

SUMMARY
iPSC-derived microglia offer a powerful tool to study microglial homeostasis and disease-associated inflammatory responses. Yet, microglia are highly sensitive to their environment, exhibiting transcriptomic deficiencies when kept in isolation from the brain. Furthermore, species-specific genetic variations demonstrate that rodent microglia fail to fully recapitulate the human condition. To address this, we developed an approach to study human microglia within a surrogate brain environment. Transplantation of iPSC-derived hematopoietic-progenitors into the postnatal brain of humanized, immune-deficient mice results in context-dependent differentiation into microglia and other CNS macrophages, acquisition of an ex vivo human microglial gene signature, and responsiveness to both acute and chronic insults. Most notably, transplanted microglia exhibit robust transcriptional responses to Aβ-plaques that only partially overlap with that of murine microglia, revealing new, human-specific Aβ-responsive genes. We therefore have demonstrated that this chimeric model provides a powerful new system to examine the in vivo function of patient-derived and genetically-modified microglia.
Graphical Abstract
INTRODUCTION
Microglia play critical roles in sculpting brain development, modulating neural plasticity, and maintaining homeostasis (Salter and Stevens, 2017; Stevens et al., 2007; Wu et al., 2015). As the primary immune cell of the central nervous system (CNS), microglia are highly responsive, reacting rapidly to local injury, neuroinflammation, and a multiplicity of brain pathologies (Nimmerjahn et al., 2005; Perry and Holmes, 2014). Recent genetic studies have further highlighted the importance of these cells in disease, with the discovery of many polymorphisms in microglial-enriched genes that are associated with a variety of neurological disorders including Alzheimer’s disease (AD), frontotemporal dementia, amyotrophic lateral sclerosis, autism, and schizophrenia (Karch et al., 2014; Salter and Stevens, 2017). However, despite these important findings, experimental platforms that enable systematic analyses of human microglia in vivo, and the effects of genetic variability on microglia function within the brain, have yet to be realized.
While transgenic mouse models have provided invaluable tools for examining the role of microglia in these disorders, rodents cannot fully recapitulate the growing complement of human genetic variability implicated in these polygenic diseases (Dawson et al., 2018; Friedman et al., 2018; Ueda et al., 2016). Fortunately, the ability to generate induced pluripotent stem cells (iPSCs) from patients, and then differentiate iPSCs into defined cell subtypes, has generated exciting opportunities to examine the relationships between complex genetic backgrounds and disease-associated phenotypes. The recent development of methods to differentiate iPSCs into microglia has further allowed researchers to begin unraveling the contribution of microglial risk genes to human disease (Pocock and Piers, 2018). Yet, while these protocols have provided researchers with the ability to generate an abundance of human microglia in vitro, a recent study has highlighted that microglia, as highly plastic cells, undergo dramatic alterations when maintained outside of the brain environment, exhibiting numerous changes in gene expression within hours to days of transfer to culture conditions (Gosselin et al., 2017). Unfortunately, many of these in vitro-related transcriptional programs are mirrored in iPSC-derived microglia (iMGLs), demonstrating an important limitation to the modeling of microglial biology in a cell culture environment. Overall, this suggests that experiments utilizing in vitro microglia to model disease states may present an incomplete picture of their genetic state or how they respond to stimuli, presenting a major roadblock to a deeper and more complete understanding of in vivo microglial biology.
To begin to address this challenge, we and others performed initial experiments to determine the feasibility of transplanting human microglia or hematopoietic stem cells (HSCs) into the brains of immunodeficient mice (Abud et al., 2017; Bennett et al., 2018; Capotondo et al., 2017; McQuade et al., 2018). Yet, to date no studies have thoroughly examined and validated the phenotype, transcriptional profile, and functional responses of engrafted human microglia to injury or disease-associated pathology, steps that are critical for determining the suitability of this approach for studying in vivo microglia biology. Toward this goal, we present the development and validation of a chimeric model system that allows researchers to examine iPSC-derived human microglia in the context of the living mammalian brain.
RESULTS
Human iPSC-derived hematopoietic progenitor cells engraft and differentiate into cells expressing mature microglia markers
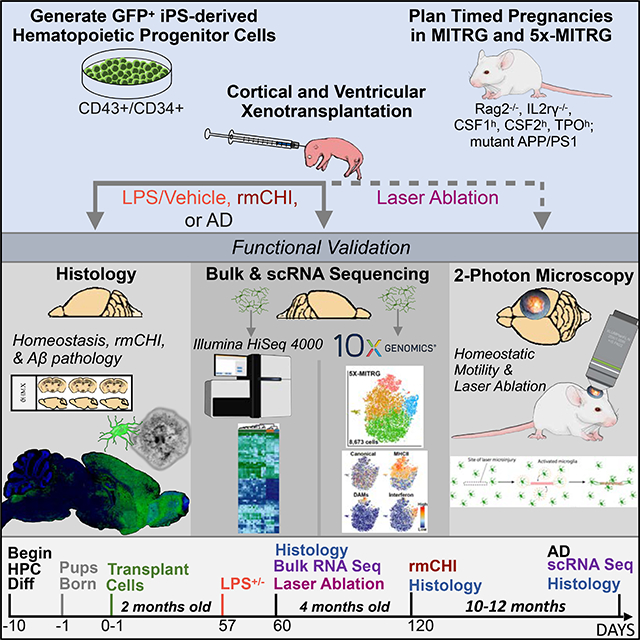
Given the developmental ontogeny of microglia, which arise from yolk sac-derived primitive hematopoietic progenitor cells (HPCs) (Prinz et al., 2017), we hypothesized that transplantation of iPSC-derived HPCs (iHPCs), as generated in McQuade et al. (2018) which express multiple genes associated with primitive HPCs (Figure 1A), into the early postnatal MITRG brain would result in robust engraftment of human cells that would differentiate into mature microglia. To test this, we transplanted GFP-expressing iHPCs directly into the lateral ventricles and overlying cortex of postnatal day 1 (P1) MITRG mice (Rongvaux et al., 2014) (Figure 1B). After allowing the mice to age for two months, immunohistochemical (IHC) analysis revealed robust engraftment of human cells throughout the forebrain that strongly expressed the homeostatic microglial marker P2RY12 (Figure 1C), IBA1 (Figure S1A), the myeloid transcription-factor PU.1 (Figure S1B), and the brain resident microglia-specific marker TMEM119, along with the human nuclei marker, Ku80 (Figure S1C–D). Further analysis revealed a high degree of concordance between the expression of GFP and P2RY12 (Figure 1D–G) as well as the complex, ramified morphology adopted by the transplanted iHPCs (Figure 1H–I) strongly suggesting that the engrafted iHPCs had differentiated into microglia.
Figure 1. iPSC-derived human HPCs differentiate into microglia and display robust engraftment within the forebrain of MITRG mice.

(A) A heatmap comparing iPSC, iHPC, and iMGL (McQuade et al., 2018) across a sampling of genes related to iPSC, hematopoietic stem cell (HSC), primitive HPC, erythromyeloid progenitor (EMP), and microglia lineages, along with lineage negative (Neg) genes, shows that iHPCs most closely resemble primitive HPCs but also express some EMP markers. (B) Schematic of transplantation paradigm. (C) Human P2RY12 staining displays the overall migration and distribution of xMGs 2 months after P1 transplantation (P2RY12, pseudocolored green; DAPI, blue). (D-G) Transplanted cells display near complete co-localization of cytosolic eGFP (green) with human P2RY12 (red). (H-I) Engrafted cells uniformly tile in the cortex and take on a highly ramified morphology. (J) Colocalization of (Green, cytoGFP), PU.1 (Red, all myeloid nuclei) and Ku80 (pseudocolored blue, human nuclei only) was used to quantify xMG engraftment. (K) Percentage of xMG engraftment in the cortex (CTX), the hippocampus (HIP), and the striatum (STR) from two separate experiments (HPCs #1 and HPCs #2). In some cases, brightness and contrast settings of confocal images were adjusted to reveal fine structures and morphology. Scale = 1mm (C), 50μm (G, H, & J), 10μm (I) See also Figures S1 & S2.
Assessment of the distribution of the xenotransplanted microglia (xMG) throughout the mouse brain revealed some variation depending on the distance from the site of transplantation. In areas that were directly targeted by human iHPC injection, robust xMG engraftment was observed along with very few host murine microglia (Figure S2A). In contrast, more distant regions displayed typical distribution and tiling of murine microglia with only an occasional xMG (Figure S2B). Quantification of microglia within the targeted regions, including the cortex, hippocampus, and striatum, demonstrated that ~80% of the PU.1+ microglia co-expressed the human-specific nuclear marker Ku80 (Figure 1J–K). Furthermore, no significant differences in engraftment were observed between different batches of iHPC transplants (F1,18=0.6923, p=0.4163), across different brain regions (F2,18=1.462, p=0.2580), or in the interaction between iHPC batch and region (F2,18=0.4417, p=0.6497) (Figure 1K) demonstrating the reproducibility of this approach.
Expression of human CSF1 is necessary and sufficient for the long-term engraftment of human microglia in the murine brain
We previously demonstrated that human microglia could survive for at least 2 months following transplantation into adult MITRG immune-deficient mice (Rag2 KO, IL2rγ KO) (Abud et al., 2017). However, we remained curious as to whether all three humanized genes present in the MITRG; hCSF1, hCSF2, or Thrombopoietin (hTPO) (Rongvaux et al., 2014), were necessary for xMG survival. Given the importance of CSF1R signaling for microglia survival, and previous reports that the murine CSF1 ligand cannot fully activate human CSF1R signaling (Elmore et al., 2014; Rathinam et al., 2011; Sieff, 1987), we hypothesized that expression of humanized CSF1 alone was necessary for robust engraftment of human microglia in the murine brain. To test this, we performed transplantations of GFP+ iHPCs into MITRG mice (Figure S2E) and MITRG mice which had the humanized hCSF2 and hTPO genes bred out (Figure S2F), revealing virtually identical engraftment of human cells in both strains and confirming that expression of hCSF2 and hTPO is not necessary for xMG survival. In stark contrast, no xMGs were detected in mice that expressed hCSF2 and hTPO but lacked hCSF1 expression (Figure S2G). Interestingly, the necessity for hCSF1 was also dose-dependent, as heterozygous hCSF1 mice exhibited partial survival of xMGs (Figure S2H). Lastly, transplantations into the parental mouse strain from which the MITRG model was developed (JAX# 017708), which includes Rag2/IL2rγ deletion and hCSF1 but lacks hCSF2 and hTPO, demonstrated that human iHPCs differentiate into microglia which engraft at levels that are consistent with transplantation into MITRG mice (Figure S2I). Taken together, these data demonstrate that human CSF1 is both necessary and sufficient to enable the long-term engraftment and survival of human microglia in the mouse brain.
Transplanted iHPCs acquire altered morphologies and phenotypic signatures in a niche-dependent manner
xMGs were observed throughout the forebrain and had adopted characteristic, region-specific distributions, morphologies, and marker expression levels. For example, within the cortex and olfactory bulb, xMGs created a mosaic network of ramified cells that tiled neatly with neighboring microglia, establishing distinct microdomains (Figure 2A–C). Within the hippocampus and overlying corpus collosum, the expected contrasting morphologies were present, with ramified GFP+/P2RY12+ cells appearing within CA1, and more elongated microglia expressing lower levels of P2RY12 within the overlying white matter of the corpus callosum (Figure 2D). In addition, relatively few xMGs were observed within the granule cell layer (Figure 2E), a hippocampal subregion that exhibits the lowest number of microglia (De Lucia et al., 2016; Shapiro et al., 2009). Within the striatum (Figure 2F), xMGs engrafted predominantly within the gray matter, avoiding the white matter bundles, again consistent with the typical distribution of microglia within this region (Savchenko et al., 2000). Interestingly, limited engraftment of GFP+/P2RY12+ xMGs could also be observed far from the transplantation sites, including parts of the cerebellum (Figure 2G) and the spinal cord, where xMGs adopted a more linear, less ramified morphology (Figure 2H).
Figure 2. Transplanted iHPCs adapt to diverse niches within the brain and differentiate into the four CNS macrophage subtypes in a context-dependent manner.

(A-H) High power images demonstrate the variety of morphological features xMGs display during homeostatic conditions, including neat tiling and complex ramifications in (A) the olfactory bulb (OB) and (B-C) the neocortex (CTX). (D) Within the corpus callosum (CC) overlying the hippocampus (Hip), xMGs exhibit a more elongated morphology with a diminished expression of P2YR12. (E-F) Consistent with normal anatomical distributions, xMGs also tend to avoid the granule cell layer of the dentate gyrus (GrDG) and the axon-bundles within the striatum (STR). (G-H) Remarkably, xMGs also migrate to parts of the cerebellum (CRB) and spinal cord (SC). (I-L) A subset of GFP+ cells exhibit an amoeboid morphology, linear organization, and lack the homeostatic microglia marker, P2RY12. These cells also encircle GLUT1+ blood vessels, localize to the perivascular space (J, K), and express CD163 (L) suggesting a perivascular macrophage (pvMϕ) phenotype. (M) Another population of GFP+/PU.1+ cells display robust engraftment in meningeal whole mounts. (N-O) These ameboid shaped meningeal macrophages (mMϕ) and can also be found in sections where parts of the meninges (MN) were preserved and are distinct from the fully ramified GFP+/hP2RY12+ microglia within the parenchyma (PAR). (P) Similar to the pvMϕ, these mMϕ are also GFP+/hP2RY12−/CD163+ (arrows). (Q-T) A few GFP+/hP2RY12−/CD163+ choroid plexus macrophage-like (cpMϕ) cells can be found within the choroid plexus (arrows). In some images, brightness and contrast settings were adjusted. Scale = 50μm (A, D-J, N-Q, S, T), 25μm (C, K), 20μm (L), 100μm (B, M, R).
In addition to these varying populations of P2RY12+ xMGs, we observed subsets of human cells that expressed GFP, yet exhibited a more ameboid morphology (Figure S1D, arrows). When further examined, these cells were found to lack P2RY12 expression, instead exhibiting morphology, localization, and markers typical of other CNS-myeloid cells (Figure 2I–T). This finding suggests that transplanted iHPCs maintain the potential to differentiate into the other yolk sac-derived CNS-myeloid cell types: perivascular (pvMϕ), meningeal (mMϕ), and choroid plexus (cpMϕ) macrophages. For example, GFP+/P2RY12− pvMϕ-like cells were found along the vasculature, in close opposition to GLUT1-expressing murine blood vessel endothelial cells (Figure 2I–K). These pvMϕ-like cells also exhibited colocalized expression of CD163 (Figure 2L), an established marker of non-microglial CNS myeloid cells (Goldmann et al., 2016). Whole meningeal mounts (Figure 2M) confirmed engraftment of more amoeboid GFP+/P2RY12− mMϕ-like cells which were also observed in sections where the meninges were preserved during cryosectioning (Figure 2N–O). As with the pvMϕ-like cells, these meningeal-localized cells also expressed CD163 (Figure 2P). Additionally, a small population of GFP+/P2RY12−/CD163+ cells was found within the choroid plexus (Figure 2Q–T). The relatively small number of cpMϕ-like cells is consistent with the understanding that cpMϕs are maintained in the adult brain through a partial turnover from definitive bone marrow hematopoietic stem cells, whereas microglia, pvMϕ, and mMϕ continually self-renew (Prinz et al., 2017). Overall, these results suggest that transplantation of iHPCs into the early postnatal brain permits the context-dependent maturation of microglia and other CNS myeloid cells.
xMGs acquire a transcriptomic signature that closely resembles in vivo human microglia
While IHC analysis revealed that xMGs expressed many microglial markers and adopted the morphology of homeostatic microglia, an in-depth transcriptomic analysis of multiple cell lines was needed to test whether xMGs fully acquire a microglial fate. Therefore, we developed methods to recover pure populations of xMGs, derived from fluorescent or non-fluorescent iPSC lines, from transplanted brains in order to perform RNA-sequencing (Figure S3). To assess xMGs in comparison to endogenous human microglia, transcriptomes were compared between xMGs, brain-derived human microglia (ExVivo), cultured human microglia (InVitro), and iMGLs differentiated from iPSCs (Table S1). To further increase the power of these comparisons, a previously published dataset (Gosselin et al., 2017) that included additional ExVivo and InVitro samples was also included (Table S1).
To explore the overall similarities and differences between these 49 human microglial samples, the studies were batch corrected (Figure S4A–B; Table S2) and principal component analysis (PCA) was performed. This analysis revealed a clear separation of microglia groups based on environment, with a high degree of clustering between the xMGs and ExVivo microglia (Figure 3A). However, as we were primarily interested in determining if transplantation had induced an in vivo transcriptomic profile, we used the dataset published by Gosselin et. al. to define a list of 3,432 brain-dependent microglial genes that were differentially expressed between the ExVivo and InVitro samples (Table S3). PCA analysis with this gene list further enhanced clustering of xMGs with ExVivo microglia, whereas iMGLs and InVitro microglia formed distinct, segregated clusters, demonstrating that in vivo differentiation of iHPCs within the murine brain induces a brain-dependent microglia transcriptome (Figure 3B).
Figure 3. Transplantation of human iHPCs into the murine brain recapitulates an in vivo human transcriptome.

(A) PCA plot, including 16,413 genes, comparing freshly isolated human microglia (ExVivo, green; n=17 patients), xMGs (purple; 3 iHPC cell lines, n=3–6 mice per line, 13 mice total), iMGLs (yellow; n=6 wells), and cultured human microglia (InVitro, orange; n=13 patients) from our lab (MBJ, circles) or the Gosselin samples (triangles). ExVivo microglia and xMGs cluster together while both in vitro groups (iMGLs and InVitro) cluster separately. (B) PCA comparing the 3,432 differentially expressed genes between the Gosselin ExVivo and InVitro microglia (FDR=0.05, LFC cutoff=±2). Again, xMGs cluster closely with ExVivo microglia, demonstrating that transplantation recovered a brain-dependent microglia signature. (C) Heatmap comparing sample groups from our lab (MBJ, Blue) and Gosselin et al. (Red) based on the top 190 brain-dependent microglia signature genes. Euclidean clustering shows samples cluster by environment (in vivo or in vitro) and xMG samples are intermixed with ExVivo microglia samples. (D) xMGs express transcription factors at levels that are comparable to ExVivo microglia, many of which were either lowly or not expressed in vitro (red text). (E) xMGs express microglia signature genes and activation markers, including P2RY12, TGFB1, and CX3CR1, at comparable levels to ExVivo microglia, suggesting that xMGs have taken on a homeostatic transcriptomic profile. xMGs also express the brain-dependent microglial gene TMEM119, which was not previously expressed in iMGLs. (F) xMGs express AD risk genes at levels that coincide with non-AD ExVivo microglia. This finding demonstrates that xMGs could be accurate surrogates for AD studies in mouse models. Heatmaps in D-F represent VST normalized expression values, averaged for all samples in a group. See also Figures S3 & S4 and Table S1.
To perform an enhanced, gene-level analysis, the brain-dependent gene list was cross-referenced to the 881-gene microglial signature published by Gosselin et al., resulting in 190 brain-dependent microglial signature genes (Figure 3C). Euclidean clustering of the samples mirrored the PCA clustering, with iMGLs and InVitro groups on one branch and the xMGs and ExVivo samples intermixed within the other branch (Figure S4C). However, while the xMG profile was highly similar to the ExVivo signature, differential gene expression (DGE) analysis between xMGs and ExVivo samples demonstrated that 97 of the 881 signature genes were differentially expressed (Table S3, Figure S4D). Interestingly, Euclidean clustering of all in vivo samples using these 97 differentially expressed genes revealed that, while xMGs clustered separately from most of the ExVivo samples, three of the youngest ExVivo samples (avg. age 16.5 months) were intermixed with the xMG samples (Figure S4E). This finding potentially suggests that xMGs have yet to fully mature at the examined time points. However, there were also many MHC Class II genes (Figure S4D) that were expressed at higher levels in the older ExVivo samples, possibly indicating an increased activation state. One explanation could be that xMGs were isolated from the healthy mouse brain whereas the ExVivo samples were isolated from patient tissue affected by epilepsy or collected from tumor margins, resulting in an activated state. Longer-term aging studies may provide the evidence needed to understand these small, albeit intriguing, differences.
Further gene-level analysis showed that transplantation into the brain environment induced a number of important microglia gene sets. Key microglia-associated transcription factors, including EGR1, FOS, FOSB, JUN, KLF2, KLF4, and SALL1 (Figure 3D), that were either not expressed or lowly expressed in vitro, were fully recovered following transplantation. We also focused on a number of microglia genes that have been proposed to be important for homeostasis or activation (Keren-Shaul et al., 2017; Krasemann et al., 2017), as it has been suggested that microglia derived from transplanted cells can exhibit an ‘activated’ profile (Bennett et al., 2018). With regards to homeostatic genes such as CX3CR1, P2RY12, SELPLG and TGFB1, xMG expression is similar to that of the ExVivo samples, with xMGs displaying slightly elevated expression (Figure 3E). In contrast, xMGs do not appear to present with increased expression of genes that have been implicated in microglia activation states including CLEC7A, ITGAX, IL21R, or SIGLEC1. Additionally, whereas in vitro iMGLs express very low levels of TMEM119, transplantation fully recovers expression, consistent with the recent demonstration that TMEM119 is a brain-dependent microglia gene (Bennett et al., 2016) (Figure 3E).
Finally, as we are interested in utilizing this model to study microglia responses to AD pathology, we compiled a list of AD risk genes to ensure that xMGs were not aberrantly expressing these transcripts. As with the previous analyses, xMGs presented with an expression profile closely mirroring that of the ExVivo samples (Figure 3F). Notably, genes of current interest in the field such as APOE, TREM2, BIN1, PLCG2, MS4A4A and MS4A6A, are expressed at ExVivo levels. These findings, in conjunction with the previously presented transcriptomic profiling, not only suggest that xMGs have the potential to be utilized in future disease studies but also, considering the human iPSC origin of these cells, offer a level of genetic similarity to human microglia that cannot currently be matched by other approaches.
xMGs survey the local environment and rapidly respond to focal brain injury
While validating the morphological, histological, and transcriptomic properties of human microglia within the rodent brain was critical, the utility of this chimeric approach is also dependent on whether xMGs can accurately recapitulate microglial function in vivo. Therefore, we utilized 2-photon microscopy to visualize the in vivo behavior of GFP+ xMGs. Live imaging of xMGs revealed an array of neatly tiled, highly ramified human cells that were actively surveying their immediate environment (Figure 4A, VideoS1), as evidenced by constant extension and retraction of processes at rates consistent with endogenous murine microglia (Figure 4B). Based on previously reported descriptions of endogenous murine microglia behavior (Nimmerjahn et al., 2005), these observations are highly consistent with the activity and morphology of homeostatic microglia.
Figure 4. xMGs survey their surroundings, rapidly respond to laser ablation, and interact with neuronal components after trauma.

Human microglia were visualized using multiphoton in vivo imaging in (A) homeostasis and (C) following focal, high intensity laser exposure. (A) Time coded colorization of homeostatic xMGs demonstrate high process motility, actively surveying their niches. Overlaid color images of t0–5 min. (right) more clearly reveals the morphological dynamics of individual cells (also see Video S1). (B) The average length of extension/retraction in 5 minutes found in xMGs (3.73±0.10μm, n=300 observations, 3 mice) was not different from those found in mouse microglia (3.51±0.08μm, n=300 observations, 3 mice) (p=0.25, U=42581). (C) Time coded colorization of microglial response to focal damage reveals that processes rapidly move toward and surround the site of injury within 8 minutes post laser ablation. At later timepoints (t25-t35), some microglia outside the direct injury region translocate (white arrow), positioning their cell bodies closer to the injury (also see Video S2). (D) The mean intensity at the injury site (local, dark green) is higher than the mean intensity 150um away (distant, light green), showing that xMG processes rapidly localize to the injury site (repeated measures two-way ANOVA, p = 0.006. Scale = 50μm). (E) The average length of extension toward the ablation site in 10 minutes by xMGs (11.76±0.90μm, n=22 observations, 3 mice) is similar to those by mouse microglia (11.01±0.87μm, n=37 observations, 3 mice) (p=0.34, U=346). (F-G) Transplanted MITRG mice underwent repeat mild closed head injury (rmCHI) and histological analysis was performed 2-months post-injury. (F) GFP+ xMGs infiltrate the injury site and express increased levels of CD68 (blue). (G) Higher-power images of GFP+ xMGs reveal β3-tubulin (red) and CD68 (blue) colocalized within xMGs (white arrows), indicative of microglial phagocytosis of neuronal components. All error bars reflect SEM. Scale = 200μm (F), 20μm (G). See also Videos S1 & S2.
Since xMGs appeared to be actively surveying the brain parenchyma, we next sought to challenge that behavior with a localized, parenchymal injury. Due to the limited availability of in vivo, functional microglia data from humans, we concentrated our analysis on focal laser ablation, an approach that has been used to induce a highly reproducible microglial response in mice (Davalos et al., 2005; Nimmerjahn et al., 2005). We again used 2-photon imaging to visualize xMGs in the immediate vicinity of the damage site as well as those distally located to the damage site and microglial response was monitored for up to 50 minutes post-injury (Figure 4C–D). In concordance with previously characterized murine microglial responses, laser damage elicited a rapid and highly localized response in which xMGs within 40μm of the damage site immediately responded by sending processes to assess the injury, while distal xMGs showed no obvious deviations from homeostatic surveillance activity (Video S2). Accordingly, quantification of mean GFP intensity within the damage site showed a steady increase for 30 minutes post-injury followed by a sustained intensity for up to 50 minutes of imaging, whereas distal GFP intensity remained consistent across all timepoints (Figure 4D). Importantly, the extension rate and magnitude of this human microglial injury response is similar to that of murine microglia (Figure 4E) (Davalos et al., 2005; Nimmerjahn et al., 2005), indicating that xMGs respond to local CNS injury in a manner that is consistent with in vivo microglia.
xMGs phagocytose neuronal components after brain trauma
In several murine models, repeat mild closed head injury (rmCHI) has been shown to induce sustained microglial activation and interaction with neurons post injury (Gold et al., 2018; Shitaka et al., 2011). Thus, to determine if xMGs can interact with murine neuronal components, mice were exposed to rmCHI two months post-transplantation and histological assessment of the injury site was performed 8-weeks later (Figure 4F–G). Low-power images reveal that GFP+ xMGs infiltrated the injury site (Figure 4F), while higher-power imaging revealed that lysosomes (CD68; blue) within GFP+ xMGs colocalized with β3-tubulin (red), indicative of phagocytosis of neuronal microtubule components and debris (Figure 4G; white arrows indicate colocalization of CD68 and β3-tubulin). Thus, xMGs can readily interact with and phagocytose degenerating neurons.
xMGs respond robustly to a systemic inflammatory challenge
Our next functional assessment focused on characterizing the xMG response to the commonly used peripheral immune challenge lipopolysaccharide (LPS). Mice transplanted with GFP+ iHPCs were aged 2 months and then administered either saline or LPS via intraperitoneal injection. Quantification of IHC images revealed a significant reduction in the homeostatic marker P2RY12 following LPS treatment and, conversely, a significant upregulation of CD45 (Figure 5A–C), consistent with an LPS-induced increase in microglial activation state. Interestingly, LPS also induced increased GFP expression, likely because of the activating effect of inflammatory stimuli on CMV-based promoters such as the CAG promoter used to drive GFP expression (Figure 5A–C) (Varley et al., 1995).
Figure 5. Differential responses of xMGs and iMGLs to lipopolysaccharide administration.

(A) xMGs treated with saline exhibit strong staining for the homeostatic microglial protein P2RY12 (pseudocolored gray) and cytosolic GFP expression (green). In contrast, LPS-treated xMGs downregulate P2RY12, whereas GFP intensity increases along with distinct alterations in microglial morphology (Scale=50μm). (B) An upregulation of CD45 immunoreactivity (gray) can been seen after LPS treatment (Scale=50μm). (C) Quantification of GFP reveals a significant increase in intensity with LPS stimulation (p=0.0107) accompanied by a significant decrease in P2RY12 intensity (p=0.0003), and a significant increase in CD45 intensity (p=0.0004). (D) DGE analysis between microglia isolated from saline and LPS-treated animals revealed 607 upregulated genes (red, right) and 287 downregulated genes (blue, left) (FDR=0.01, LFC cutoff=±2; Table S4). Many upregulated genes (labeled, right) have been implicated in immune activation while a number of downregulated genes (labeled, left) have been described as markers of microglia homeostasis. (E) Pearson correlation matrix comparing the entire transcriptome of each in vivo (xMG) saline and LPS treated sample with each in vitro (iMGL) saline and LPS treated sample. Heatmap colors correspond to R2 values and samples are clustered via Euclidean distancing. (F) Venn diagram comparing in vivo and in vitro LPS-induced, differentially expressed genes (FDR=0.01, LFC cutoff=±2), demonstrating few conserved changes. (G) Gene ontology overrepresentation analysis and GO term clustering reveals unique regulation depending on LPS treatment environment, with limited overlap between in vivo (blue) and in vitro (pink) groups. Bar plots represent mean±SEM. See also Figure S5 and Tables S1, S4, & S5.
In addition to the observed morphological and protein-level changes, xMGs were isolated from saline and LPS-treated mice in order to examine transcriptomic alterations. DGE analysis revealed that LPS treatment resulted in 607 up-regulated and 287 down-regulated genes (Figure 5D, Table S4). As anticipated, several of the highly upregulated genes (e.g. TMEM176A/B, IL21R, SPP1, MSR1, TLR2) are involved in cytokine recognition, phagocytosis, and pathogen responses while many downregulated genes (e.g. P2RY12, P2RY13, ITGAM, SELPLG) are typical markers of microglia homeostasis. Further assessment of gene ontology via gene set enrichment analysis (Subramanian et al., 2005) demonstrated a coordinated upregulation of pathways related to immune response, antigen presentation, and translation initiation (Figure S5A–C, Table S4). Additionally, examination of the differentially expressed xMG signature genes from Figure S4D showed that many genes were normalized toward ExVivo levels following LPS treatment, including some of the previously noted MHC Class II genes (Figure S5D, red text). This finding lends some preliminary support to our aforementioned hypothesis that, rather than being immature, xMGs transplanted into the healthy mouse brain may present with a less activated profile than microglia isolated from disease-affected human tissues.
The human microglia response to systemic inflammation is not accurately modeled by LPS in vitro
Many studies regarding microglia responses to LPS have been performed in vitro, by treating murine or human microglia directly with LPS (Abud et al., 2017; Pulido-Salgado et al., 2018). We therefore decided to recapitulate this approach by treating iMGLs made from the same GFP+ cell line as the LPS-exposed xMGs, with LPS in vitro. RNA-seq and DGE analysis revealed 888 up-regulated genes and 679 down-regulated genes following in vitro LPS treatment (Table S5). Compared to the in vivo results of 607 up-regulated and 287 down-regulated genes, it was immediately clear that the response to LPS in a dish was more dramatic than the response in vivo. Correlation analysis between saline and LPS samples from both environments demonstrated limited correlation between in vitro and in vivo groups, with the least correlated groups being in vivo LPS and in vitro LPS (Figure 5E). Unsurprisingly, the lack of correlation further manifested as a limited overlap between the DGE lists, with the LPS groups only sharing 22.7% of the up-regulated genes and an abysmal 4% of down-regulated genes (Figure 5F). To determine if these gene-level differences also manifested at the pathway level, we performed gene ontology (GO) analysis, which similarly revealed very little overlap between the two environments (Figure 5G, Table S5).
Transitions in the expression profile of xMGs are orchestrated by well-established microglial transcriptional regulators
An overarching goal of our approach was to develop a predictive experimental model that could be used to study human microglia in vivo. Therefore, we next sought to perform an unbiased analysis of the two environmental transitions that we had exposed human microglia to: one from in vitro to in vivo and another from in vivo to peripheral stimulation with LPS. We began by performing DGE analysis, to assess how these transitions altered the transcriptional state of xMGs, and then utilized k-means clustering (Lloyd, 1982) to identify 8 clusters of genes with expression profiles that were unique to each of the states (Figure 6A, Table S6). Exemplifying the powerful predictive potential of this approach, a subsequent literature-based search of all differentially expressed genes revealed that a large majority of these genes are highly relevant to microglial biology (Figure 6A). Likewise, many of these genes have been implicated in AD (Hickman et al., 2018). Next, we performed GO analysis to determine which gene classes changed in response to each transition (Figure 6B). Comparison of GO terms revealed that when xMGs undergo transplantation, genes related to synapse assembly, exploratory behavior, and cell-cell adhesion are strongly induced, all of which would be predicted to increase following xMG integration into the brain. In contrast, GO terms associated with genes that were upregulated following peripheral LPS stimulus were instead consistent with immune activation, interferon response, antigen presentation, and a hallmark of in vivo microglia activation: zinc homeostasis (Kauppinen et al., 2008).
Figure 6: xMG gene expression profiles are indicative of known microglial activation signatures.

(A) Heatmap of differentially expressed genes from the same GFP iPSC line differentiated into microglia in vitro versus GFP-xMGs in vivo, versus in vivo GFP-xMGs activated with LPS. Black lines on the right indicate genes that have previously been implicated in Alzheimer’s Disease and/or microglial biology. (B) Network graph of enriched GO terms generated from (A), when comparing up-regulated genes from in vitro, to in vivo, to in vivo with LPS stimulation. Nodes represent individual GO terms (gene sets), and edges represent the relatedness between them. (C) Summary of predicted transcription factors and their association with genes up- or down-regulated between states. Heatmaps represent their expression levels at each state. See also Tables S1 & S6.
To further test if the observed expression signatures were influenced by known microglial gene regulatory factors, we performed an unbiased search to identify transcription factor (TF) motifs enriched within the promoter regions of all genes that exhibited differential expression with each transition state (Figure 6C, Table S6). Using this approach, we were able to identify several TF motifs that were highly enriched during the in vitro to in vivo transition and known to be critical regulators of microglial homeostatic function (e.g. RUNX2, MEF, JUN, FOS, KLF). Similar analysis on the xMG+LPS samples identified unique TF motifs that have been implicated in the transformation of microglia to an activated state (e.g. IRF8, STAT3, RelA, CEBPA, MTF-1) (Table S6).
xMGs adjacent to beta-amyloid plaques exhibit a Disease Associated Microglia (DAM)-like phenotype
Since we confirmed that xMGs were capable of appropriately responding to both exogenous and endogenous inflammatory insults, we next sought to determine if xMGs could be utilized in a disease modeling application. As it was recently reported that murine microglia transition into distinct and highly specialized subpopulations upon chronic exposure to pathological beta-amyloid (Aβ) in AD mouse models (Keren-Shaul et al., 2017; Krasemann et al., 2017), we sought to verify whether a similar phenotype may occur in xMGs. We, therefore, transplanted GFP+ iHPCs into the newly developed 5X-MITRG mouse model and allowed the mice to age for 9 months. Subsequent histological analysis revealed a clear and marked difference in the expression of numerous DAM markers in xMGs near Aβ plaques (Figure 7). Similar to mouse DAMs and neurodegeneration-associated microglia (MGnD), xMGs in contact with plaques downregulated the homeostatic marker P2RY12 (Figure 7A–H), adopted an activated, ameboid morphology, and occasionally phagocytosed fibrillar Aβ (Video S3). Additionally, plaque-associated xMGs upregulated the DAM markers CD9 (Figure 7I–P), MERTK (Figure 7Q), APOE (Figure 7R), CD11C (Figure 7S), and TREM2 (Figure 7T).
Figure 7. xMGs down-regulate homeostatic markers and upregulate activation markers around Aβ plaques.

Representative images of xMGs in the hippocampus and subiculum of the 5X-MITRG at 9-months. (A-H) The homeostatic marker P2RY12 is downregulated in plaque-associated xMGs while more distal xMGs continue to express P2RY12. (cytosolic GFP, green; P2RY12, pseudocolored red; fibrillar amyloid (AmyloGlo), gray). (I-P) Plaque-associated xMGs also display and increase in the DAM gene CD9, while distal cells do not (cytosolic GFP, green; CD9, red; fibrillar amyloid (AmyloGlo), gray). (Q-T) Additionally, plaque-associated xMGs upregulate other DAM markers, including MERTK (Q), human APOE (R), CD11C (S), and TREM2 (T). (U-X) 5X-MITRG mice were transplanted with xMGs possessing either WT TREM2 (U) or a homozygous R47H mutation (V) and aged to 9 months. Quantification of xMG migration towards Aβ plaques (blue, Amyglo) revealed a significant decrease in plaque-associated R47H-expressing xMGs (green, hNuclei/Ku80; red, IBA1), but no significant difference in total plaque burden (X). ** p<0.001, scale=500um (A-D), 100um (E-L), 20um (M-S; U-V), 10um (T). See also Video S3.
We next sought to further test the utility of this model to study disease-relevant mutations by generating xMGs from a modified iPSC line. Deletion of TREM2 impairs the migration of murine microglia to Aβ plaques (Jay et al., 2017; Ulrich et al., 2014; Wang et al., 2015; Wang et al., 2016) and the AD-associated R47H mutation in TREM2 has been shown to impair the association between microglia and plaques in human AD subjects (Yuan et al., 2016) and the 5XfAD mouse model of AD (Song et al., 2018). To determine whether TREM2 plays a similar role in the migration of xMGs to Aβ plaques, we transplanted 5X-MITRG mice with HPCs derived from isogenic, CRISPR-edited iPSCs that carry either wild-type or R47H mutant TREM2. 9-months later, brains were examined by confocal microscopy and the association between plaques and xMGs examined by IMARIS 3D-quantification of confocal Z-stacks. As expected, this analysis revealed significantly less R47H xMGs clustering around Aβ plaques when compared to WT xMGs (Figure 7U–W) despite there being no significant difference in overall plaque burden across mice (Figure 7X). Taken as a whole, these data demonstrate that xMGs do, indeed, adopt relevant activation states in response to amyloid pathology and that the impact of disease-relevant microglial mutations can be accurately examined with this model.
Single-cell RNA-seq reveals transcriptomic alterations in response to AD pathology that are unique to human xMGs
While our histological analyses appeared promising, we sought to further characterize the xMG response to amyloid pathology at the transcriptomic level by isolating xMGs from the brains of aged MITRG (n=2) and 5X-MITRG (n=4) mice. Single-cell RNA sequencing (scRNA-seq) resulted in 10,184 MITRG and 8,673 5X-MITRG cells and average gene counts of 1,583 and 1,593 per cell, respectively. tSNE clustering of xMGs from both MITRG and 5X-MITRG mice (Figure 8A–B), from male and female mice (Figure S6A–B), showed similar groups of MHC Class II cells (Orange; HLA-DRB1, HLA-DPB1, HLA-DQA1, and CD74), cells responding to Type I Interferon (Blue; IFI6, IRF7, ISG15, STAT1, and IFIT3), a group resembling murine DAMs (Salmon; CD9, TREM2, LPL, and ITGAX), as well as a cluster that was defined by a lack of the other cluster markers, which we deemed homeostatic (Green). Additionally, the MITRG xMGs displayed a cluster defined by high levels of inflammatory chemokines (Purple; CCL2, CXCL10, CCL8, and CXCL11) while the 5X-MITRG xMGs did not have a clearly defined chemokine cluster but, instead, displayed a cluster of cells defined by genes related to secretory function. Interestingly, canonical microglia markers (P2RY12, P2RY13, CX3CR1, and TMEM119) showed a relatively even distribution across most clusters, but decreased expression within the DAM clusters (Figure 8A–B, Figure S6C–D, Table S7). Additionally, despite general similarities between the clusters present in the MITRG and 5X-MITRG xMGs, differences existed in the proportion of microglia within each cluster (Figure 8C), with the DAM and MHCII clusters being larger in the 5X-MITRG and the Interferon cluster being reduced.
Figure 8. Single-cell sequencing of xMGs from MITRG and MITRG-5X mice reveals altered population distributions and human-specific genetic responses.

(A) tSNE plot of 10,184 xMGs isolated from 10.5-month-old MITRG mice (n=2) reveals multiple clusters defined by genes related to MHC class II (Orange; HLA-DRB1, HLA-DPB1, HLA-DQA1, and CD74), Type I Interferon (Blue; IFI6, IRF7, ISG15, STAT1, and IFIT3), DAMs (Salmon; CD9, TREM2, LPL, and ITGAX), Inflammatory chemokines (Purple; CCL2, CXCL10, CCL8, and CXCL11), and a “homeostatic” cluster (Green) that was mainly defined by a lack of activation markers. Additionally, canonical microglia markers (P2RY12, P2RY13, CX3CR1, and TMEM119) showed a uniform distribution across all clusters besides DAMs. (B) tSNE plot of 8,673 xMGs isolated from 10.5 and 12-month-old 5X-MITRG mice (n=4) reveals similar clusters to the MITRG xMGs. Exceptions include the addition of a secretory cluster (Olive; ANAX3, AGR2, PLAC8, and PLA2G7) the loss of a clearly defined chemokine cluster. (C) Bar plot comparing the percentage of total cells making up each cluster in WT MITRG and 5X-MITRG tSNE plots. (D) DAM genes, reported by Keren-Shaul et al. (2017), from 5XfAD murine microglia were filtered to contain only genes defined by Ensembl or NCBI Homologene to have 1:1 human orthologs. Remaining genes were then compared to the differentially expressed genes between DAM and homeostatic xMGs (FDR≤0.01) demonstrating that limited overlap exists between the human xMG and mouse DAM genes. (E) Protein-level validation of human-specific DAM genes HLA-DRB1 (top) and LGALS3 (bottom) in both 5X-MITRG and human AD brain sections. See also Figure S6 and Table S7.
We next sought to further examine the expression levels of AD risk genes within the 5X-MITRG xMGs. Analysis of the expression levels of 50 AD risk genes revealed that 39 of these genes were expressed in at least one cluster and many were differentially expressed in specific clusters (FDR≤0.01; Figure S6E). Furthermore, analysis of the amino acid sequences of these genes demonstrated that a number of AD risk genes display low amino acid homology when compared to their human counterparts and/or ambiguous gene homologs, and with CD33 lacking any reliable homolog in the mouse (Figure S6E).
This observation led us to investigate whether the xMG approach was capable of revealing novel human-specific aspects of the microglia response to Aβ. To do so, we performed DGE analysis between the DAM and homeostatic clusters in the 5X-MITRG and compared these genes to the DAM genes published by Keren-Shaul et al. (2017), as this was the most directly comparable study published to date. This comparison revealed a substantial number of both up (363 genes) and down (378 genes) regulated genes that were unique to human xMGs (Figure 8D). However, it is important to note that while the study published by Keren-Shaul et al. (2017) is the only currently published study examining the 5XfAD murine microglia response to Aβ pathology at the single-cell level, other studies have attempted to elucidate a similar phenotype via bulk sequencing, microarray, and qPCR in APP/PS1 and 5XfAD mice (Kamphuis et al., 2016; Krasemann et al., 2017; Yin et al., 2017). Therefore, we also performed a comparison of our xMG DAM genes to an aggregated gene list composed of all the unique genes specified in these studies plus the Keren-Shaul study (Figure S6F; Table S7). While this comparison yielded slightly altered genes lists, there was still a high degree of discordance between the datasets, with 342 upregulated and 336 downregulated genes uniquely attributed to human xMGs.
In order to confirm that these differences were indeed relevant to the human microglial response to Aβ, we selected two genes, HLA-DRB1 and LGALS3 (galectin 3), which have both been implicated in AD (Allen et al., 2015; Boza-Serrano et al., 2019), for histological validation at the protein level in both xenotransplanted mice and AD-patient tissue. As shown in Figure 8E, both proteins are highly expressed in xMGs adjacent to murine Aβ plaques and this expression pattern bears a striking resemblance to the plaque-associated microglial labeling in AD-patient tissue. While an in-depth validation of all the unique xMG genes discovered in this analysis goes beyond the scope of the current article, this model clearly provides ample opportunity to investigate novel aspects of the in vivo microglial response to amyloid pathology. We, therefore, propose that xMGs are not just capable of recapitulating general in vivo microglia behavior, but that utilizing xMGs in conjunction with disease models provides a method of uncovering new genetic insights into human neurological disease.
Discussion
Over the last decade, the field has increasingly recognized the importance of microglia in both homeostatic brain function and neurological disease. As our understanding has grown, so too has the need to develop more predictive models of human microglia to study these highly adaptable cells. Traditionally, this has meant collecting and culturing microglia from postmortem or surgically resected tissue or trusting that rodent microglia faithfully recapitulate human microglial phenotypes. While these approaches continue to be fundamental to our understanding of microglial biology, recent advancements have led to a more widely applicable toolkit. With the development of the first protocols to generate microglia from patient-derived stem cells in vitro, researchers are now able to model and manipulate the complex, polygenic landscapes that potentially underlie human microglia function and dysfunction. However, the field has also recently recognized that, despite these exciting new capabilities, the plasticity that makes microglia so fascinating represents a double-edged sword that complicates our attempts at in vitro modeling.
To address this challenge, we turned to transplantation of human iPSC-derived HPCs into the humanized, immune-deficient mouse brain. This paradigm induces robust microglial engraftment and differentiation of the complete complement of CNS macrophage subtypes. Additionally, microglia arising from this approach recapitulate the complex expression profile that is characteristic of their endogenous human counterparts, dynamically react to local and peripheral insults, and robustly respond to Aβ pathology on both the morphological and transcriptomic levels. Taken together, these results support the conclusion that xMGs serve as suitable surrogates for endogenous human microglia, enabling new and informative in vivo studies of this important cell type.
Despite the many potential advantages of this chimeric model, it is important to note that xMGs expectedly come with caveats of their own, the most prominent being the requirement of an immune-deficient recipient organism. With recent studies demonstrating the interactive relationship between microglia and peripheral immune cells, especially in the context of neurodegeneration (Marsh et al., 2016), this deficiency is important to acknowledge. While this problem is by no means unique to this model, as in vitro models inevitably suffer from the same pitfall, determining the most effective method of rectifying this shortcoming will require many additional studies.
Our hope is that the assessments herein provide sufficient demonstrations of the utility of this model that will encourage further studies examining the complex in vivo interactions between human microglia and neurological disease. The experiments utilizing the 5X-MITRG model and subsequent single-cell analysis have already revealed a substantial genetic response to Aβ plaques that appears to be unique to human microglia. Additionally, our combination of this approach with an isogenic TREM2 mutant line, has highlighted the potential use of this model to interrogate the impact of individual risk genes on human microglia function. By leveraging CRISPR technology and the growing availability of patient iPSC lines, our hope is that this approach can be broadly used to examine the impact of mutations, genetic diversity, and polygenic risk on human microglial function, thereby improving our understanding of these complex and fascinating cells.
Star Methods
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to Mathew Blurton-Jones (mblurton@uci.edu). Requests for the 5X-MITRG mouse model will require co-approval from the original creator of the MITRG parental line (Richard Flavell, Yale University).
Experimental Model and Subject Details
Animals
All animal procedures were conducted in accordance with the guidelines set forth by the National Institutes of Health and the University of California, Irvine Institutional Animal Care and Use Committee. The MITRG mouse was purchased from Jackson Laboratories (stock #017711); this BALB/c/129 model includes two knockouts alleles, Rag2− (Rag2tm1.1Flv), γc− (Il2rgtm1.1Flv), and three humanized knock-in alleles, M-CSFh (Csf1tm1(CSF1)Flv), IL-3/GM-CSFh (Csf2/Il3tm1.1(CSF2,IL3)Flv), TPOh (Thpotm1.1(TPO)Flv). The related and parental M-CSFh mouse line was also purchased form Jackson Laboratories (stock # 017708) and contains Rag2 and Il2rg deletions and humanized M-CSFh. The 5xFAD-MITRG model was created by backcrossing the MITRG mouse with 5xFAD mice which overexpress co-integrated transgenes for Familial Alzheimer’s Disease (FAD) mutant APP (Swedish, Florida, and London) and mutant FAD PS1 (M146L and L286V). Progeny of these pairings were then genotyped and backcrossed with MITRG mice to return the 5 MITRG genes to homozygosity and maintain the APP/PS1 transgenic loci in the hemizygous state, resulting in the MITRG5x (Rag2−; γc−; M-CSFh; IL-3/GM-CSFh; TPOh; Tg(APPSwFlLon,PSEN1*M146L*L286V)6799Vas). To generate mice that lacked M-CSFh but included GM-CSFh and TPOh, MITRG mice were crossed with M-CSFh mice and then F1 progeny were crossed and genotyped to select mice that lacked either both or one copy of M-CSFh. For 2 photon living-imaging of mouse microglia, the CX3CR1-GFP (B6.129P2(Cg)-Cx3cr1tm1Litt/J; stock # 005582) mouse was used. All mice were age and sex matched and group housed on a 12h/12h light/dark cycle with food and water ad libitum.
Acquisition and maintenance of iPSC lines
iPSC lines were purchased from Coriell and the European Bank for induced pluripotent Stem Cells. The GFP cell line (Coriell, AICS-0036) was generated by CRISPR modification of the line WTC11 to insert a monoallelic mEGFP into the AAVS1 safe harbor locus (chromosomal location 19q13.4-qter) under the control of a CAGG promoter. The WT iPSC line (EBisc #BIONi010-C) was modified by CRISPR gene editing to generate the TREM2 R47H isogenic iPSC line (EBiSC #BIONi010-C-7). Maintenance of all iPSC lines involved culturing in feeder-free conditions in complete mTeSR E8 medium (Stemcell Technologies), in a humidified incubator (5% CO2, 37°C), with medium changed daily. Passaging was performed every 7–8 days using ReLeaSR (Stemcell Technologies) and cells were plated onto 6-well plates (Corning), coated with growth factor-reduced Matrigel (1mg/mL; BD Biosciences), in mTeSR E8 medium, supplemented with 0.5uM Thiazovivin (Stemcell Technologies) for the first 24 hours post-passage. All cell lines were tested for mycoplasma on a monthly basis, and confirmed to be negative, and cell line karyotyping was performed every ten passages.
Differentiation of Hematopoietic Progenitor Cells and in vitro Microglia from iPSCs
iHPCs and iPS-microglia were differentiated according to the protocol published by McQuade et al. (McQuade et al., 2018). To begin iHPC differentiation, iPSCs were passaged in mTeSR-E8 to achieve a density of 80 colonies of 100 cells each per 35mm well. On day 0, cells were transferred to Medium A from the STEMdiff™ Hematopoietic Kit (Stem Cell Technologies). On day 3, flattened endothelial cell colonies were exposed to Medium B and cells remained in medium B for 7 additional days while iHPCs began to lift off the colonies. On day 10, non-adherent CD43+ iHPCs were collected by removing medium and cells with a serological pipette. At this point, d10-d11 iHPCs can be frozen in Bambanker (Wako). Cells used for early-postnatal iHPC transplantation were thawed in iPS-Microglia medium (DMEM/F12, 2X insulin-transferrin-selenite, 2X B27, 0.5X N2, 1X glutamax, 1X non-essential amino acids, 400 μM monothioglycerol, and 5 μg/mL human insulin freshly supplemented with 100ng/mL IL-34, 50ng/mL TGFβ1, and 25 ng/mL M-CSF (Peprotech)) and allowed to recover for 24 hours, then resuspended at 62,500 cells/uL in 1X DPBS (low Ca2+, low Mg2+). Cells utilized for in vitro experiments continued microglial differentiation for 28 days. During the last 3 days in culture, 100ng/mL CD200 (Novoprotein) and 100 ng/mL CX3CL1 (Peprotech) were added to mature microglia in a brain-like environment.
Method Details
Early Postnatal Intracerebroventricular Transplantation of iHPCs
P0 to P1 MITRG mice were placed in a clean cage over a heating pad with a nestlet from the home cage to maintain the mother’s scent. Pups were then placed on ice for 2–3 minutes to induce hypothermic anesthesia. Free-hand transplantation was performed using a 30-gauge needle affixed to a 10μL Hamilton syringe, mice received 1μL of iHPCs suspended in sterile 1X DPBS at 62.5K cells/μL at each injection site (8 sites) totaling 500K cells/pup. Bilateral injections were performed at 2/5th of the distance from the lambda suture to each eye, injecting into the lateral ventricles at 3mm and into the overlying anterior cortex at 1 mm, and into the posterior cortex in line with the forebrain injection sites, and perpendicular to lambda at a 45° angle (Figure 1B). Transplanted pups were then returned to their home cages and weaned at P21.
Adult intracranial transplants
All mouse surgeries and use were performed in strict accordance with approved NIH and AALAC-certified institutional guidelines. Direct bilateral intracranial injections of WT iMGL into the cortex and hippocampus were performed on adult MITRG mice. Briefly, adult mice (3 months old) were anesthetized under continuous isoflurane and secured to a stereotaxic frame (Kopf), and local anesthetic (Lidocaine 2%) was applied to the head before exposing the skull. Using a 30-guage needle affixed to a 10μL Hamilton syringe, mice received 2 μL of mature iMGL suspended in sterile 1X DPBS at 50,000 cells/μL at each injection site. Transplantation was conducted bilaterally in the cortex and hippocampus at the following coordinates relative to bregma: anteroposterior, −2.06 mm; dorsoventral, −1.75 mm (hippocampus), −0.95 mm; mediolateral, ±1.75 mm. Cells were injected at a rate of 50,000/ 30s with 4min in between injections. The needle was cleaned with consecutive washes of PBS, 70% (vol/vol) ethanol, and PBS in between hemispheres and animals. Animals were allowed to recover on heating pads before being placed in their home cages and received 2 mg/mL Acetaminophen (Mapap) diluted in water for five days. Animals were perfused 2 months following surgery with 1X PBS followed by 4% paraformaldehyde, entire brains were removed for immunohistochemistry.
Immunohistochemistry and Confocal Microscopy
Animals were administered Euthasol and monitored for loss of consciousness. Once animals no longer responded to toe pinch, mice were intracardially perfused with ice cold 1X DPBS. If xMGs were being isolated from ½ brains, the remaining half brain was drop fixed in 4% (w/v) PFA for 48 hours, otherwise, the mice were intracardially perfused with 4% PFA and post-fixed for 24 hours. Samples were then cryoprotected in 30% (w/v) sucrose until the tissue sank in the solution. Brains were then cut either coronally or sagittally at a section thickness of 40um on a sliding microtome cooled to −79°C. Tissue sections were collected as free-floating sections in PBS with and 0.05% sodium azide. For staining, tissue was blocked for 1 hour in 1X PBS, 0.2% Triton X-100, and 10% goat or donkey serum. Immediately following blocking, sections were placed in primary antibodies diluted in 1X PBS and 1% goat or donkey serum and incubated overnight on a shaker at 4°C. Sections were then incubated in conjugated secondary antibodies for 1 hour, before washing and mounting on microscope slides. Immunofluorescent sections were then visualized and captured using an Olympus FX 1200 confocal microscope. In some cases, brightness and contrast settings of confocal images were slightly adjusted to reveal fine structures and morphology. Importantly, no such changes were made to any images used for quantification.
Quantification Percent xMG Engraftment
For analysis of xMG engraftment and its reproducibility, two separate preparations of iHPCs (HPCs #1; n=4 & HPC #2; n=4) were transplanted as described above, on separate days, and perfused at 2 months. Brains were section sagittally and stained with Ku80 (human nuclear marker) and PU.1 (myeloid nuclear marker). The Allen Brain Atlas was used to identify regions where, 4, 40x images per region were captured (Somatosensory Cortex: CTX; Hippocampus: HIP; and Striatum: STR), all images were acquired using identical settings with an Olympus FV1200 scanning laser confocal microscope. Using the “Spots” function in IMARIS software (Bitplane) total Ku80+ and PU.1+ spots were quantified separately. Then total human cells (all Ku80+ spots) and total mouse cells (all PU.1 positive spots minus total number of Ku80+ spots) were determined. Total percentage of human cells was then found by dividing total human cells by mouse cells and multiplying by 100. To determine any significant interactions between transplantation preparation and brain region a 2-way ANOVA was run using Prism 7 (Figure 1).
Antibody Immunoreactivity after systemic LPS Treatment
For analysis of antibody immunoreactivity all images for a given antibody were acquired using identical settings with an Olympus FV1200 scanning laser confocal microscope, then analyzed using the image processing package FIJI, a distribution of NIH Image J software. Images were first split into single channels (GFP and P2RY12) and thresholded to create a mask around cells. The resulting mask was then overlaid onto the original images and mean intensity values were measured within the ROI. For normalization, human cells were hand counted in the GFP channel and mean intensity was divided by total cell counts, then divided by the saline mean values. For analysis of CD45 immunoreactivity, IMARIS “Surfaces” function was used to detect CD45 immunoreactivity, then mean intensity values within CD45+ surfaces were measured. For normalization, intensity values for each animal were divided by the saline mean values. Normalized intensity values for all antibodies were tested for statistical significance (p<0.05) though unpaired t test using Prism 7 (n=5 LPS; 4 Saline).
Plaque Proximity and Total Plaque Area
To examine the impact of the TREM2 R47H mutation on plaque migration, iHPCs derived from isogenic wild-type and R47H mutant iPSCs (EBiSC # BIONi010-C and #BIONi010-C-7) were transplanted into P1 5X-MITRG mice. 9-months later, immunohistochemistry was performed, and confocal Z-stacks collected within the Piriform Cortex at 40x magnification using identical confocal settings (n=3 mice per TREM2 genotype). Human microglia numbers and locations were detected and quantified through Ku80 immunofluorescence using the “Spots” function in IMARIS software. IMARIS based quantification of human microglial proximity to plaques was done by first identifying Aβ plaques using the “Surfaces” function, then using the “Spots close to Surfaces” Matlab extension (threshold set to 10 μm). Total plaque area was measured from sum of surfaces for each image. Proximity to plaque and total plaque area were tested for statistical significance (p<0.05) though Welch’s t test using Prism 7.
Tissue dissociation for bulk RNA-seq
Following perfusion with ice cold PBS, whole or half brains were dissected, and the cerebellum was removed. Tissue was stored briefly in 1X HBSS until subsequent perfusions were completed. Tissue dissociation was then performed utilizing the Adult Brain Dissociation Kit (Miltenyi) and the gentleMACS Octo Dissociator with Heaters (Miltenyi) according to manufacturer guidelines with modifications. Briefly, tissue was cut into ~1mm3 pieces and placed into the C-tubes with the kit’s enzymes and samples were dissociated using the preprogrammed protocol. Following enzymatic digestion, samples were strained through a 70um filter and pelleted by centrifugation. Myelin and debris were removed by resuspending the pellet in 6mL 23% Percoll, overlaid with 2mL of 1X DPBS, and spinning at 400xg for 25 minutes at 4°C, with acceleration and brake set to 0. The supernatant was discarded, and the cell pellet was processed for flow cytometry.
Fluorescence activated cell sorting of xMGs
For sorting of xMGs expressing endogenous GFP, dissociated cell pellets were resuspended in 400uL of FACS buffer (2% BSA in 1X DPBS) containing either Zombie Violet (Biolegend, 1:100) or Propidium Iodide (PI) (Biolegend, 1:400) as a viability marker. For animals transplanted with non-fluorescent cell lines, cell pellets were resuspended in 100uL of FACS buffer containing 5ug of mouse and human Fc block (BD) and incubated for 5 minutes on ice. 100uL of FACS buffer including the following antibodies was then added to the sample for a final staining volume of 200uL: Rat anti-HLA-ABC-PE clone YTH862.2 (Novus, 1:100) and mouse anti-human CD11b clone ICRF44 (Biolegend, 1:200). Samples were then spun down and resuspended in 400uL of FACS buffer containing either Zombie Violet (Biolegend, 1:100) or PI (Biolegend, 1:400) as a viability marker. Samples were then sorted on a FACSARIA Fusion II (BD Biosciences) directly into 900uL of Trizol (Thermo Fisher), immediately vortexed to ensure rapid lysis, and placed on dry ice.
Bulk RNA Isolation from xMGs
FACS sorted xMGs, ranging from 40,000–172,000 cells, were stored at −80°C until RNA isolation. Samples were then thawed on ice and RNA was isolated by adding 140uL of TET (10mM Tris 8.0/0.01mM EDTA/0.05% Tween20) followed by 140uL Chloroform:Isolamyl alcohol 24:1 (Sigma). Samples were centrifuged at 15,000xg for 10 min. at 4°C and the aqueous phase was collected and added to 1.5uL of GlycolBlue (Thermo Fisher). 55uL (~1/10 supernatant volume) of 3M sodium acetate and 550uL (~1 supernatant volume) of isopropanol (Sigma) were added and the samples were mixed by inverting then stored at −20°C overnight. Samples were then spun at 15,000xg for 30 min. at 4°C, supernatants were removed from pellets, and 500uL of 75% ethanol was added to the pellet. Samples were then spun at 15,000xg for 30 min. at 4°C, and supernatants were removed from pellets. RNA pellets were dried at RT to remove any residual ethanol, resuspended in 14uL of RNase-free H2O, and used immediately or aliquoted and stored at −80°C for downstream analysis.
Isolation of microglia from human tissue
Human microglia were isolated from adult, normal appearing cortical tissue resected from non-malignant cases of temporal lobe epilepsy. Tissue was mechanically dissociated followed by gentle enzymatic digestion, using DNase and trypsin, to generate a single cell suspension. Cells were then passed through a nylon mesh filter and separated from debris via ultracentrifugation. Pelleted cells were resuspended in PBS (0.4% BSA), counted, stained with cell surface markers, and purified by flow cytometry (BD Biosciences FACs ARIA Fusion). Microglia were defined as live (Zombie Aqua Fixable Viability stain, Biolegend), CD11b+ (Anti-human PE, Biolegend) CD45low (Anti-human APC/Cy7, Biolegend), CX3CR1high (Anti-human FITC, Biolegend) single cells. ExVivo samples were immediately pelleted, resuspended in Trizol LS (Thermo Fisher Scientific), and stored at −80°C until subsequent RNA isolation.
InVitro samples were pelleted, resuspended, counted, and plated at 2×106 cells/mL in MEM supplemented with heat-inactivated FBS (5%), P/S (0.1% v/v) and glutamine (0.1% v/v.). Microglia were grown for 3 days, collected and plated at 1×105 cells/mL, and maintained in culture for 6 days during which time cells received two treatments of TGFb (20 ng/mL) on days 3 and 5. Human fetal brain tissue was obtained from the Fetal Tissue Repository (Albert Einstein College of Medicine, Bronx, NY). Total RNA was isolated using standard Trizol LS (Thermo Fisher Scientific) protocols and stored at −80°C.
RNA analysis, Library Construction and Bulk RNA-seq
RNA integrity (RIN) values were determined using an Agilent Bioanalyzer 2100 series and RNA concentrations were assayed by Qubit and the average RIN value for RNA samples used in sequencing was 9.4. 10ng of RNA was used for library construction using ClonTech SMART-seq V4 Ultra Low Input kit (Takara Bio) which utilized poly-A selection to enrich for mRNAs. The quality of the DNA libraries was assayed using the Agilent 2100 bioanalyzer high sensitivity DNA assay and the DNA high sensitivity Qubit. The libraries were quantified by Kapa qPCR, normalized to 2nM and then multiplexed for sequencing on the Illumina HiSeq 4000 platform with single read 100 base chemistry.
Tissue dissociation for scRNA-seq
Following perfusion with ice cold PBS containing 5ug/ml actinomycin D (act D), half brains were dissected, and the cerebellum was removed. Tissue was stored briefly in RPMI 1640 containing 5ug/mL act D, 10uM triptolide, and 27.1ug/mL anisomycin until subsequent perfusions were completed. Tissue dissociation was then performed utilizing the Tumor Dissociation kit, human (Miltenyi) and the gentleMACS OctoDissociator with Heaters (Miltenyi) according to manufacturer guidelines with modifications. Briefly, tissue was cut into ~1mm3 pieces and placed into the C-tubes with the kit’s enzymes, 5ug/mL act D, 10uM triptolide, and 27.1ug/mL anisomycin and samples were dissociated using the preprogrammed protocol. Following enzymatic digestion, samples were strained through a 70um filter and pelleted by centrifugation. Myelin and debris were removed by resuspending the pellet in 8mL 23% Percoll, overlaid with 2mL of 1X DPBS, spinning at 400xg for 25 minutes at 4°C, with acceleration and brake set to 0, and discarding the myelin band and supernatant.
Magnetic Isolation of xMGs for scRNA-seq
Dissociated cell pellets were resuspended in 160uL FACS buffer (0.5% BSA in 1X DPBS) + 40uL Mouse cell removal beads (Miltenyi) and incubated at 4°C for 15 minutes. Samples were then separated using LS columns and the MidiMACs separator (Miltenyi) and the human cells were collected in the flow through. Cells were pelleted via centrifugation (10 minutes, 400xg) and resuspended to ~1,000 cells per microliter in FACS buffer, according to counts performed on a hemocytometer.
Single-cell sequencing via 10X
Following magnetic isolation, the scRNA-seq library preparation was performed according to the Chromium Single Cell 3’ Reagents kit v2 user guide. Briefly, a volume of single cells to target a capture of 10,000 cells was used in the generation of GEMS (gel bead in emulsions). The workflow was followed according to the 10X protocol and sequenced on an Illumina HiSeq 4000. All samples were multiplexed and sequenced to a depth of 60–100K reads per cell.
Intraperitoneal LPS injections
A neuroinflammatory state was induced via systemic LPS treatment. Animals received i.p. injections of either 2mg/kg LPS (eBioscience) (n=5 mice) or PBS (n=4 mice) once every 24 hours over a 72-hour period (3 injections total). 12-hours after the final LPS treatment, animals were euthanized, perfused with PBS, and half brains were collected for either IHC or transcriptomic analyses, as described above.
In vitro iMGL LPS Stimulation
Day 38 iMGLs were plated at 200,000 cells/well on a Matrigel-coated (1mg/well) 24-well-plate containing 2mL of microglia medium (McQuade et al., 2018). Cells were treated with either LPS (100ng/mL; eBioscience) or DPBS (Ca2+ and Mg2+ free) and allowed to incubate for 24 hours at 37°C before immediate lysis for RNA isolation (n=3 wells per treatment group).
Cranial Window Implantation
Mice were anesthetized with isoflurane (Patterson Veterinary) in O2 (2% for induction, 1–1.5% for maintenance). To provide perioperative analgesia, minimize inflammation, and prevent cerebral edema, Carprofen (10 mg/kg, s.c., Zoetis) and Dexamethasone (4.8mg/kg, s.c. Phoenix) were administered immediately following induction. Ringer’s lactate solution (0.2mL/20g/hr, s.c, Hospira) was given to replace fluid loss. Sterile eye ointment (Rugby) was used to prevent corneal drying. Surgical tools were sterilized using a hot glass bead sterilizer (Germinator 500). Following hair removal, Povidone-iodine (Phoenix) and Lidocaine Hydrochloride Jelly (2%, Akorn) was used to disinfect and numb the scalp, respectively. The scalp and underlying connective tissue were removed to expose the parietal and interparietal bone. Lidocaine hydrochloride injectable (2%, Phoenix) was used for muscle analgesia and the right temporal muscle detached from the superior temporal line. The skull was dried using ethanol (70% in DI water) and a thin layer of Vetbond Tissue Adhesive (3M) applied. Custom-printed ABS headplates were attached using Contemporary Ortho-Jet liquid and powder (LANG) at an angle parallel to the skull. A small craniotomy (3mm diameter) was performed over the right hemisphere 2.5mm anterior and 3mm lateral lambda. Hemostatic gelfoam sponges (Pfizer) pre-soaked in sterile saline (CareFusion AirLife Modudose) were used to absorb dural bleeding. Surgery was terminated if dural tears or parenchymal hemorrhage was observed. A 4mm glass coverslip (World Precision Instruments) was placed over the exposed brain and its edges attached to the skull first with a thin layer of Vetbond and second with dental acrylic. Following surgery, mice recovered in their home cage over a warm heating pad until normal behavior resumed (~15–30 minutes). Postoperative care consisted of daily Carprofen injections (10mg/kg, s.c.) for one week.
Two-Photon Imaging and Laser Ablation
Fluorescence was gathered with a resonant two-photon microscope (Neurolabware, Los Angeles, CA) with 900 nm excitation light (Mai Tai HP, Spectra-Physics, Santa Clara, CA). A 16x Nikon water immersion lens (0.8 NA) or a 20x water immersion lens (Olympus, 1.0 NA) was used. Emissions were filtered using a 510/84nm and 607/70 nm BrightLine bandpass filter (Semrock, Rochester, NY). Image stacks (2.89μm step size) were gathered every 30–300s using Scanbox acquisition software (Scanbox, Los Angeles, CA) at a depth of 150–250μm below the meninges, corresponding to cortical layer 2/3. Laser ablation consisted of scanning over a small region (70×10μm, 1.2–1.5W) at magnification 25 for 1 minute.
Homeostatic and Response to Laser-Induced Ablation Analysis
All image stacks were processed and analyzed using the image processing package FIJI. Images were temporally binned down to 5 min. intervals and the resulting z stack used to analyze homeostatic activity and response to laser-induced ablation. To measure extension and retraction rates, the first 100 notable changes in process length was measured manually for each mouse. To discriminate ablation-specific motility from homeostatic extension/retraction, we restricted our measurements to long process extensions (>20 μm) growing toward the laser ablation site. The increase in microglia processes toward the laser-ablated site is reflected by an increase in the amount of GFP fluorescence. Therefore, we measured the local microglial response to laser-induced ablation by taking the average intensity of GFP within a circle (r = 50μm) centered at the epicenter of damage. These values were compared to the average intensity of GFP within a circle (r = 50μm) centered 100μm away from the epicenter of damage. The intensity at any timepoint (tx) was normalized using the intensity at t5 min.
Repeat Mild Closed Head Injury (rmCHI)
2 month old, EGFP HPC transplanted MITRG mice underwent repeat mild closed head injury (rmCHI) as described by Gold et al. (Gold et al., 2018). Briefly, transplanted MITRG mice were randomly assigned to either sham or hit groups Sham animals received 5 days of Isoflorane anesthesia only, with no injury, 5-Hit animals received 5 days of Isoflorane anesthesia with 1 injury each day. Injuries were performed using a controlled cortical impact device (TBI-0310 Head Impactor, Precision Systems and Instrumentation, LLC, Fairfax Station, VA) Injury parameters are as follows: speed: 5 m/s; depth: 1mm; dwell time: 50ms. Following injury, animals rested until euthanized for histology 8 weeks later.
Quantification and Statistical Analysis
Bulk RNA-seq Data Analysis
FASTQ files were preprocessed using BBDuk (Bushnell, 2018) to filter out ribosomal RNA and PhiX reads, trim Illumina adapters, and to quality trim any base pairs below a PHRED score of 10. FASTQC (Andrews, 2014) was then performed to verify the quality of the sequencing files and all files were determined to be of sufficient quality for downstream processing. Reads were then pseudoaligned to the human GRCh38.p12 transcriptome (Ensembl release 94) (Schneider et al., 2017; Zerbino et al., 2018) using Kallisto (Bray et al., 2016), transcripts were summarized to the gene level via tximport (Soneson et al., 2015), and differential gene expression analysis was performed using DESeq2 (Love et al., 2014) after removing genes with summed counts <10. For Kallisto pseudoaligment of samples without available library information, an average fragment length of 250 and a standard deviation of 120 were used.
For TF motif analysis, RNA-seq reads were checked for quality using FASTQC (Andrews, 2014). Reads were then trimmed using Trimmomatic (Bolger et al., 2014) to remove Illumina adapters and regions of reads with PHRED quality scores below 30, as well as the leading and trailing 15 base pairs of each read. Reads with a minimum length of 30 after trimming were retained. Reads were then aligned to the GRCh38.p12 transcriptome (Ensembl release 94) using Bowtie2 (Langmead and Salzberg, 2012) and summarized at the gene level using tximport (Soneson et al., 2015). Differential expression analysis was performed using DESeq2 (Love et al., 2014).
scRNA-seq Data Analysis
FASTQ files were aligned to a dual-species transcriptome made up of the human GRCh38.p12 transcriptome (Ensembl release 95) and the mouse GRCm38 transcriptome (Ensembl release 95) using CellRanger (v2.2.0) with default commands and an expected cell count of 5,500. Following alignment, all barcodes that were identified in the mouse alignment were removed from the human dataset.
Bulk RNA-seq Data Visualization
Data was normalized and converted to a log2 scale for visualization using DESeq2’s varianceStabilizingTransformation followed by batch correction using the removeBatchEffect function from limma (Ritchie et al., 2015). Heatmaps were generated using the R “Pheatmap” package (Kolde, 2018) while volcano and bar plots were generated using the “ggplot2” package (Wickham, 2016).
scRNA data Visualization and Differential Gene Analysis
UMI count tables, for 13,597 MITRG xMGs and 11,054 5X-MITRG xMGs, were read into Seurat (v2.3.4) (Butler et al., 2018) for preprocessing and clustering analysis. First, cells were log normalized, centered, and scaled (Default settings) followed by PCA using all genes in the dataset. PCs 1:10 were used for clustering with a resolution parameter of 0.3. Clusters identified as doublets, dividing, or gene poor (representing damaged cells) were then discarded before further analysis. Cells passing these QC parameters were then merged by genotype (e.g. Male and Female 5X-MITRG) using the MergeSeurat function.
Secondary QC cutoffs were then applied to include only cells with less than 32% ribosomal genes, 7% mitochondrial genes, greater than 800 genes but less than double the median gene count, and greater than 500 UMI but less than double the median UMI count. This resulted in 10,184 MITRG and 8,673 5X-MITRG cells with average UMI counts of 3,201 for MITRG and 3,242 for 5X-MITRG, and average genes counts of 1,583 and 1,593, respectively. Data for these cells were log normalized, centered, and scaled, using the ‘negbinom’ general linear model, while regressing out library size differences, percent mitochondrial genes, and sex. A second round of PCA was then performed on the cleaned data and interrogation of the PCs revealed a set of 41 highly variable genes (TableS7) contained across all samples, regardless of genotype or sex. These genes were present across multiple PCs and had a large effect on clustering but, as a gene set, had no discernable biological relevance. Therefore, we treated these genes as a technical artifact and removed the set from the variable gene list before repeating subsequent PCA analysis.
PCA was then performed using a set of variable genes selected according to expression and dispersion cutoffs (low expression cutoff = 0.01, high expression cutoff = 3, low dispersion cutoff = 1.1). Subsequent tSNE clustering was performed using PCs 1:14 for xMGs isolated from the MITRG mouse and PCs 1:14 for xMGs isolated from the 5X-MITRG mouse, both at a resolution of 0.45. tSNE plots were generated using the same PCs used for clustering and variable genes were determined between clusters using the Wilcoxon Rank Sum Test.
Comparison of Human and Mouse DAM Genes
Differential genes analysis was performed between the DAM and Homeostatic clusters for 5X-MITRG xMGs with an FDR cutoff of 0.01 and the requirement that the gene be expressed in at least 10% of the cluster. Murine DAM genes were obtained from Keren-Shaul et al. (Keren-Shaul et al., 2017) (Table S7) and filtered to remove pseudogenes, genes without a verifiable official name (e.g. GM*), as well as ambiguous genes without 1:1 mouse to human homologs, as specified by either Ensembl or NCBI Homologene. The subsequent comparison utilizing qPCR, microarray, and bulk RNA-seq microglia gene lists (Kamphuis et al., 2016; Krasemann et al., 2017; Yin et al., 2017) was performed by merging the unique genes from all four lists and filtering the list for genes with 1:1 homologs.
xMG Response to Laser Ablation
All image stacks were processed and analyzed using the image processing package FIJI, a distribution of NIH Image J software. Stacks were temporally binned (5 min) and the average GFP intensity within a circle (r = 50μm) centered at site of damage was used to assess local microglial response to laser ablation. A second circle (r = 50μm) at least 100μm away from the site of damage was used to assess distal changes in intensity following laser ablation. The intensity at any timepoint (tx) was normalized to the intensity in that area at t5min. To determine differences in GFP intensity within and between groups we used a repeated measures two-way ANOVA corrected for multiple comparison (Sidak).
Gene set enrichment analysis
Pathway analysis was performed using the Broad Institute’s Gene Set Enrichment Analysis (GSEA) desktop application version 3.0 (Subramanian et al., 2005). Differential expression results, generated by comparing saline and LPS-treated samples, were taken from DESeq2 and ranked according to the sign of the fold change * -log10(adjusted p-value). The entire list of ranked genes present in both samples was then input into GSEA’s pre-ranked analysis module and the module was set to perform 1000 permutations using the weighted enrichment statistic. The gene list was compared to MSigDB’s gene ontology database (c5.all.v6.2.symbols.gmt) consisting of the entire set of Gene Ontology terms (Ashburner et al., 2000; The Gene Ontology, 2017) and results were output, excluding any gene sets less than 15 genes or more than 5000 genes in length.
Gene Ontology Analysis
Gene ontology (GO) analysis was performed using PANTHER (Mi et al., 2017). Upregulated or downregulated genes (|log fold change| >= 2) were compared to genes with a log fold change between 2 and −2 using a statistical overrepresentation test. Significantly (p<0.05) overrepresented GO terms were clustered and visualized using the EnrichmentMap plugin (Merico et al., 2010) in Cytoscape 3.6.1 (Shannon et al., 2003).
Transcription Factor Binding Prediction and Co-Occurrence
Potential transcription factor binding sites were identified by searching the region 500bp upstream of genes using FOCIS (Webster, 2013) for binding motifs from the JASPAR database (2018 release) (Khan et al., 2018). Upregulated (log fold change >= 2) or downregulated (log fold change <= −2) genes were compared to genes with a log fold change between 2 and −2 as background. Transcription factors with putative binding sites upstream of the same gene were considered to be co-occurring. Co-occurrence frequency of a transcription factor pair was described using the Jaccard index.
Literature search
Pubmed searches were performed using the term “microglia” or “Alzheimer” plus the name of each differentially expressed gene and transcription factor of interest. At least one publication result matching both terms was considered a positive result.
Statistical Analysis
All statistical analyses were performed in either R programming language or utilizing GraphPad Prism.
Data and Code Availability
The bulk and single-cell RNA-seq datasets generated during this study are available through GEO SuperSeries accession number GSE133434 or individual series accession numbers GSE133432 or GSE133433, respectively. The bulk RNA-seq datasets generated by Gosselin et al. (Figure 3) are available through NCBI dbGaP, accession number phs001373.v1.p1. The bulk RNA-seq datasets generated by Abud et al. (Figure 3) are available through GEO, series accession number GSE89189. The bulk RNA-seq datasets generated by McQuade et al. (Figure 1) are available through GEO, series accession number GSE117829.
Additional Resources
Not applicable
Supplementary Material
Video S2. In vivo, 2-photon recording of xMG response to laser ablation (Related to Figure 4).
Video S1. In vivo, 2-photon recording of homeostatic xMGs (Related to Figure 4).
Video S3. Z-stack of Aβ internalization by an xMG (Related to Figure 7).
Table S3. DGE analysis related to microglia signature gene lists (Related to Figure 3).
Table S5. DGE analysis for in vitro LPS treatment (Related to Figure 5).
Table S6. DGE and Z-scores related to transcription factor motif analysis (Related to Figure 6).
Table S4. DGE analysis and GSEA analysis for in vivo LPS treatment (Related to Figure 5).
Table S7. scRNA-seq DGE and Venn Diagram comparisons (Related to Figure 8).
Table S2. Gene expression for assessment of xMG in vivo transcriptomic signature (Related to Figure 3).
Acknowledgements
This work was supported by NIH grants AG048099, AG055524, AG016573, AG056303 (M.B.J.), 1DP2GM119164 (R.S.), AG061895 (H.D.), EY028212 (S.P.G.), Brain Initiative R01 DA048813 (S.G., M.B.J, R.S.), MNI New Investigator Start-up Fund (L.M.H.), NIA Training Grant AG00096 (J.H.), Training Grant NS082174 (A.M.), F31EY028046 (D.F.V.), MSTP Training grant T32GM008620 and F30 Training grant EY029596-02 (R.A.), CIRM RT3-07893 (M.B.J.), the Women’s Alzheimer’s Movement: UCI MIND/WAM 01-2018 (M.B.J. & S.P.G.), and the Susan Scott Foundation (M.B.J.). C.Z.H. is supported by Cancer Research Institute Irvington Fellowship. R.S. is a Pew Biomedical Scholar. Experiments using the GFP-expressing iPSC line AICS-0036 were made possible through the Allen Cell Collection, available from Coriell Institute for Medical Research. iPSC line 10C (BIONi010-C) was purchased from the European Bank for Induced Pluripotent Stem Cells (EBiSC), funded by IMI Grant Agreement No. 115582. The UCI Genomics High Throughput Facility Shared Resource of the Cancer Center was supported by NIH grants P30CA-062203, 1S10RR025496-01, 1S10OD010794-01, and 1S10OD021718-01. The authors would like to thank Dr. Melanie Oakes, Seung-Ah Chung, Valentina Ciobanu, Christina Lin and Charlotte Spassoff for their outstanding work with RNA-seq; Vanessa Scarfone and Jennifer Atwood flow cytometry training; Dr. Samuel Marsh and the lab of Dr. Beth Stevens for providing expert guidance and cell isolation protocols for single-cell sequencing; Drs. Edsel Abud and Samuel Marsh for piloting initial HPC transplants into adult mice; and Dr. Laura McIntyre for P1 transplantation training.
Footnotes
Declaration of Interests
M.B.J. is a co-inventor of patent WO/2018/160496, related to the differentiation of human pluripotent stem cells into microglia.
References
- Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CHH, Newman SA, Yeromin AV, Scarfone VM, Marsh SE, Fimbres C, et al. (2017). iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 94, 278–293 e279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen M, Kachadoorian M, Carrasquillo MM, Karhade A, Manly L, Burgess JD, Wang C, Serie D, Wang X, Siuda J, et al. (2015). Late-onset Alzheimer disease risk variants mark brain regulatory loci. Neurol Genet 1, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S (2014). FastQC: a quality control tool for high throughput sequence data.
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett FC, Bennett ML, Yaqoob F, Mulinyawe SB, Grant GA, Hayden Gephart M, Plowey ED, and Barres BA (2018). A Combination of Ontogeny and CNS Environment Establishes Microglial Identity. Neuron 98, 1170–1183 e1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A, Tucker A, et al. (2016). New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A 113, E1738–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boza-Serrano A, Ruiz R, Sanchez-Varo R, Garcia-Revilla J, Yang Y, Jimenez-Ferrer I, Paulus A, Wennstrom M, Vilalta A, Allendorf D, et al. (2019). Galectin-3, a novel endogenous TREM2 ligand, detrimentally regulates inflammatory response in Alzheimer’s disease. Acta Neuropathol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Bushnell B (2018). BBMap.
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capotondo A, Milazzo R, Garcia-Manteiga JM, Cavalca E, Montepeloso A, Garrison BS, Peviani M, Rossi DJ, and Biffi A (2017). Intracerebroventricular delivery of hematopoietic progenitors results in rapid and robust engraftment of microglia-like cells. Sci Adv 3, e1701211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, and Gan WB (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8, 752–758. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Golde TE, and Lagier-Tourenne C (2018). Animal models of neurodegenerative diseases. Nat Neurosci 21, 1370–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lucia C, Rinchon A, Olmos-Alonso A, Riecken K, Fehse B, Boche D, Perry VH, and Gomez-Nicola D (2016). Microglia regulate hippocampal neurogenesis during chronic neurodegeneration. Brain Behav Immun 55, 179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL, et al. (2014). Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82, 380–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, Cao Y, Lee SH, Haddick PCG, Ngu H, et al. (2018). Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep 22, 832–847. [DOI] [PubMed] [Google Scholar]
- Gold EM, Vasilevko V, Hasselmann J, Tiefenthaler C, Hoa D, Ranawaka K, Cribbs DH, and Cummings BJ (2018). Repeated Mild Closed Head Injuries Induce Long-Term White Matter Pathology and Neuronal Loss That Are Correlated With Behavioral Deficits. ASN Neuro 10, 1759091418781921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmann T, Wieghofer P, Jordao MJ, Prutek F, Hagemeyer N, Frenzel K, Amann L, Staszewski O, Kierdorf K, Krueger M, et al. (2016). Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol 17, 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, Jaeger BN, O’Connor C, Fitzpatrick C, Pasillas MP, et al. (2017). An environment-dependent transcriptional network specifies human microglia identity. Science 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman S, Izzy S, Sen P, Morsett L, and El Khoury J (2018). Microglia in neurodegeneration. Nat Neurosci 21, 1359–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, Lamb BT, and Landreth GE (2017). Disease Progression-Dependent Effects of TREM2 Deficiency in a Mouse Model of Alzheimer’s Disease. J Neurosci 37, 637–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphuis W, Kooijman L, Schetters S, Orre M, and Hol EM (2016). Transcriptional profiling of CD11c-positive microglia accumulating around amyloid plaques in a mouse model for Alzheimer’s disease. Biochim Biophys Acta 1862, 1847–1860. [DOI] [PubMed] [Google Scholar]
- Karch CM, Cruchaga C, and Goate AM (2014). Alzheimer’s disease genetics: from the bench to the clinic. Neuron 83, 11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppinen TM, Higashi Y, Suh SW, Escartin C, Nagasawa K, and Swanson RA (2008). Zinc triggers microglial activation. J Neurosci 28, 5827–5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290 e1217. [DOI] [PubMed] [Google Scholar]
- Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, Bessy A, Cheneby J, Kulkarni SR, Tan G, et al. (2018). JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res 46, D260–D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolde R (2018). pheatmap: pretty heatmaps.
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, et al. (2017). The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47, 566–581 e569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd S (1982). Least squares quantization in PCM. IEEE Transactions on Information Theory 28, 129–137. [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh SE, Abud EM, Lakatos A, Karimzadeh A, Yeung ST, Davtyan H, Fote GM, Lau L, Weinger JG, Lane TE, et al. (2016). The adaptive immune system restrains Alzheimer’s disease pathogenesis by modulating microglial function. Proc Natl Acad Sci U S A 113, E1316–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuade A, Coburn M, Tu CH, Hasselmann J, Davtyan H, and Blurton-Jones M (2018). Development and validation of a simplified method to generate human microglia from pluripotent stem cells. Mol Neurodegener 13, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico D, Isserlin R, Stueker O, Emili A, and Bader GD (2010). Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One 5, e13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, and Thomas PD (2017). PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res 45, D183–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, and Helmchen F (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. [DOI] [PubMed] [Google Scholar]
- Perry VH, and Holmes C (2014). Microglial priming in neurodegenerative disease. Nat Rev Neurol 10, 217–224. [DOI] [PubMed] [Google Scholar]
- Pocock JM, and Piers TM (2018). Modelling microglial function with induced pluripotent stem cells: an update. Nat Rev Neurosci 19, 445–452. [DOI] [PubMed] [Google Scholar]
- Prinz M, Erny D, and Hagemeyer N (2017). Ontogeny and homeostasis of CNS myeloid cells. Nat Immunol 18, 385–392. [DOI] [PubMed] [Google Scholar]
- Pulido-Salgado M, Vidal-Taboada JM, Barriga GG, Sola C, and Saura J (2018). RNA-Seq transcriptomic profiling of primary murine microglia treated with LPS or LPS + IFNgamma. Sci Rep 8, 16096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam C, Poueymirou WT, Rojas J, Murphy AJ, Valenzuela DM, Yancopoulos GD, Rongvaux A, Eynon EE, Manz MG, and Flavell RA (2011). Efficient differentiation and function of human macrophages in humanized CSF-1 mice. Blood 118, 3119–3128. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, Saito Y, Marches F, Halene S, Palucka AK, et al. (2014). Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol 32, 364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MW, and Stevens B (2017). Microglia emerge as central players in brain disease. Nat Med 23, 1018–1027. [DOI] [PubMed] [Google Scholar]
- Savchenko VL, McKanna JA, Nikonenko IR, and Skibo GG (2000). Microglia and astrocytes in the adult rat brain: comparative immunocytochemical analysis demonstrates the efficacy of lipocortin 1 immunoreactivity. Neuroscience 96, 195–203. [DOI] [PubMed] [Google Scholar]
- Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen HC, Kitts PA, Murphy TD, Pruitt KD, Thibaud-Nissen F, Albracht D, et al. (2017). Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res 27, 849–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro LA, Perez ZD, Foresti ML, Arisi GM, and Ribak CE (2009). Morphological and ultrastructural features of Iba1-immunolabeled microglial cells in the hippocampal dentate gyrus. Brain Res 1266, 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shitaka Y, Tran HT, Bennett RE, Sanchez L, Levy MA, Dikranian K, and Brody DL (2011). Repetitive closed-skull traumatic brain injury in mice causes persistent multifocal axonal injury and microglial reactivity. J Neuropathol Exp Neurol 70, 551–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieff CA (1987). Hematopoietic growth factors. J Clin Invest 79, 1549–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, Love MI, and Robinson MD (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WM, Joshita S, Zhou Y, Ulland TK, Gilfillan S, and Colonna M (2018). Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J Exp Med 215, 745–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology, C. (2017). Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res 45, D331–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda Y, Gullipalli D, and Song WC (2016). Modeling complement-driven diseases in transgenic mice: Values and limitations. Immunobiology 221, 1080–1090. [DOI] [PubMed] [Google Scholar]
- Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H, Stewart FR, Piccio L, Colonna M, and Holtzman DM (2014). Altered microglial response to Abeta plaques in APPPS1–21 mice heterozygous for TREM2. Mol Neurodegener 9, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varley AW, Coulthard MG, Meidell RS, Gerard RD, and Munford RS (1995). Inflammation-induced recombinant protein expression in vivo using promoters from acute-phase protein genes. Proc Natl Acad Sci U S A 92, 5346–5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S, et al. (2016). TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med 213, 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster DE (2013). FOCIS.
- Wickham H (2016). ggplot2: Elegant Graphics for Data, 2nd edn (Springer Nature; ). [Google Scholar]
- Wu Y, Dissing-Olesen L, MacVicar BA, and Stevens B (2015). Microglia: Dynamic Mediators of Synapse Development and Plasticity. Trends Immunol 36, 605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Raj D, Saiepour N, Van Dam D, Brouwer N, Holtman IR, Eggen BJL, Moller T, Tamm JA, Abdourahman A, et al. (2017). Immune hyperreactivity of Abeta plaque-associated microglia in Alzheimer’s disease. Neurobiol Aging 55, 115–122. [DOI] [PubMed] [Google Scholar]
- Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, and Grutzendler J (2016). TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron 92, 252–264. [DOI] [PubMed] [Google Scholar]
- Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, Billis K, Cummins C, Gall A, Giron CG, et al. (2018). Ensembl 2018. Nucleic Acids Res 46, D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S2. In vivo, 2-photon recording of xMG response to laser ablation (Related to Figure 4).
Video S1. In vivo, 2-photon recording of homeostatic xMGs (Related to Figure 4).
Video S3. Z-stack of Aβ internalization by an xMG (Related to Figure 7).
Table S3. DGE analysis related to microglia signature gene lists (Related to Figure 3).
Table S5. DGE analysis for in vitro LPS treatment (Related to Figure 5).
Table S6. DGE and Z-scores related to transcription factor motif analysis (Related to Figure 6).
Table S4. DGE analysis and GSEA analysis for in vivo LPS treatment (Related to Figure 5).
Table S7. scRNA-seq DGE and Venn Diagram comparisons (Related to Figure 8).
Table S2. Gene expression for assessment of xMG in vivo transcriptomic signature (Related to Figure 3).
Data Availability Statement
The bulk and single-cell RNA-seq datasets generated during this study are available through GEO SuperSeries accession number GSE133434 or individual series accession numbers GSE133432 or GSE133433, respectively. The bulk RNA-seq datasets generated by Gosselin et al. (Figure 3) are available through NCBI dbGaP, accession number phs001373.v1.p1. The bulk RNA-seq datasets generated by Abud et al. (Figure 3) are available through GEO, series accession number GSE89189. The bulk RNA-seq datasets generated by McQuade et al. (Figure 1) are available through GEO, series accession number GSE117829.