

ABSTRACT
Genetic screens have identified two sets of genes that act at distinct steps of basal autophagy in higher eukaryotes: the pan-eukaryotic ATG genes and the metazoan-specific EPG genes. Very little is known about whether these core macroautophagy/autophagy genes are differentially employed during multicellular organism development. Here we analyzed the function of core autophagy genes in autophagic removal of SQST-1/SQSTM1 during C. elegans development. We found that loss of function of genes acting at distinct steps in the autophagy pathway causes different patterns of SQST-1 accumulation in different tissues and developmental stages. We also identified that the calpain protease clp-2 acts in a cell context-specific manner in SQST-1 degradation. clp-2 is required for degradation of SQST-1 in the hypodermis and neurons, but is dispensable in the body wall muscle and intestine. Our results indicate that autophagy genes are differentially employed in a tissue- and stage-specific manner during the development of multicellular organisms.
Abbreviations: ATG: autophagy related; CLP: calpain family; EPG: ectopic PGL granules; ER: endoplasmic reticulum; ESCRT: endosomal sorting complex required for transport; GFP: green fluorescent protein; LGG-1/LC3: LC3, GABARAP and GATE-16 family; MIT: microtubule interacting and transport; PGL: P granule abnormality protein; SQST-1: sequestosome-related; UPS: ubiquitin-proteasome system
KEYWORDS: Aggrephagy, ATG gene, C. elegans, clp-2, EPG gene
Introduction
Macroautophagy/autophagy involves the formation of a double membrane autophagosome, which engulfs a portion of cytosol and delivers it to vacuoles/lysosomes for degradation [1,2]. Autophagosome formation starts with the initiation and nucleation of a crescent-shaped phagophore that further expands and closes [1,2]. Our molecular understanding of autophagosome formation mainly originates from the study of autophagy-related (ATG) genes identified from yeast genetic screens [1,2]. The Atg proteins encoded by these genes form different complexes that act at distinct steps of autophagosome biogenesis [1,2]. The Atg1 complex and the Vps34-containing PtdIns3K complex are required for the initial formation of phagophores. The ubiquitin-like Atg8 conjugation system mediates phagophore expansion.
The autophagy pathway in higher eukaryotes is more complex than that in yeast [3–5]. In yeast, autophagosomes are generated at the single phagophore assembly site (PAS), and nascent autophagosomes directly fuse with the vacuoles [1,2]. In more complex eukaryotes, PtdIns3P-enriched subdomains of the ER act as cradles for autophagosome formation [6]. The ER forms extensive contact with the phagophore during its expansion [7–10]. Upon closure, the autophagosome detaches from the ER and undergoes a maturation process by fusing with endosomes before fusion with lysosomes to generate functional autolysosomes [7,11]. A group of EPG genes, whose homologs are present in mammals but absent in yeast, was identified from worm genetic screens and found to act at steps unique to the autophagy pathway in higher eukaryotes [3,12]. EPG-3/VMP1 is required for disassembly of the phagophore/ER contact during autophagosome formation by modulating the activity of ATP2A/SERCA [9]. Depletion of EPG-3 causes stable association of phagophores with the ER [9]. EPG-5 functions as a tether to mediate the specific fusion of autophagosomes with late endosomes/lysosomes [13]. Recent studies demonstrated that these core autophagy genes are not equally employed under different autophagy induction conditions [5]. For example, ULK1 is dispensable for autophagosome formation induced by prolonged glucose starvation [14].
Multicellular organisms contain different cell types with intracellular membrane structures that differ in composition and organization. Distinct developmental signaling and environmental cues also impinge on autophagy to maintain cellular homeostasis. It has been shown that the autophagic machinery is modulated in different cell types [5]. For example, genes involved in the Atg8 lipidation system function differentially in different cell types in Drosophila [15]. Atg7 and Atg3, which act as E1 and E2 enzymes respectively for conjugation of PE to Atg8, are required for autophagy in fat and salivary gland cells. However, in midgut, autophagy does not require Atg7 and Atg3, but requires the E1 enzyme Uba1 [15]. Similarly, ULK1/2 is dispensable for autophagy in neurons in mice [16]. It has been shown that in the absence of some ATG genes, compensatory mechanisms are utilized for autophagosome formation [17]. Mouse cells lacking Atg5 or Atg7 still form autophagosomes in the presence of certain stressors such as etoposide [17]. The involvement of core autophagy genes in autophagosome formation in different cell types and developmental stages has not been thoroughly investigated during multicellular organism development.
During C. elegans embryogenesis, SQST-1, the SQSTM1/p62 mammalian homolog, is selectively removed by autophagy [12]. In wild-type embryos, SQST-1::GFP is weakly expressed and diffusely localized in the cytoplasm. In autophagy mutant embryos, SQST-1 accumulates into large numbers of protein aggregates in multiple tissues [12]. This autophagic degradation of protein aggregates is known as aggrephagy. Subsequently, both ATG and EPG genes are required for autophagic degradation of SQST-1 during embryogenesis [3,12]. C. elegans contains 2 homologs of ATG4 (atg-4.1 and atg-4.2) and 2 homologs of ATG16 (atg-16.1 and atg-16.2). atg-4.1 and atg-16.2 act redundantly with atg-4.2 and atg-16.1, respectively, to play a major role in autophagy [18,19]. C. elegans also contains two ATG8 homologs, LGG-1 and LGG-2, which act at distinct steps of the aggrephagy pathway in degradation of SQST-1 [20]. LGG-1 is required for degradation of SQST-1 at all developmental stages, while the requirement for LGG-2 is temporal [20]. Loss of function of LGG-2 results in defective degradation of SQST-1 aggregates at embryonic stages, while SQST-1 is removed normally at larval stages [20].
Here we characterized the involvement of core autophagy genes in autophagic removal of SQST-1 and found that different autophagy mutants exhibit different patterns of accumulation of SQST-1 aggregates in different tissues and developmental stages. We also identified a member of the C. elegans calpain family, clp-2, that is involved in degradation of SQST-1 in a tissue- and stage-specific manner. Our results indicate that autophagy genes are employed in a cell context-dependent manner in the aggrephagy pathway during multicellular organism development.
Results
The involvement of autophagy genes in removal of SQST-1 during C. elegans embryogenesis
A variety of protein aggregates, including components of oocyte-derived P granules (PGL-1 and PGL-3), and SQST-1, are degraded by the basal level of autophagy during C. elegans embryogenesis [12,21]. Genetic screens have identified a set of core autophagy genes that are essential for removal of both PGL granules and SQST-1 aggregates during embryogenesis [3,12]. We investigated whether different tissues show differential accumulation of SQST-1 aggregates in embryos carrying mutations in these genes. For our analysis, we selected genes acting at different steps in the aggrephagy pathway [22], including epg-9 (encoding the ATG101 homolog, a component of the UNC-51/Atg1 complex), epg-8 (encoding the Atg14 homolog, a component of the Vps34 complex), atg-18 (in the Atg2-Atg18 complex), lgg-1 and atg-3 (components of the ubiquitin-like conjugation systems), atg-9, and the metazoan-specific autophagy genes epg-3, epg-4 and epg-5 (Figure 1A). epg-9(bp320), epg-8(bp251), epg-3(bp933), epg-4(bp425), atg-9(bp564), epg-5(tm3425) and atg-18(gk378) mutants are putative genetic nulls. lgg-1(bp500) encodes a protein that lacks the C terminal 30 amino acids and appears to be null in autophagy [12]. atg-3(bp412) is a hypomorphic mutant, as the genetic null is lethal.
Figure 1.
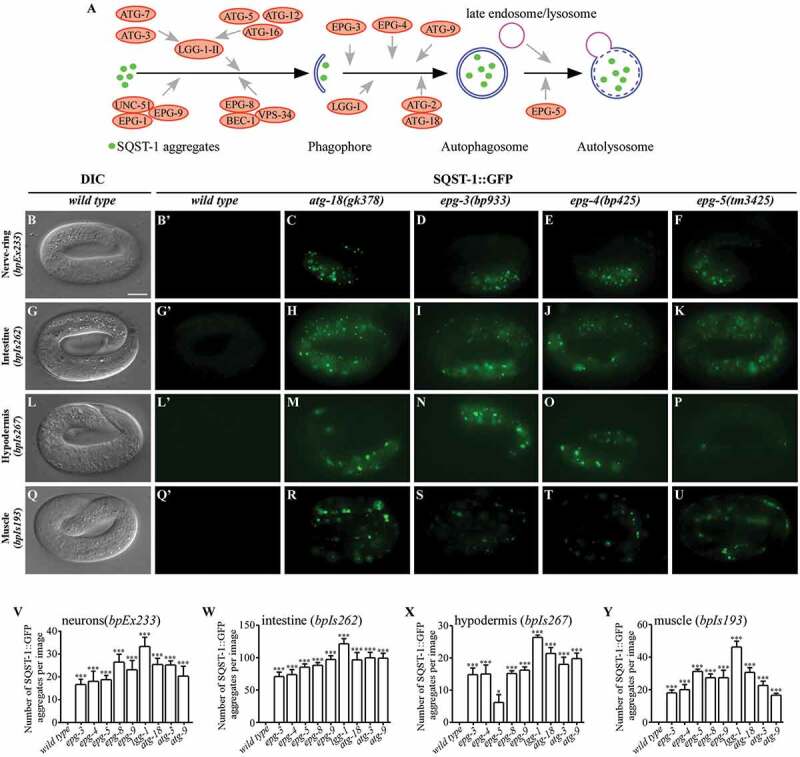
Autophagy genes are differentially involved in degradation of SQST-1::GFP aggregates during C. elegans embryogenesis. (A) Schematic illustration of the role of autophagy proteins in the aggrephagy pathway in C. elegans. (B), (G), (L) and (Q) In wild-type embryos, SQST-1::GFP is weakly expressed and diffusely localized in the cytoplasm. SQST-1::GFP was expressed in neuronal cells (B, bpEx233), intestinal cells (G, bpIs262), hypodermal cells (L, bpIs267), and body wall muscle cells (Q, bpIs193). DIC images of the embryos are shown in the left panels. (B’), (G’), (L’) and (Q’) are the GFP subpanels corresponding to (B), (G), (L) and (Q), respectively. All embryos analyzed in this study were at the 4-fold stage. (C-F) SQST-1::GFP aggregates accumulate in neurons in atg-18(gk378) (C), epg-3(bp933) (D), epg-4(bp425) (E) and epg-5(tm3425) (F) embryos carrying the transgene bpEx233, which is expressed specifically in neurons. (H-K) Accumulation of SQST-1::GFP aggregates in intestinal cells (bpIs262) in atg-18(gk378) (H), epg-3(bp933) (I), epg-4(bp425) (J) and epg-5(tm3425) mutants (K). (M-P) Accumulation of SQST-1::GFP aggregates in hypodermal cells (bpIs267) in atg-18(gk378) (M), epg-3(bp933) (N), epg-4(bp425) (O) and epg-5(tm3425) (P) mutants. Fewer SQST-1::GFP aggregates accumulate in epg-5(tm3425) mutants (P). (R-U) Accumulation of SQST-1::GFP aggregates in body wall muscle cells (bpIs193) in various autophagy mutants. The number and intensity of SQST-1::GFP aggregates in atg-18(gk378) (R) and epg-5(tm3425) (U) embryos is greater than that in epg-3(bp933) (S) and epg-4(bp425) (T) mutants. (V-Y) Quantification of the number of SQST-1::GFP aggregates per image in neurons (bpEx233) (V), intestinal cells (bpIs262) (W), hypodermal cells (bpIs267) (X) and body wall muscle cells (bpIs193) (Y) in wild-type and different autophagy-mutant embryos. In all tissues, lgg-1(bp500) mutants show the strongest phenotype. Five independent images of five embryos (n = 5) for each strain were quantified. Data are shown as mean ± SEM, *P ≤ 0.05; ***P ≤ 0.001. Scale bar: 10 μm. C. elegans embryos remain the same size during embryogenesis. Thus, bars for embryos are only shown once in each figure.
The bpIs151 (Psqst-1::SQST-1::GFP) reporter, in which SQST-1 is driven by its own promoter and expressed in all tissues, was previously used for conducting genetic screens [12]. The accumulation of SQST-1::GFP aggregates was similar in all the mutants examined except for lgg-1(bp500), which had a slightly stronger defect (Figure S1A-E and data not shown). The size of the embryo remains the same from the 1-cell stage to the end of embryogenesis (558 cells). As development proceeds, cells become smaller and more tightly packed, and their organization is dynamically changed for morphogenesis. To aid the analysis of the expression of SQST-1 in specific tissues, we generated transgenic lines in which expression of SQST-1::GFP is driven by tissue-specific promoters, including body wall muscle (bpIs193, Phlh-1::SQST-1::GFP), hypodermis (bpIs267, Phyp7::SQST-1::GFP), intestine (bpIs262, Pges-1::SQST-1::GFP), and all neurons (bpEx233, Prgef-1::SQST-1::GFP). In wild-type embryos carrying these transgenes, SQST-1 was weakly expressed and diffusely localized in the cytoplasm (Figure 1B,G,L,Q). SQST-1::GFP aggregates accumulated at the embryonic stage in all the autophagy mutants examined, but there were clear differences in the different mutants, as measured by the number and fluorescence intensity of the aggregates. In neurons (bpEx233) and intestinal cells (bpIs262), the number and intensity of SQST-1::GFP aggregates in epg-5(tm3425), epg-3(bp933) and epg-4(bp425) mutants were slightly weaker compared to other mutants (Figures 1C–F,H–K,V,W, S1F and S1G). When SQST-1::GFP was expressed in hypodermal cells (bpIs267) in epg-5(tm3425) mutants, it formed fewer, weaker aggregates compared to other mutants (Figures 1M–P,X and S1H). In body wall muscle cells (bpIs193), the accumulation of SQST-1::GFP aggregates in atg-3(bp412), epg-8(bp251), atg-9(bp564), epg-3(bp933) and epg-4(bp425) mutants was similar, and slightly weaker than in atg-18(gk378), epg-9(bp320) and epg-5(tm3425) mutants (Figures 1R–U,Y and S1I). Among all the autophagy mutants, lgg-1(bp500) mutants showed the strongest phenotype for all transgenes examined (Figures 1V–Y and S1F-I). Thus, autophagy genes are differentially involved in degradation of SQST-1 by basal autophagy in different tissues during embryogenesis.
Tissues- and stage-specific functions of epg-3 and epg-4 during post-embryonic development
After hatching, C. elegans development requires external nutrients. We characterized the accumulation of SQST-1::GFP aggregates in L2 larvae, L4 larvae and day one adults. In wild-type animals, SQST-1::GFP was weakly expressed and diffusely localized in the cytoplasm. None or very few aggregates were formed in the neuron (Figure 2A,U), intestine (Figures 2F,V, S2A, S2E, S2I, S2M and S2Q), hypodermis (Figures 2K,W, S3A, S3E, S3I, S3M and S3Q), and muscle (Figure 2P,X).
Figure 2.
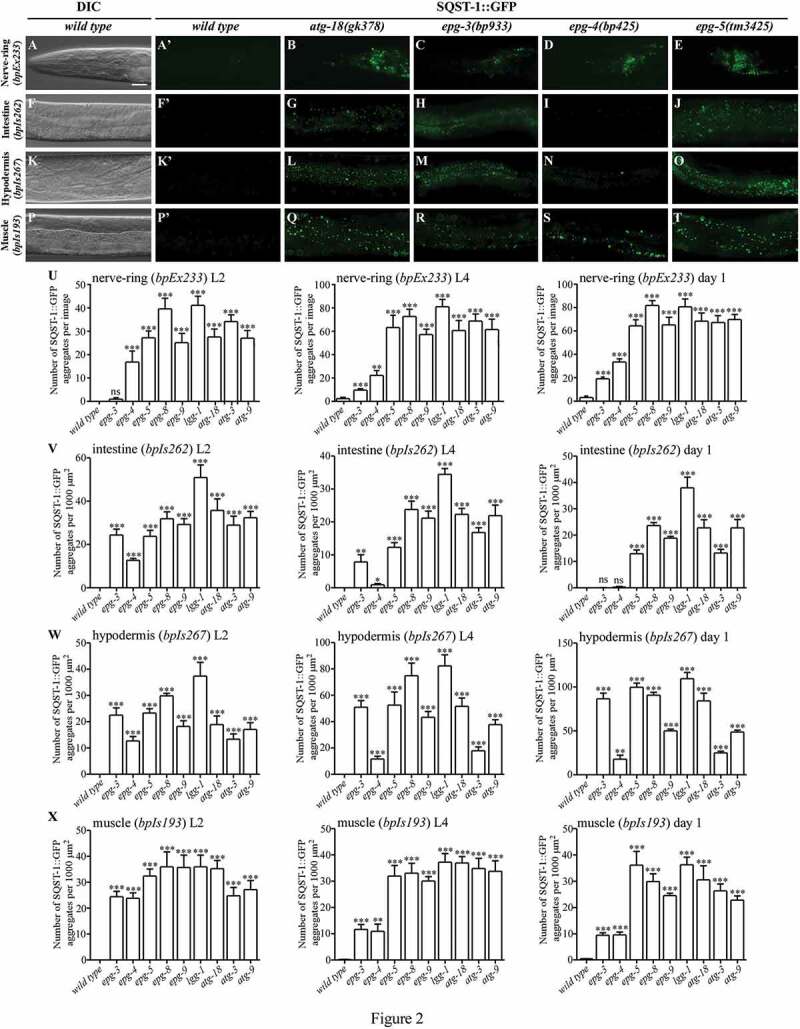
Loss of function of core autophagy genes causes differential accumulation of SQST-1::GFP aggregates at different larval and adult stages. (A), (F), (K) and (P) In wild-type animals, SQST-1::GFP is weakly expressed and diffusely localized in the cytoplasm. Only a few aggregates are formed. SQST-1::GFP was expressed in neurons (A, bpEx233), intestinal cells (F, bpIs262), hypodermal cells (K, bpIs267), and body wall muscle cells (P, bpIs193). L4 larvae are shown. DIC images of the animals are shown in the left panels. (A’), (F’), (K’) and (P’) are the GFP subpanels corresponding to (A), (F), (K) and (P), respectively. (B-E) A large number of SQST-1::GFP aggregates accumulate in different autophagy mutants in the nerve-ring (bpEx233) at the L4 larval stage. The number of SQST-1::GFP aggregates in epg-3(bp933) (C) and epg-4(bp425) (D) mutants is lower than in atg-18(gk378) (B) and epg-5(tm3425) (E). (G-J) In intestinal cells (bpIs262) at the L4 larval stage, atg-18(gk378) mutants (G) have slightly more SQST-1::GFP aggregates than epg-5(tm3425) mutants (J). The number of SQST-1::GFP aggregates in epg-3(bp933) (H) is lower than in atg-18(gk378) (G) and epg-5(tm3425) (J) mutants. Only a few aggregates are formed in epg-4(bp425) mutants (I). (L-O) In hypodermal cells (bpIs267) at the L4 larval stage, SQST-1::GFP aggregates accumulate in atg-18(gk378) (L), epg-3(bp933) (M) and epg-5(tm3425) (O) mutants at a similar level, while far fewer SQST-1::GFP aggregates are formed in epg-4(bp425) (N) mutants. (Q-T) At the L4 larval stage, SQST-1::GFP aggregates accumulate in the body wall muscle cells (bpIs193) of all mutants. The number of aggregates in atg-18(gk378) (Q) and epg-5(tm3425) (T) mutants is higher than that in epg-3(bp933) (R) and epg-4(bp425) (S) mutants. (U-X) Quantification of the number of SQST-1::GFP aggregates per 1000 μm2 in wild-type and autophagy mutants at the L2 larval, L4 larval and day 1 adult stages in neurons (bpEx233) (U), intestinal cells (bpIs262) (V), hypodermal cells (bpIs267) (W), and body wall muscle cells (bpIs193) (X). Five independent animals (n = 5) for each strain were quantified. Data are shown as mean ± SEM, ns: not significant, P > .05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. Scale bar: 20 μm (A-T).
In animals carrying the transgene bpEx233, SQST-1::GFP aggregates accumulated in neurons in the nerve-ring and ventral nerve cord (VNC) in autophagy mutants. epg-8(bp251) and lgg-1(bp500) mutants contained slightly more SQST-1::GFP aggregates than other mutants (Figure 2U). In epg-3(bp933) mutants, SQST-1::GFP aggregates were absent or scarce at the L2 larval stage, but were evident at the L4 larval and adult stages (Figure 2C,U). epg-4(bp425) and epg-3(bp933) mutants accumulated fewer aggregates than other mutants (Figure 2B–E,U).
In intestinal cells (bpIs262), epg-9(bp320), epg-5(tm3425), epg-8(bp251), atg-9(bp564), atg-3(bp412), atg-18(gk378) and lgg-1(bp500) mutants showed dramatic accumulation of SQST-1 aggregates at all stages (Figures 2G,J,V, S2B, S2F, S2J, S2N, S2R and S2U). Among these mutants, lgg-1(bp500) exhibited the strongest defect (Figures 2V and S2U). In epg-3(bp933) mutants, the accumulation of SQST-1::GFP aggregates was strong at the L1 and L2 larval stages, comparable to epg-9(bp320) mutants (Figures 2V, S2B, S2C, S2F, S2G, and S2U). At the L3 and L4 larval stages, the number of SQST-1::GFP aggregates in epg-3(bp933) mutants gradually became lower than in other autophagy mutants (Figures 2H,V, S2K, S2O and S2U) and no aggregates formed at the adult stage (Figures 2V, S2S and S2U). SQST-1::GFP aggregates formed in epg-4(bp425) mutant intestine at the L1 larval stage, then became weaker at the L2 larval stage, and were not detectable at the adult stage (Figures 2I,V, S2D, S2H, S2L, S2P, S2T and S2U).
In hypodermal cells (bpIs267) at the L2 larval and L4 larval stages, both the number and fluorescence intensity of SQST-1::GFP aggregates were higher in epg-8(bp251) and lgg-1(bp500) mutants than in the other mutants (Figures 2W and S3Y). At the adult stage, the SQST-1::GFP fluorescence intensity in epg-8(bp251) and lgg-1(bp500) mutants were stronger than that in epg-3(bp933), epg-5(tm3425) and atg-18(gk378) mutants, although the number of SQST-1::GFP aggregates was comparable (Figures 2W, S3U-W and S3Y). atg-3(bp412) mutants contained less SQST-1::GFP aggregates, but had some big SQST-1::GFP aggregates, and the total fluorescence intensity of SQST-1::GFP aggregates in atg-3(bp412) was similar to that in atg-9(bp564), epg-9(bp320), atg-18(gk378) and epg-3(bp933) mutants at the L2, L4 larval and adult stages (Figures 2W, S3U, S3X and S3Y). Noticeably, the accumulation of SQST-1::GFP aggregates in epg-4(bp425) mutants was similar to that in epg-9(bp320) and epg-3(bp933) mutants at the L1 and L2 larval stages, but became much weaker in later developmental stages (Figures 2L–O,W, S3A to T and S3Y).
In body wall muscle cells (bpIs193), SQST-1::GFP aggregates accumulated in all mutants from the L1 to adult stages (Figures 2X and S3Z). The number and GFP intensity of SQST-1::GFP aggregates were lower in epg-4(bp425) and epg-3(bp933) mutants than in other mutants (Figures 2Q–T,X and S3Z). Taken together, these results provide evidence that the core autophagy genes appear to have differential roles in aggrephagy in different tissues and developmental stages.
Accumulation of GFP::LGG-1 puncta in epg-3, epg-4 and epg-5 mutants
The C. elegans homolog LGG-1 is widely used as a marker to monitor autophagy activity [23]. Mutants with loss of function of genes that act at distinct steps of autophagy show characteristic patterns of formation of LGG-1 puncta. In epg-3, epg-4 and epg-5 mutant embryos, LGG-1 puncta dramatically accumulate [12]. We further analyzed the expression of GFP::LGG-1 in wild-type and epg-3, epg-4, epg-5 and atg-18 mutants in the hypodermis and intestine at the L4 larval and day 1 adult stages. In wild-type worms, GFP::LGG-1 was diffuse and formed a few small puncta (Figure S4A, F, K and P). In hypodermal cells at the L4 larval and day 1 adult stages, GFP::LGG-1 puncta dramatically increased in number in epg-3(bp933) and epg-5(tm3425) mutants (Figure S4B, D, G, I, E1 and F1), while they were only slightly increased in epg-4(bp425) and atg-18(gk378) mutants (Figure S4C, E, H, J, E1 and F1). In the intestine at the L4 larval stage, GFP::LGG-1 puncta obviously increased in number in epg-3(bp933) and epg-5(tm3425) mutants, and were slightly increased in epg-4(bp425) and atg-18(gk378) mutants (Figure S4L-O and G1). In intestinal cells at the day 1 adult stage, GFP::LGG-1 puncta evidently accumulated in epg-5(tm3425) mutants, but not in epg-3(bp933), epg-4(bp425) and atg-18(gk378) mutants (Figure S4Q-T and H1). We also examined the expression of a GFP::LGG-1G116A reporter in various autophagy mutants. In wild-type animals, GFP::LGG-1G116A was diffusely localized, apart from a few small puncta in the intestine and hypodermis (Figure S4U and Z). The number of GFP::LGG-1G116A puncta was slightly increased in hypodermal cells in epg-3(bp933), epg-5(tm3425) and atg-18(gk378) mutants at the L4 larval stage, but not in epg-4(bp425) mutants (Figure S4V-Y and I1). In the intestine, the number of GFP::LGG-1G116A puncta was slightly increased in epg-3(bp933) and epg-5(tm3425) mutants, but not in epg-4(bp425) and atg-18(gk378) mutants (Figure S4A1-D1 and J1). Therefore, we presumed that the majority of LGG-1 puncta that accumulated in epg-3, epg-4 and epg-5 mutants are autophagic structures, but not LGG-1 aggregates. These results are consistent with the idea that at late larval and adult stages, epg-3 is not essential for autophagy in the intestine and epg-4 is dispensable in the intestine and hypodermis.
Expression pattern of translation reporters for autophagy genes
We next investigated whether the tissue- and time-dependent function of autophagy genes is associated with their expression pattern. The expression patterns of functional translation reporters for the autophagy genes lgg-1, atg-18, epg-4, epg-5 and epg-9 were analyzed. These reporters were expressed in almost all cells at the embryonic stages (Figure S5A-E). At the larval and adult stages, they were also widely distributed, including in hypodermis, body wall muscles, intestinal cells and cells in the head and tail regions (Figure S5F-T) [12,24]. The intensity of the GFP signal, however, varied in different cell types. For example, levels of EPG-9::GFP were stronger in neurons than in hypodermis, intestine and muscle at the post-embryonic stages (Figure S5R-T), while EPG-4::GFP was expressed more in the intestine than in muscle and hypodermal cells at the L4 larval stage (Figure S5U and V). Thus, the difference in degradation of SQST-1 cannot be attributed to differential expression of autophagy genes.
Genetic screens to identify tissue-specific autophagy genes involved in degradation of SQST-1
To identify genes that act in a tissue- or stage-specific manner in aggrephagy, we performed genetic screens to isolate mutants that show accumulation of SQST-1::GFP aggregates. Using bpIs151 (Psqst-1::SQST-1::GFP) as a reporter, we screened ~5000 genomes and isolated 32 mutations that showed accumulation of SQST-1 aggregates from embryonic to adult stages (Table S1 and data not shown). Further mapping and non-complementation experiments revealed that these mutations were new alleles of known autophagy genes (Table S1). We also isolated one mutation, bp852, in which a large number of SQST-1::GFP aggregates accumulated in mutant larvae and adults, but not at the embryonic stage (Table S1, Figure S6A, B and data not shown). We also performed genetic screens using the hypodermis-specific SQST-1::GFP reporter (bpIs267). We isolated ~124 mutations from ~12,000 genomes screened that showed accumulation of SQST-1 aggregates. Further characterization revealed that 38 mutations, which caused accumulation of SQST-1::GFP aggregates from embryos to adults, were new alleles of known autophagy genes (Table S1 and data not shown). We also identified mutations that resulted in accumulation of spherical SQST-1 aggregates in larvae and adults, but not in embryos (Figures 3A,C, and Table S1). Among these mutations, bp987 and bp1009 failed to complement with bp852, indicating that they affect the same gene. Other mutations caused accumulation of irregularly shaped SQST-1::GFP aggregates at the larval and adult stages (Figure 3B,D,M,N and Table S1). These mutations include new alleles of scav-3, which encodes the C. elegans homolog of the human lysosomal integral membrane protein SCARB2/LIMP-2 [25]. Loss of function of scav-3 disrupts lysosome function [25]. These results indicate that degradation of SQST-1 requires factors that specifically function at post-embryonic stages.
Figure 3.
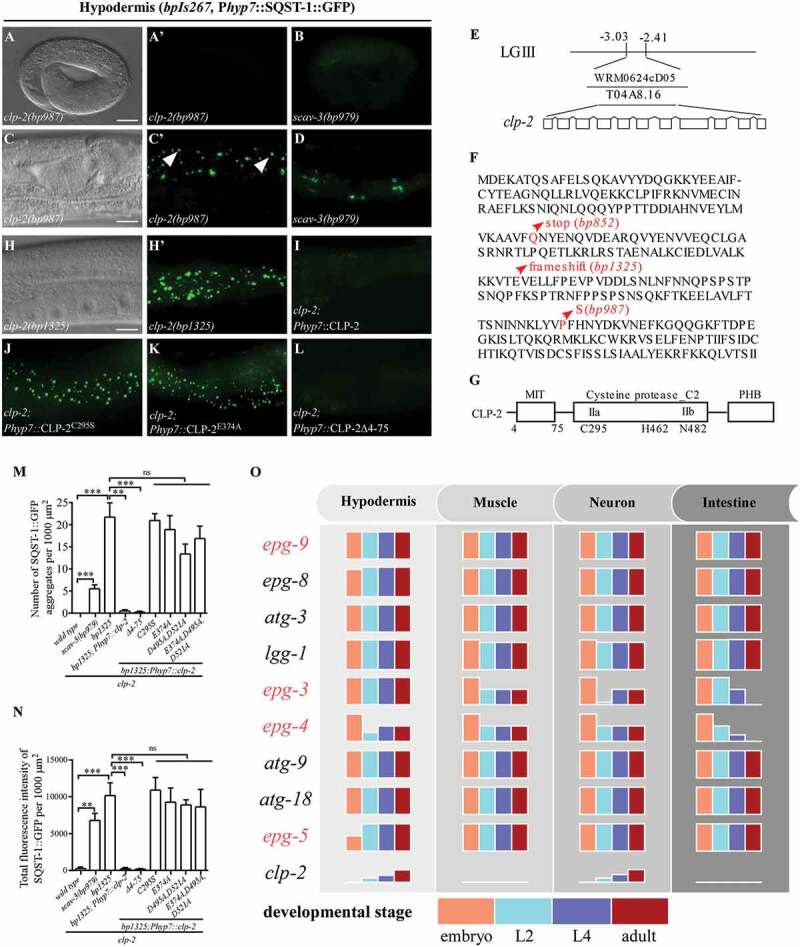
Loss of function of clp-2 causes tissue- and stage-specific accumulation of SQST-1 aggregates. (A and C) bp987 mutants show accumulation of SQST-1::GFP aggregates in hypodermal cells (bpIs267) at the larval and adult stages (C, day 1 adult is shown here), but not at the embryonic stage (A). DIC images of the same animals are shown in the left panels. (A’) and (C’) are the GFP subpanels corresponding to (A) and (C), respectively. Arrows indicate the spherical SQST-1::GFP aggregates. (B and D) scav-3(bp979) mutants accumulate irregularly shaped SQST-1::GFP aggregates at the larval and adult stages (D), but not at embryonic stage (B). (E) Mapping and cloning of clp-2. Fosmid WRM0624cD05, containing gene T04A8.16/clp-2, rescued the defective degradation of SQST-1::GFP aggregates in bp852 mutants. LG III: chromosome III. (F) Protein sequence of CLP-2. The mutations in clp-2 alleles are indicated. clp-2(bp852) mutates the glutamine at position 99 to a premature stop codon. clp-2(bp987) mutates the highly conserved proline at position 229 to serine. clp-2(bp1325), generated through CRISPR-Cas9, has a 2-nucleotide deletion at exon 3 (AAGAAAGTCACAGAGGTGGA, underlined nucleotides are deleted), which causes a frame shift. (G) Schematic illustration of CLP-2. CLP-2 possesses one MIT domain at its N terminus, a cysteine protease C2 domain containing two subdomains (IIa and IIb), and a PHB (PalB homology domain with some domain III homology) domain at its C terminus. C295, H462 and N482 comprise the catalytic center. (H-L) Expression of WT CLP-2 (I) and CLP-2 with a deletion of the MIT domain (L) rescues the defective degradation of SQST-1::GFP aggregates in clp-2(bp1325) mutants (H). Expression of the catalytically dead form CLP-2C295S (J) or the Ca2+-binding-defective form CLP-2E374A (K) fails to rescue clp-2(bp1325). DIC image of the same animal is shown in the left panel. (H’) is the GFP subpanel corresponding to (H). (M and N): Quantification of the number (M) and the fluorescence intensity (N) of SQST-1::GFP aggregates per 1000 μm2 in wild-type, scav-3(bp979), clp-2(bp1325), and clp-2(bp1325) mutants carrying the following CLP-2 transgenes: WT CLP-2, CLP-2 with a deletion of the MIT domain, CLP-2C295S, CLP-2E374A, CLP-2D495A,D521A, and CLP-2E374A,D495A,D521A. Three independent animals at the day 1 adult stage (n = 3) for each strain were quantified. Data are shown as mean ± SEM, ns: not significant, P > .05; **P ≤ 0.01; ***P ≤ 0.001. (O) Summary of the tissue- and developmental stage-specific involvement of autophagy genes in the aggrephagy pathway in C. elegans. Accumulation of SQST-1 aggregates at different stages (embryonic, L2, L4 and adult) is shown as different colored columns. The height of each column stands for the degree of SQST-1 aggregate accumulation. At the embryonic stage, all the core autophagy genes are essential for SQST-1 degradation in different cell types, while epg-5 mutants show a milder defect in hypodermis. At post-embryonic stages, epg-3 and epg-4 have stage-dependent functions in different tissues. clp-2 is involved in SQST-1 degradation in hypodermis and neurons in a stage-dependent manner. Genes are ordered according to the steps in which they function in the aggrephagy pathway. The metazoan-specific genes, epg-9, epg-3, epg-4 and epg-5, are indicated in red. The step at which clp-2 function is unknown; thus, clp-2 is placed at the end. Scale bar: 10 μm (A and B); 20 μm (C, D and H-L).
bp852 contains a mutation in the C. elegans calpain homolog CLP-2
We cloned bp852 and found that it contained a glutamine-to-stop codon mutation at position 99 of clp-2 (Figure 3E and F). bp987 mutated the highly conserved proline at position 229 to serine (Figure 3F). bp1009 carried a point mutation in intron 2, which may affect the splicing of clp-2 (data not shown). clp-2(bp1325, bp1326, bp1327 and bp1328), generated by CRISP-Cas9, showed similar SQST-1::GFP accumulation phenotypes (Figures 3A, C, H, S6B, S6C and data not shown). clp-2(bp1325) contained a 2-nucleotide deletion that introduced a frame shift at codon 159 and is likely to be a genetic null (Figure 3F). clp-2 encodes a member of the calpain family of Ca2+-activated proteases and contains a MIT (microtubule interacting and transport) domain at its N terminus, a cysteine protease C2 domain containing subdomains (IIa and IIb) and a PHB (PalB homology domain with some domain III homology) domain at its C terminus. C295, H462 and N482 comprise the catalytic center (Figure 3G) [26].
To determine whether clp-2 regulates degradation of SQST-1 by autophagy or by the ubiquitin-proteasome system (UPS), we examined the accumulation of SQST-1::GFP aggregates in clp-2 double mutants with other autophagy genes. The number of SQST-1::GFP aggregates in clp-2(bp1325); atg-18(bp594) and clp-2(bp1325); bec-1(bp613) double mutants was similar to that in atg-18(bp594) and bec-1(bp613) single mutants (Figure S6C-H). clp-2(bp1325) also failed to enhance the autophagy defect in epg-5(bp450) mutants (data not shown). These results indicate that clp-2 functions in the same pathway as bec-1, atg-18 and epg-5 in degradation of SQST-1.
To test the role of clp-2 in UPS, we used a UPS reporter system based on an inducible subunit of the proteasome, Paip-1::GFP, whose expression is activated by UPS dysfunction [27]. We found that expression of Paip-1::GFP was induced by defective proteasome degradation after rpt-3(RNAi) but not in clp-2 mutants (Figure S6I-K). We also determined whether impaired UPS function affects the accumulation of SQST-1::GFP aggregates in clp-2(bp1325) mutants. rpt-3(RNAi) caused no accumulation of SQST-1::GFP aggregates in wild-type animals, and did not enhance the defect in epg-5(tm3425) mutants. However, rpt-3(RNAi) partially suppressed the accumulation of SQST-1::GFP aggregates in clp-2(bp1325) mutants (Figure S6L-Q), consistent with the observation that mild disruption of the UPS function induces autophagy activity [27,28]. These results indicate that loss of clp-2 has no effect on UPS function.
The transgene Phyp7::CLP-2, in which WT CLP-2 was specifically expressed in hypodermal cells, fully rescued the accumulation of SQST-1::GFP aggregates in hypodermal cells in clp-2(bp1325) mutants (Figure 3I,M,N), suggesting that clp-2 acts cell-autonomously in regulating autophagy. Phyp7::CLP-2C295S, in which the residue critical for the catalytic activity was mutated, failed to rescue the defect in clp-2(bp1325) mutants (Figure 3J,M,N), suggesting that protease activity is required for its role in autophagy. Calpain contains two non-EF-hand Ca2+-binding sites [29]. We mutated the conserved residues required for Ca2+ binding in CLP-2, E374A in one site and D495A D521A in the other site. The Ca2+-binding-defective CLP-2 mutants failed to rescue the autophagy defect in clp-2(bp1325) mutants (Figure 3K,M,N), which indicates that clp-2 regulates autophagy in a Ca2+-dependent manner. The MIT domain of CAPN7, the homolog of CLP-2 in mammals, is essential for the association of CAPN7 with ESCRT-III-related proteins [30]. ESCRT-III is required for autophagosome maturation [4]. Mutant CLP-2 with a deletion of the MIT domain (4–72 aa) retained the ability to rescue the autophagy defect in clp-2 mutants (Figure 3L,M,N). Expression of a variety of markers for endosomal vesicles, including early endosomes, late endosomes and lysosomes, remained unchanged in clp-2 mutants (Figure S6R-W). Thus, CLP-2 regulates autophagy independent of its MIT domain and its potential interaction with the ESCRT-III complex.
We constructed a Pclp-2::gfp::clp-2 reporter, containing a 2-kb promoter, the entire open reading frame of clp-2, and an in-frame insertion of gfp at the N terminus of clp-2, to determine its expression pattern. GFP::CLP-2 was widely expressed from embryonic to adult stages, including in hypodermal cells, cells in the head region and tail region, muscle cells and intestinal cells (Figure S7A-E). GFP::CLP-2 was diffusely localized in the cytoplasm and also formed a few punctate structures (Figure S7F-I).
The C. elegans genome contains 10 calpain genes (clp-1 to −7, clp-8/F44F1.3, clp-9/T11A5.6 and clp-10/W05G11.4) [26]. Loss of function of clp-1, -3, −4, −5, −6, −7, −8, −9 and −10 neither caused accumulation of SQST-1::GFP aggregates (Figure S8B-J), nor enhanced the defective SQST-1::GFP degradation in clp-2 mutants (Figure S8A and S8L-T). Simultaneous loss of function of clp-8, clp-9 and clp-10, which encode orthologs of human CAPN15 [26], caused no defect in SQST-1 degradation (Figure S8K) and did not enhance the defect in clp-2(bp1325) mutants (Figure S8U). Therefore, CLP-2 is the only member of the calpain family involved in degradation of SQST-1.
Loss of clp-2 activity causes tissue- and stage-specific accumulation of SQST-1 aggregates
We next examined the role of clp-2 in degradation of SQST-1 in different tissues and developmental stages. In clp-2 mutant embryos, SQST-1::GFP aggregates were not detected in hypodermal cells (bpIs267) and neurons (bpEx233 and bpIs151) (Figures 3A, S9A, S9B and data not shown). A few aggregates were formed in L1 mutant larvae. The number of aggregates increased in L2 to L4 larvae and reached a peak in adults in neurons (Figure S9C-J and data not shown). No SQST-1::GFP aggregates accumulated in intestinal cells (bpIs262) and body wall muscle cells (bpIs193) from the embryonic to adult stages (Figure S9K-N and data not shown). Thus, loss of function of clp-2 causes accumulation of SQST-1 aggregates in a stage-dependent and tissue-specific manner.
Discussion
Tissue- and developmental stage-specific involvement of autophagy genes
Multicellular organisms contain different cell types with distinct morphology and intracellular membrane systems. Autophagosome formation and maturation exhibit different spatiotemporal features. For example, autophagosomes in neurons are preferentially generated at the distal tip of the axon, and then undergo retrograde trafficking to the soma, accompanied by stepwise maturation into autolysosomes [31]. Here we systematically examined the accumulation of the autophagy substrate SQST-1 in different tissues and developmental stages in C. elegans and found that core autophagy genes are employed in a context-dependent manner in the aggrephagy pathway (Figure 3O).
The C. elegans embryo is enclosed by a hard eggshell and its development is completely independent of external nutrients. Autophagy occurs at a basal level that mediates the degradation a variety of protein aggregates [12,21]. The core autophagy genes, including ATG genes and the metazoan-specific epg-3, −4 and −5 genes, are required for degradation of SQST-1 in all tissues in embryos. At the larval and adult stages, when C. elegans development requires external nutrients, loss of function of the core autophagy genes causes differences in SQST-1 accumulation. Among all the autophagy mutants analyzed, lgg-1(bp500) mutants showed the strongest defect. LGG-1 acts at several steps in the aggrephagy pathway [20,22]. LGG-1 directly interacts with SQST-1, and with the scaffold protein EPG-7, which promotes the degradation efficiency of SQST-1 along with multiple ATG proteins for autophagosome formation [20]. The lgg-1(bp500) mutation deletes the C terminal 30 amino acids, including the glycine necessary for phosphatidylethanolamine (PE) conjugation, and thus appears to be null in autophagy [12]. The lgg-1(tm3489) mutation, which deletes 175 nucleotides in exon 1 of lgg-1 (causing a frame shift at codon 38), is lethal, like the null alleles for other ATG genes involved in LGG-1 conjugation, including atg-3, atg-7, atg-5 and atg-10. Null mutants for other autophagy genes, including atg-9, atg-2, atg-18 and epg-1, are viable. The two conjugation systems may be involved in other biological processes in addition to autophagy. The ubiquitin-like conjugation systems may participate in degradation of SQST-1 through processes that are independent of autophagosome formation, such as endosome-mediated degradation.
Different from other core autophagy genes essential for autophagosome formation, the defect in SQST-1 degradation in epg-3 and epg-4 mutants is weaker at larval and adult stages. epg-3 is essential for SQST-1 removal in the hypodermis, while the defect is mild in the body wall muscle, neuron and intestine. epg-4 mutants show weak defects in all tissues. Both EPG-3 and EPG-4 are ER-localized transmembrane proteins [9,12]. Autophagosome biogenesis in multicellular organisms has been shown to occur at the contact sites between the ER and other organelles, such as the ER-mitochondrion contact sites (called MAMs), and also the ER-PM (plasma membrane) contact sites [32–34]. During phagophore expansion into autophagosomes, the phagophore dynamically contacts with the ER [7–10]. Multiple membrane sources, including PM, mitochondria, ERGIC (ER-Golgi intermediate compartment), COPII vesicles, and ATG9 vesicles have been suggested to contribute to phagophore expansion [32]. Autophagosome biogenesis in different cell types or developmental stages may involve different ER membrane contact sites and membranes sources for phagophore expansion. EPG-3/VMP1 regulates the activity of the ER-localized Ca2+ channel ATP2A/SERCA to modulate the ER-phagophore contact for phagophore expansion [9]. The autophagy defect in VMP1/EPG-3-depleted cells is partially suppressed by depletion of PLN/SLN, which form an inhibitory complex with ATP2A/SERCA [9]. The differential regulatory mechanism for ATP2A/SERCA activity could result in a distinct autophagy defect caused by loss of epg-3 function in different tissues. The molecular mechanism underlying the role of epg-4 in autophagosome formation remains unknown. Mice deficient in Ei24, the mammalian epg-4 homolog, also exhibit a weaker defect in accumulation of SQSTM1- and ubiquitin-positive aggregates in neuronal cells and hepatocytes than Atg5 and Atg7 KO mice [35]. Another explanation for the differential accumulation of SQST-1 during development is that levels of SQST-1 may vary in different tissues and developmental stages, and the demand for autophagy activity is therefore different. Autophagy genes involved in controlling the size of autophagosomes and/or the rate of autophagosome formation and maturation could result in differential accumulation of SQST-1 in different tissues. lgg-2 mutants, in which the autophagosome size is small, show defective degradation of SQST-1 during embryonic stages, while SQST-1 is normally removed at larval stages [20].
epg-5 encodes a RAB7 effector that mediates the fusion of autophagosomes with late endosomes/lysosomes, and thus is required for formation of degradative autolysosomes [13]. The accumulation of SQST-1 aggregates in epg-5 mutants can be ameliorated by increased formation of the SYX-17-SNAP-29-VAMP-7 SNARE complex that mediates autophagosome-lysosome fusion [36]. epg-5 mutants exhibit a weaker autophagy defect in hypodermal cells at the embryonic stage than other autophagy mutants. In the absence of EPG-5, other tethering factors such as the HOPS complex [37], or mechanisms that facilitate formation of the SNARE complex that drives autophagosome maturation, such as by reducing O-GlcNAcylation of SNAP-29, may be involved in autophagosome maturation in hypodermal cells.
Calpains regulate autophagy in a cell context-specific manner
Proteases have been shown to modulate autophagy activity at several steps by mediating cleavage of different substrates [2,38–43]. The cysteine protease ATG4 cleaves the C terminus of Atg8/LC3/LGG-1 to expose the glycine residue for lipidation [2]. Proteases in lysosomes degrade autophagy cargos for recycling, and loss of function of these proteases leads to accumulation of non-degradative autolysosomes [38]. Calpains, which are calcium-dependent non-lysosomal cysteine proteases, have also been shown to regulate autophagy by cleaving autophagy proteins. CAPN1 cleaves multiple human ATG proteins in an in vitro assay [39]. CAPN1 and CAPNS1 cleave ATG5 in human cell lines, and consequently negatively regulate basal autophagy activity by reducing the levels of ATG12–ATG5 conjugates [40]. CAPNS1 has also been reported to positively regulate both basal and starvation-induced autophagy activity in MEF cells [41]. Ras-induced activation of CAPN1 mediates degradation of BECN1 (beclin 1) [42]. Anoxia/reoxygenation (A/R)-induced CAPN2 activation degrades ATG7 and BECN1 and impairs mitophagy, a process involved in selective degradation of damaged mitochondria [43]. The activity of calpains is thus tightly controlled. Different calpains are differentially modulated by levels of intracellular Ca2+. CAPN1 is activated by micromolar concentrations of Ca2+, while millimolar concentrations of Ca2+ is required for CAPN2 activation [40]. Therefore, calpains may regulate autophagy in a cell context-specific manner, depending on the Ca2+ level.
During C. elegans development, clp-2 regulates autophagic degradation of protein aggregates in a stage- and tissue-specific manner. Loss of function of clp-2 causes accumulation of SQST-1 aggregates in hypodermis and neurons at larval and adult stages, while degradation of SQST-1 is normal in the body wall muscle and intestine. CLP-2 is likely to regulate autophagy through cleavage of its substrates, as the protease activity and the Ca2+-binding activity of CLP-2 are required for its role in autophagy. CAPN1 and CAPN2 cleave ATG5 at serine residue 35 [44]; however, this site is not conserved in C. elegans ATG-5. The substrates involved in autophagy that are cleaved by CLP-2 have yet to be determined. Other calpains are not involved in removal of SQST-1 during C. elegans development. It remains possible that these calpains are involved in autophagy regulation under specific pathological or stress conditions. Our study reveals the differential function of core autophagy genes and the presence of tissue-specific factors in autophagy during C. elegans development.
Materials and methods
Worm strains
The following strains were used in this work: N2 Bristol (wild-type), him-5(e1490), him-8(e1489), epg-3(bp933), atg-3(bp412), lgg-1(bp500), atg-9(bp564), epg-8(bp251), epg-9(bp320), epg-4(bp425), clp-2(bp852), clp-2(bp1325), clp-2(bp1326), clp-2(bp1327), clp-2(bp1009), clp-2(bp987), scav-3(bp979), epg-5(tm3425), epg-5(bp450), atg-18(gk378), atg-18(bp594), tra-3(ok2207), clp-7(ok2750), t11a5.6/clp-9(ok1866), w05g11.4/clp-10(ok2713), bec-1(bp613), clp-1(bp1316), clp-3(bp1477), clp-4(bp1317), clp-6(bp1318), clp-8(bp1321), bpIs267(Py37a1b.5::sqst-1::gfp, unc-76), bpIs262(Pges-1::sqst-1::gfp, unc-76), bpIs151(Psqst-1::sqst-1::gfp, unc-76), bpIs193(Phlh-1::sqst-1::gfp, unc-76), adIs2122(Plgg-1::gfp::lgg-1, rol-6), zcIs1(Paip::gfp), bpIs163(Pepg-4::epg-4::gfp, rol-6), bpIs183(Pepg-9::epg-9::gfp, rol-6), bpIs299(Patg-18::atg-18::gfp, unc-76), bpEx340(Pepg-5::epg-5::gfp, unc-76), bpEx230(Pclp-2::gfp::clp-2, unc-76), bpEx231(Plgg-1::gfp::lgg-1(g116a), rol-6), bpEx233(Prgef-1::sqst-1::gfp, odr-1::rfp), qxIs257(Pced-1::nuc-1::mcherry, unc-76), qxIs456 (Pced-1::mcherry::rab-5, unc-76) and qxIs66 (Pced-1::gfp::rab-7, unc-76).
Mapping and cloning of clp-2
Genetic and SNP (single nucleotide polymorphism) markers were used for mapping. bp852 was placed on chromosome III, between −3.03 cM and −2.41 cM. Fosmids in this region were used for transformation rescue experiments. Mutations in clp-2(bp852, bp987 and bp1009) were identified by sequencing genomic DNA.
Imaging, quantification and statistical analysis
A Zeiss Axio Imager M2 microscope, a Zeiss LSM710 and a Zeiss LSM880 were used for imaging. Comparable images were captured with the same exposure time and magnification. Similar regions in animals at the same developmental stage were analyzed. The fluorescence intensity and the number of SQST-1::GFP aggregates were quantified using ImageJ. Images captured at 400 x magnification were used for analyzing hypodermis, intestine and body wall muscle cells at the larval and adult stages and at 1000 x magnification for analyzing expression at the embryonic stage. For analyzing the GFP::LGG-1 and GFP::LGG-1G116A puncta, images were acquired at 63x by a Zeiss LSM710. Five (n = 5) or three (n = 3) independent animals for each strain were collected for quantification of the fluorescence intensity and the number of SQST-1::GFP aggregates. Three independent animals (n = 3) for each strain were collected for quantification of GFP::LGG-1 and GFP::LGG-1G116A punctate structures. For quantification of SQST-1::GFP fluorescence intensity and the number of SQST-1::GFP aggregates in body wall muscle cells at the larval and adult stages, worms were co-injected with 60 ng/ul pRF4(rol-6(su1006)) [45] to generate Rol transgenic lines, which facilitate image capture. The two-tailed unpaired Student’s t test was performed for statistical analysis and the results are shown as mean ± SEM. ns: not significant, P > .05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Generation of CRISPR-Cas9 knockout mutants in C. elegans
CRISPR-Cas9 sgRNAs were designed via http://crispr.mit.edu and cloned into the CRISPR-Cas9 vector pDD162 (Addgene, 47549). Cas9-sgRNA plasmids (50 ng/µl) were co-injected with pRF4(rol-6(su1006)) (50 ng/µl) in relevant strains. Rol F1 progeny were isolated and their progeny were examined. sgRNA target sequences were as follows:
clp-1: CCTCCAAGGATATCACCAC; clp-2: AAAAAGAAAGTCACAGAGG, GATACTTCGAAGGCAGATC; clp-3: GAGAGCTCGAATCACTCGC; clp-4: CAGGGATCACTGGAGCCAC; clp-6: GATCAGGGCAGTGAGTTGA; clp-8/F44F1.3: TGCATGGTCTGTATTTCGG.
The mutations in each gene were as follows: clp-1(bp1316): 13-nucleotide deletion (CCGGTGGTGATAT); clp-3(bp1477): 4-nucleotide deletion at exon 6 (GAGCTCGAATCACTCGCCGG, underlined nucleotides are deleted); clp-4(bp1317): 19-nucleotide deletion (CTCCGGTGGCTCCAGTGAT); clp-6(bp1318): 26-nucleotide deletion (GACGAGCTGCTCCGTCAACTCACTGC) and 4-nucleotide insertion (TCGT); clp-2(bp1325): 2-nucleotide deletion at exon 3 (AAGAAAGTCACAGAGGTGGA, underlined nucleotides are deleted). All mutations resulted in a frame-shift and introduced a premature stop codon.
RNA interference (RNAi) in C. elegans
RNAi bacterial clones, rpt-3 and the control L4440, were cultured on NGM agar plates containing 1 mM IPTG (Amresco, 0487-100G). The L2/L3 larval animals were plated onto RNAi feeding plates and day 1 adult worms were analyzed for Paip-1::GFP expression and SQST-1::GFP accumulation phenotype. RNAi clones were confirmed by sequencing.
Plasmid construction
To construction tissue-specific promotors driven SQST-1 reporters, SQST-1 cDNA was amplified from N2 cDNA library and inserted into pPD49.26 vectors (Addgene, 1686) with tissue-specific promoters using NheI and KpnI with the 2x Master Assembly Mix (Taihe Biotechnology, C5891-50), including body wall muscle cells (Phlh-1::gfp, 2850 bp promoter sequence amplified from WRM063aH05, nucleotides 20,437 to 23,286), neuronal cells (Prgef-1::gfp, 3542 bp promoter sequence amplified from WRM0623bF10, nucleotides 14,183 to 17,725), hypodermal cells (Py37a1b.5::gfp, also known as Phyp7::gfp, a vector provided by the Xiaochen Wang lab (Institute of Biophysics, Chinese Academy of Sciences)), containing 2892 bp promoter sequence), and intestinal cells (Pges-1::gfp, a vector provided by the Xiaochen Wang lab, containing 3313 bp promoter sequence).
To generate Py37a1b.5::CLP-2, CLP-2 cDNA was amplified from N2 cDNA library, and inserted into ppD49.26 Py37a1b.5::gfp vector, which had been digested by KpnI and SacI to remove the gfp fragment, by 2x Master Assembly Mix. Mutant CLP-2C295S, CLP-2∆4–75, CLP-2E374A, CLP-2D495A, D521A and CLP-2E374A, D495A, D521A were generated by PCR-based mutagenesis using Py37a1b.5::CLP-2 as a template.
To generate Pclp-2::gfp::clp-2 reporter by 2x Master Assembly Mix, genomic DNA containing 2-kb promoter and the entire open reading frame of clp-2 was cloned into the ppD49.26 vector and gfp was then inserted at the N terminus of CLP-2. All constructs were confirmed by sequencing.
Funding Statement
This work was supported by the National Natural Science Foundation of China [31561143001]; National Natural Science Foundation of China [31630048]; National Natural Science Foundation of China [31421002]; National Natural Science Foundation of China [31790403]; the Chinese Ministry of Science and Technology [2017YFA0503401]; the Strategic Priority Research Program of the Chinese Academy of Sciences [XDB19000000]; Key Research Program of Frontier Sciences of the Chinese Academy of Sciences [QYZDY-SSW-SMC006].
Acknowledgments
Work in the authors’ lab is supported by the National Natural Science Foundation of China (NSFC) (31561143001, 31630048, 31421002 and 31790403 to H.Z.), the Chinese Ministry of Science and Technology (2017YFA0503401), the Strategic Priority Research Program of the Chinese Academy of Sciences (CAS) (grant XDB19000000) and Key Research Program of Frontier Sciences, CAS (grant QYZDY-SSW-SMC006).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accesssed here.
References
- [1].Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10(7):458–467. [DOI] [PubMed] [Google Scholar]
- [2].Feng Y, He D, Yao Z, et al. The machinery of macroautophagy. Cell Res. 2014;24(1):24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lu Q, Wu F, Zhang H.. Aggrephagy: lessons from C. elegans. Bio-Chem J. 2013;452(3):381–390. [DOI] [PubMed] [Google Scholar]
- [4].Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14(12):759–774. [DOI] [PubMed] [Google Scholar]
- [5].Zhang H, Baehrecke EH. Eaten alive: novel insights into autophagy from multicellular model systems. Trends Cell Biol. 2015;25(7):376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Axe EL, Walker SA, Manifava M, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182(4):685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hayashi-Nishino M, Fujita N, Noda T, et al. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11(12):1433–1437. [DOI] [PubMed] [Google Scholar]
- [8].Yla-Anttila P, Vihinen H, Jokita E, et al. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy. 2009;5(8):1180–1185. [DOI] [PubMed] [Google Scholar]
- [9].Zhao YG, Chen Y, Miao G, et al. The ER-localized transmembrane protein EPG-3/VMP1 regulates SERCA activity to control ER-isolation membrane contacts for autophagosome formation. Mol Cell. 2017;67(6):974–989 e6. [DOI] [PubMed] [Google Scholar]
- [10].Zhao GY, Liu N, Miao GY, et al. The ER contact proteins VAPA/B interact with multiple autophagy proteins to modulate autophagosome biogenesis. Curr Biol. 2018;28(8):1234–1245. [DOI] [PubMed] [Google Scholar]
- [11].Ktistakis NT, Tooze SA. Digesting the expanding mechanisms of autophagy. Trends Cell Biol. 2016;26(8):624–635. [DOI] [PubMed] [Google Scholar]
- [12].Tian Y, Li Z, Hu W, et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell. 2010;141(6):1042–1055. [DOI] [PubMed] [Google Scholar]
- [13].Wang Z, Miao G, Xue X, et al. The Vici syndrome protein EPG5 is a Rab7 effector that determines the fusion specificity of autophagosomes with late endosomes/lysosomes. Mol Cell. 2016;63(5):781–795. [DOI] [PubMed] [Google Scholar]
- [14].Cheong H, Lindsten T, Wu J, et al. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci USA. 2011;108(27):11121–11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chang TK, Shravage BV, Hayes SD, et al. Uba1 functions in Atg7- and Atg3-independent autophagy. Nat Cell Biol. 2013;15(9):1067–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Joo JH, Wang B, Frankel E, et al. The noncanonical role of ULK/ATG1 in ER-to-Golgi trafficking is essential for cellular homeostasis. Mol Cell. 2016;62(6):491–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nishida Y, Arakawa S, Fujitani K, et al. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461(7264):654–658. [DOI] [PubMed] [Google Scholar]
- [18].Wu F, Li Y, Wang F, et al. Differential function of the two Atg4 homologues in the aggrephagy pathway in Caenorhabditis elegans. J Biol Chem. 2012;287(35):29457–29467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhang H, Wu F, Wang X, et al. The two C. elegans ATG-16 homologs have partially redundant functions in the basal autophagy pathway. Autophagy. 2013;9(12):1965–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wu F, Watanabe Y, Guo XY, et al. Structural basis of the differential function of the two C. elegans Atg8 homologs, LGG-1 and LGG-2, in autophagy. Mol Cell. 2015;60(6):914–929. [DOI] [PubMed] [Google Scholar]
- [21].Zhang Y, Yan L, Zhou Z, et al. SEPA-1 mediates the specific recognition and degradation of P granule components by autophagy in C. elegans. Cell. 2009;136(2):308–321. [DOI] [PubMed] [Google Scholar]
- [22].Lu Q, Yang P, Huang X, et al. The WD40 repeat PtdIns(3)P-binding protein EPG-6 regulates progression of omegasomes to autophagosomes. Dev Cell. 2011;21(2):343–357. [DOI] [PubMed] [Google Scholar]
- [23].Zhang H, Chang JT, Guo B, et al. Guidelines for monitoring autophagy in Caenorhabditis elegans. Autophagy. 2015;11(1):9–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kaletsky R, Yao V, Williams A, et al. Transcriptome analysis of adult Caenorhabditis elegans cells reveals tissue-specific gene and isoform expression. PLoS Genet. 2018;14(8):e1007559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li Y, Chen B, Zou W, et al. The lysosomal membrane protein SCAV-3 maintains lysosome integrity and adult longevity. J Cell Biol. 2016;215(2):167–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Joyce PI, Satija R, Chen M, et al. The atypical calpains: evolutionary analyses and roles in Caenorhabditis elegans cellular degeneration. PLoS Genet. 2012;8(3):e1002602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Keith SA, Maddux SK, Zhong Y, et al. Graded proteasome dysfunction in Caenorhabditis elegans activates an adaptive response involving the conserved SKN-1 and ELT-2 transcription factors and the autophagy-lysosome pathway. PLoS Genet. 2016;12(2):e1005823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Guo B, Huang X, Zhang P, et al. Genome-wide screen identifies signaling pathways that regulate autophagy during Caenorhabditis elegans development. EMBO Rep. 2014;15(6):705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Moldoveanu T, Hosfield CM, Lim D, et al. A Ca(2+++) switch aligns the active site of calpain. Cell. 2002;108(5):649–660. [DOI] [PubMed] [Google Scholar]
- [30].Maemoto Y, Ono Y, Kiso S, et al. Involvement of calpain-7 in epidermal growth factor receptor degradation via the endosomal sorting pathway. Febs J. 2014;281(16):3642–3655. [DOI] [PubMed] [Google Scholar]
- [31].Maday S, Wallace KE, Holzbaur EL. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol. 2012;196(4):407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhao YG, Zhang H. Formation and maturation of autophagosomes in higher eukaryotes: a social network. Curr Opin Cell Biol. 2018;53:29–36. [DOI] [PubMed] [Google Scholar]
- [33].Hamasaki M, Furuta N, Matsuda A, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495(7441):389–393. [DOI] [PubMed] [Google Scholar]
- [34].Nascimbeni AC, Giordano F, Dupont N, et al. ER-plasma membrane contact sites contribute to autophagosome biogenesis by regulation of local PI3P synthesis. Embo J. 2017;36(14):2018–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhao YG, Zhao H, Miao L, et al. The p53-induced gene Ei24 is an essential component of the basal autophagy pathway. J Biol Chem. 2012;287(50):42053–42063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Guo B, Liang Q, Li L, et al. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat Cell Biol. 2014;16(12):1215–1226. [DOI] [PubMed] [Google Scholar]
- [37].Jiang P, Nishimura T, Sakamaki Y, et al. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol Biol Cell. 2014;25(8):1327–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kaminskyy V, Zhivotovsky B. Proteases in autophagy. Biochim Biophys Acta. 2012;1824(1):44–50. [DOI] [PubMed] [Google Scholar]
- [39].Norman JM, Cohen GM, Bampton ETW. The in vitro cleavage of the hAtg proteins by cell death proteases. Autophagy. 2014;6(8):1042–1056. [DOI] [PubMed] [Google Scholar]
- [40].Xia HG, Zhang L, Chen G, et al. Control of basal autophagy by calpain1 mediated cleavage of ATG5. Autophagy. 2010;6(1):61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Demarchi F, Bertoli C, Copetti T, et al. Calpain is required for macroautophagy in mammalian cells. J Cell Biol. 2006;175(4):595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yoo BH, Wu X, Li Y, et al. Oncogenic ras-induced down-regulation of autophagy mediator Beclin-1 is required for malignant transformation of intestinal epithelial cells. J Biol Chem. 2010;285(8):5438–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kim JS, Nitta T, Mohuczy D, et al. Impaired autophagy: A mechanism of mitochondrial dysfunction in anoxic rat hepatocytes. Hepatology. 2008;47(5):1725–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yousefi S, Perozzo R, Schmid I, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8(10):1124–1132. [DOI] [PubMed] [Google Scholar]
- [45].Mello CC, Kramer JM, Stinchcomb D, et al. Efficient gene transfer in C.elegans: extrachromosomal maintenance and integration of transforming sequences. Embo J. 1991;10(12):3959–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.