89 | |
| GCG | 0.24 | 0.15 | 0.14 | 0.31 | 0.3 | 0.42 | 0.37 | 0.42 | 0.38 | 0.42 | |
| Tyr | UAU | 1.19 | 1.22 | 1.01 | 1.16 | 1.14 | 0.88 | 0.9 | 0.89 | 0.86 | 0.85 |
| UAC | 0.81 | 0.78 | 0.99 | 0.84 | 0.86 | 1.12 | 1.1 | 1.11 | 1.14 | 1.15 | |
| His | CAU | 1.39 | 1.39 | 1.27 | 1.03 | 1.16 | 0.89 | 0.84 | 0.84 | 0.81 | 0.78 |
| CAC | 0.61 | 0.61 | 0.73 | 0.97 | 0.84 | 1.11 | 1.16 | 1.16 | 1.19 | 1.22 | |
| Gln | CAA | 1.24 | 1.39 | 1 | 1.2 | 0.68 | 0.59 | 0.53 | 0.53 | 0.49 | 0.5 |
| CAG | 0.76 | 0.61 | 1 | 0.8 | 1.32 | 1.41 | 1.47 | 1.47 | 1.51 | 1.5 | |
| Asn | AAU | 1.34 | 1.35 | 1.16 | 0.9 | 1.05 | 0.96 | 0.95 | 0.94 | 0.93 | 0.9 |
| AAC | 0.66 | 0.65 | 0.84 | 1.1 | 0.95 | 1.04 | 1.05 | 1.06 | 1.07 | 1.1 | |
| Lys | AAA | 1.2 | 1.31 | 1.13 | 1.21 | 1.05 | 0.96 | 0.85 | 0.87 | 0.85 | 0.83 |
| AAG | 0.8 | 0.69 | 0.87 | 0.79 | 0.95 | 1.04 | 1.15 | 1.13 | 1.15 | 1.17 | |
| Asp | GAU | 1.24 | 1.28 | 1.18 | 1.19 | 1.08 | 1.08 | 0.94 | 0.93 | 0.91 | 0.88 |
| GAC | 0.76 | 0.72 | 0.82 | 0.81 | 0.92 | 0.92 | 1.06 | 1.07 | 1.09 | 1.12 | |
| Glu | GAA | 1.27 | 1.44 | 1.49 | 1.32 | 1.09 | 0.94 | 0.85 | 0.84 | 0.82 | 0.82 |
| GAG | 0.73 | 0.56 | 0.51 | 0.68 | 0.91 | 1.06 | 1.15 | 1.16 | 1.18 | 1.18 | |
| Cys | UGU | 1.47 | 1.56 | 0.99 | 0.95 | 0.95 | 0.9 | 0.94 | 0.91 | 0.91 | 0.91 |
| UGC | 0.53 | 0.44 | 1.01 | 1.05 | 1.05 | 1.1 | 1.06 | 1.09 | 1.09 | 1.09 | |
| Trp | UGG | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1.00 | 1 | 1 |
| Arg | CGU | 1.52 | 1.45 | 0.61 | 0.97 | 0.7 | 0.59 | 0.49 | 0.48 | 0.48 | 0.48 |
| CGC | 0.63 | 0.59 | 0.39 | 0.26 | 0.74 | 0.96 | 1.08 | 1.10 | 1.06 | 1.17 | |
| CGA | 0.32 | 0.29 | 0.8 | 0.4 | 0.57 | 0.61 | 0.69 | 0.65 | 0.63 | 0.68 | |
| CGG | 0.1 | 0.19 | 0.32 | 0.44 | 0.74 | 0.98 | 1.22 | 1.21 | 1.26 | 1.25 | |
| AGA | 2.63 | 2.67 | 2.97 | 2.47 | 1.84 | 1.52 | 1.24 | 1.29 | 1.23 | 1.22 | |
| AGG | 0.79 | 0.81 | 0.91 | 1.46 | 1.42 | 1.35 | 1.28 | 1.27 | 1.34 | 1.21 | |
| Gly | GGU | 2.16 | 2.34 | 0.89 | 0.82 | 0.78 | 0.76 | 0.68 | 0.65 | 0.65 | 0.65 |
| GGC | 0.77 | 0.71 | 0.47 | 0.56 | 1.03 | 1.11 | 1.32 | 1.35 | 1.35 | 1.41 | |
| GGA | 0.91 | 0.83 | 2.03 | 1.95 | 1.19 | 1.19 | 1.02 | 1.00 | 0.95 | 0.96 | |
| GGG | 0.16 | 0.12 | 0.6 | 0.68 | 1 | 0.94 | 0.99 | 1.01 | 1.05 | 0.97 |
Note: The most preferred codons are in bold.
Abbreviation: RSCU, relative synonymous codon usage.
This article is being made freely available through PubMed Central as part of the COVID-19 public health emergency response. It can be used for unrestricted research re-use and analysis in any form or by any means with acknowledgement of the original source, for the duration of the public health emergency.
Two types of snakes are common in Southeastern China including the city of Wuhan (Figure 4). Geographical distributions of B. multicinctus include Taiwan, the Central and Southern China, Hong Kong, Myanmar (Burma), Laos, and Northern Vietnam. 24 N. atra is found in Southeastern China, Hong Kong, Northern Laos, Northern Vietnam, and Taiwan. 25 Snakes were also sold at the Huanan Seafood Wholesale Market where many patients worked or had a history of exposure to wildlife or farm animals.
Figure 4.

Geographic distribution of Bungarus multicinctus and Naja atra in China. The geographic distribution of Bungarus multicinctus and Naja atra are highlighted in colors. Yellow color represents the common geographic distribution of Bungarus multicinctus and Naja atra. Green color represents additional geographic distribution of Bungarus multicinctus. The location of Wuhan city where the 2019‐nCoV outbreak occurs is indicated in red. Maps were obtained from Craft MAP website (http://www.craftmap.box-i.net/)
4. DISCUSSION
In this study, we have performed an evolutionary analysis using 272 genomic sequences of coronaviruses obtained from various geographic locations. Our results show that the novel coronavirus sequence obtained from the viral pneumonia outbreak occurring in the city of Wuhan forms a separate group that is highly distinctive to SARS‐CoV. The SARS‐CoV first emerged in China in 2002 and then spread to 37 countries/regions in 2003 and caused a travel‐related global outbreak with 9.6% mortality rate. 26 More importantly, results from our analysis reveal a homologous recombination may occurred between the bat coronavirus and an origin‐unknown coronavirus within the viral spike glycoprotein gene. Sequence homology analysis of the partial spike glycoprotein genes (1‐783 bp) from the 2019‐nCoV was done through BLAST at the NCBI website. Interestingly, no similar sequence was found with known sequence in the database, suggesting that a putative recombination parent virus was still unknown. Previous study suggested that the recombination of SARS in the spike glycoprotein genes might have mediated the initial cross‐species transmission event from bats to other mammals. 27 Bootscanning plot analysis (data not shown) suggested that the major parents of the 2019‐nCoV originated from Clade A (bat‐SL‐CoVZC45 and bat‐SL‐CoVZXC21) but formed a monophyletic cluster different from them. Overall, the ancestral origin of the 2019‐nCoV was more likely from divergent host species rather than SARS‐CoV.
The host range of some animal coronaviruses was promiscuous. 7 They caught our attention only when they caused human diseases such as SARS, MERS, and 2019‐nCoV pneumonia. 4 , 9 , 28 It is critical to determine the animal reservoir of the 2019‐nCoV to understand the molecular mechanism of its cross‐species spread. Homologous recombination within viral structural proteins between coronaviruses from different hosts may be responsible for “cross‐species” transmission. 27 Information obtained from RSCU analysis provides some insights to the question of wildlife animal reservoir although it requires further validation by experimental studies in animal models. Currently, the 2019‐nCoV has not been isolated from animal species although it was obtained from one patient. Identifying and characterizing the animal reservoir for 2019‐nCoV will be helpful for investigation of the recombination and for a better understanding of its person‐to‐person spread among human populations.
The 2019‐nCoV has caused a total of 217 confirmed cases of pneumonia in China as of 20 January 2020 with new patients also reported in Hong Kong, Thailand, Singapore, South Korea, and Japan. Unlike SARS‐CoV, the 2019‐nCoV appeared to initially cause a mild form of viral pneumonia and have limited capability for person‐person spread. This might be due to the recombination occurred within the receptor‐binding glycoprotein. However, there is a concern about its adaptation in humans that may acquire the capability to replicate more efficiently and spread more rapidly via close person‐person contact.
In summary, results derived from our evolutionary analysis suggest that 2019‐nCoV has most similar genetic information with bat coronovirus and has most similar codon usage bias with snake. Additionally, a homologous recombination may occured within the viral receptor‐binding spike glycoprotein, which may determine cross‐species transmission. These novel findings warrant future investigation to experimentally determine if homologous recombination within the spike glycoprotein determine the tropism of the 2019‐nCoV in viral transmission and replication. New information obtained from our evolutionary analysis is highly significant for effective control of the outbreak caused by the 2019‐nCoV‐induced pneumonia.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTIONS
Writing: WJ and XL. Data collection: JZ, WW, and XZ. Data analysis: WJ and XL.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
This study was supported by Project of Guangxi Health Committee (No. Z20191111) and Natural Science Foundation of Guangxi Province of China (No. 2017GXNSFAA198080) to Dr Xiaofang Zhao. This study was sponsored by K.C. Wong Magna Fund in Ningbo University. The authors would like to thank Prof Yongzhen Zhang (Fudan University) for deposing the sequence of the newly identified coronavirus 2019‐nCoV to GeneBank, which was used in this study.
Ji W, Wang W, Zhao X, Zai J, Li X. Cross‐species transmission of the newly identified coronavirus 2019‐nCoV. J Med Virol. 2020;92:433–440. 10.1002/jmv.25682
All authors contributed equally to this work.
REFERENCES
- 1. Chan PK. Outbreak of avian influenza A (H5N1) virus infection in Hong Kong in 1997. Clin Infect Dis. 2002;34:S58‐S64. [DOI] [PubMed] [Google Scholar]
- 2. Guan Y. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science. 2003;302:276‐278. [DOI] [PubMed] [Google Scholar]
- 3. Yu XJ, Liang MF, Zhang SY, et al. Fever with thrombocytopenia associated with a novel bunyavirus in China. N Engl J Med. 2011;364:1523‐1532. 10.1056/NEJMoa1010095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lu H, Stratton CW, Tang YW. Outbreak of pneumonia of unknown etiology in Wuhan China: the mystery and the miracle. J Med Virol. 2020. 10.1002/jmv.25678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. World Health Organization . Coronavirus. 2019. https://www.who.int/health-topics/coronavirus
- 6. Zhang N, Wang L, Deng X, et al. Recent advances in the detection of respiratory virus infection in humans. J Med Virol. 2020. 10.1002/jmv.25674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. MacLachlan NJ, Dubovi EJ. In: MacLachlan NJ, Dubovi EJ, eds. Fenner's Veterinary Virology. 5th ed. Cambridge, MA: Academic Press; 2017:393‐413. [Google Scholar]
- 8. Howley DMKP. Fields Virology. 1 Lippincott Williams & Wilkins; 2013:830P. [Google Scholar]
- 9. World Health Organization . Consensus Document on the Epidemiology of Severe Acute Respiratory Syndrome (SARS). Geneva: World Health Organization; 2003. [Google Scholar]
- 10. Wang H, Liu S, Zhang B, Wei W. Analysis of synonymous codon usage bias of Zika virus and its adaption to the hosts. PLoS One. 2016;11:e0166260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bahir I, Fromer M, Prat Y, Linial M. Viral adaptation to host: a proteome‐based analysis of codon usage and amino acid preferences. Mol Syst Biol. 2009;5:311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772‐780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hall TA. BioEdit: a user‐friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95‐98. https://doi.org/citeulike-article-id:691774 [Google Scholar]
- 14. Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics. 2014;30:1312‐1313. 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lole KS, Bollinger RC, Paranjape RS, et al. Full‐length human immunodeficiency virus type 1 genomes from subtype C‐infected seroconverters in India, with evidence of intersubtype recombination. J Virol. 1999;73:152‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu X, Zhang Y, Fang Y, Wang Y. Patterns and influencing factor of synonymous codon usage in porcine circovirus. Virol J. 2012;9:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wright F. The ‘effective number of codons’ used in a gene. Gene. 1990;87:23‐29. [DOI] [PubMed] [Google Scholar]
- 18. Ji W, Niu D‐D, Si H‐L, Ding N‐Z, He C‐Q. Vaccination influences the evolution of classical swine fever virus. Infect Genet Evol. 2014;25:69‐77. [DOI] [PubMed] [Google Scholar]
- 19. Howe H, Holton K, Nair S, Schlauch D, Sinha R, Quackenbush J. In: Ochs MF, Casagrande JT, Davuluri RV, eds. Biomedical Informatics for Cancer Research. Boston, MA: Springer; 2019:267‐277. [Google Scholar]
- 20. Tolou HJ, Couissinier‐Paris P, Durand JP, et al. Evidence for recombination in natural populations of dengue virus type 1 based on the analysis of complete genome sequences. J Gen Virol. 2001;82:1283‐1290. [DOI] [PubMed] [Google Scholar]
- 21. Clavel F, Hoggan MD, Willey RL, Strebel K, Martin MA, Repaske R. Genetic recombination of human immunodeficiency virus. J Virol. 1989;63:1455‐1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bollyky PL, Rambaut A, Harvey PH, Holmes EC. Recombination between sequences of hepatitis B virus from different genotypes. J Mol Evol. 1996;42:97‐102. [DOI] [PubMed] [Google Scholar]
- 23. Colina R, Casane D, Vasquez S, et al. Evidence of intratypic recombination in natural populations of hepatitis C virus. J Gen Virol. 2004;85:31‐37. [DOI] [PubMed] [Google Scholar]
- 24. Wikipedia . Many‐banded krait. 2020. https://en.wikipedia.org/wiki/Many-banded_krait.
- 25. Wikipedia . Chinese cobra. 2020. https://en.wikipedia.org/wiki/Chinese_cobra.
- 26. Smith RD. Responding to global infectious disease outbreaks: lessons from SARS on the role of risk perception, communication and management. Soc Sci Med. 2006;63:3113‐3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Graham RL, Baric RS. Recombination, reservoirs, and the modular spike: mechanisms of coronavirus cross‐species transmission. J Virol. 2010;84:3134‐3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cunha CB, Opal SM. Middle East respiratory syndrome (MERS) A new zoonotic viral pneumonia. Virulence. 2014;5:650‐654. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information