

ABSTRACT
PRKN/parkin activation through phosphorylation of its ubiquitin and ubiquitin-like domain by PINK1 is critical in mitophagy induction for eliminating the damaged mitochondria. Deubiquitinating enzymes (DUBs) functionally reversing PRKN ubiquitination are critical in controlling the magnitude of PRKN-mediated mitophagy process. However, potential DUBs that directly target PRKN and antagonize its pro-mitophagy effect remains to be identified and characterized. Here, we demonstrated that USP33/VDU1 is localized at the outer membrane of mitochondria and serves as a PRKN DUB through their interaction. Cellular and in vitro assays illustrated that USP33 deubiquitinates PRKN in a DUB activity-dependent manner. USP33 prefers to remove K6, K11, K48 and K63-linked ubiquitin conjugates from PRKN, and deubiquitinates PRKN mainly at Lys435. Mutation of this site leads to a significantly decreased level of K63-, but not K48-linked PRKN ubiquitination. USP33 deficiency enhanced both K48- and K63-linked PRKN ubiquitination, but only K63-linked PRKN ubiquitination was significantly increased under mitochondrial depolarization. Further, USP33 knockdown increased both PRKN protein stabilization and its translocation to depolarized mitochondria leading to the enhancement of mitophagy. Moreover, USP33 silencing protects SH-SY5Y human neuroblastoma cells from the neurotoxin MPTP-induced apoptotic cell death. Our findings convincingly demonstrate that USP33 is a novel PRKN deubiquitinase antagonizing its regulatory roles in mitophagy and SH-SY5Y neuron-like cell survival. Thus, USP33 inhibition may represents an attractive new therapeutic strategy for PD patients.
Abbreviations: CCCP: carbonyl cyanide 3-chlorophenylhydrazone; DUB: deubiquitinating enzymes; MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; OMM: outer mitochondrial membrane; PD: Parkinson disease; PINK1: PTEN induced kinase 1; PRKN/PARK2: parkin RBR E3 ubiquitin protein ligase; ROS: reactive oxygen species; TM: transmembrane; Ub: ubiquitin; UBA1: ubiquitin like modifier activating enzyme 1; UBE2L3/UbcH7: ubiquitin conjugating enzyme E2 L3; USP33: ubiquitin specific peptidase 33; WT: wild type.
KEYWORDS: Apoptosis, mitophagy, PRKN/parkin, ubiquitination, USP33 deubiquitinase
Introduction
Mutations of human PINK1 (PTEN induced kinase 1) and the ring-like E3 ubiquitin ligase PRKN have been linked to early-onset familial PD [1]. PINK1-PRKN axis plays a crucial role in maintaining mitochondrial homeostasis through triggering mitochondrial elimination (mitophagy) [2], during which PINK1-dependent phosphorylation of both PRKN and its ubiquitin is critical in PRKN activation which subsequently ubiquitinates several downstream targets, in particular “outer membrane of mitochondria (OMM)” proteins [2,3]. As ubiquitination and deubiquitination processes are delicately balanced, deubiquitinating enzymes (DUBs) that counteract PRKN through reversing ubiquitination are critical in regulating the magnitude of PRKN-mediated mitophagy as unregulated mitophagy is likely to result in a massive cell death.
Extensive efforts in search of PRKN-specific DUBs resulted in the identification of USP15 and USP30 that effectively antagonized PRKN in the depolarized mitochondria by opposing PRKN-mediated ubiquitination of OMM proteins, but not the ubiquitination status of PRKN itself [2,4]. USP8 was shown to directly deubiquitinate PRKN, however, its role was mainly removing K6-linked Ub conjugates and moreover, its silencing inhibited rather than promoted the extent of mitophagy by delaying PRKN translocation to depolarized mitochondria [5].
To search for unknown DUBs of PRKN, we performed immunoprecipitation (IP) and mass spectrometry analyses using the lysates of Flag-PRKN overexpressed U2OS cells, and identified deubiquitinase USP33/VDU1 in the IP precipitates. USP33, a VHL (von Hippel-Lindau tumor suppressor) protein-interacting deubiquitinating enzyme [6], has been shown to deubiquitinate multiple proteins involved in diverse cellular functions: ADRB/β-adrenergic receptor recycling [7], vertebrate commissural axon guidance [8], macroautophagy/autophagy and innate immune response [9], centrosome amplification [10] and cancer cell migration [11,12]. Endogenous USP33 is localized to the endoplasmic reticulum-associated structure [13], but its cellular functions are not clearly elucidated. This study provides the evidence for the first time that USP33 is localized at the outer membrane of mitochondria, and deubiquitinates PRKN through their physical interaction. USP33 knockdown promotes the recruitment of PRKN to depolarized mitochondria leading to an enhanced mitophagy versus a significantly decreased apoptotic cell death induced by a neurotoxin: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). USP33 selectively removes K6, K11, K48 and K63-linked ubiquitin conjugates from PRKN. Furthermore, USP33 deubiquitinates PRKN at Lys435, and on depolarized mitochondria, mainly regulates K63-linked PRKN ubiquitination. Our findings convincingly established that USP33, as the deubiquitinase of PRKN, antagonizes PRKN-mediated role in mitophagy and SH-SY5Y neuroblastoma cell survival under stress conditions.
Results
USP33 interacts with PRKN
To identify and characterize the DUBs interacting with PRKN, tandem affinity purification coupled with mass spectrometry was performed on lysates of U2OS cells ectopically expressing FLAG-PRKN. We identified two DUBs in the PRKN-pull down complex: USP15 and USP33 (Table S1). It has been reported that USP15 antagonizes PRKN-mediated mitophagy without any impact on PRKN ubiquitination and its mitochondrial translocation [4]. We next examined the physical interaction of USP33 with PRKN by coimmunoprecipitation (co-IP) assay. Since EDTA in the lysis buffer might produce false-positive results in the mass spectrometry analysis [14], we then validated the specificity of interactions between PRKN and USP33 using EDTA-free lysis buffer in our co-IP assays. In the Flag-PRKN affinity-isolation precipitates using anti-Flag antibody, endogenous USP33 was detected with anti-USP33 antibody (Figure 1A). Likewise, endogenous PRKN was observed in HA-USP33 pull-down complex using anti-HA antibody (Figure 1B). In vitro GST pull-down assay was also performed using purified GST-USP33 and His-PRKN proteins expressed in E. coli BL21. Purified GST-USP33 protein immobilized on glutathione Sepharose beads was incubated with His-PRKN (Figure S1). Consistent with the IP results, His-PRKN was pulled down with GST-USP33. These observations support the interaction of USP33 with PRKN.
Figure 1.
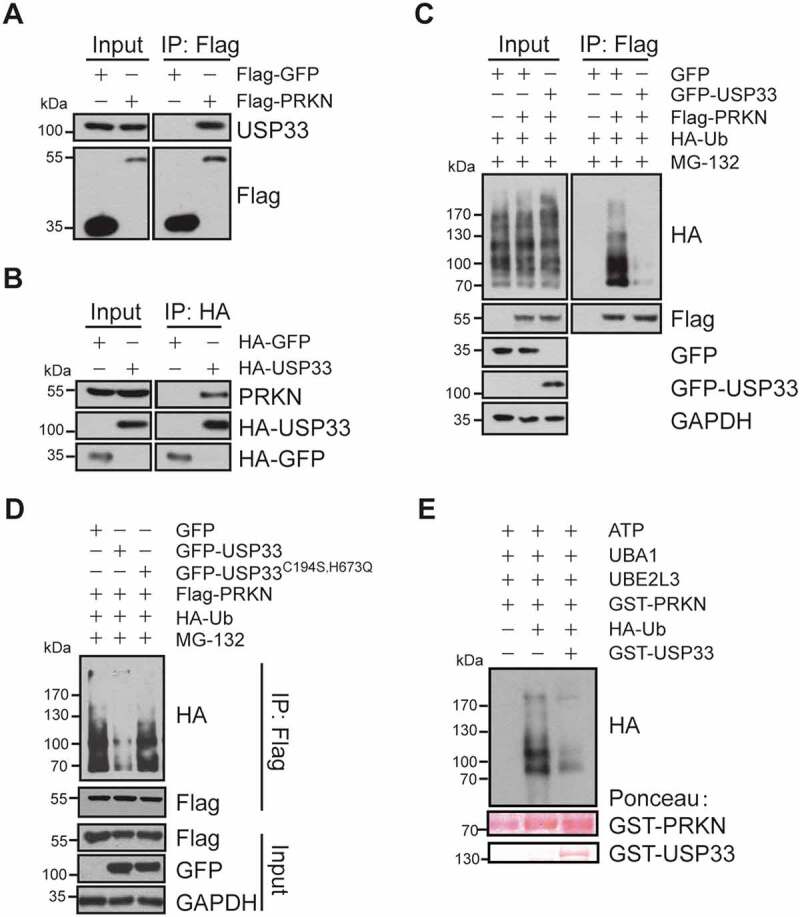
USP33 interacts with PRKN. (A) Endogenous USP33 protein was detected in anti-Flag immunoprecipitate from cell lysate of HEK293 cells overexpressing Flag-PRKN by western blotting. (B) Endogenous PRKN protein was verified in anti-HA immunoprecipitate from the lysate of HA-USP33 transfected HEK293 cells. HA-GFP was used for negative control. (C) Decreased ubiquitination level of PRKN in HEK293 cells overexpressing GFP-USP33. HEK293 cells were co-transfected with Flag-PRKN, HA-Ub and GFP-USP33, then treated with MG-132 for 3 h. Flag-PRKN was pulled-down with anti-Flag antibody and the PRKN ubiquitination level was detected with anti-HA antibody by western blotting. (D) Defective deubiquitination activity of USP33 mutant (USP33C194S,H673Q mutant) on PRKN. HEK293 cells were co-transfected with Flag-PRKN, HA-Ub and GFP-USP33 or GFP-USP33C194S,H673Q mutant, then treated with 10 μM MG-132 for 3 h. PRKN ubiquitination level was examined in Flag immunoprecipitates with anti-HA by western blotting. (E) USP33 removes PRKN Ub conjugates by in vitro ubiquitination assay. GST-PRKN and GST-USP33 levels stained with Ponceau were included.
A list of PRKN deletion mutants were next constructed to determine PRKN domains interacting with USP33. The full-length of USP33 showed a strong binding with the PRKN UBL and RING2 domains, but weakly with RING1 domain (Figure S2A). In contrast, among the various USP33 domains, including zinc finger and ubiquitin-specific protease (USP), DUSP1 and DUSP2, only USP domain showed interaction with PRKN (Figure S2B). The USP domain has been shown to possess the catalytic activity for removing K48- and K63-linked Ub from polypeptides [15], indicating that USP33 may regulate the ubiquitination level of PRKN.
USP33 deubiquitinates PRKN
Demonstration of the interaction between PRKN and USP33 led us to examine whether or not USP33 deubiquitinates PRKN. The results showed that PRKN ubiquitination level was markedly reduced in GFP-USP33 overexpressed HEK293 cells (Figure 1C). Conversely, USP33 knockdown in U2OS-Flag-PRKN cells led to a robust increase in PRKN ubiquitination tested with anti-PRKN antibody (Figure S3). In contrast, overexpression of the catalytically inactive USP33C194S,H673Q mutant [9] failed to show any effect on PRKN ubiquitination (Figure 1D) indicating the DUB activity of USP33 is necessary for PRKN ubiquitination.
In vitro ubiquitination assay was subsequently performed to address if USP33 could directly deubiquitinate PRKN using the previously reported self-ubiquitination reaction system including UBA1 (ubiquitin like modifier activating enzyme 1), UBE2L3/UbcH7 (ubiquitin conjugating enzyme E2 L3), ATP, HA-Ub and affinity-purified recombinant GST-PRKN [16]. When GST-USP33 proteins was included in the reaction system, PRKN was efficiently deubiquitinated in vitro by USP33 (Figure 1E).
USP33 is a novel mitochondrial transmembrane DUB
An earlier study detected the localization of USP33 protein at endoplasmic reticulum-associated structures and Golgi apparatus [13]. We then wished to determine if USP33 is localized to mitochondria using exogenously expressing GFP-USP33 in U2OS cells since commercial anti-USP33 antibody for immunofluorescence application is not currently available. In GFP-USP33-positive U2OS cells, USP33 was found to colocalize with a mitochondrial marker TOMM20 (Figure 2A), which is further validated by western blotting analysis showing the presence of USP33 protein in the mitochondrial fractions isolated from HEK293 cells (Figure 2B). The mitochondrial fraction appears to be nearly free of nuclear, cytoplasmic and ER contaminations as the markers of LMNB1 (lamin B1) for nuclear protein, TUBB/β-tubulin for cytoplasm and CANX (calnexin) for ER were at undetectable levels.
Figure 2.
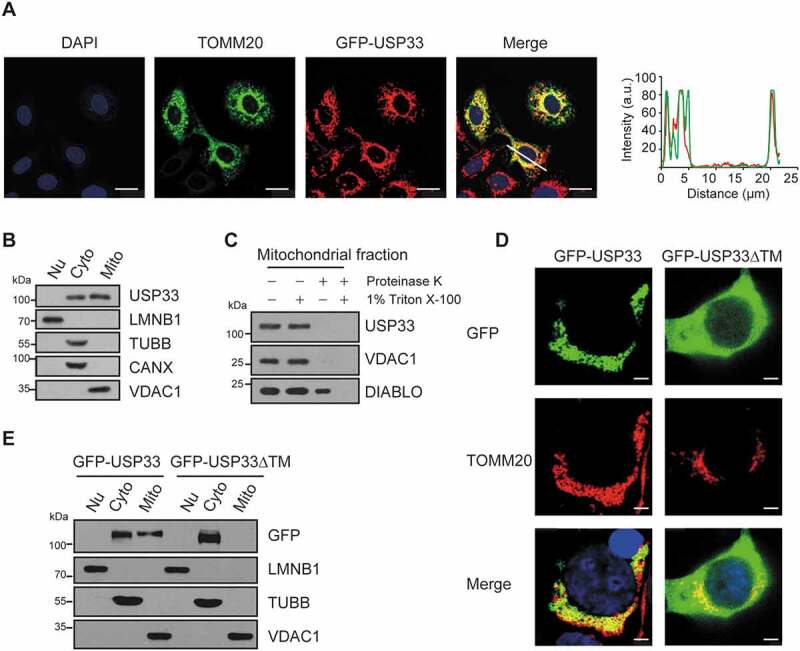
USP33 is a novel OMM DUB. (A) GFP-USP33 colocalizes with mitochondrial marker TOMM20 by immunofluorescence staining. Images were captured using Leica TCS SP8 Confocal microscope. Line drawing graph was quantified by ImageJ software (http://rsbweb.nih.gov/ij/). a.u., arbitrary units. Scale bar: 25 μm. (B) USP33 level in different fractions (Nu: nucleus; Cyto: cytoplasm; Mito: mitochondria) of HEK293 cells by western blotting. LMNB1 (lamin B1): nuclear marker; TUBB/β-tubulin: cytoplasmic marker; CANX (calnexin): ER marker; VDAC1: mitochondrial marker. (C) Protease K assay showing localization of USP33 at OMM. VDAC1: mitochondrial outer membrane marker, DIABLO/SMAC: intermembrane space marker. (D) Colocalization of GFP-USP33, but not GFP-USP33ΔTM (residues 549-569 deletion), with mitochondrial protein TOMM20 by immunostaining analysis. Scale bar: 2.5 μm. (E) GFP-USP33 is present in both mitochondrial and cytosolic fractions, whereas GFP-USP33ΔTM only in cytosolic fraction of HEK293 cells transiently transfected with WT or mutant USP33.
For further validation, a protease K protection assay was utilized to determine the sub-localization of USP33 in mitochondria. When mitochondrial fraction was treated with 100 ng/ml proteinase K for 30 min on ice in the absence of 1% Triton X-100, USP33 and the outer mitochondrial membrane protein VDAC1 were digested, whereas the mitochondrial intermembrane space protein DIABLO/SMAC was still present. In contrast, in the presence of 1% Triton X-100, all three proteins were digested. These findings suggest that USP33 is localized at OMM (Figure 2C).
It has been reported that some OMM proteins, like TOMM20 and TOMM70, are anchored to mitochondria through transmembrane (TM) region flanking with positively charged residues, called “signal-anchor” sequences [17]. The putative TM region of USP33 was predicted by TMpred program analysis (http://www.ch.embnet.org/software/TMPRED_form.html), and amino acid residues 549-569 in its DUB catalytic domain were identified to be the potential TM. To validate this finding, a USP33 mutant with 21 amino acid residues deletion in the TM (GFP-USP33ΔTM) was generated. Western blotting result clearly showed the presence of GFP-USP33 in both mitochondrial and cytosolic fractions, whereas GFP-USP33ΔTM was found only in the cytosolic fraction. This observation was further substantiated by immunofluorescence staining showing the colocalization of mitochondria with GFP-USP33, but not with GFP-USP33ΔTM (Figure 2D,E). Collectively, these results illustrate that USP33 is a mitochondrial DUB with specific sub-localization to OMM.
USP33 knockdown enhances mitophagy through promoting the translocation of PRKN to depolarized mitochondria
One of the earlier events during PRKN-mediated mitophagy is the translocation of PRKN with USP33 prompted us to investigate whether or not the functional status of USP33 impacts the translocation of PRKN to depolarized mitochondria [18,19]. Using previously reported GFP-PRKN overexpressed cell model system [5], we found that upon treatment with 20 μM mitochondrial uncoupler carbonyl cyanide 3-chlorophenylhydrazone (CCCP), USP33 knockdown led to a much earlier translocation of GFP-PRKN to mitochondria than siRNA control, with the starting time of ~15 min and 20 min in USP33-depleted and control cells, respectively. Upon continuous treatment, both siControl and USP33-depleted cells showed a rapid increase in the percentage of cells positive for GFP-PRKN recruitment to depolarized mitochondria. Remarkably, USP33 silencing induced a significantly higher percentage of positive cells at each time-point of 15 to 60 min compared to controls. After 75 min of treatment, the percentage of positive cells in both groups reached a plateau with approximately 80% of the cells showing GFP-PRKN recruitment to mitochondria (Figure 3A,C). In our USP33 knockdown assays, a mixture (1:1) of siRNA-1 and siRNA-2 specifically targeting USP33 was used. To exclude the possible siRNA off-target effect, each siRNA was independently validated for its capability in inhibiting USP33 expression as well as affecting PRKN recruitment to mitochondria and mtDNA clearance during mitophagy (Figure S4). The USP33 knockdown efficiency was determined by western blotting (Figure 3B). These findings clearly demonstrated that USP33 silencing accelerates CCCP-induced PRKN translocation to depolarized mitochondria.
Figure 3.
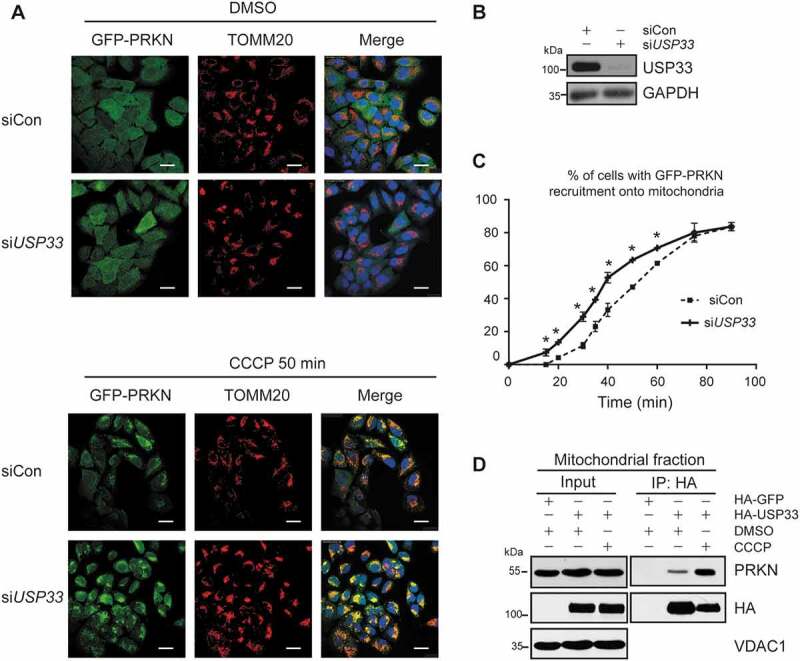
USP33 depletion promotes PRKN recruitment to depolarized mitochondria. U2OS-GFP-PRKN cells were transfected with control siRNA (siCon) or USP33 siRNA1 + 2 (siUSP33) for 48 h and then treated with 20 μM CCCP. The cells were collected at indicated time-points for immunostaining with anti-TOMM20. (A) The appearance of GFP-PRKN puncta superposed on the mitochondrial TOMM20 was visualized. Scale bar: 25 μm. (B) Knockdown of USP33 validated by western blotting. (C) Percentage of cells with GFP-PRKN puncta colocalized with TOMM20 was quantified in siControl and USP33-depleted cells at 5-min interval. Over 150 cells were examined at each time-point, and data represents mean ± SD from three independent experiments. One-way ANOVA was used for statistical analysis. *P < 0.05. (D) Interaction between PRKN and USP33 in mitochondrial fraction of HEK293 cells with or without CCCP (20 μM) treatment for 2 h.
Mitochondrial homeostasis is well-maintained under physiological conditions through mitophagy-mediated clearance of damaged mitochondria induced by reactive oxygen species (ROS) [2,10,20]. We then used co-IP assay to examine the interaction between PRKN and USP33 in mitochondrial fraction under CCCP treatment. HA-USP33 was transiently expressed in HEK293 cells with or without 20 μM CCCP for 2 h, and mitochondrial fraction was isolated. Interaction of USP33 with PRKN at mitochondria exists at a relatively low level but is substantially increased under CCCP treatment (Figure 3D), supporting that USP33 controls PRKN-mediated mitophagy at a proper level.
Given that the depolarized mitochondria can be eliminated by mitophagy in a PRKN-dependent manner after prolonged CCCP treatment [5], we wished to determine whether the enhanced mitochondrial translocation of PRKN by USP33 silencing is accompanied by an increased mitophagy manifested by the changed levels in TOMM20 protein and mitochondrial DNA content. USP33 expression was depleted in U2OS-GFP-PRKN cells followed by treatment with 20 μM CCCP for 6 and 12 h (Figure 4C). TOMM20 protein level at 6 h of treatment showed a minimal decrease in both siControl and USP33 knockdown cells. In contrast, at 12 h of treatment, a substantial reduction in TOMM20 expression in both groups was observed. Specifically, the reduced expression was more pronounced in USP33-depleted cells relative to siControl cells (Figure 4C). Immunofluorescence staining with TOMM20 also demonstrated a significant decrease in the percentage of cells with TOMM20 fluorescence staining in USP33-deleted cells after 12 h of CCCP treatment (Figure 4A,B). It should be noted that GFP-PRKN protein level in USP33-deficient cells was also enhanced relative to USP33 proficient cells after CCCP treatment (Figure 4A,C).
Figure 4.
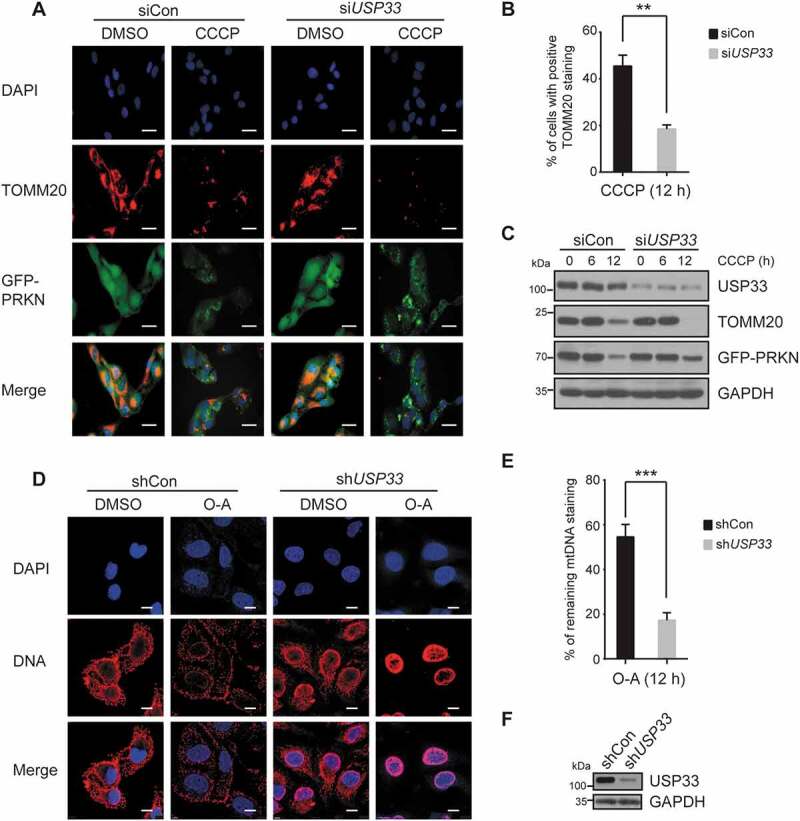
USP33 depletion enhances PRKN-mediated mitochondrial clearance. U2OS-GFP-PRKN cells were transfected with siCon or USP33 siRNA1 + 2 (siUSP33) for 48 h, treated with 20 μM CCCP for 6 or 12 h, and immunostained with anti-TOMM20. TOMM20 staining was visualized (A) and percentage cells with TOMM20 staining was quantified (B). A minimum of 200 cells at 12 h was analyzed. The data represents mean ± SD from three independent experiments. **P < 0.01, Student’s t test. Images were captured using Leica DM5000 microscope. Scale bar: 25 μm. (C) Expression levels of USP33, TOMM20 and PRKN determined by western blotting. (D) Images of mtDNA content and (E) quantification of mtDNA fluorescence density in U2OS-GFP-PRKN cells upon treatment of 10 μM oligomycin and 4 μM antimycin A for 12 h. Cells were infected with adenoviral scrambled control (shCon) or USP33 shRNA 1 + 2 (shUSP33) for 72 h before treatment. Images were captured using Leica TCS SP8 Confocal microscope. Scale bar: 10 μm. The mtDNAs from >300 cells per group were quantified by Image-Pro Plus software. The data represents mean ± SD from three independent experiments. ***P < 0.001, Student’s t test. (F) Knockdown of USP33 validated by western blotting.
Mitochondrial clearance efficiency was next examined in USP33-depleted cells by measuring mtDNA content with an anti-DNA antibody. USP33 expression was first silenced in U2OS-GFP-PRKN cells using adenoviral shRNA, and then treated for 12 h with 10 μM oligomycin and 4 μM antimycin A (O-A), two mitochondrial uncouplers to induce mitophagy through inhibiting complex III of the electron transport system and Fo-ATPase, respectively [21,22]. mtDNA fluorescence density in USP33-depleted cells was observed to be largely diminished in USP33-depleted cells, but only marginally in shControl cells, upon 12 h of prolonged treatment (Figure 4D–F). U2OS cells with an endogenous level of PRKN were employed to examine the effect of USP33 depletion on mitochondrial clearance in comparison with USP15 and USP30 that were previously shown to inhibit in vivo mitophagy by counteracting PRKN-mediated ubiquitination of its substrates [2,4]. Similar to USP30 and USP15, USP33 silencing led to a marked decrease in TOMM20 protein level determined by western blotting and immunostaining assays (Figure S5). These findings indicate that loss of USP33 expression accelerates the clearance of impaired mitochondria by mitophagy.
USP33 removes K6, K11, K48 and K63-linked ubiquitin conjugates from PRKN
PRKN is an E3 ubiquitin ligase undergoing self-ubiquitination. It was demonstrated that all seven internal lysine residues (Lys6, 11, 27, 29, 33, 48 and 63) of ubiquitin (Ub) can serve as the potential sites for chain-elongation and mediate different endpoints such as proteasome degradation pathway or non-degradation functions [23]. To identify the preferentially targeted Ub linkages conjugated to PRKN by USP33, a series of HA-tagged Ub mutants were generated that had only one single lysine (K) with all other lysine residues mutated to arginine. Each of these mutant constructs was transfected with Flag-PRKN plasmid into HEK293 cells. These cells were subsequently transfected with GFP-USP33 or GFP-vector alone after 24 h. Immunoprecipitates with anti-Flag antibody were subjected to western blotting with anti-HA antibody to evaluate the level of each K linkage-conjugated PRKN. USP33 overexpression was found to preferentially remove the PRKN-Ub conjugates at K6, K11, K48 and K63 (Figure 5A, Figure S6), illustrating that USP33 has a marked preference for certain ubiquitin linkages.
Figure 5.
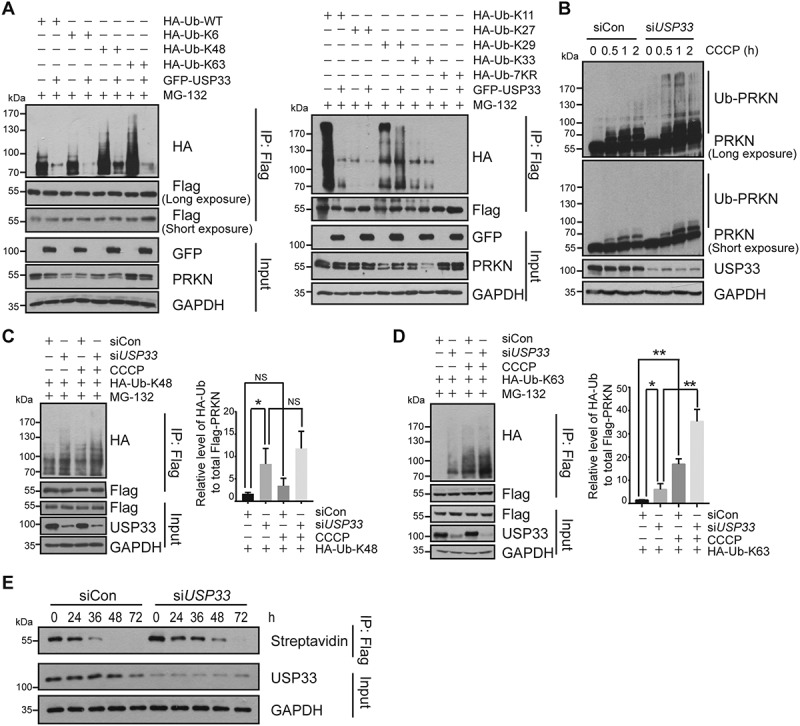
USP33 preferentially removes K6, K11, K48 and K63-linked ubiquitin conjugates from PRKN. (A) HEK293 cells were co-transfected with a series of indicated expression plasmids and harvested after treated with 10 μM MG-132 for 3 h. The levels of different K-linked PRKN ubiquitination in Flag immunoprecipitates upon USP33 overexpression were determined by western blotting with anti-HA antibody. (B) U2OS-Flag-PRKN cells were transfected with siCon or USP33 siRNA1 + 2 (siUSP33) for 48 h, then treated with 20 μM CCCP for different times (0, 0.5, 1, 2 h). PRKN ubiquitination during mitophagy induced by CCCP was examined by western blotting. (C and D) K48 and K63-linked PRKN ubiquitination in U2OS cells after USP33 depletion with or without CCCP treatment for 45 min. The band density of PRKN ubiquitination level was quantified by Image J software. The data represent the mean ± SD from three independent experiments. A one-way ANOVA and Student’s t test were performed for statistical analysis. *P < 0.05, **P < 0.01. NS, not significant. (E) Stability of PRKN protein after USP33 depletion in U2OS-Flag-PRKN cells using metabolic labeling assay determined by immunoprecipitation followed by western blotting.
Effect of USP33 on PRKN ubiquitination during mitophagy process was also examined under CCCP treatment. We first tested the PRKN ubiquitination in total cell lysates of control and USP33-silenced U2OS-Flag-PRKN cells after CCCP treatment. In siRNA control cells, CCCP treatment led to a transient increase in the level of PRKN self-ubiquitination at different time-points of 0.5, 1 and 2 h (Figure 5B), which is consistent with previous report [24]. In contrast, USP33 depletion resulted in a much higher level of PRKN self-ubiquitination at each time-point upon CCCP relative to control (Figure 5B). It should be noted that CCCP treatment combined with USP33 depletion markedly enhanced the ubiquitination level of PRKN and TOMM20, whereas PINK1 or USP30 protein levels were either slightly increased or unchanged upon USP33 silencing (Figure S7), suggesting that USP33 modulates PRKN activity and subsequent ubiquitination of its substrates.
It is well established that K48- and K63-linked Ub chains mainly target the proteins for proteasome degradation and diverse signaling pathways, respectively, whereas the functional roles of K6- and K11-linked Ub chains are not very clear [23]. Since both K48- and K63-Ub linkages were identified on polyubiquitinated PRKN post CCCP treatment, we wished to verify which Ub linkage is preferentially removed from PRKN by USP33 in the absence or presence of CCCP treatment. USP33 knockdown was observed to significantly enhance the levels of both K48- and K63-linked polyubiquitination on PRKN (Figure 5C,D). Interestingly, only CCCP treatment led to a substantial increase in K63-, but not K48-linked PRKN polyubiquitination. In particular, K63-linked PRKN polyubiquitination was more significantly enhanced by CCCP treatment or USP33 depletion relative to K48-linked ubiquitination (Figure 5C,D). In support, USP33 overexpression preferentially reduced the levels of PRKN-Ub conjugates at K48 and K63 with or without CCCP treatment (Figure S8). These findings suggest that the augmented PRKN ubiquitination by CCCP treatment is mainly mediated by K63-Ub chain.
USP33 deubiquitinates both K48- and K63-linked Ub chain from PRKN and its depletion enhances PRKN protein level under mitochondrial depolarization (Figure 4A,C). To verify if USP33 status impacts PRKN protein stability, a methionine surrogate AHA-based metabolic labeling assay [25,26] was performed using U2OS-Flag-PRKN cells. The result showed that in comparison with siControl, USP33 depletion markedly enhances the stability of PRKN protein (Figure 5E), which is consistent with the findings from cycloheximide (CHX) chase assay (Figure S9), suggesting that the overall effect of USP33 depletion stabilizes PRKN protein mainly through the enhanced K63-linked PRKN polyubiquitination rather than K48-associated proteasome degradation pathway.
USP33 regulates PRKN self-ubiquitination at Lys435 mainly through K63-linked Ub chains and its silencing protects SH-SY5Y neuroblastoma cells from the neurotoxin MPTP
To determine which lysine amino acid on PRKN protein is specifically targeted by USP33, all the 18 lysine residues on PRKN was screened by point-mutation assay (lysine mutated to arginine). Our study revealed that arginine substitution at Lys48 or Lys435 markedly reduced the level of PRKN ubiquitination (Figure S10). We next tested if these two lysine sites are also targeted by USP33. As expected (Figure 6A), the WT PRKN showed a markedly enhanced ubiquitination level after USP33 silencing but PRKNK48R displayed only a marginal increase. Remarkedly, Lys435 mutation led to an unchanged level of PRKN ubiquitination in both siControl and USP33-depleted cells, indicating that USP33 primarily deubiquitinates PRKN at Lys435.
Figure 6.
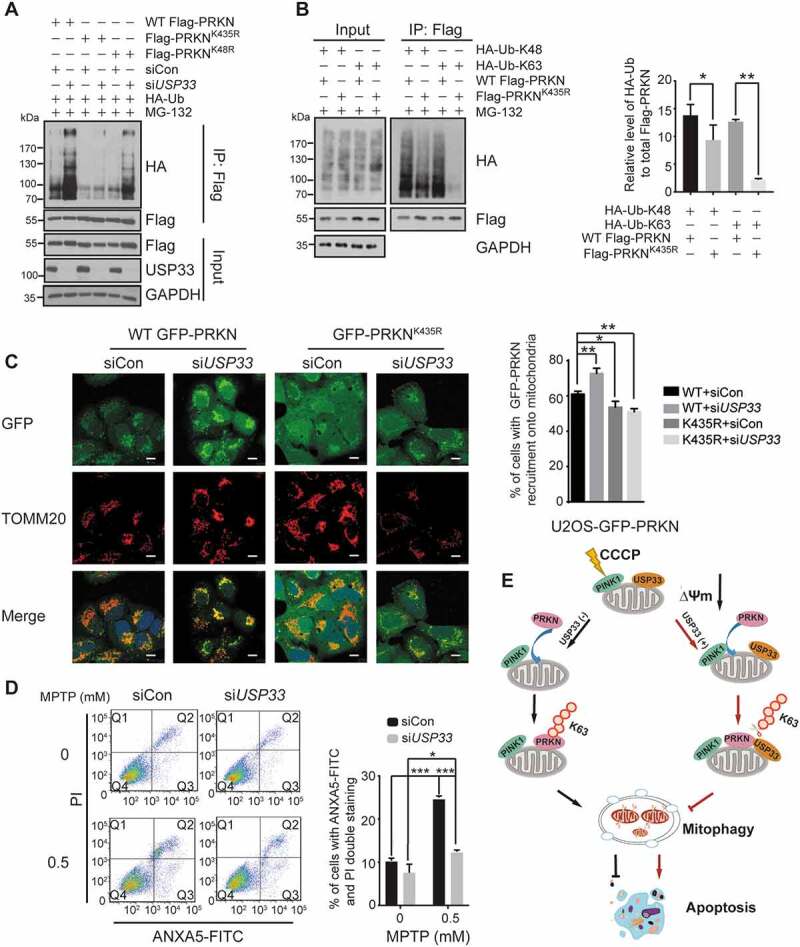
USP33 regulates PRKN ubiquitination at Lys435 and its silencing protects SH-SY5Y cells from the neurotoxin MPTP. (A) Lysine sites on PRKN deubiquitinated by USP33. U2OS cells were transfected with the indicated constructs for 24 h, then with siCon or USP33 siRNA1 + 2 (siUSP33) for 48 h. PRKN ubiquitination level in Flag immunoprecipitates was examined with anti-HA antibody by western blotting. (B) Levels of K48- and K63-linked Ub at Lys435. HEK293 cells were transfected with Flag-tagged WT PRKN or K435R mutant with HA-Ub-K48 or HA-Ub-K63 for 36 h, and harvested after 10 μM MG-132 treatment. The densities of PRKN ubiquitination were quantified using Image J software. The data represent the mean ± SD from three independent experiments. A one-way ANOVA and Student’s t test were performed for statistical analysis. *P < 0.05, **P < 0.01. (C) An enhanced recruitment of WT PRKN, but not K435R mutant, to depolarized mitochondria in USP33-depleted U2OS cells. U2OS cells stably expressing WT or K435-mutated PRKN were used. The images were captured using Leica TCS SP8 Confocal microscope. Scale bar: 10 μm. Percentage of cells with WT GFP-PRKN or GFP-PRKNK435R puncta colocalized with TOMM20 was quantified in siControl and USP33-depleted cells after 60 min of CCCP treatment. Over 100 cells were examined for each time-point, and data represents mean ± SD from three independent experiments. *P < 0.05, **P < 0.01, Student’s t test. (D) SH-SY5Y cells were transfected with siCon or USP33 siRNA1 + 2 (siUSP33) for 48 h, and then treated with 0.5 mM MPTP for 24 h. The cells were collected and stained with ANXA5-FITC and propidium iodide (PI) using the ANXA5/annexin V-FITC/PI Detection Kit (Beyotime, C1062M). The percentage of apoptotic cells were quantified by flow cytometry (BD FACSAria II). The data represents mean ± SD from three independent experiments. *P < 0.05 and ***P < 0.001, Student’s t test. (D) Model depicting the antagonizing effect of USP33 on PRKN-mediated mitophagy. USP33 as an OMM protein interacts with PRKN under mitochondrial depolarization and regulates the magnitude of PRKN-induced mitophagy mainly through removing K63-linked Ub chain from PRKN. USP33 deficiency leads a persistent elevation of K63-linked Ub conjugates on PRKN consequently stabilizing PRKN protein and augmenting mitophagy induction. Overall, USP33 depletion promotes an efficient clearance of damaged mitochondria and potently protects neuron-like cells from neurotoxin-induced apoptotic cell death.
Our findings that USP33 overexpression decreases the levels of PRKN-Ub conjugates at K6, K11, K48 and K63 sites led us to determine which K-linked Ub chain is specifically conjugated to PRKN Lys435. We mainly focused on K48 and K63 since these two types of chain linkages have been well-recognized as the key performers in mediating either protein stability or functional pathways. Flag-tagged WT PRKN or its mutant (K435R) in combination with HA-tagged K48 or K63 Ub was transfected into HEK293 cells. The results from immunoprecipitation with anti-Flag followed by western blotting with anti-HA showed that Lys435 mutation results in a slight decrease in K48-linked Ub conjugates, but a distinctly reduced level of K63-linked Ub conjugates on PRKN (Figure 6B). Our collective findings suggest that PRKN ubiquitination at Lys435 is regulated by USP33 through K63-linked Ub chains.
The importance of Lys435 in USP33-mediated deubiquitination of PRKN and subsequent mitophagy induction was further tested in U2OS cells ectopically expressing WT or K435R mutated PRKN. A significant enhancement in the percentage of cells with positive GFP-PRKN recruitment to depolarized mitochondria was observed in WT GFP-PRKN cells, but not in GFP-PRKNK435R cells treated with 20 μM CCCP for 60 min (Figure 6C). Additionally, mtDNA clearance efficiency was also significantly augmented in WT GFP-PRKN cells instead of K435R mutant cells upon 12 h of treatment with 10 μM oligomycin and 4 μM antimycin A (O-A) (Figure S11). These data strongly support that USP33 regulates mitophagy mainly through deubiquitinating PRKN at Lys435.
USP33 depletion augments PRKN ubiquitination leading to a more efficient and orderly clearance of damaged mitochondria by mitophagy thereby probably conferring cellular survival through maintenance of mitochondrial quality. To test this possibility, apoptotic cell death was measured in siControl and USP33-depleted SH-SY5Y cells exposed to 0.5 mM MPTP, a neurotoxin inducing mitochondrial-dependent cell apoptosis because of excessive intracellular ROS production [27]. In the absence of MPTP treatment, SH-SY5Y cells transfected with control siRNA showed a relatively higher level of apoptotic cell death (10.17%), which is similar with previous reports [28]. However, USP33 knockdown significantly decreased the apoptotic cell death (7.56%, **P < 0.01 compared to control). Additionally, under MPTP treatment, both siRNA control and USP33-depleted cells showed a significantly enhanced apoptotic cell death with 23.72% and 12.26%, respectively. Moreover, extent of apoptotic cell death observed in USP33 knockdown cells was significantly lower relative to siRNA control (Figure 6D). This finding clearly indicates a protective role of USP33 silencing on neurotoxin-exposed SH-SY5Y cells.
Discussion
PINK1-PRKN axis plays an essential role in clearing the damaged mitochondria selectively through mitophagy. This process involves the recruitment of PRKN to depolarized mitochondria and PINK1-mediated Ub phosphorylation on PRKN which is critical for PRKN activation and subsequent mono- and poly-Ub assembly on its substrates in a self-reinforcing cycle [20]. DUBs have the potential to suppress the effect of PRKN on removal of damaged mitochondria by mitophagy by regulating the extent of ubiquitination of both PRKN itself and its substrates. Here, we provided the conclusive evidence for the first time that USP33 located at the outer membrane of mitochondria serves as a DUB specific for PRKN. As expected, depletion of USP33 under CCCP treatment promotes mitophagy through accelerating and enhancing the translocation of PRKN to depolarized mitochondria. Furthermore, we found that USP33 specifically deubiquitinates PRKN at Lys435 and preferentially removes K6, K11, K63 and K48-linked ubiquitin conjugates from PRKN. USP33 silencing enhanced PRKN ubiquitination and subsequent protein stabilization during mitophagy, thereby protected the SH-SY5Y neuroblastoma cells from neurotoxin-induced apoptotic cell death. Overall, our study demonstrates that USP33 is a novel PRKN DUB potently antagonizing its effect on mitophagy through directly deubiquitinating PRKN.
Several K-linked chains (K6, K11, K48 and K63) can be formed on PRKN [5]. Previous finding that USP33 deubiquitinates Ub chains linked via K63 and K48 [7] led us to clarify which type of Ub chain linkages conjugated on PRKN is preferentially removed by USP33. Our findings demonstrated that USP33 mainly regulates K63-linked ubiquitination of PRKN, and its silencing induces a significantly enhanced level of K63-linked ubiquitin chain formation on PRKN and subsequent PRKN protein stabilization. This result is consistent with the observations that USP33 depletion enhances translocation of PRKN to damaged mitochondria and subsequent mitophagy induction.
PRKN Lys435 was identified to be the main ubiquitination site regulated by USP33, which is supported by the observations that PRKNK435R cells are insensitive to USP33 depletion during CCCP-induced mitophagy. It should be noted that PRKNK435R cells under siRNA control treatment also show a slightly defective mitophagy, which possibly results from K435R being very close to the ubiquitin acceptor site of Cys431 [19] and therefore affecting the catalysis of ubiquitin transfer.
Neurons are post-mitotic cells with high demand for energy production and therefore, tend to accumulate oxidative damage over time in their mitochondria [29]. PINK1-PRKN-mediated mitophagy is critical in protecting neurons through maintaining mitochondrial quality and restraining innate immunity. DUBs that are capable of maintaining the balance of ubiquitination/deubiquitination of PRKN and its substrates are potentially significant in counteracting PRKN-mediated mitophagy process. USP30 serves as the deubiquitinase of OMM substrates of PRKN, and its silencing has been shown to functionally protect dopaminergic neurons against the mitochondrial toxin paraquat in PRKN and PINK1 mutant flies through restoring mitochondrial integrity [2]. We provided the novel evidence that USP33 directly deubiquitinates PRKN, and its silencing promotes PRKN translocation to depolarized mitochondria and mitophagy induction. Moreover, USP33 inhibition protects SH-SY5Y neuron-like cells from apoptotic cell death in both neurotoxin MPTP treated and untreated conditions, suggesting a potential protective role through interfering its expression under in vivo condition.
Mitophagy is a high-quality clearance system fundamentally for preserving cellular homeostasis and for critical processes, such as inflammation and cell death or diseases, including cancer and neurodegeneration [20]. USP33 was reported to have higher expression levels in the central nervous system (CNS), predominantly in the regions affected in PD, such as amygdala and substantia nigra [30]. Our findings suggest that USP33 inhibition may have therapeutic value in PD patient treatment through enhancing mitochondrial quality control.
Materials and methods
Chemicals and antibodies
The following chemical reagents were used: CCCP (C2759), antimycin A (A0149), MG-132 (C2211), MPTP (M0896) from Sigma-Aldrich; oligomycin A (Selleck Chemicals, S1478); Click-iT™ AHA (L-azidohomoalanine; C10102), Click-iT™ DIBO biotin (C10412), ER-Tracker™ Red (E34250) from Invitrogen. Antibodies: TUBB/β-Tubulin (T5293), FLAG (F3165), USP33 (WH0023032M1) and USP30 (HPA016952) from Sigma-Aldrich; LMNB1/Lamin B1 (sc-374015) and TOMM20/TOM20 (sc-11415) from Santa Cruz Biotechnology; Streptavidin-HRP (3999S), HA (3724S), VDAC1 (D73D12) and DIABLO/SMAC (2954S) from Cell Signaling Technology; USP15 (14354-1-AP) and CANX/calnexin (10427-2-AP) from Proteintech Group; PRKN/parkin (Abcam, ab15954), PINK1 (Novus Biologicals, BC100-494SS), GAPDH (Millipore, MB374), GFP (Abclonal, AE012), and anti-DNA (Progen, 61014).
Vector construction
Full-length human USP33 (isoform 1) and PRKN genes were PCR-amplified from a cDNA pool reversely transcribed from total RNA of U2OS cells, and inserted into pcDNA3.0 vector (Youbio, VT2051). Ubiquitination site-mutated PRKN was generated using site-mutagenesis kit (Beyotime, D0206). All inserts were confirmed by DNA sequencing, and their expression was validated by western blotting.
shRNA and siRNA
We chose the previously reported USP33 siRNA oligo sequences [10], and used Rfect siRNA transfection reagent (Bio-trans, 21014) to transfect these siRNA into U2OS cells.
USP33 siRNA1: CAAUGUUAAUUCAGGAUGATT;
USP33 siRNA2: GGCUUGGAUCUUCAGCCAUTT;
USP30 siRNA1: GCUGCUUGUUGGAUGUCUUTT;
USP30 siRNA2 GGGAUGUUGUGUGUGACAATT;
USP15 siRNA1: GGAACACCUUAUUGAUGAATT;
USP15 siRNA2: GCAGAUGGAAGGCCAGAUATT;
control siRNA: UUCUCCGAACGUGUCACGUTT.
A mixture (1:1) of siRNA1 and siRNA2 targeting USP33 was used for knockdown assays. The adenoviral shRNA (1 and 2) for USP33 and scrambled control shRNA had the same targeting sequences as respective siRNAs.
Immunofluorescence microscopy
For colocalization of USP33 with mitochondria, U2OS cells transiently transfected with GFP-USP33 were fixed, permeabilized and immunostained with rabbit anti-human TOMM20 followed by TEXAS Red-conjugated secondary antibody (Vector Laboratories, TI-1000). The images were captured with Leica TCS SP8 Confocal microscope and line graph densities were quantified by ImageJ software (NIH).
For PRKN recruitment to mitochondria and mitochondrial clearance analyses, USP33 was silenced in U2OS-GFP-PRKN cells and treated with 20 μM CCCP or 10 μM oligomycin and 4 μM antimycin A. After fixation and permeabilization, cells were immunostained with anti-TOMM20 and anti-DNA antibody followed by fluorescence-conjugated secondary antibody. The images were captured with a Leica confocal laser scanning microscope. The percentage of positive cells showing PRKN recruitment or TOMM20 staining were calculated. A total of >150 cells were examined for each time-point, and data represents mean ± SD from three independent experiments. The mtDNA fluorescence density was calculated according to previously reported method [21]. The data represents mean ± SD from >300 cells, and three independent experiments were performed.
Mitochondrial fractionation isolation and protease K treatment assay
Qiagen Qproteome mitochondria isolation kit (Qiagen, 37612) was used for isolating mitochondrial fraction following the manufacture’s instruction. For proteinase K treatment assay, purified mitochondrial fraction was digested with 100 ng/ml proteinase K (Amresco, 0706) in the absence or presence of 1% Triton X-100 (Sigma-Aldrich, V900502) on ice for 30 min. The levels of USP33, VDAC1 and SMAC proteins were examined by western blotting.
Co-immunoprecipitation (co-IP) assay and LC-MS/MS analysis
U2OS-Flag-PRKN cells were lysed (150 mM NaCl, 1% NP-40 [Amresco, J619], 50 mM Tris-HCl, pH 7.5, 5 mM EDTA, 0.1 mM PMSF [Beyotime, ST505], 20 mM NaF, 0.5 mM benzamidine [Sigma-Aldrich, 12072], 1 mg/ml aprotinin [Amresco, E429], 0.1 mM NaVO3, 2 mM microcystin [Millipore, 475815] and 1 mg/ml leupeptin [Sigma-Aldrich, L2884]). After centrifugation, the supernatant was incubated with anti-Flag-M2 beads (Sigma-Aldrich, M8823) for 2 h at 4°C. After being washed three times with lysis buffer and heated at 80°C for 10 min in 1× Laemmli sample buffer, the binding complex was separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by silver staining and LC-MS/MS analysis.
The EDTA-free lysis buffer was used in the following co-IP assays for validation of protein-protein interactions.
GST affinity-isolation assay
His-PRKN and GST-USP33 proteins were expressed in E. coil BL21 (DE3) cells. The purified proteins were dissolved in lysis buffer and 20 μL of 50% GST gel suspension for each sample were used. After rotation at 4°C for 3 h, the agarose gel was washed three times with lysis buffer, heated in 2× sodium dodecyl sulfate (SDS) sample buffer, and the affinity-isolated protein was analyzed by western blotting with the indicated antibodies.
In vivo and in vitro ubiquitination assays
HEK293 cells were transfected with the indicated Flag-PRKN and HA-Ub for 24 h, and then transfected with GFP-USP33 or USP33 siRNA1 + 2 (siUSP33). After 36–48 h, the cells were treated with 10 μM MG-132 for 3 h, lysed and immunoprecipitated with Flag M2 beads followed by western blotting using anti-HA to detect the level of PRKN ubiquitination.
Purified GST-PRKN with or without GST-USP33 proteins were incubated in a mixture of ubiquitin ligation reaction (final volume, 30 μl) containing 5 mM MgCl2, 50 mM Tris-HCl (pH 7.5), 2 mM DTT, 4 mM ATP, 1 μg ubiquitin (Boston Biochem, U-110-01M), 200 ng E1-UBA1/UbE1 (Boston Biochem, E-305-025), and 400 ng E2-UBE2L3/UbcH7 (Boston Biochem, E2-640-100) at 30°C for 2 h. The reaction was terminated by boiling for 5 min in a 1× SDS sample buffer, and the proteins were resolved by SDS-PAGE followed by western blotting with the indicated antibodies.
Metabolic labeling assay for PRKN protein stability
L-azidohomoalanine (AHA)-based metabolic labeling assay was performed following previously reported procedures [25,26] with some modifications. In brief, U2OS-Flag-PRKN cells were transfected with siCon or USP33 siRNA1 + 2 (siUSP33) for 48 h. After washed with warm PBS (Beyotime, C0221A), cells were cultured in L-methionine-free DMEM (Invitrogen, 21013) for 30 min to deplete the intracellular methionine reserves, and then labeled with AHA in 10% FBS DMEM (L-methionine-free) for 4 h. Cells were continually cultured in 10% FBS DMEM at 37°C in 5% CO2 after washed with warm PBS and harvested at different time points. Click reaction was performed by incubating the cells with DIBO biotin (Invitrogen, C10412) at final concentration of 20 μM in PBS containing 1% FBS for 15 min at 37°C. After washed three times with PBS containing 1% FBS, the cells were lysed (150 mM NaCl, 0.5% NP-40, 50 mM Tris-HCl, pH 7.5, 10% glycerin [Sigma-Aldrich, G2289], 1× protease inhibitor cocktail [Sigma-Aldrich, P8340]) and immunoprecipitation with anti-Flag was performed. PRKN protein level in the immunoprecipitated complex was determined by western blotting.
Statistical analysis
All experimental data were presented as the mean ± SD. One-way ANOVA or Student’s t test were used to determine significance using SPSS version 20.0 for Windows (SPSS Inc.). *P < 0.05, **P < 0.01 or ***P < 0.001 indicated that the difference was statistically significant.
Funding Statement
This work was supported by the National Key Research and Development Program of China under Grant no. 2018YFA0108501, the National Basic Research Program of China (973 Program) under Grant no. 2015CB910601, the National Natural Science Foundation of China under Grant nos. 31570815, 31770869 and Open Project of Key Laboratory of Genomic and Precision Medicine, CAS.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Gegg ME, Schapira AH.. PINK1-parkin-dependent mitophagy involves ubiquitination of mitofusins 1 and 2: implications for Parkinson disease pathogenesis. Autophagy. 2011;7(2):243–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bingol B, Tea JS, Phu L, et al. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature. 2014;510(7505):370–375. [DOI] [PubMed] [Google Scholar]
- [3].Gladkova C, Maslen SL, Skehel JM, et al. Mechanism of parkin activation by PINK1. Nature. 2018;559(7714):410–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cornelissen T, Haddad D, Wauters F, et al. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum Mol Genet. 2014;23(19):5227–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Durcan TM, Tang MY, Perusse JR, et al. USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. Embo J. 2014;33(21):2473–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Li Z, Na X, Wang D, et al. Ubiquitination of a novel deubiquitinating enzyme requires direct binding to von Hippel-Lindau tumor suppressor protein. J Biol Chem. 2002;277(7):4656–4662. [DOI] [PubMed] [Google Scholar]
- [7].Shenoy SK, Modi AS, Shukla AK, et al. Beta-arrestin-dependent signaling and trafficking of 7-transmembrane receptors is reciprocally regulated by the deubiquitinase USP33 and the E3 ligase Mdm2. Proc Natl Acad Sci U S A. 2009;106(16):6650–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yuasa-Kawada J, Kinoshita-Kawada M, Wu G, et al. Midline crossing and Slit responsiveness of commissural axons require USP33. Nat Neurosci. 2009;12(9):1087–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Simicek M, Lievens S, Laga M, et al. The deubiquitylase USP33 discriminates between RALB functions in autophagy and innate immune response. Nat Cell Bio. 2013;15(10):1220–1230. [DOI] [PubMed] [Google Scholar]
- [10].Li J, D’Angiolella V, Seeley ES, et al. USP33 regulates centrosome biogenesis via deubiquitination of the centriolar protein CP110. Nature. 2013;495(7440):255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Huang Z, Wen P, Kong R, et al. USP33 mediates Slit-Robo signaling in inhibiting colorectal cancer cell migration. Int J Cancer. 2015;136(8):1792–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wen P, Kong R, Liu J, et al. USP33, a new player in lung cancer, mediates Slit-Robo signaling. Protein Cell. 2014;5(9):704–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Thorne C, Eccles RL, Coulson JM, et al. Isoform-specific localization of the deubiquitinase USP33 to the Golgi apparatus. Traffic. 2011;12(11):1563–1574. [DOI] [PubMed] [Google Scholar]
- [14].Beasley SA, Hristova VA, Shaw GS. Structure of the Parkin in-between-ring domain provides insights for E3-ligase dysfunction in autosomal recessive Parkinson’s disease. Proc Natl Acad Sci U S A. 2007;104(9):3095–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nijman SMB, Luna-Vargas MPA, Velds A, et al. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123(5):773–786. [DOI] [PubMed] [Google Scholar]
- [16].He J, Xia M, Yeung PKK, et al. PICK1 inhibits the E3 ubiquitin ligase activity of Parkin and reduces its neuronal protective effect. Proc Natl Acad Sci U S A. 2018;115(30):E7193–E7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kanaji S, Iwahashi J, Kida Y, et al. Characterization of the signal that directs Tom20 to the mitochondrial outer membrane. J Cell Biol. 2000;151(2):277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Akabane S, Uno M, Tani N, et al. PKA regulates PINK1 stability and Parkin recruitment to damaged mitochondria through phosphorylation of MIC60. Mol Cell. 2016;62(3):371–384. [DOI] [PubMed] [Google Scholar]
- [19].Trempe JF, Sauve V, Grenier K, et al. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science. 2013;340(6139):1451–1455. [DOI] [PubMed] [Google Scholar]
- [20].Durcan TM, Fon EA. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015;29(10):989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ito K, Turcotte R, Cui J, et al. Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science. 2016;354(6316):1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–229. [DOI] [PubMed] [Google Scholar]
- [24].Chan NC, Salazar AM, Pham AH, et al. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20(9):1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhang J, Wang J, Ng S, et al. Development of a novel method for quantification of autophagic protein degradation by AHA labeling. Autophagy. 2014;10(5):901–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chai Q, Webb SR, Wang Z, et al. Study of the degradation of a multidrug transporter using a non-radioactive pulse chase method. Anal Bioanal Chem. 2016;408(27):7745–7751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Xu D, Duan H, Zhang Z, et al. The novel tetramethylpyrazine bis-nitrone (TN-2) protects against MPTP/MPP+-induced neurotoxicity via inhibition of mitochondrial-dependent apoptosis. J Neuroimmune Pharmacol. 2014;9(2):245–258. [DOI] [PubMed] [Google Scholar]
- [28].Wu W, Jingbo S, Xu W, et al. S-trityl-L-cysteine, a novel Eg5 inhibitor, is a potent chemotherapeutic strategy in neuroblastoma. Oncol Lett. 2018;16(1):1023–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kalia LV, Lang AE. Parkinson’s disease. Lancet. 2015;386(9996):896–912. [DOI] [PubMed] [Google Scholar]
- [30].Fekete C, Freitas BC, Zeold A, et al. Expression patterns of WSB-1 and USP-33 underlie cell-specific posttranslational control of type 2 deiodinase in the rat brain. Endocrinology. 2007;148(10):4865–4874. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.