

ABSTRACT
Loss of IFNB/interferon-β in mice causes a Parkinson disease-like phenotype where many features, including SNCA/α-synuclein and MAPT/tau accumulation, can be attributed to a late-stage block in autophagic flux. Recently, we identified a mechanism that can explain this phenotype. We found that IFNB induces expression of Mir1, a microRNA that can reduce the levels of TBC1D15, a RAB GTPase-activating protein. Induction of this pathway decreases RAB7 activity and thereby stimulates macroautophagy/autophagy. The relevance of these key players is deeply conserved from humans to Caenorhabditis elegans, highlighting the importance of this ancient autophagy regulatory pathway.
KEYWORDS: Autophagy, Huntington disease, interferon-beta, miR-1, Parkinson disease, proteinopathies, TBC1D15
Autophagy is a pathway that enables degradation of cytoplasmic contents by wrapping them within double-layered vesicles called autophagosomes. These autophagosomes fuse with lysosomes, which facilitates degradation of their lumenal contents. The transport and fusion steps are governed by numerous proteins, including RAB GTPases, such as RAB7, HOPS and SNARE proteins.
The accumulation of neurotoxic aggregation-prone proteins is a hallmark of many age-related neurodegenerative diseases, such as Parkinson disease, Alzheimer disease, Huntington disease, and amyotrophic lateral sclerosis. Mendelian mutations associated with many neurodegenerative diseases compromise autophagy, and these potentiate the accumulation of various aggregate-prone proteins, because such proteins, including mutant HTT (huntingtin), MAPT/tau and SNCA/α-synuclein, are autophagic substrates. Autophagy is particularly important for the degradation of oligomeric and higher-order assemblies that characterize these neurodegenerative disease-causing proteins, as these are inaccessible to the proteasome, or chaperone-mediated autophagy.
Previously, we reported that mice lacking the Ifnb1 (interferon beta 1, fibroblast) gene, an essential cytokine in anti-viral immune responses and T-cell regulation, develop a phenotype resembling Parkinson disease with the accumulation of intraneuronal SNCA and MAPT aggregates, and loss of dopaminergic neurons, associated with cognitive and motor disturbances. We found that lack of IFNB input causes a late-stage block in neuronal autophagy compatible with impaired autophagosome degradation. Conversely, treatment with recombinant IFNB promotes autophagy flux, and coherently we demonstrated that in vivo brain expression of Ifnb reduces SNCA-induced neurotoxicity in a rat model of Parkinson disease. However, it was not clear how IFNB regulates autophagy.
Recently, we identified a mechanism explaining how IFNB influences autophagy by focussing our studies on the microRNA, mir-1, which is depleted in a Drosophila melanogaster model of Alzheimer disease, and MIR1, which is reduced in the cerebrospinal fluid of patients with Parkinson disease [1]. microRNAs rapidly downregulate translation of multiple target genes by imperfect binding to motifs in their 3ʹ UTR region. MIR1 is a highly conserved miRNA and despite 600 million years of evolutionary distance, the sequence of the miR-1/MIR1 seed region is fully conserved in worms and humans. Thus, we made use of a C. elegans in vivo model to ectopically express an aggregation-prone fragment of mutant HTT (HTTQ40), which causes Huntington disease-like pathology. Loss of mir-1 leads to the formation of significantly more HTTQ40 protein aggregates when compared to the wild-type worms, and expressing wild-type mir-1 in mutant worms reduces the aggregation burden. Similarly, loss of mir-1 in worms enhances exogenous SNCA aggregate formation and causes behavioral abnormalities. These defects are associated with our finding that mir-1 is a positive autophagy regulator.
Consistent with the worm data, we found that overexpression of MIR1 decreases mutant HTT exon 1 aggregation in HeLa cells, and this could be attributed to enhanced autophagic flux. From the TargetScan database we identified human TBC1D15 (TBC1 domain family member 15) and a C. elegans homolog, TBC-7, as potential targets of MIR1/mir-1, both of which are putative negative regulators of autophagy. These proteins are RAB-GTPase activating proteins (GAPs), which promote termination signaling of RAB GTPases, and TBC1D15 targets RAB7, a known regulator of autophagy. Thus, we tested and confirmed that TBC1D15/TBC-7 is a genuine target of MIR1/mir-1 using C. elegans- and human-specific reagents. For example, MIR1 targets the 3ʹ UTR of TBC1D15 in HeLa cells and consequently reduces the amount of TBC1D15 protein.
Next, we investigated how TBC1D15 influences autophagy flux. Overexpression of TBC1D15 in HeLa cells causes a late-stage block in autophagy compatible with defective autophagosome degradation, and TBC1D15 knockdown mimics the autophagy induction observed with MIR1 overexpression. The increase of autophagy induced by MIR1 is blocked by overexpression of TBC1D15 that lacks the endogenous 3ʹ target sequence for MIR1. Finally, we showed that RAB7 is indeed a target of TBC1D15, as overexpression of TBC1D15 significantly reduces the amount of active GTP-bound RAB7. As expected, loss of RAB7 leads to a late-stage block in autophagy, and co-expression of a constitutive active RAB7Q67L mutant counteracts the TBC1D15-mediated autophagy block.
A previous study observed that IFNB regulates MIR1 in hepatic cells. This provided the clue and impetus to test whether this cytokine could regulate autophagy and aggregate-prone protein clearance via the MIR1-TBC1D15-RAB7 pathway. We found that IFNB upregulates Mir1 while reducing the amount of TBC1D15 protein in primary cortical neuron cultures from mice, and in HeLa cells, where the effect of the cytokine on TBC1D15 levels depends on the presence of an intact 3ʹ UTR MIR1 binding site. The effects of IFNB on autophagic flux and mutant HTT accumulation depend on MIR1 and TBC1D15, as they are abrogated by a MIR1 hairpin inhibitor or TBC1D15 overexpression.
Collectively, our data provide new mechanistic insight into how IFNB promotes autophagy via MIR1-TBC1D15-RAB7, thereby limiting accumulation of aggregation-prone proteins such as SNCA and HTT (Figure 1). The deep evolutionary conservation from worm to human emphasizes the biological importance of MIR1, and it will be interesting to explore the possible therapeutic potential of this pathway.
Figure 1.
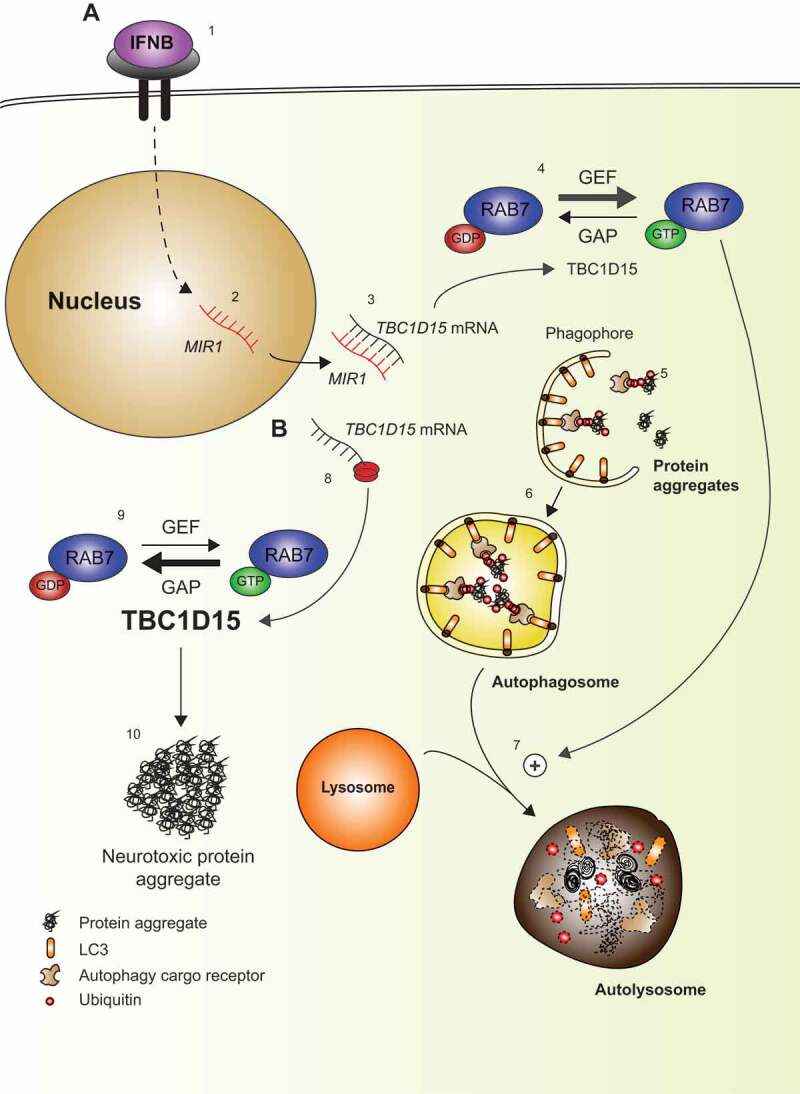
Schematic overview. (A) IFNB binds to IFNAR1/IFN-α/β-receptor and activates downstream signaling (1) including transcription of MIR1 (2), which binds to the 3ʹ UTR of TBC1D15 and thereby repress its translation (3). TBC1D15 is a GTPase activating protein (GAP), which shuts down the activity of RAB7 by promoting GDP-binding. Thus, with low levels of TBC1D15, the abundance of active GTP-bound RAB7 will be favored due to guanine nucleotide exchange factor (GEF) activity (4). Protein aggregates are targeted by autophagy cargo receptors and subsequently couple to LC3-II in the phagophore membrane (5), which eventually forms a vesicular autophagosome (6). The autophagosomes fuse with lysosomes for clearance of the protein aggregates in a process promoted by active RAB7 (7). (B) If TBC1D15 translation is increased, e.g., by low MIR1 (8) this shuts down RAB7 activity (9), and under stress conditions neurotoxic protein aggregates will form due to reduced autophagy (10).
Funding Statement
This work was supported by UK Dementia Research Institute at the University of Cambridge (funded by the MRC, Alzheimer’s Research UK and the Alzheimer’s Society), the National Institute for Health Research Cambridge Biomedical Research Centre, the Wellcome Trust [095317/Z/11/Z]; the Spoelberch Foundation and an anonymous donation to the Cambridge Centre for Parkinson-Plus to D.C.R., NHMRC [Senior Research Fellowship GNT1137645 and Project Grant GNT1156481 to R.P.]; veski Innovation Fellowship (VIF23 to R.P.), The Danish Council for Independent Research [DFF - 6110-00461 to P.E.]; Lundbeck Foundation [R210-2015-3372 to P.E.]; and Parkinsonforening in Denmark (to P.E.). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care.
Disclosure statement
No potential conflict of interest was reported by the authors.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Reference
- [1].Nehammer C, Ejlerskov P, Gopal S, et al. Interferon-beta-induced miR-1 alleviates toxic protein accumulation by controlling autophagy. Elife. 2019;8:e49930. [DOI] [PMC free article] [PubMed] [Google Scholar]