

ABSTRACT
ULK1 (unc-51 like autophagy activating kinase 1) is the key mediator of MTORC1 signaling to macroautophagy/autophagy. ULK1 functions as a protein complex by interacting with ATG13, RB1CC1/FIP200, and ATG101. How the ULK1 complex is regulated to trigger autophagy induction remains unclear. In this study, we have determined roles of Atg8-family proteins (ATG8s) in regulating ULK1 activity and autophagy. Using human cells depleted of each subfamily of ATG8, we found that the GABARAP subfamily positively regulates ULK1 activity and phagophore and autophagosome formation in response to starvation. In contrast, the LC3 subfamily negatively regulates ULK1 activity and phagophore formation. By reconstituting ATG8-depleted cells with individual ATG8 members, we identified GABARAP and GABARAPL1 as positive and LC3B and LC3C as negative regulators of ULK1 activity. To address the role of ATG8 binding to ULK1, we mutated the LIR of endogenous ULK1 to disrupt the ATG8-ULK1 interaction by genome editing. The mutation drastically reduced the activity of ULK1, autophagic degradation of SQSTM1, and phagophore formation in response to starvation. The mutation also suppressed the formation and turnover of autophagosomes in response to starvation. Similar to the mutation of the ULK1 LIR, disruption of the ATG13-ATG8 interaction suppressed ULK1 activity and autophagosome formation. In contrast, RB1CC1 did not show any specific binding to ATG8s, and mutation of its LIR did not affect ULK1 activity. Together, this study demonstrates differential binding and opposite regulation of the ULK1 complex by GABARAPs and LC3s, and an important role of the ULK1- and ATG13-ATG8 interactions in autophagy induction.
Abbreviations: ATG5: autophagy related 5; ATG7: autophagy related 7; ATG8: autophagy related 8; ATG13: autophagy related 13; ATG14: autophagy related 14; ATG16L1: autophagy related 16 like 1; ATG101: autophagy related 101; BAFA1: bafilomycin A1; BECN1: beclin 1; Cas9: CRISPR associated protein 9; CRISPR: clustered regularly interspaced short palindromic repeats; EBSS: earle’s balanced salt solution; DAPI: 4ʹ-6-diamidino-2-phenylindole; GABARAP: GABA type A receptor-associated protein; GABARAPL1: GABA type A receptor-associated protein like 1; GABARAPL2: GABA type A receptor-associated protein like 2; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GFP: green fluorescence protein; gRNA: guide RNA; KI: kinase inactive mutant; KO: knockout; LC3A: microtubule associated protein 1 light chain 3 alpha; LC3B: microtubule associated protein 1 light chain 3 beta; LC3C: microtubule associated protein 1 light chain 3 gamma; LIR: LC3-interacting region; MTORC1: mechanistic target of rapamycin kinase complex 1; PBS: phosphate buffered saline; PCR: polymerase chain reaction; PE: phosphatidylethanolamine; PtdIns3P: phosphatidylinositol-3-phosphate; qPCR: quantitative PCR; RB1CC1/FIP200: RB1 inducible coiled-coil 1; RPS6KB1: ribosomal protein S6 kinase B1; SEM: standard error of the mean; SQSTM1/p62: sequestosome 1; TALEN: transcription activator-like effector nuclease; TUBA: tubulin alpha; ULK1: unc-51 like autophagy activating kinase 1; WB: western blotting; WIPI2: WD repeat domain phosphoinositide interacting 2; WT: wild type.
KEYWORDS: ATG8, GABARAP, LC3, LIR, ULK1
Introduction
Autophagy is an evolutionarily conserved process where intracellular components are sequestered in double-membrane vesicles and degraded by lysosomes. Autophagy is induced in response to diverse cellular stresses, such as nutrient starvation, accumulation of damaged mitochondria, and misfolded protein aggregates. Despite recent progress in our understanding, the molecular mechanism underlying autophagy induction remains elusive. The main theme in the field is that MTORC1 (mechanistic target of rapamycin kinase complex 1) regulates autophagy initiation via ULK1 (unc-51 like autophagy activating kinase 1), a serine/threonine protein kinase. ULK1 interacts with ATG13, RB1CC1/FIP200, and ATG101 to form a complex that plays the central role in triggering the molecular events required for initiation of autophagosome formation [1–5]. Under nutrient-enriched conditions, MTORC1 phosphorylates ULK1 to suppress the activity of ULK1. The suppression is released under nutrient starvation or MTORC1 inhibition, the condition that is minimally required to trigger autophagosome formation. When active, ULK1 phosphorylates PI3KR4-associated BECN1/Beclin 1 and ATG14 to stimulate the class III phosphatidylinositol 3-kinase (PtdIns3K) [6–8]. PtdIns3K catalyzes the conversion of phosphatidylinositol to phosphatidylinositol-3-phosphate (PtdIns3P). PtdIns3P recruits ZFYVE1/DFCP1 and WIPI2 to mediate the formation of the phagophore, the early membrane structure that develops into an autophagosome [9,10].
One of the key molecular events of autophagosome formation is the conjugation of ATG8 family proteins with phosphatidylethanolamine (PE) [11,12]. Mammals express 2 subfamilies of ATG8 proteins defined by amino acid sequence similarity: the LC3 subfamily consisting of LC3A, LC3B, LC3B2, and LC3C (referred to as LC3 proteins or simply LC3s); the GABARAP subfamily consisting of GABARAP, GABARAPL1, and GABARAPL2 (referred to as GABARAP proteins or GABARAPs) [13]. ATG8 interacts with the LC3-interacting region (LIR) motif-containing proteins and recruits them to the autophagic machinery for degradation. However, the interactions do not appear to always function for cargo degradation. Recent studies have shown that recombinant or overexpressed ULK1, ATG13 and RB1CC1 bind ATG8 proteins through their LIRs [14–17]. Kraft et al. (2012) showed that the ULK1 LIR is important for ULK1 to associate with autophagosomes, implying that the ULK1-ATG8 interaction might play a role in autophagosome formation [15]. Joachim et al. (2015) showed that the ULK1 LIR is important for the ULK1-mediated phosphorylation of ATG13 Ser318, a phosphorylation important for mitophagy [18], in a full medium condition using overexpressed ULK1 and ATG13 [16]. Although those studies have provided important insights, the role of ATG8 proteins in regulating ULK1 and autophagy has remained largely unknown.
In this study, we used genome editing techniques to manipulate expression of ATG8 proteins or disrupt the interactions between the ULK1 complex and ATG8s. Through the analysis, we determined that GABARAPs play crucial roles in activating ULK1 and phagophore formation in response to starvation. Interestingly, those roles of GABARAPs were opposed by LC3s. Furthermore, we determined that the ULK1-ATG8 interaction is crucial for ULK1 activation, phagophore formation, and autophagosome turnover in response to starvation.
Results
GABARAPs and LC3s differentially bind to the ULK1 complex proteins
Previous studies have shown that recombinant or overexpressed ULK1, ATG13, and RB1CC1 bind ATG8 proteins via their LIR motifs [14–17]. To characterize the interactions at endogenous levels, we attempted to isolate endogenous ATG8 proteins by immunoprecipitation using anti-ATG8 antibodies or by co-immunoprecipitation using anti-ULK1 or anti-ATG13 antibodies. This approach was not successful in enriching or co-immunoprecipitating ATG8 proteins, probably due to hindrance of antibody binding by protein-protein interactions or weak affinities of the association between ATG8 and the ULK1 complex in our experimental condition. As an alternative approach, we expressed MYC-tagged ATG8 proteins in HEK293T cells and analyzed endogenous ULK1 and its associated proteins co-immunoprecipitated with MYC-ATG8s. Each of the 3 GABARAPs showed interaction with ULK1 (Figure 1A), a result consistent with the previous studies using overexpressed and recombinant proteins [14,15]. Among GABARAPs, GABARAPL1 showed the strongest binding affinity towards ULK1 and ATG13. We could not detect a specific interaction between endogenous ATG13 and MYC-GABARAPL2, indicating that GABARAPL2 might selectively bind to ULK1. In contrast, RB1CC1 did not specifically interact with any ATG8 protein. Among the LC3 subfamily members, only LC3C showed specific interactions with the ULK1 complex proteins (Figure 1A). The immunoprecipitation enriched GABARAP and LC3C more efficiently than other ATG8s probably due to difference in accessibility to antibody binding for immunoprecipitation.
Figure 1.
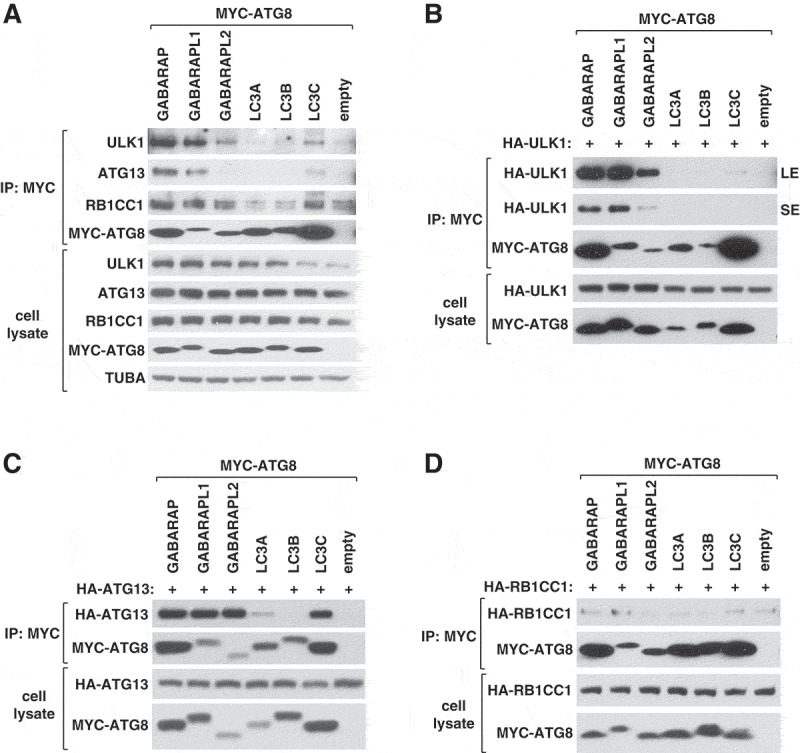
GABARAPs and LC3s interact with endogenous ULK1 and ATG13 with differential binding affinities. (A) GABARAP, GABARAPL1, GABARAPL2 and LC3C, but not LC3A or LC3B, interact with endogenously expressed ULK1 complex proteins. MYC-tagged ATG8 proteins were transiently expressed in HEK293T cells. Endogenous ULK1, ATG13, and RB1CC1 co-immunoprecipitated with MYC-ATG8 were analyzed by western blotting (WB). (B–D) ATG8 proteins interact with ULK1 and ATG13 with varying affinities. MYC-tagged ATG8 proteins were co-expressed with HA-tagged ATG13, ULK1, or RB1CC1 in HEK293T cells. The amounts of HA-tagged proteins co-immunoprecipitated with MYC-ATG8 were analyzed by WB. LE, long exposure; SE, short exposure.
To further clarify the differential binding of ATG8 proteins towards the ULK1 complex proteins, we tested individual components of the ULK1 complex for their interactions with ATG8s. Consistent with the previous reports [14,15], HA-ULK1 was co-immunoprecipitated with GABARAPs. However, ULK1 did not show substantial interaction with any of the tested LC3 proteins (Figure 1B). This result is different from that of Alemu et al. [14], where ULK1 was shown to interact with LC3C and GABARAPs at high levels, with LC3A moderately, and with LC3B weakly. HA-ATG13 was co-immunoprecipitated with all 3 GABARAPs. When accounting for the total amounts of immunoprecipitated GABARAPs, stronger binding affinities of ATG13 were observed for GABARAPL1 and GABARAPL2 (Figure 1C). Since no interaction was detected between endogenous ATG13 and MYC-GABARAPL2 (Figure 1A), the interaction between overexpressed ATG13 and MYC-GABARAPL2 might indicate a special state of ATG13 manifested in its overexpressed form. Unlike ULK1, ATG13 in overexpression interacted with LC3C and relatively weakly with LC3A (Figure 1C). RB1CC1 did not show specific binding to any of the tested ATG8 proteins (Figure 1D).
GABARAPs and LC3s have opposite effects on ULK1 activity
The finding that ATG8s have differential binding affinities toward the ULK1 complex proteins has raised a question of whether the ATG8 members might have distinct functions in regulating the ULK1 complex. To address this question, we analyzed the effects of depletion of ATG8s on the activity of ULK1 toward the phosphorylation of ATG14 at Ser29 (p-ATG14), which we recently identified as a key event for phagophore and autophagosome formation [7]. First, we used HeLa cells where 3 LC3s, 3 GABARAPs, or 6 ATG8s were depleted, which were generously provided by Dr. Lazarou [19]. Hereafter, these cells are referred to as LC3 TKO, GABARAP TKO, and hexa KO cells, respectively. The cells were incubated in full medium or amino acid-deprived medium for 1 h, and the amount of p-ATG14 was analyzed by western blotting. Interestingly, LC3 TKO cells, deficient of LC3A, LC3B, and LC3C, showed a robust increase of p-ATG14 in both full and starvation medium conditions compared to wild-type (WT) cells (Figure 2A,B). We observed similar changes with BECN1 phosphorylation at Ser30 (p-BECN1), another key event for phagophore formation that is a direct target of ULK1 [8] (Figure 2A). This result suggests that at least one of those 3 LC3 members might negatively regulate the activity of ULK1. The LC3 subfamily includes LC3B2 that was not knocked out in the LC3 TKO cells. Therefore, we do not exclude the possibility that LC3B2 might also have a negative effect on ULK1 activity.
Figure 2.
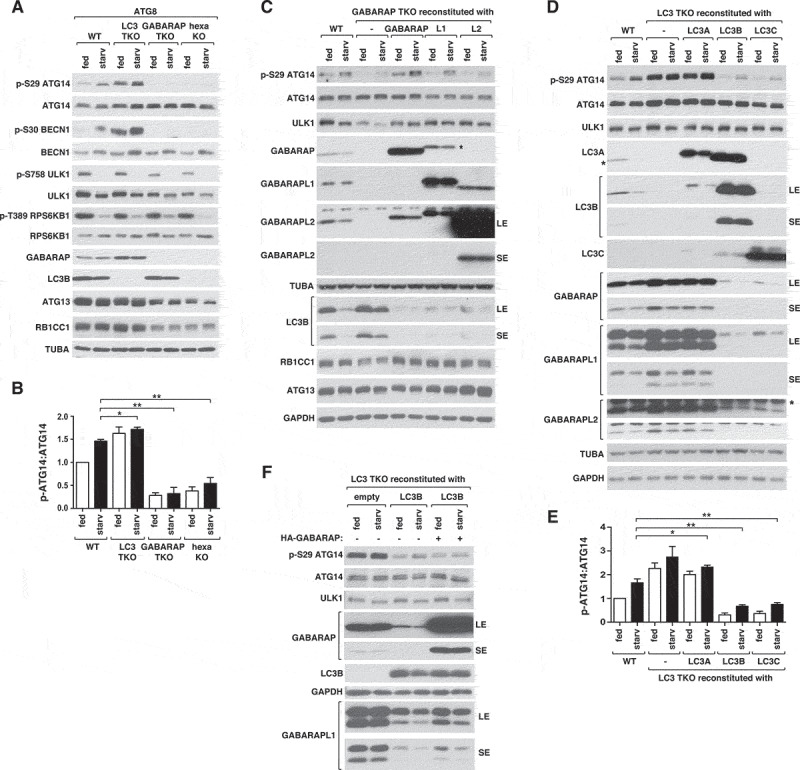
GABARAPs and LC3s have opposite effects on ULK1 activity. (A) GABARAPs are necessary for basal and starvation-induced activation of ULK1, whereas LC3s have negative effects on ULK1 activity. Wild-type (WT) or ATG8-depleted HeLa cells were cultured in full medium (fed) or EBSS (starv) for 1 h. ULK1 activity was assessed by WB using polyclonal antibodies specific to ATG14 phosphorylation at Ser29 and BECN1 phosphorylation at Ser30. (B) Quantitative analysis of ATG14 Ser29 phosphorylation in (A). Bar values are mean ± SEM (*p < 0.05; **p < 0.01; Student t-test, n = 3). (C) GABARAP and GABARAPL1 have positive roles for ULK1 activity. Individual GABARAPs without any tag at either N- or C-terminus were stably expressed in GABARAP TKO HeLa cells and treated as described in (A). GABARAPL1 marked by * indicates cross-reactivity with anti-GABARAP antibody. (D) LC3B and LC3C have negative effects on ULK1 activity. Individual members of LC3s without any tag were stably expressed in LC3 TKO HeLa cells and treated as described in (A). The bands marked by * are LC3B that cross-reacted with anti-LC3A antibody and a protein that is non-specifically recognized by anti-GABARAPL2. (E) Quantitative analysis of ATG14 Ser29 phosphorylation in (D). Bar values are mean ± SEM (*p < 0.05; **p < 0.01 relative to starved WT cells; Student t-test, n = 3). (F) The negative effect of LC3B on ULK1 activity does not require downregulation of GABARAP expression. LC3 TKO HeLa cells stably reconstituted with an empty vector or untagged LC3B were transiently transfected to express untagged GABARAP. The fed and starvation conditions were as described in (A).
In contrast, GABARAP TKO cells, deficient of GABARAP, GABARAPL1, and GABARAPL2, showed almost complete suppressions of p-ATG14 and p-BECN1 (Figure 2A,B), indicating that GABARAPs might be critical for the activity of ULK1. The hexa KO cells, depleted of 3 GABARAPs and 3 LC3s, showed a similar result as GABARAP TKO cells, indicating that the effect of GABARAP depletion is dominant over that of LC3 depletion in regulating ULK1. Because hexa KO cells were shown to have a slowed rate of autophagosome formation [19], we examined whether the activity of ULK1 might be recovered in the hexa KO cells after prolonged starvation. However, p-ATG14 remained suppressed for up to 6 h of amino acid starvation without showing any recovery in the hexa KO cells (Figure S1A). Depletion of ATG8s did not cause any noticeable difference in the phosphorylations of ULK1 Ser758 (Ser757 for mouse) and RPS6KB1 Thr389, that are target sites of MTORC1 [5] (Figure 2A). This indicates that the changes caused by ATG8 depletion are unlikely due to alteration of MTORC1 activity.
GABARAP and GABARAPL1 positively regulate starvation-induced ULK1 activation
To determine which member of the GABARAP subfamily regulates ULK1 activity, we reintroduced individual GABARAPs in GABARAP TKO cells and analyzed p-ATG14 by western blotting. The level of p-ATG14 was restored by re-expression of either GABARAP or GABARAPL1, and partially by re-expression of GABARAPL2 (Figure 2C and Figure S1B). This result suggests that either GABARAP or GABARAPL1 is sufficient for the activity of ULK1. In support of the redundant function of GABARAPs, depletion of GABARAP, GABARAPL1, or GABARAPL2 individually in HCT116 cells did not reduce p-ATG14 (Figure S1C). We noted that GABARAP TKO cells and hexa KO cells showed reduction of the expression levels of ULK1, ATG13, and RB1CC1 (Figure 2A). This indicates that GABARAPs are important for maintaining the expression or stability of the ULK1 complex proteins. Supporting this notion, reconstitution of GABARAP TKO cells with GABARAP, GABARAPL1, or GABARAPL2 restored the expression levels of ULK1 and RB1CC1 (Figure 2C). Interestingly, ATG13 level was drastically increased by re-introducing GABARAPL2 but moderately by GABARAPL1 and barely by GABARAP (Figure 2C and Figure S1B), implying a specific relation between ATG13 and GABARAPL2. Although the re-introduction of any GABARAP protein was sufficient to restore ULK1 expression, p-ATG14 was fully restored only by re-expressing GABARAP or GABARAPL1. This indicates that the role of GABARAP and GABARAPL1 in promoting ULK1 activity might not simply be due to their role in maintaining the expression levels of the ULK1 complex proteins. We found that GABARAP TKO cells did not show any drastic alteration of the expression level of LC3B, whereas the reconstitution of GABARAPs in the TKO cells deprived the expression level of LC3B (Figure 2C). This result indicates that overexpressed GABARAPs can have negative effects on the expression level of LC3B.
LC3B and LC3C negatively regulate the activity of ULK1
We also analyzed the roles of LC3s in regulating ULK1 activity by re-introducing individual LC3s in LC3 TKO cells. Re-expression of LC3A did not have any drastic effect on p-ATG14 (Figure 2D,E and Figure S1D). In contrast, re-expression of LC3B or LC3C in LC3 TKO cells reduced p-ATG14 for both basal and starvation conditions. These results suggest a negative role of LC3B and LC3C in regulating ULK1 activity. To further examine the negative effect of LC3B and LC3C, we generated HCT116 cells where LC3B or LC3C was depleted by CRISPR-Cas9-mediated gene targeting. Depletion of either LC3B or LC3C did not increase ULK1 activity (Figure S1E-G), suggesting their complementary function in regulation of ULK1.
Since the effect of GABARAP depletion on the activity of ULK1 was dominant over that of LC3 depletion (Figure 2A,B), we considered that the negative effects of LC3s might depend on GABARAPs. Related to this notion, we found that the cells depleted of LC3s showed higher levels of GABARAPs (Figure 2A,D). Conversely, reconstitution of LC3B or LC3C in LC3 TKO cells almost completely deprived the expression levels of GABARAPs (Figure 2D). This effect of LC3s on GABARAP expression levels was not due to changes in mRNA levels (Figure S1H). Therefore, we considered a possibility that LC3B and LC3C might negatively regulate ULK1 by reducing the expression or stability of GABARAPs. However, restoring the expression level of GABARAP in LC3B-reconstituted LC3 TKO cells failed to recover p-ATG14 (Figure 2F). This result indicates that LC3B might negatively regulate ULK1 independently of GABARAP.
LC3B negatively regulates GABARAPL1 lipidation
Interestingly, depletion of the 3 LC3 members largely increased the lipidated form of GABARAPL1 (Figure 2D). We could not find such an effect on GABARAP and GABARAPL2 in our experimental condition with HeLa and HCT116 cells. Consistent with our result, only a very little amount of the lipidated forms of GABARAP and GABARAPL2 were detected in animal cells [12]. The pattern of GABARAPL1 lipidation correlated with the activity of ULK1 (Figure 2D). The lipidation of GABARAPL1 was largely increased by depletion of LC3B but not LC3C (Figure S1E and S1F). Supporting the negative role of LC3B on the GABARAPL1 lipidation, reconstitution of LC3 TKO cells with LC3B suppressed the GABARAPL1 lipidation (Figure 2F). The increase of GABARAPL1 lipidation by LC3B depletion was not accompanied with upregulation of ULK1 activity (Figure S1E), indicating that GABARAPL1 lipidation is not sufficient to activate ULK1.
GABARAPs are required for proper control of autophagy initiation and progression in response to starvation
The finding that GABARAPs and LC3s differentially regulate the activity of ULK1 led us to test whether the differential effects are corroborated by changes in the formation of phagophores. In WT cells, amino acid starvation for 1 h increased the number of WIPI2-positive puncta, which represent phagophores [10] (Figure 3A,B). In full medium, LC3 TKO cells showed fewer WIPI2 puncta than WT cells. However, in response to starvation, LC3 TKO cells showed a greater increase in the number of WIPI2 puncta relative to WT cells (Figure 3B). In contrast, GABARAP TKO cells showed higher numbers of WIPI2 puncta relative to WT cells in full medium (Figure 3A,B). This observation is consistent with the recent report showing that GABARAP deficiency increased GFP-WIPI1 puncta [20]. However, GABARAP TKO cells did not show a further increase in WIPI2 puncta formation in response to starvation. This result suggests that GABARAPs might play a role in suppressing phagophore formation in nutrient-enriched conditions to sensitize the autophagy induction machinery to respond to starvation. Relative to GABARAP TKO cells, the hexa KO cells showed lower numbers of WIPI2 puncta in full medium (Figure 3A,B). The difference between GABARAP TKO cells and the hexa KO cells suggests that LC3s might play a role in phagophore formation in nutrient rich conditions when GABARAPs are deficient.
Figure 3.
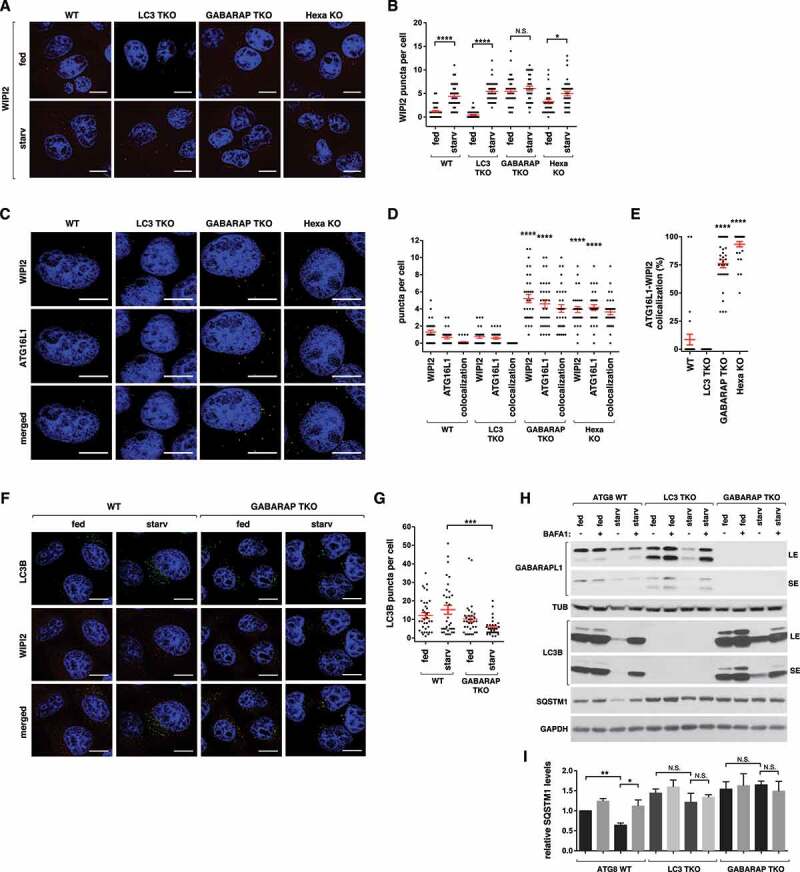
GABARAPs are required for proper control of autophagy initiation and flux. (A) Wild-type (WT) or ATG8-depleted HeLa cells were cultured in full medium or EBSS for 1 h. Endogenous WIPI2 (red) was detected by immunostaining using anti-WIPI2 antibody (EMD-Millipore, MABC91). Nuclei were stained with DAPI (blue). Scale bar: 10 μm. (B) Quantitative analysis of (A). Red bars are mean ± SEM (*p < 0.05; ****p < 0.0001; N.S., not significant; Student t-test, n > 30). (C) Endogenous WIPI2 (red) and ATG16L1 (green) were detected in HeLa cells described in (A) by immunostaining cells cultured in full medium. Scale bar: 10 μm. (D) Quantitative analysis of (C). Red bars are mean ± SEM. The statistical significance (****p < 0.0001; Student t-test, n > 30) is relative to WIPI2 and ATG16L1 puncta in WT cells. (E) Percent of WIPI2 puncta that colocalize with ATG16L1 puncta per cell. The values were obtained from the quantitative analysis of (D). (F) WT or GABARAP TKO HeLa cells were cultured in full medium or EBSS for 3 h. Endogenous LC3B (green) was detected by immunostaining using anti-LC3B antibody (MBL, PM036). Nuclei were stained with DAPI (blue). Scale bar: 10 μm. (G) Quantitative analysis of (F). Red bars are mean ± SEM (***p < 0.001; Student t-test, n > 30) (H) GABARAPs are important for autophagic flux of LC3B and autophagic degradation of SQSTM1. WT, LC3 TKO, or GABARAP TKO HeLa cells were cultured in full medium or EBSS for 3 h in the presence or absence of 100 nM bafilomycin A1 (BAFA1). (I) Quantitative analysis of SQSTM1 levels in (H) (*p < 0.05; **p < 0.01; N.S., not significant; Student t-test).
It is unclear how the number of WIPI2 puncta is increased in GABARAP TKO cells and how GABARAP depletion decouples phagophore formation from ULK1 activity. One possibility is that GABARAP depletion might lead to accumulation of phagophores due to blockage of the phagophore-to-autophagosome progression. We found that the majority of WIPI2 puncta colocalized with ATG16L1 puncta in GABARAP TKO cells and hexa KO cells (Figure 3C–E), a result consistent with the previous finding that depletion of GABARAPs induced accumulation of ATG5-ATG16L1 puncta [21]. In contrast, WIPI2 puncta barely colocalized with ATG16L1 in WT and LC3 TKO cells. A previous study showed that ATG16L1, which localizes to phagophores, is absent from mature autophagosomes [22]. Therefore, our result suggests that most of the WIPI2-ATG16L1 puncta in cells depleted of GABARAPs might represent incomplete autophagosomes. Supporting this notion, GABARAP TKO cells showed reduced formation of LC3B puncta, which reflect autophagosomes and autolysosomes, in response to starvation (Figure 3F,G). The reduction of LC3B puncta in GABARAP TKO cells in response to starvation might be due to autophagosomal breakdown in the lysosome, which occurs in the TKO cells even though the rate is slower compared to WT cells, with reduced de novo biogenesis of phagophore and autophagosome.
Upon starvation, ULK1 forms puncta that mark sites of autophagosome nucleation at a step prior to WIPI2 recruitment [23–25]. The ULK1 complex is only transiently associated with phagophores [24], forming puncta structures that are detectable much less in the number than that of WIPI2 puncta. WT cells and LC3 TKO cells showed similar levels of ULK1 puncta during starvation (Figure S2A-S2D). Relative to those cells, GABARAP TKO cells and hexa KO cells displayed reduced ULK1 puncta during starvation. This result suggests that GABARAPs play a role in ULK1 puncta formation in response to starvation. Since WIPI2 puncta occurred in the absence of GABARAPs (Figure 3A,B), we predict that GABARAPs might function to coordinate phagophore formation to occur at a site designated by ULK1 puncta. Without GABARAPs, WIPI2 puncta might form but in discordance with ULK1 resulting in phagophores that are defective in progression into autophagosomes. One of the defects might be the lack of GABARAP-dependent release of ATG16L1 from WIPI2 puncta as described above.
GABARAPs and LC3s are important for autophagic degradation of SQSTM1
To further clarify the roles of ATG8s in regulating autophagy, we assessed the autophagic degradation of ATG8 and SQSTM1 in ATG8-depleted cells. Compared to WT cells, LC3 TKO cells showed higher levels of the lipidated form of GABARAPL1 (Figure 3H), as also shown above in Figure 2D. LC3 TKO cells showed autophagic degradation of GABARAPL1 in response to 3 h of starvation, which was comparable to the degradation of LC3B in WT cells. However, autophagic degradation of SQSTM1 was suppressed in LC3 TKO cells compared to WT cells (Figure 3H,I and Figure S2E). This result suggests that LC3s are important for starvation-induced autophagic degradation of SQSTM1. Although LC3 TKO cells did not show any significant reduction of SQSTM1 during 3 h starvation, Nguyen et al. (2016) showed reduction of SQSTM1 during 8 h starvation [19]. This indicates that LC3s might play roles to facilitate the autophagic degradation of SQSTM1, but it might not be essential for the degradation. Since LC3 depletion did not suppress the formation of ULK1 puncta and phagophore, LC3s might play a role at later stages of autophagy, such as autophagosome maturation, or sequestration of autophagic substrates, such as SQSTM1. GABARAP TKO cells displayed a complete suppression of autophagic degradation of SQSTM1 and a reduced autophagic degradation of LC3B in response to starvation. This result is consistent with the positive roles of GABARAPs in regulation of ULK1 activity and autophagosome formation during starvation.
ATG8 binding to ULK1 is important for ULK1 activity
We next asked whether ATG8s regulate ULK1 activity by binding to ULK1. To test this, we introduced point mutations, D356A/F357A, into the LIR of endogenous ULK1 using CRISPR-cas9-assisted genome editing technique to disrupt the ULK1-ATG8 interaction (Figure S3A). We used diploidic HCT116 colon cancer cells that showed a high efficiency of genome editing in our previous studies [7,8]. The phosphorylation of ATG14 at Ser29 was drastically diminished in ULK1 LIR mutant cells relative to WT cells (Figure 4A,B). Similar to the phosphorylation of ATG14, the starvation-dependent phosphorylation of BECN1 Ser30 was reduced in ULK1 LIR mutant cells (Figure 4C). The ULK1 LIR mutations did not show any correlative changes in the phosphorylations of ULK1 at Ser758 and Ser556 (Figure 4A), indicating that the inhibitory effects of the mutations might not be due to MTORC1 and AMP-activated protein kinase (AMPK). We also generated HEK293T cells harboring the ULK1 LIR mutations by the CRISPR-Cas9-based method (Figure S3B), obtaining a similar result for ATG14 phosphorylation as with HCT116 cells (Figure S3C).
Figure 4.
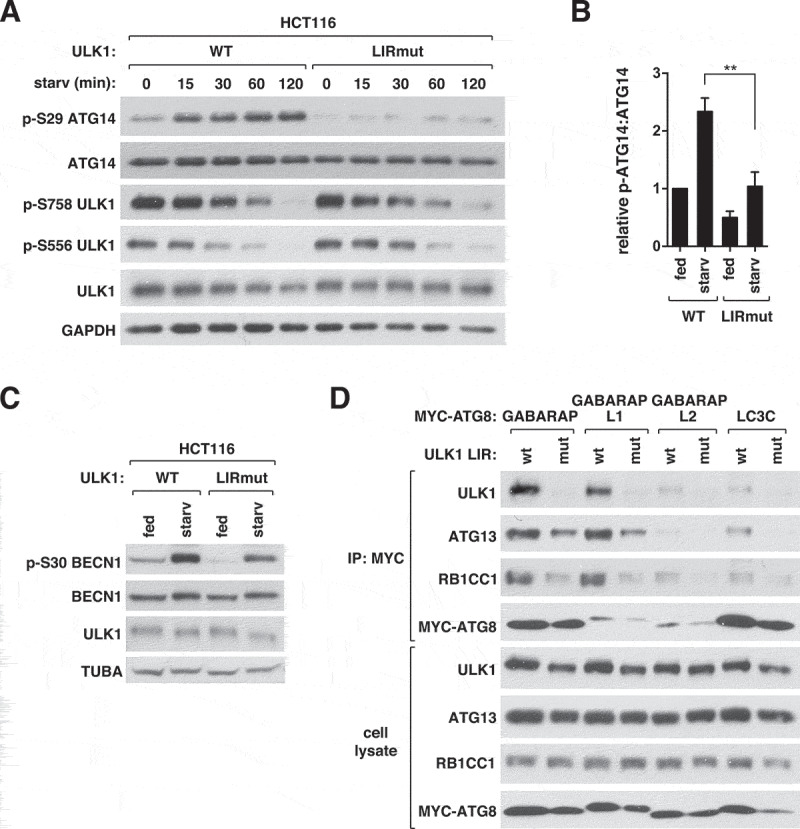
ATG8 binding to ULK1 is important for ULK1 activity. (A) ATG8 binding to ULK1 is important for the phosphorylation of ATG14 Ser29 by ULK1 in response to starvation. HCT116 cells whose genome was modified to express LIR-mutated ULK1 (LIRmut) and unmodified HCT116 cells (WT) were cultured in EBSS for the indicated periods of time. (B) Quantitative analysis of ATG14 Ser29 phosphorylation at 60 min of starvation in (A). Bar values are mean ± SEM (**p < 0.01; Student t-test, n = 3). (C) ATG8 binding to ULK1 is important for the phosphorylation of BECN1 Ser30 by ULK1 in response to starvation. WT or ULK1 LIR mutant HCT116 cells were cultured in full medium or EBSS for 1 h. (D) ATG8 binding to ULK1 stabilizes the interactions of ATG13 and RB1CC1 with ATG8. MYC-ATG8s were transiently expressed in WT or ULK1 LIR mutant HEK293T cells and immunoprecipitated using anti-MYC antibody.
The LIR mutation did not alter the interaction of ULK1 with ATG13, RB1CC1, and ATG101, indicating that the integrity of the ULK1 complex was unlikely disturbed by the mutation (Figure S3D). This result is in agreement with the previous report showing that the LIR of Atg1 (yeast homolog of ULK1) is dispensable for the interaction between Atg1 and Atg13 in yeast [15]. The ULK1 LIR mutation did not alter the binding of ATG13 to ATG14, an interaction required for ULK1 to phosphorylate ATG14 [7] (Figure S3E). However, the mutation reduced the interactions of ATG8s with ATG13 and RB1CC1 (Figure 4D), indicating that the ATG8-ULK1 interaction stabilizes the interactions between ATG8s and the whole ULK1 complex.
ATG8 binding to ULK1 is important for phagophore and autophagosome formation
We next analyzed the effects of the ULK1 LIR mutation on autophagic degradation of SQSTM1 and LC3B. The LIR mutant cells had higher levels of SQSTM1 and LC3B-II relative to WT cells, and showed suppressed autophagic degradation of SQSTM1 and LC3B-II in response to starvation (Figure 5A–C). We also analyzed autophagosome formation in ULK1 LIR mutant cells by immunostaining endogenous LC3B. The steady state number of autophagosomes in full medium was higher in the mutant cells relative to WT cells (Figure 5D,E). However, unlike WT cells, the mutant cells did not show a significant increase in LC3B puncta number upon starvation. BAFA1, which inhibits autophagosome-lysosomal fusion, largely increased the number of LC3B puncta in both basal and starvation conditions for WT cells. Such increases were suppressed in the mutant cells, suggesting that ATG8 binding to ULK1 is important for the turnover of autophagosomes. A more striking effect of the mutation was observed with WIPI2 puncta formation. Starvation-induced WIPI2 puncta formation was almost completely suppressed by the LIR mutation (Figure 5F,G). The mutation also almost completely suppressed ULK1 puncta formation (Figure S4A and S4B), suggesting that ATG8 binding to ULK1 might play a role in facilitating ULK1-mediated nucleation events that trigger autophagosome formation during amino acid starvation.
Figure 5.
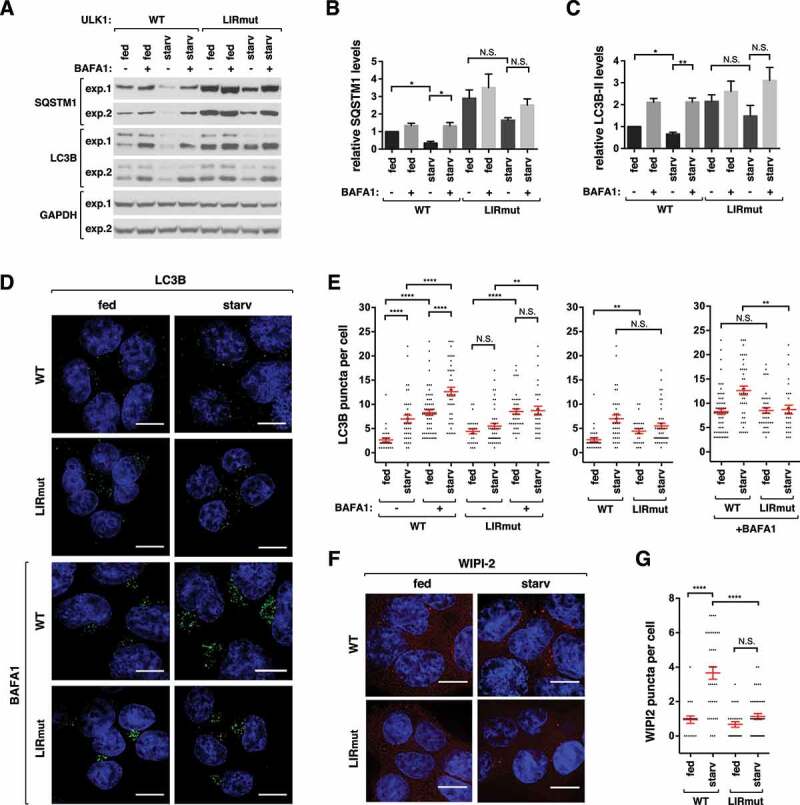
ATG8 binding to ULK1 is important for autophagy. (A) ATG8 binding to ULK1 is important for autophagic degradation of SQSTM1 and autophagic flux of LC3B. WT or ULK1 LIR mutant HCT116 cells were cultured in full media or EBSS for 2 h in the presence or absence of 100 nM BAFA1. Blots from 2 independent experiments are shown. (B-C) Quantitative analysis of SQSTM1 and LC3B-II from (A). The protein levels were normalized based on the levels of GAPDH. Bar values are mean ± SEM (*p < 0.05; **p < 0.01; Student t-test, n = 3). (D) ATG8 binding to ULK1 is important for starvation induced LC3B puncta formation. WT or ULK1 LIR mutant HCT116 cells were cultured in full medium or EBSS for 2 h in the presence or absence of 100 nM BAFA1. LC3B (green) was detected by immunostaining of endogenous LC3B using anti-LC3B antibody (MBL, PM036). Nuclei were stained with DAPI (blue). Scale bar: 10 μm. (E) Quantitative analysis of (D). Red bars are mean ± SEM (**p < 0.01; ****p < 0.0001; Student t-test, n > 25). (F) ATG8 binding to ULK1 is important for starvation induced WIPI2 puncta formation. WT or ULK1 LIR mutant HCT116 cells were cultured in full medium or EBSS for 1 h. WIPI2 (red) was detected by immunostaining endogenous WIPI2 using anti-WIPI2 antibody (EMD-Millipore, MABC91). Scale bar: 10 μm. (G) Quantitative analysis of (F). Red bars are mean ± SEM (****p < 0.0001; Student t-test, n > 20).
Interestingly, the LIR mutation increased the lipidated form of GABARAPL1 (Figure S4C). Since the level of LC3B, which negatively regulates GABARAPL1 lipidation, was increased in the mutant cells (Figure 5A), the higher level of the lipidated GABARAPL1 may not be related to LC3B. Rather, we interpret the increase of the lipidated form as being due to reduction of autophagic degradation of GABARAPL1 in the mutant cells. Alternatively, there might be some unknown mechanism through which the ULK1-ATG8 interaction negatively regulates GABARAPL1 lipidation.
ATG8 binding to ATG13, but not to RB1CC1, is important for ULK1 activity
We wondered whether the binding of ATG8 to RB1CC1 or ATG13 has a similar role as the binding to ULK1. We generated HCT116 cells where point mutations D701A/F702A/I705A were introduced into the LIR of endogenous RB1CC1 by CRISPR-Cas9-assisted genome editing technique (Figure S5A). Unlike ULK1 LIR mutant cells, RB1CC1 LIR mutant cells did not show a reduction of p-ATG14 (Figure 6A). RB1CC1 LIR mutation also did not affect starvation induced ULK1 puncta formation (Figure S5B and S5C) and autophagic degradation of SQSTM1 and LC3B (Figure S5D). These results suggest that the binding of ATG8 to RB1CC1, even if it exists, is not important for the activation of ULK1 and autophagy induction.
Figure 6.
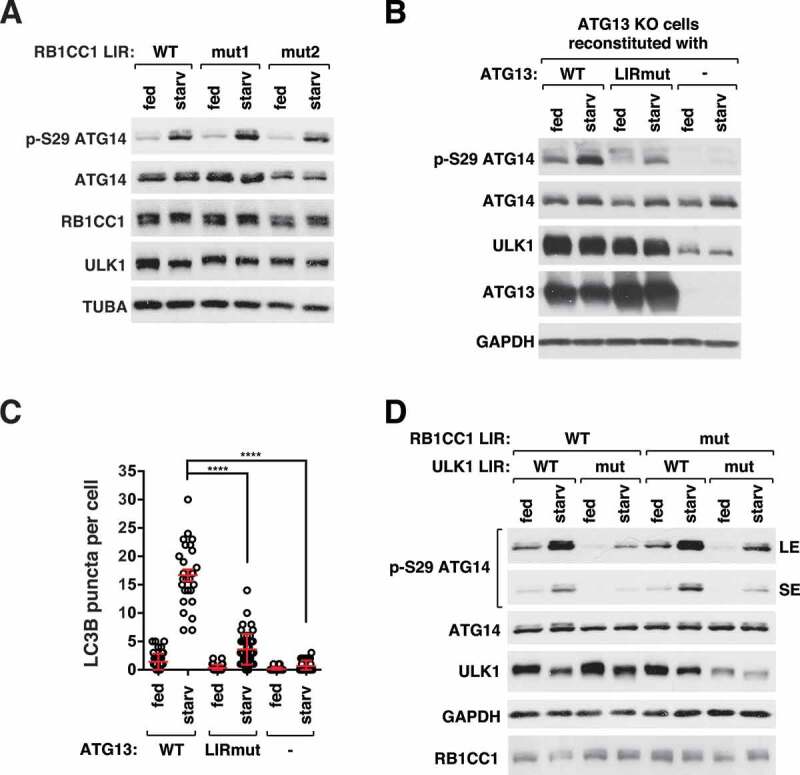
ATG8 binding to ATG13, but not RB1CC1, is important for ULK1 activity. (A) RB1CC1 LIR mutation does not affect ULK1 activity. WT or RB1CC1 LIR mutant HCT116 cells were cultured in full medium or EBSS for 1 h. Two LIR mutant clones (mut1 and mut2) were analyzed. (B) ATG8 binding to ATG13 is important for ULK1 activity. ATG13-depleted HCT116 cells stably reconstituted with ATG13 WT, ATG13 LIR mutant (I447A/D448A) or empty vector were cultured in full medium or EBSS for 1 h. (C) ATG8 binding to ATG13 is important for starvation-induced formation of LC3B puncta. Red bars are mean ± SEM (****p < 0.0001; Student t-test, n > 25). (D) Disruption of both the ULK1-ATG8 interaction and the RB1CC1-ATG8 interaction does not completely suppress ULK1 activity. The ULK1 LIR mutation was introduced in RB1CC1 LIR mutant cells by CRISPR-Cas9-assisted genome editing. Cells were cultured in full medium or EBSS for 1 h.
Our attempt to introduce a mutation into the endogenous ATG13 LIR by genome editing was not successful. As an alternative approach, we reconstituted ATG13 KO HCT116 cells with a WT or a LIR mutant ATG13 construct harboring point mutations I447A/D448A that disrupt the binding of ATG8 to ATG13 (Figure S5E). As with ULK1 LIR mutant cells, ATG13 LIR mutant cells showed reduction of p-ATG14 (Figure 6B) and LC3B puncta formation (Figure 6C and Figure S5F) in response to starvation. The mutational effects occurred without disruption of the interaction of ATG13 with ULK1 or ATG14 (Figure S5G and S5H), suggesting that the reduced p-ATG14 in the mutant cells is not due to ATG13 dissociation from ULK1 or ATG14. Together, these results demonstrate that ATG8 binding to both ULK1 and ATG13 is important for the activity of ULK1 and autophagosome formation. The 2 interactions might play a cooperative role to achieve an optimal activation of ULK1.
One question arose about whether the RB1CC1-ATG8 interaction, albeit barely detectable, might play a role in regulating ULK1 when the ULK1-ATG8 interaction was disrupted. To address this question, we introduced the ULK1 LIR mutation into the RB1CC1 LIR mutant cells. The dual LIR mutated cells did not show any additional suppressive effect on p-ATG14 in response to starvation compared to ULK1 LIR mutant cells (Figure 6D). This result further confirms that the RB1CC1-ATG8 interaction, even if it occurs, might not contribute to the activity of ULK1. Since the ULK1 complex in the dual LIR mutant cells retains only the ATG13 LIR, the remaining ATG8-dependent activity of ULK1 might be due to the contribution from ATG13-mediated interaction with ATG8. Interestingly, the dual mutation, unlike the ULK1 LIR mutation alone, reduced the expression level of ULK1, indicating that the RB1CC1-ATG8 interaction might contribute to the stability of ULK1.
GABARAPL1 binding to ULK1 and ATG13 is regulated by starvation and PE conjugation
As the activity of ULK1 is regulated by cellular nutritional conditions [1,3,4], we tested whether the ULK1-ATG8 interaction is regulated by amino acid starvation. Greater amounts of ULK1 and ATG13 were co-immunoprecipitated with GABARAPL1 from starved cells compared to nutrient-enriched cells (Figure 7A). Such a change of interaction was not obviously seen with GABARAP and GABARAPL2. Knowing the regulatory interaction, we tested whether the interaction of ULK1 with GABARAPs depends on ULK1 kinase activity. To test this, we transiently expressed GABARAPs with WT or kinase inactive mutant (KI) ULK1 in HEK293T cells, and analyzed the ULK1- and ATG13-GABARAP interactions by co-immunoprecipitation assay. The binding of GABARAPs to ULK1 and ATG13 was drastically suppressed when KI ULK1 was expressed compared to WT ULK1 (Figure 7B). KI ULK1 was co-immunoprecipitated with recombinant ATG13 and RB1CC1 with a similar efficiency as WT ULK1 (Figure 7C), indicating that the reduced binding of GABARAPs to KI ULK1 is not due to dissociation of ULK1 from ATG13 or RB1CC1.
Figure 7.
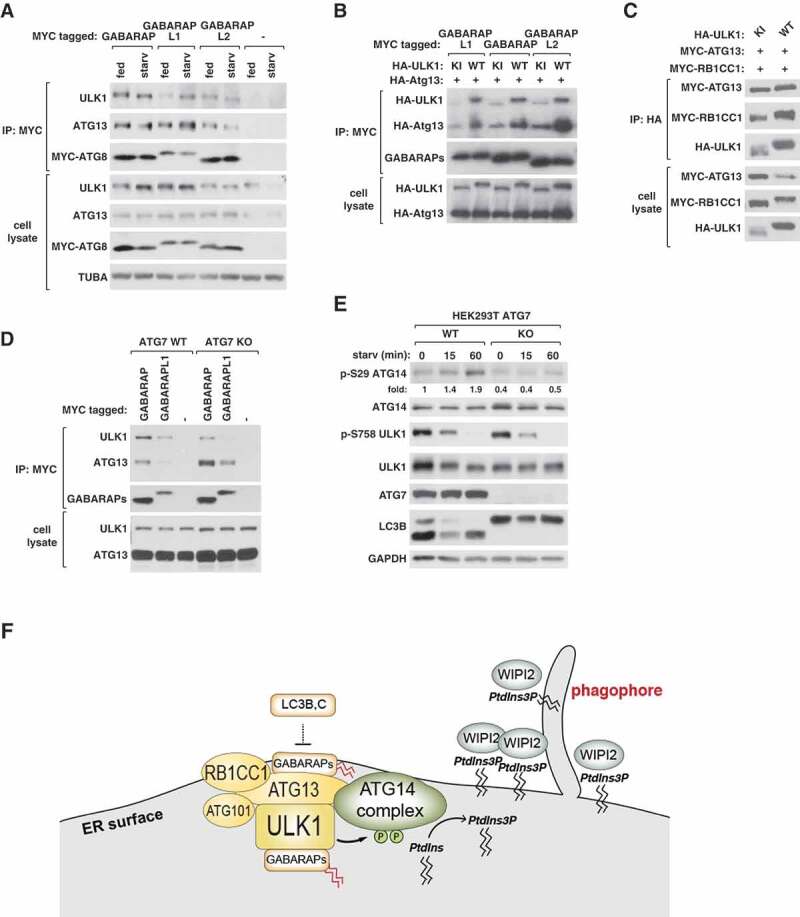
GABARAP bindings to ULK1 and ATG13 are regulated by starvation and PE conjugation. (A) Amino acid starvation enhances the interaction of GABARAPL1 with ULK1 and ATG13. MYC-GABARAPs were transiently expressed in HEK293T cells. Cells were cultured in full medium or EBSS for 1 h. The amounts of endogenous ULK1 and ATG13 interacting with MYC-GABARAPs were analyzed by co-immunoprecipitation and WB. (B) The interactions of GABARAPs with ULK1 and ATG13 depend on the kinase activity of ULK1. MYC-tagged GABARAPs were transiently expressed in HEK293T cells together with HA-tagged ATG13 and HA-tagged WT or kinase inactive (KI) ULK1. The amounts of HA-ULK1 and HA-ATG13 co-immunoprecipitated with MYC-GABARAPs were analyzed by WB. (C) ULK1 kinase activity is not required for the interaction between ULK1, ATG13, and RB1CC1. HEK293T cells were transiently transfected to express the indicated proteins. (D) ATG7 depletion suppresses the interaction of GABARAPs with ULK1 and enhances the interaction with ATG13. MYC-GABARAPs were transiently expressed in ATG7 WT and ATG7-depleted (KO) HEK293T cells. Cells were cultured in full medium or EBSS for 1 h before co-immunoprecipitation was conducted as described in (A). (E) ATG7 is important for starvation-induced activation of ULK1. ATG7 WT or KO HEK293T cells were treated as described in (A). Numbers below the p-ATG14 blot represent fold changes of the band intensities relative to the intensity of the first lane band. (F) Diagram showing the interactions and relations between the autophagy initiation machinery and ATG8s.
Our previous study has shown that ATG5 or ATG7 depletion suppresses the activity of ULK1 [7]. Therefore, we wondered whether the PE conjugation of ATG8s regulates their interaction with the ULK1 complex thereby altering the activity of ULK1. To test this, we depleted ATG7 in HEK293T cells by CRISPR-Cas9 genome targeting and analyzed the interaction between the ULK1 complex and ATG8s by co-immunoprecipitation assay. GABARAP and GABARAPL1 were tested because of their high affinity of interaction with the ULK1 complex. In the absence of ATG7, less ULK1 was co-immunoprecipitated with GABARAPs (Figure 7D). This result suggests that the PE conjugation of GABARAPs might stabilize the interaction between ULK1 and GABARAPs. Interestingly, the depletion of ATG7 increased the amount of ATG13 co-immunoprecipitated with GABARAPs. Thus, the PE conjugation likely has opposite effects on the bindings of ATG8s to ULK1 and ATG13. ATG7 depletion in HEK293T cells suppressed the activation of ULK1 in response to starvation (Figure 7E), a result consistent with our previous report [7]. This result suggests that the PE conjugation might regulate the activity of ULK1 via altering the interactions of ATG8s with ULK1 and ATG13.
Discussion
The ATG8 family proteins, despite their relatively small sizes of 13–16 kDa, show a high versatility of interaction with diverse groups of proteins containing LIR motifs [26]. While the main role of the interactions may be to mediate the recruitment of the autophagy machinery to cargo proteins, the binding of ATG8 to the ULK1 complex appears to have a distinct function. Through disrupting the endogenous ULK1-ATG8 interaction, we determined that the interaction is important for ULK1 activation and autophagy induction in response to starvation. We demonstrated that GABARAPs and LC3s have opposite effects on ULK1 activity and that ATG8 binding to ULK1 and ATG13, but not binding to RB1CC1, is important for the activity of ULK1 (Figure 7F).
It is unclear how ATG8 binding to the ULK1 complex regulates the activity of ULK1. It is possible that, by binding to ULK1, ATG8s allosterically regulate ULK1 stability and function. In support of this possibility, depletion of GABARAPs reduced the expression level and activity of ULK1 (Figure 2A,C). Furthermore, the dual mutation of ULK1 and RB1CC1 LIRs reduced the expression level of ULK1 (Figure 6D). These results imply that GABARAPs play a positive role in regulating the expression level of ULK1 by binding to the ULK1 complex. However, ULK1 activity, but not the expression level of ULK1, was reduced by mutation of ULK1 LIR alone (Figure 4A). Thus, it appears that the role of the ATG8-ULK1 interaction in regulating ULK1 activity is distinct from the role of GABARAPs in regulating the expression level of ULK1.
Despite the positive role of GABARAPs in regulating ULK1 activity, the GABARAP TKO cells showed higher numbers of steady-state WIPI2 puncta compared to WT cells (Figure 3A,B). Most of the WIPI2-positive structures were also positive for ATG16L1 in GABARAP TKO cells (Figure 3C–E). Given that ATG16L1 is not present on mature autophagosomes [23], it is likely that the WIPI2 puncta represent immature autophagosomes. GABARAPs might not be suppressing the formation of these WIPI2-positive structures but rather required for the progression of these structures into fusion-competent autophagosomes. Perhaps, GABARAPs are critical for the release of autophagosome nucleation factors such as ATG16L1 for autophagosome maturation, and the release of these factors is a prerequisite for autophagosome-lysosome fusion. In this model, GABARAP-deficient cells have minimal ULK1 activity and generate autophagic membranes at a low rate, but these autophagic structures accumulate because they are not turned over. Due to low ULK1 activity and retention of nucleation factors such as ATG16L1 on immature autophagosomes, new phagophores are not formed in response to starvation (Figure 3A,B).
Unlike GABARAP TKO cells, ULK1 LIR mutant cells did not exhibit a steady state accumulation of WIPI2 puncta (Figure 5F,G). Thus, the predicted role of GABARAPs in developing fusion-competent autophagosomes might be independent of their binding to ULK1. It is unclear how the ULK1 LIR mutant cells accumulate LC3B puncta despite the suppression of WIPI2 puncta formation. BAFA1 induced the accumulation of LC3B puncta to a much less extent in ULK1 LIR mutant cells compared to WT cells (Figure 5D,E), indicating that the ATG8-ULK1 interaction is important for autophagosome turnover. With minimal ULK1 activity, it is possible that autophagosomes form in ULK1 LIR mutant cells at a low basal rate, but accumulate because the autophagosome-lysosome fusion is impaired (at a step downstream of WIPI2 release). This might be the reason that the mutant cells exhibit a higher number of steady-state WIPI2-negative LC3B puncta and a diminished increase in the number of LC3B puncta in response to starvation (Figure 5D,E). A recent report also supports this notion by showing that ULK1 is involved in autophagosome-lysosome fusion [27]. Nguyen et al. (2016) showed that ATG8s are essential for autophagosome-lysosome fusion [19], and we predict that this role of ATG8s depends on their interaction with ULK1.
It is intriguing that LC3B and LC3C have negative effects on ULK1 activity (Figure 2). This finding raises the concern that using LC3B overexpression construct, which is broadly used to assay autophagy, could potentially suppress ULK1 and autophagy. It is unclear how LC3B can negatively regulate ULK1 as we could not detect its binding to the ULK1 complex (Figure 1). One possibility is that the LC3B-ULK1 interaction might occur in cells but be disrupted in vitro during co-immunoprecipitation. Perhaps, overexpressed LC3B or LC3C might occupy the LIRs of the ULK complex, thus preventing GABARAPs, the positive regulators of ULK1, from binding to the complex. All ATG8 proteins contain a series of α-helices at their N-termini [12]. The residues in the N-terminal α-helical regions are relatively basic in LC3s and acidic or neutral in GABARAPs [28]. This difference in the N-terminal charged residues might play roles in their differential binding and regulation of ULK1.
The opposite effects of GABARAPs and LC3s on the activity of ULK1 imply that the ATG8 proteins might play roles in maintaining the cellular balance between autophagy induction and suppression. The opposite roles of GABARAPs and LC3s in autophagy are also manifested by the inverse relation between their expression levels (Figure 2). The inverse relation is unlikely due to autophagy, since autophagic degradation of GABARAPL1 occurred in LC3 TKO cells (Figure 3H). The relation is also unlikely due to gene transcription (Figure S1H). An additional layer of their inverse relation appears to exist in the negative role of LC3B for the PE conjugation of GABARAPL1 (Figure 2 and Figure S1E). Perhaps, LC3B is the major client for the PE conjugation machinery. GABARAPL1 would be efficiently modified with PE only when the expression level of LC3B is low. Since the PE conjugation machinery positively regulates ULK1 activity [7] (Figure 7E), LC3B-mediated negative regulation of the GABARAPL1 lipidation might contribute to ULK1 suppression. LC3B depletion increased GABARAPL1 lipidation but not ULK1 activity (Figure S1E). Thus, the PE conjugation of GABARAPs might be required but not sufficient to activate ULK1 in response to starvation.
Although the 3 GABARAPs have compensatory roles in regulation of ULK1 activity, their differential binding affinities toward ULK1 and ATG13 suggest that they might have distinct functions (Figure 1). This notion is supported by our finding that the prominent level of PE-conjugated form was detected only with GABARAPL1 but not with GABARAP and GABARAPL2 (Figure 2). GABARAPL1 was also distinct from other GABARAPs in its enhanced binding affinity for the ULK1 complex in response to amino acid starvation (Figure 7A). This could be explained if PE conjugation and ULK1-mediated phosphorylations do not contribute equally to the interactions between different GABARAPs and the ULK1 complex. Interestingly, PE conjugation appears to have opposite effects on the binding of GABARAPs to ULK1 and ATG13 (Figure 7D). ULK1 and ATG13 interact with each other, and therefore it is technically difficult to tease apart how each member contributes to the regulation of ATG8-ULK1 complex interactions. The opposite effects of ATG7 depletion on the interactions of GABARAPs with ULK1 and ATG13 (Figure 7D) imply that the PE conjugation might provide a physical constraint on the membrane-anchored ATG8 thus triggering a conformational rearrangement of the ULK1 complex inducing differential binding of ULK1 and ATG13 to GABARAPs. The PE conjugation might also enable ATG8 to couple the kinase activity of ULK1 with ATG8-mediated tethering and hemifusion of membranes [11].
Our findings give rise to several outstanding questions about the roles of ATG8s in autophagy. How do GABARAP binding and the PE conjugation positively regulate ULK1 activity? What is the underlying mechanism for the inverse relation between LC3s and GABARAPs? How do LC3s exert their negative effect on the activity of ULK1? What is the molecular basis for the starvation-dependent interaction between the ULK1 complex and GABARAPs? Addressing these questions will bring us closer to a comprehensive understanding of the molecular processes of autophagy.
Materials and methods
Reagents and antibodies
The following antibodies were used for this study: antibodies for ULK1 (sc-10900 for IP, sc-33182 for WB, sc-390904 for immunostaining), SQSTM1 (sc-28359), BECN1/Beclin 1 (sc-11427), ATG14 for IP (sc-164767), GAPDH (sc-25778), and TUB (sc-12462) from Santa Cruz Biotechnology; antibodies for ATG13 for IP (13,468), phospho-ATG14 Ser29 (13155), phospho-ULK1 Ser757 (6888), phospho-ULK1 Ser555 (5869), ATG14 (96752), GABARAPL1 (26632), LC3A (4599), LC3B (2775), LC3C (14723), ATG16L1 (8089) from Cell Signaling Technology; antibodies for MYC (9E10; OP10) and WIPI2 (MABC91) from EMD-Millipore; anti-HA antibody (MMS-101P) from Biolegend; antibodies for GABARAPL2 (PM038) and LC3B for immunostaining (PM036) from MBL; anti-GABARAP antibody (AP1821a) from Abgent; anti-ATG13 antibody for WB was generated as described previously [1]. The following reagents were used in this study: DMEM (11995), Lipofectamine 3000 (L3000015), DSP (dithiobis[succinimidyl propionate]) (22586), puromycin (A11138-03), and zeocin (R250-01) from Thermo Fisher Scientific; Earle’s Balanced Salt Solution (EBSS; 2888) and bafilomycin A1 (B1793-10UG) from Sigma-Aldrich; protein G-agarose (P9202) from GenDEPOT.
Genome editing to generate KO cells and LIR mutant cells
LC3 TKO, GABARAP TKO, hexa KO, and their parental control HeLa cells were kindly provided by Dr. Lazarou (Monash University, Australia). The CRISPR-cas9 assisted gene knockout method was used to generate HCT116 and HEK293T cells depleted of LC3B, LC3C, or ATG7. The TALEN-assisted genome editing method was used to generate HCT116 cells depleted of GABARAPs and ATG13. The detailed procedures are described in our recent reports [7,8], and the target sequences are listed in Table S1 and Table S2. To introduce LIR mutations into ULK1 and RB1CC1 of the genome of HCT116 cells, we used the CRISPR-Cas9-assisted genome editing technique following the procedure we described in our previous report [8]. The gRNA sequences listed in Table S1 were cloned into pSpCas9(BB)-2A-GFP (Addgene, PX458; deposited by Dr. Feng Zhang). Mutant clones were screened by PCR method using a pair of primers as listed in Table S3. Candidate mutant clones from the screen were amplified by PCR using primers listed in Table S4, cloned into pRK5 vector, and sequenced to confirm the mutations. Due to the lack of LC3C-specific antibodies commercially available, we confirmed LC3C KO mutant clones by PCR amplification of the mutated region using primers listed in Table S4 followed by DNA sequencing.
mRNA analysis by qPCR
Total RNA was isolated using TRIzol Reagent (ThermoFischer Scientific, 15596018) and cDNA was synthesized using a reverse transcriptase kit (GenDEPOT, R5600) following the manufacturers’ protocols. Quantitative PCR (qPCR) was performed using iQ SYBR Green Supermix (Bio-Rad, 170–8885) and the Bio-Rad CFX 96 real-time system. Primers used in the reaction are listed in Table S5. The CT value for each GABARAP target mRNA was normalized to the corresponding GAPDH mRNA.
DNA construction
The cDNA clones for human GABARAP, GABARAPL1, GABARAPL2, LC3A, LC3B, and LC3C were obtained from Open Biosystems (Huntsville, AL). The cDNA was amplified and cloned into pRK5-myc, pRK5-HA, and pCSII-MCS-EF vectors, the vector systems described in our previous reports [29,30]. The LIR motif mutant of ATG13 was made by using the site-directed mutagenesis kit (Stratagene, 200523). The primers we used are listed in Table S6. The mutant construct was cloned into the pCSII-MCS-EF lentiviral vector.
Generation of ATG8- and ATG13-reconstituted cells
MYC-tagged WT or LIR mutant ATG13 construct and untagged ATG8 constructs in pCSII-MCS-EF lentiviral vector were used to generate reconstituted cells. Lentivirus was prepared as we described previously [30]. Target cells were infected with virus, and transduced cells were selected under zeocin as we described previously [30].
Cell culture and transfection
HEK293T, HCT116, and HeLa cells were cultured in DMEM (Thermo Fisher Scientific, 11,995) supplemented with 10% fetal bovine serum, penicillin and streptomycin at 37°C in 5% CO2. For transient expression, cells were transfected with recombinant DNA using Lipofectamine 3000 (Thermo Fisher Scientific) following the manufacturer’s protocol. Cells were harvested 2 days post-transfection for all transient expression experiments. EBSS was used for amino acid starvation.
Co-immunoprecipitation and western blotting
Whole cell extracts were prepared in lysis buffer containing 40 mM HEPES, pH 7.4, 120 mM NaCl, 1 mM EDTA, 50 mM NaF, 1.5 mM Na3VO4, 10 mM beta-glycerophosphate, 1% Triton X-100 (Sigma-Aldrich, X100) supplemented with protease inhibitors (Roche, 05056489001). Immunoprecipitates were obtained using protein G agarose beads (GenDEPOT, P9202-800) and appropriate antibodies. Immunoprecipitated proteins were washed 4 times using lysis buffer, loaded onto Tris-glycine gels (Thermo Fisher Scientific, XP04125BOX), transferred onto immunoblot polyvinylidene difluoride (PVDF) membranes (Bio-Rad, 1,620,177) and detected using ECL reagents (GenDEPOT, 20-300B). For the immunoprecipitation experiment coupled with crosslinking, cells were washed with cold phosphate-buffered saline (PBS; VWR Life Science, 0780-50L) twice then treated with dithiobis(succinimidyl propionate) at the indicated concentration for 10 min in cold PBS on ice. Unreacted DSP was quenched by incubating cells in 40 mM Tris-HCl, pH 7.4 for 10 min on ice. Cell lysate was prepared and immunoprecipitation was conducted as described above.
Immunofluorescence microscopy
Cells were cultured on glass coverslips, fixed with 4% formaldehyde in PBS for 15 min at room temperature, and permeabilized with 0.25% Triton X-100 for 5 min at room temperature. Permeabilized cells were incubated in PBS supplemented with 0.05% Tween 20 (Sigma-Aldrich, 274,348) and 2% BSA (EMD-Millipore, 2960-500GM) for 60 min and then incubated with antibodies overnight at 4°C. After the primary antibody labeling, cells were incubated with Alexa Flour 488-conjugated anti-rabbit IgG (ThermoFisher Scientific, A-21,441) or Alexa Flour 555-conjugated anti-mouse IgG (Life Technologies, A-31,570). Nuclei were stained with DAPI (4ʹ-6-diamidino-2-phenylindole, ThermoFisher Scientific, D1306). Images from stained cells were obtained using a Deltavision PersonelDV microscope (Applied Precision) and analyzed by softWoRx version 6.1.3 (GE Healthcare).
Statistical analysis
Western blot protein bands were quantitatively analyzed for their intensities using ImageJ software. Puncta images were quantitatively analyzed by counting the number in each cell, setting the same threshold for control and experimental groups using ImageJ. Outcomes were summarized by using the mean and the standard error of the mean (SEM) as indicated in the figure legends. Statistical significance of difference between groups was analyzed by the Student t test using Prism 6 (Version 6.0d, GraphPad Software).
Funding Statement
This study was supported by the functional proteomics of aging training grant T32AG029796 (to DSG and NMO); GM097057 and GM130353 (to DHK); National Institute of General Medical Sciences [GM097057] and [GM130353]; National Institute on Aging [T32AG029796].
Acknowledgments
We thank M. Lazarou for providing ATG8 hexa KO, LC3 TKO, and GABARAP TKO cells; Erik Toso and Michael Kyba for generation of ATG8 KO cells and LIR mutant cells; Colby Starker, Becca Greenstein, and Dan Voytas for TALEN construction.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Jung CH, Jun CB, Ro SH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009. April;20(7):1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mercer CA, Kaliappan A, Dennis PB.. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy. 2009. July;5(5):649–662. [DOI] [PubMed] [Google Scholar]
- [3].Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009. April;20(7):1981–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ganley IG, Lam Du H, Wang J, et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009. May 1;284(18):12297–12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011. February;13(2):132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013. July;15(7):741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Park JM, Jung CH, Seo M, et al. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy. 2016. March 3;12(3):547–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Park JM, Seo M, Jung CH, et al. ULK1 phosphorylates Ser30 of BECN1 in association with ATG14 to stimulate autophagy induction. Autophagy. 2018;14(4):584–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Axe EL, Walker SA, Manifava M, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008. August;182(4):685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Polson HE, de Lartigue J, Rigden DJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010. May;6(4):506–522. [DOI] [PubMed] [Google Scholar]
- [11].Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007. July 13;130(1):165–178. [DOI] [PubMed] [Google Scholar]
- [12].Kabeya Y, Mizushima N, Yamamoto A, et al. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004. June 1;117(Pt 13):2805–2812. [DOI] [PubMed] [Google Scholar]
- [13].Shpilka T, Weidberg H, Pietrokovski S, et al. Atg8: an autophagy-related ubiquitin-like protein family. Genome Biol. 2011;12(7):226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Alemu EA, Lamark T, Torgersen KM, et al. ATG8 family proteins act as scaffolds for assembly of the ULK complex: sequence requirements for LC3-interacting region (LIR) motifs. J Biol Chem. 2012. November;287(47):39275–39290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kraft C, Kijanska M, Kalie E, et al. Binding of the Atg1/ULK1 kinase to the ubiquitin-like protein Atg8 regulates autophagy. Embo J. 2012. September;31(18):3691–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Joachim J, Jefferies HB, Razi M, et al. Activation of ULK kinase and autophagy by GABARAP trafficking from the centrosome is regulated by WAC and GM130. Mol Cell. 2015. December 17;60(6):899–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rogov VV, Stolz A, Ravichandran AC, et al. Structural and functional analysis of the GABARAP interaction motif (GIM). EMBO Rep. 2017. August;18(8):1382–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Joo JH, Dorsey FC, Joshi A, et al. Hsp90-cdc37 chaperone complex regulates ulk1- and atg13-mediated mitophagy. Mol Cell. 2011. August 19;43(4):572–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nguyen TN, Padman BS, Usher J, et al. Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol. 2016. December 19;215(6):857–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Vaites LP, Paulo JA, Huttlin EL, et al. Systematic analysis of human cells lacking ATG8 proteins uncovers roles for GABARAPs and the CCZ1/MON1 regulator C18orf8/RMC1 in macroautophagic and selective autophagic flux. Mol Cell Biol. 2018;38(01):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Weidberg H, Shvets E, Shpilka T, et al. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. Embo J. 2010. June 2;29(11):1792–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Mizushima N, Kuma A, Kobayashi Y, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003. May;116(Pt 9):1679–1688. [DOI] [PubMed] [Google Scholar]
- [23].Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010. August 18;6(6):764–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Karanasios E, Stapleton E, Manifava M, et al. Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J Cell Sci. 2013. November 15;126(Pt 22):5224–5238. [DOI] [PubMed] [Google Scholar]
- [25].Kishi-Itakura C, Koyama-Honda I, Itakura E, et al. Ultrastructural analysis of autophagosome organization using mammalian autophagy-deficient cells. J Cell Sci. 2014. September 15;127(Pt 18):4089–4102. [DOI] [PubMed] [Google Scholar]
- [26].Kirkin V, Lamark T, Johansen T, et al. NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy. 2009. July;5(5):732–733. [DOI] [PubMed] [Google Scholar]
- [27].Wang C, Wang H, Zhang D, et al. Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat Commun. 2018. 08;9(1):3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sugawara K, Suzuki NN, Fujioka Y, et al. The crystal structure of microtubule-associated protein light chain 3, a mammalian homologue of Saccharomyces cerevisiae Atg8. Genes Cells. 2004. July;9(7):611–618. [DOI] [PubMed] [Google Scholar]
- [29].Kim DH, Sarbassov DD, Ali SM, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002. July 26;110(2):163–175. [DOI] [PubMed] [Google Scholar]
- [30].Vander Haar E, Lee SI, Bandhakavi S, et al. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007. March;9(3):316–323. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.