

ABSTRACT
Macroautophagy/autophagy functions as a quality control mechanism by degrading misfolded proteins and damaged organelles and plays an essential role in maintaining neural homeostasis. The phosphoinositide phosphatidylinositol-3-phosphate (PtdIns3P) effector Atg18 is essential for autophagosome formation in yeast. Mammalian cells contain four Atg18 homologs, belonging to two subclasses, WIPI1 (WD repeat domain, phosphoinositide interacting 1), WIPI2 and WDR45B/WIPI3 (WD repeat domain 45B), WDR45/WIPI4. The role of Wdr45b in autophagy and in neural homeostasis, however, remains unknown. Recent human genetic studies have revealed a potential causative role of WDR45B in intellectual disability. Here we demonstrated that mice deficient in Wdr45b exhibit motor deficits and learning and memory defects. Histological analysis reveals that wdr45b knockout (KO) mice exhibit a large number of swollen axons and show cerebellar atrophy. SQSTM1- and ubiquitin-positive aggregates, which are autophagy substrates, accumulate in various brain regions in wdr45b KO mice. Double KO mice, wdr45b and wdr45, die within one day after birth and exhibit more severe autophagy defects than either of the single KO mice, suggesting that these two genes act cooperatively in autophagy. Our studies demonstrated that WDR45B is critical for neural homeostasis in mice. The wdr45b KO mice provide a model to study the pathogenesis of intellectual disability.
Abbreviations: ACSF: artificial cerebrospinal fluid; AMC: aminomethylcoumarin; BPAN: beta-propeller protein-associated neurodegeneration; CALB1: calbindin 1; CNS: central nervous system; DCN: deep cerebellar nuclei; fEPSP: field excitatory postsynaptic potential; IC: internal capsule; ID: intellectual disability; ISH: in situ hybridization; KO: knockout; LTP: long-term potentiation; MBP: myelin basic protein; MGP: medial globus pallidus; PtdIns3P: phosphoinositide phosphatidylinositol-3-phosphate; WDR45B: WD repeat domain 45B; WIPI1: WD repeat domain, phosphoinositide interacting 1; WT: wild type.
KEYWORDS: Autophagy, axon swelling, intellectual disability, Wdr45, Wdr45b
Introduction
Autophagy involves the formation of a double-membrane autophagosome and its eventual delivery to lysosomes for degradation of sequestrated materials [1,2]. The formation of an autophagosome involves multiple sequential membrane remodeling events, including the initiation of a phagophore and its subsequent expansion and closure. A group of evolutionarily conserved Atg genes have been identified that act at distinct steps of autophagosome formation. The Vps34 kinase complex generates the phosphoinositide PtdIns3P that is essential for autophagosome formation [3]. In yeast, the WD40 repeat-containing PtdIns3P-binding protein Atg18 functions as an essential PtdIns3P effector in autophagy. Atg18 forms a complex with Atg2 and is involved in regulating the trafficking of the multi-membrane-spanning protein Atg9 [4]. Yeast Atg18 also binds to PtdIns(3,5)P2 on the vacuole to regulate its morphology [5]. PtdIns3P on sealed autophagosomes also promotes Mon1-Ccz1-mediated recruitment of Ypt7 or directly facilitates Ypt7 recruitment to the autophagosome, thus facilitating the fusion of autophagosomes with the vacuole [6–8].
The autophagic machinery is much more complex in multicellular organisms. An additional layer of complexity is conferred by the presence of multiple homologs of the same yeast Atg proteins. Genetic screens in C. elegans have identified two WD40 repeat-containing PtdIns3P-binding proteins, ATG-18 and EPG-6, that act non-redundantly for degradation of protein aggregates [9]. The genetic phenotypes in epg-6 mutants, such as the accumulation of ATG-9 puncta, resemble those in atg-2 mutants, while atg-18 acts upstream of epg-6 and atg-2 in the genetic hierarchical pathway [9]. In mammalian cells, there are four Atg18 homologs, WIPI1, WIPI2, WDR45B and WDR45. Based on phylogenetic analysis, these proteins can be classified into 2 subgroups, WIPI1, WIPI2 and WDR45B, WDR45 [10]. WIPI2 acts at early steps of autophagosome formation, and its function may be negatively regulated by WIPI1 [10]. Wdr45 deficiency causes a defect in autophagy in neurons [11]. The role of WDR45B in autophagy and its physiological function remain largely unknown.
The basal constitutive level of autophagy removes misfolded proteins and/or damaged organelles, thus functioning as a quality control mechanism [12]. Autophagy is crucial for maintaining neuron and axon homeostasis under physiological conditions [13]. Human genetic studies revealed that de novo mutations in WDR45 cause beta-propeller protein-associated neurodegeneration (BPAN), which is a subtype of neurodegeneration with brain iron accumulation [14–16]. Samples from affected BPAN patients display lower autophagic activity and accumulation of aberrant early autophagic structures [16]. Mice deficient in Wdr45 recapitulate some phenotypic features of BPAN patients, including accumulation of axonal spheroids in the central nervous system (CNS) and impairment of learning and memory [11]. Mice wdr45 KO also show impaired autophagic flux, which may contribute to the neural dysfunction [11]. The autophagy defect in wdr45 KO mice is much weaker compared to mice deficient in other essential autophagy genes and the autophagy defect appears to be restricted to neurons [11]. The high sequence similarity between WDR45 and WDR45B suggests that these two proteins may act redundantly in autophagy. Several human genetic studies have revealed a potential causative role of WDR45B in intellectual disability (ID) [17–19]. ID is a common disorder affecting at least 1% of the general population [17]. Suleiman et al. showed that 6 patients from 3 independent families possess homozygous mutations in WDR45B and display a similar phenotype of ID [19]. The molecular mechanism underlying the pathogenesis of ID remains unknown.
Here we generated wdr45b KO mice, and found that they exhibit motor abnormalities and cognitive impairment. Swollen axons accumulate in wdr45b KO mice. Absence of Wdr45b causes autophagy defects which result in accumulation of SQSTM1 and ubiquitin-positive aggregates and autophagosomes in affected neural cells. Double KO mice, wdr45b and wdr45, show much more severe autophagy defects than either of the single KO mice. Our studies revealed the important function of Wdr45b in autophagy and neural homeostasis during mouse development.
Results
Wdr45b-deficient mice exhibit abnormal motor behavior and impaired learning and memory
To study the in vivo function of WDR45B, we generated conventional wdr45b KO (wdr45b-/-) mice by CRISPR-Cas9 (Figure 1A). Mice wdr45b KO have a 1-bp insertion in exon 1 (Figure 1A), resulting in a premature stop codon at amino acid 24. Mice wdr45b KO were born normally, and showed growth retardation from 20 days after birth (Figure 1B and Figure. S1A).
Figure 1.
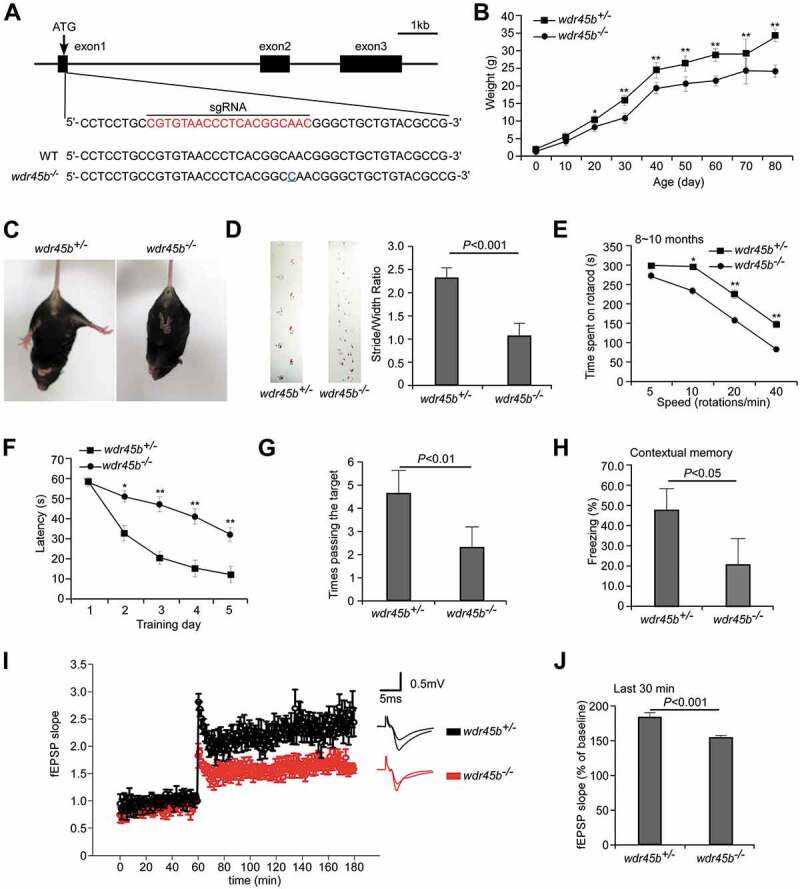
wdr45b-/- mice show motor defects and cognitive impairment. (A) Scheme for generation of wdr45b KO mice by the CRISPR-Cas9 system. The sgRNA-targeting sequence is highlighted in red. Sequencing of genomic DNA identified mutant mice with a 1-bp insertion in the targeting site (blue underline), which causes a frame-shift mutation. (B) Body weight curves of wdr45b+/- and wdr45b-/- males. Mean ± SD of 6 mice is shown. *, P < 0.05; **, P < 0.01. (C) wdr45b-/- mice show a limb-clasping reflex. (D) In gait analyses, the step width of wdr45b-/- mice at 8 ~ 10 months is significantly shorter than that of control littermates (forefeet, green; hindfeet, red). Mean ± SEM (n = 8) is shown. (E) Rotarod performance of wdr45b+/- and wdr45b-/- mice at 8 ~ 10 months of age at speeds of 5, 10, 20 and 40 rotations/min. Mean ± SEM (n = 10) mice is shown. *, P < 0.05; **, P < 0.01. (F and G) In the Morris water maze test, wdr45b-/-mice at 8 ~ 10 months of age show increased latency to locate the hidden platform during the learning stage (F), and they pass the target site fewer times in the spatial probe test (G), compared to control mice. Mean ± SEM (n = 9) is shown. *, P < 0.05; **, P < 0.01. (H) In the contextual fear conditioning test, the freezing time of wdr45b-/- mice at 8 ~ 10 months of age is significantly reduced, compared to control mice. Mean ± SD (n = 10) is shown. (I and J). LTP is attenuated in wdr45b-/- mice at 10 months of age (I). The average responses from the last 30 min in the control and wdr45b-/- mice are shown in J. Mean ± SEM (n = 9) is shown.
The wdr45b-/- mice exhibited abnormal limb-clasping reflexes from 6 months old (Figure 1C). This response was observed in 6 out of 10 knockout mice at 6 ~ 12 months and 6 out of 6 knockout mice at 12 ~ 16 months, but none of the control mice. The wdr45b-/- mice also showed tremor, as shown in Movie 1. 2 out of 10 wdr45b-/- mice aged at 6 ~ 12 months, and 5 out of 6 wdr45b-/- mice aged at 12 ~ 16 months had seizures, while control mice never had this phenotype (Movies 2 and 3). The footprint test showed that the ratios of stride lengths to paw-based widths of wdr45b KO mice were significantly smaller than those of heterozygous controls, revealing an ataxia phenotype (Figure 1D). In the rotarod assay, wdr45b KO mice stayed on the rotarod for significantly less time than controls, and the difference became more dramatic with age (Figure 1E and Figure. S1B). There was no significant difference in grip strength in control and wdr45b KO mice (Figure. S1C). Thus, wdr45b KO mice showed impaired motor coordination.
Because mutations in WDR45B cause intellectual impairment in humans [17–19], we performed multiple behavioral tests to determine the cognitive ability of wdr45b-/- mice. In order to avoid any influence of the estrous cycle in female mice, we chose to study male mice. The Morris water maze test is a two-phase experiment, which examines both the spatial learning and memory function of mice. Mice wdr45b KO had normal vision and similar swimming speed compared to control mice (Figure. S1D and data not shown). In the training test, wdr45b KO mice showed a longer latency to escape onto the hidden platform from the second day, and the difference became more significant in the following days, indicating a learning impairment (Figure 1F). In the probe trial on the 6th day, wdr45b KO mice stayed for significantly less time than controls in the target quadrant (Figure. S1E) and swam fewer times across the site where the hidden platform was previously placed (Figure 1G), suggesting a memory defect. A fear conditioning test was conducted to measuring the ability of the mice to remember the association between a conditioned stimulus (70 dB sound) and an adverse stimulus (an electric shock on the foot). After one-day training, wdr45b KO mice showed a significant decrease in freezing time in the contextual test compared to control mice (Figure 1H), but no obvious difference in the cued test (Figure. S1F). Taken together, these results show that learning and memory are impaired in wdr45b KO mice.
Long-term potentiation (LTP) is considered as one of the major cellular mechanisms that underlies learning and memory [20]. The CA1 area of the hippocampus is crucial to the formation of spatial memories in mice [21]. To determine the association of cognitive impairments with changes in hippocampal synaptic plasticity in wdr45b KO mice, we measured the induction of LTP at Schaffer collateral-CA1 synapses by θ-burst stimulation in hippocampal slices. Compared to control mice, the field excitatory postsynaptic potential (fEPSP) amplitude in wdr45b KO mice was significantly lower, indicating reduced ability to induce LTP (Figure 1I,J).
Wdr45b-deficient mice exhibit extensive axonal degeneration
We next performed histological analysis of various brain regions of wdr45b KO mice. The cerebella of KO mice were normal in foliation at early ages, but exhibited evident atrophy in size from 8 months old (Figure. S2A). Eosinophilic spheroids, which are indicators of swollen axons, accumulated in the deep cerebellar nuclei (DCN) regions in wdr45b KO mice. The number of spheroids increased with age at first, and then started to decrease when the mice reached 6 months old. Only a few large spheroids were found in sections from mice aged 12 months old (Figure 2A-E and Figure. S2B and C). Anti-CALB1 (calbindin 1) staining, which labels Purkinje cell bodies and axons, was further performed to examine the abnormal axonal structures. CALB1-positive swollen axons, enwrapped by MBP (myelin basic protein)-labeled myelin, accumulated in the DCN regions in wdr45b KO mice, accompanied by reactive astrogliosis (Figure 2F,G). In wdr45b-/- mice, astrocyte hyperproliferation was also observed in other brain regions, including cortex and thalamus, and more dramatically in the cingulate cortex and internal capsule (IC) (Figure 2H,I and Figure. S2D and E), indicating extensive neural damage. In the DCN regions, EM analysis showed accumulation of many swollen and degenerated axons, which were not found in control mice (Figure 2J-N and Figure. S2F-H). The affected axons were characterized by defects including large numbers of abnormal organelles, such as mitochondria and lysosomes; aberrant membrane structures such as lamellar bodies; dark axoplasm; empty sheaths; and demyelination (Figure 2K-M and Figure. S2F-H). Interestingly, we found accumulation of autophagosomes in the swollen axons of wdr45b KO mice (Figure 2M,O). This is indicative of defective autophagic flux, possibly at a later step in autophagosome maturation, in the absence of Wdr45b.
Figure 2.
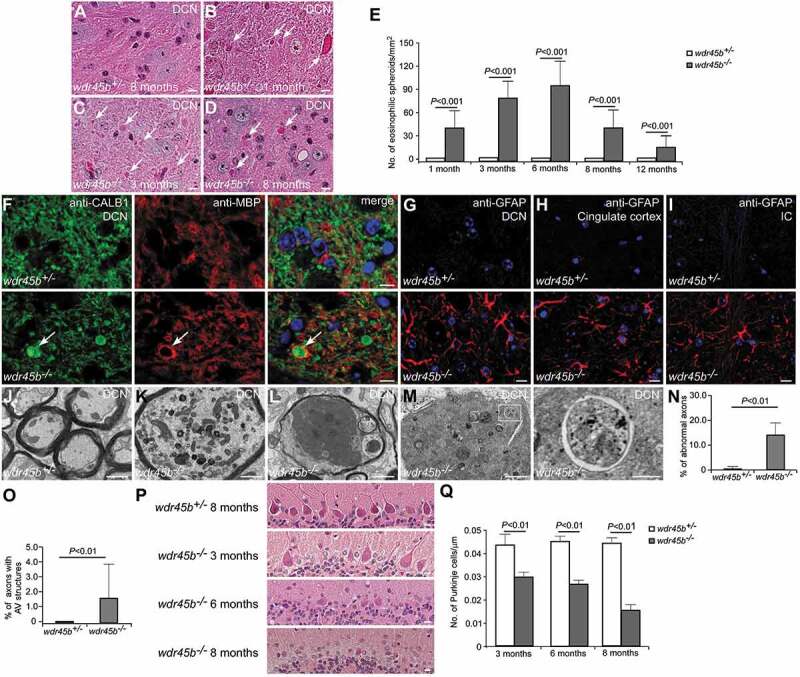
Axon swelling and cerebellar atrophy in wdr45b KO mice. (A-D) H&E staining shows eosinophilic spheroids (arrows) in the DCN of wdr45b-/- mice at 1, 3 and 8 months (B-D). No spheroids are found in control mice (A). Bars: 10 μm. (E) The number of eosinophilic spheroids in the DCN of wdr45b+/- and wdr45b-/- mice at 1, 3, 6, 8 and 12 months is shown as mean ± SD (n = 3). (F) Immunostaining with CALB1 (green) and MBP (red) antibodies shows that CALB1-positive swollen axons (arrows) are enwrapped by MBP-labeled myelin in the DCN regions of wdr45b-/- mice. (G-I) Reactive astrogliosis is found in the DCN region (G), cingulate cortex (H) and IC (I) of wdr45b-/- mice. IC, internal capsule. Bar: 10 μm (F-I). (J-M) EM images show normal myelinated axons in the DCN of wdr45b+/- mice at 5 months (J), while those in wdr45b-/- mice are swollen and some contain large numbers of mitochondria (K), dark axoplasm (L) and double-membrane autophagosomes (M). The right panel shows enlargement of the boxed area in the left panel in M. Bars: 1 μm (J, K, L and left panel in M); 200 nm (right panel in M). (N and O) The percentages of the axons that are abnormal (N) or contain autophagosome structures (O) in wdr45b+/- mice and wdr45b-/- mice are shown as mean ± SD (n = 50 micrographs of 548 μm2). AV, autophagosome. (P and Q) Purkinje cell numbers gradually decrease with age in wdr45b-/- mice (P). Mean ± SD (n = 3) is shown in Q. Bars: 10 μm.
wdr45b KO mice also showed a progressive loss of Purkinje cells, which became evident from 3 months of age (Figure 2P,Q). The loss of Purkinje cells was detected first in the 4th, 5th and 6th lobes of the cerebellum, but became evident in other lobes after 8 months of age.
Wdr45b-deficient mice exhibit autophagy defects in the brain
Although WDR45B shares extensive amino acid similarity with the autophagy protein WDR45, its function in the autophagy pathway in vivo remains unknown. We examined the degradation of two autophagy substrates, SQSTM1 and ubiquitinated proteins, in control and wdr45b KO mice. Levels of SQSTM1, MAP1LC3 (hereafter referred to as LC3)-I and LC3-II, as well as LC3-II:I ratios, were increased in wdr45b KO cerebella (Figure 3A). Sqstm1 mRNA levels and proteasome activity (Figure. S3A-C) remained unchanged in wdr45b KO mouse brain compared to control littermates. SQSTM1- and ubiquitin-positive aggregates accumulated in the cerebra at the age of 3 months. These aggregates were evident in cingulate cortex, thalamus and medial globus pallidus (MGP), and were even more abundant in the white matter regions, such as corpus callosum, internal capsule and fimbria (Figure 3B-D and Figure. S3D-G). Accumulation of SQSTM1- and ubiquitin-positive aggregates in the cerebella of KO mice, including the DCN regions and Purkinje cells, occurred as early as 1-month-old when swollen axons were evident (Figure 3E,F). The regions with reactive GFAP expression, such as the cingulate cortex, internal capsule, thalamus and DCN, correlate with SQSTM1 accumulation in wdr45b KO mice (Figure 2G-I and Figure. S2E).
Figure 3.
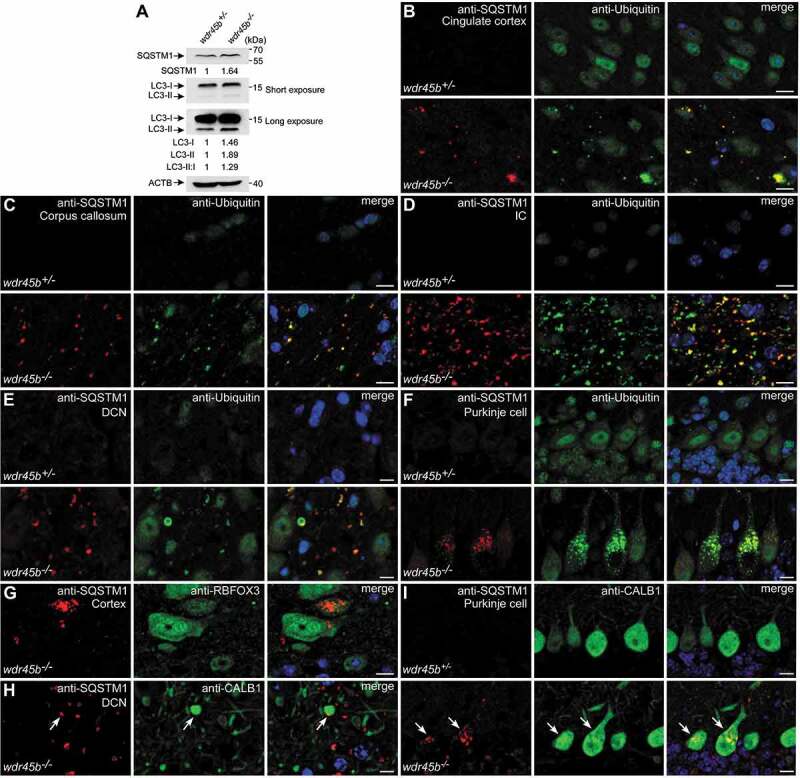
wdr45b-/- mice display autophagy defect. (A) Immunoblotting results show that levels of SQSTM1, LC3-I, LC3-II and LC3-II:I are increased in the cerebella of wdr45b-/- mice compared to wdr45b+/- mice. Quantification of SQSTM1, LC3-I and LC3-II levels normalized by ACTB levels and LC3-II:I ratios is shown. (B-F) Accumulation of SQSTM1 (red) and ubiquitin (green) positive signals in the cingulate cortex (B), corpus callosum (C), IC (D), DCN (E) and Purkinje cells (F) of wdr45b-/- mice. IC, internal capsule. (G) Costaining of SQSTM1 (red) and RBFOX3 (green) shows that SQSTM1 aggregates accumulate in neurons in the cortex of wdr45b-/- mice. (H and I) Costaining of SQSTM1 (red) and CALB1 (green) shows that SQSTM1 accumulates in the swollen axons (H) and cell bodies (I) of Purkinje cells in wdr45b-/- mice, as indicated by arrows. Bars: 10 μm (B-I).
In wdr45b KO mice, SQSTM1 aggregates accumulated in neurons labeled by anti-RBFOX3/NeuN in cortex, but were absent in neurons in other regions, including thalamus, internal capsule and corpus callosum (Figure 3G and data not shown). SQSTM1 aggregates were not detected in GFAP-positive astrocytes (Figure. S3H). We also observed accumulation of SQSTM1 and ubiquitin in the enlarged Purkinje cell axons and cell bodies, which were labeled by CALB1 (Figure 3H,I and Figure. S3I). SQSTM1 aggregates were not detected in other tissues examined in wdr45b KO mice, including livers and spleens (Figure. S3J and K). Thus, WDR45B is important for autophagy specifically in some types of neural cell but not in other neural cells or non-neural tissues.
Constitutive wdr45 KO mice show milder defects compared to wdr45b KO mice
Mice with CNS-specific knockout of wdr45 were examined previously [11]. SQSTM1 and ubiquitin-positive aggregates accumulate in wdr45 KO mice [11]. To better compare the phenotypes of mice without Wdr45 or Wdr45b, we generated constitutive wdr45 KO mice. Swollen axons, indicated by spheroids, accumulated in the DCN regions of wdr45 KO mice, and these spheroids were larger than those in wdr45b KO mice at 12 months of age (Figure. S4A-C). This is probably because no significant loss of Purkinje cells was detected in wdr45 KO mice at 12 months (Figure. S4D and E), and the Purkinje cell axon swelling gradually worsened, resulting in more enlarged spheroids in the DCN region. Taken together, these results indicate that the pathological alterations in wdr45b KO mice are stronger than in wdr45 KO mice.
In wdr45 KO mice, the accumulation of SQSTM1 and ubiquitin aggregates was less severe than in wdr45b KO mice. SQSTM1 aggregates were largely detected in neurons in thalamus, cortex and hippocampus in cerebra, and in the DCN regions, but not in the other cerebral regions or Purkinje cell bodies in wdr45 KO mice (Figure. S4F and G and data not shown). SQSTM1 aggregates accumulate in neurons (Figure. S4H). Therefore, wdr45 KO mice display a weaker defect in autophagy than wdr45b KO mice.
Wdr45b and Wdr45 are differentially expressed in the mouse
Wdr45b and Wdr45 mRNA levels were determined in various tissues of wild type (WT) mice at the age of 1 month by quantitative PCR. Both Wdr45b and Wdr45 were expressed in all tissues examined, including cerebrum, cerebellum, liver, kidney, spleen, lung, white fat and pancreas (Figure. S4I). Wdr45b and Wdr45 were expressed at a comparable level in many of the tissues, but differential expression was also observed (e.g. in cerebrum, liver, lung and pancreas).
We next examined the spatial distribution of Wdr45b and Wdr45 mRNAs by performing RNAscope in situ hybridization (ISH) assays with WT mice. Specific RNAscope probes for the genes were labeled with HRP or AP and developed into two different colors. Wdr45b and Wdr45 were widely expressed in the cerebra and cerebella (Figure. S4J-P). Compared to other brain regions, Wdr45b and Wdr45 signals were higher in hippocampus, which is important for learning and memory functions (Figure. S4M). Levels of Wdr45b were slightly higher than Wdr45 in hippocampus (Figure. S4M and P). Wdr45b levels were also higher than Wdr45 in the white matter regions, such as corpus callosum and internal capsule (Figure. S4J, K and P). In the cerebella, Wdr45b was also expressed at elevated levels compared to Wdr45, both in the DCN and Purkinje cell bodies (Figure. S4N-P). Together, these results show that Wdr45b and Wdr45 are ubiquitously expressed but display differential patterns in certain tissues, while Wdr45b levels are higher in some brain regions.
Wdr45b-/- wdr45-/Y double KO mice exhibit more severe autophagy defect
To determine the relationship between Wdr45b and Wdr45 in autophagy, we constructed wdr45b-/- wdr45-/Y double KO mice. In contrast to the wdr45b and wdr45 single KO mice that were born normally, the double KO mice were born smaller (Figure 4A,B) and 6 out of 6 mice died 6–8 h after birth. The double KO mice show suckling defects, similar to atg5 and atg7 KO mice [22,23]. Thus, mice deficient in Wdr45b and Wdr45 show synthetic defects in development. Neither wdr45b nor wdr45 single KO mice showed accumulation of SQSTM1 aggregates in brain sections at 1 day after birth. In the double KO mice at postnatal day 1, however, SQSTM1 aggregates accumulated in many brain regions, including thalamus, hypothalamus, hippocampus and cerebellum (Figure 4C,D and Figure. S5A-F). Immunostaining of wdr45b-/-wdr45-/Y mouse brain sections with SQSTM1 and RBFOX3 antibodies revealed accumulation of SQSTM1 in neurons (Figure 4E). These results indicate that the autophagy defect in wdr45b KO mice is exacerbated in the CNS by simultaneous knockout of Wdr45. No accumulation of SQSTM1 aggregates was detected in the liver and kidney in the double KO mice (Figure. S5G and H), indicating that WDR45B and WDR45 may not be essential for autophagy in these tissues.
Figure 4.
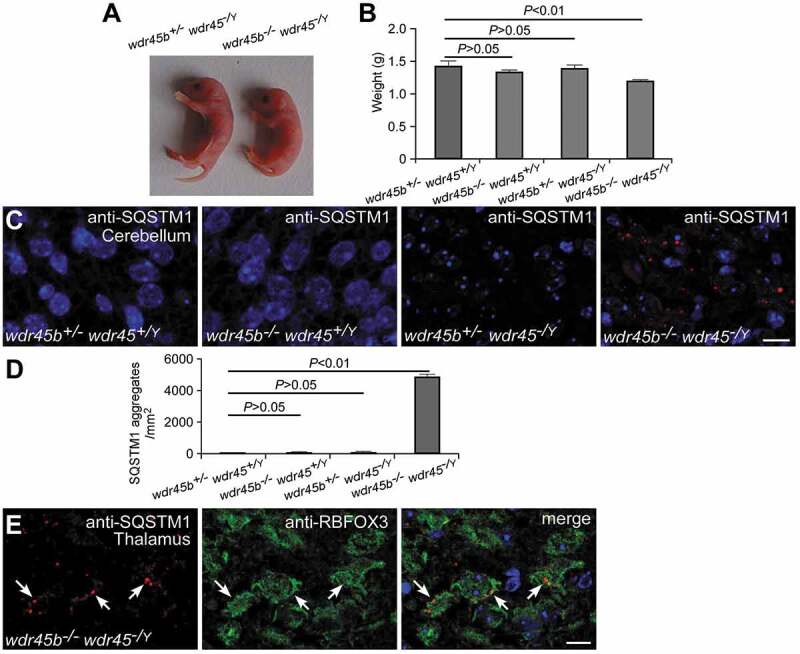
wdr45b and wdr45 double KO mice. (A and B) wdr45b and wdr45 double KO mice are born smaller than their single KO littermates (A). Mean ± SD (n = 4) is shown in B. (C and D) Accumulation of SQSTM1 in the cerebellum of wdr45b and wdr45 double KO mice, but not in control and single KO mice (C). Quantification of SQSTM1 aggregates is shown as mean ± SD (n = 3) in D. Bar: 10 μm. (E) Costaining of SQSTM1 (red) and RBFOX3 (green) shows that SQSTM1 aggregates accumulate in the neurons of the cortex in wdr45b and wdr45 double KO mice, as indicated by arrows. Bar: 10 μm.
Discussion
Wdr45b and Wdr45 exhibit a similar and tissue-specific function in autophagy
Like wdr45 KO mice, wdr45b KO mice develop late-onset neurodegeneration, as characterized by axonal swelling and behavioral abnormalities, with learning and memory impairment. In wdr45b KO mice, the number of swollen axons first increases during development, and then decreases from the age of 8 months, while in wdr45 KO mice the number of swollen axons increases with age and exceeds the level in wdr45b KO mice at later stages. The number of Purkinje cells is gradually reduced with age in wdr45b KO mice, but not in wdr45 KO mice, suggesting that the missing neurons in wdr45b KO mice lead to the reduction in axon swelling. Moreover, wdr45b KO mice exhibit severe motor defects, including limb-clasping reflexes and ataxic walking patterns, which are absent in wdr45 KO mice [11]. The dysregulated movement coordination in wdr45b KO mice is probably due to the loss of Purkinje cells. Thus, wdr45b KO mice suffer from much stronger neural defects than wdr45 KO mice.
Single knockout of Wdr45b or Wdr45 impairs autophagic flux with accumulation of SQSTM1- and ubiquitin-positive aggregates in neurons and swollen axons. The regions in brain with accumulation of SQSTM1- and ubiquitin-positive aggregates appear not to be overlapping in wdr45b KO and wdr45 KO mice. SQSTM1- and ubiquitin-positive aggregates accumulate in more extensive regions of the cerebrum, including cortex (especially cingulate cortex), corpus callosum, internal capsule, thalamus and MGP in wdr45b KO mice, while they mainly accumulate in thalamus, cortex and hippocampus in wdr45 KO mice. The accumulation of these autophagic substrates is strongly detected in cerebral white matter regions, such as corpus callosum, fimbria and internal capsule, in wdr45b KO mice, which is not obvious in wdr45 KO mice. This indicates that Wdr45b may be essential for maintaining autophagy in axons and/or oligodendrocytes that produce myelin to enwrap axons. In cerebella, SQSTM1- and ubiquitin-positive aggregates accumulate in both the DCN regions and Purkinje cell bodies in wdr45b KO mice, while they are present in the DCN regions, but not in Purkinje cells, in wdr45 KO mice. These results demonstrate that autophagy function has distinct requirements for WDR45B and WDR45 in different types of neural cells.
The autophagy defect in wdr45b and wdr45 single KO mice is much weaker than in atg5 KO mutants. The wdr45b and wdr45 double KO mice die at postnatal day 1. SQSTM1 and ubiquitin-positive aggregates are not detected in postnatal day 1 single KO mice, but accumulate in various brain regions of double KO mice. This indicates that WDR45B and WDR45 may act redundantly in the same step of the autophagy pathway. Alternatively, WDR45B and WDR45 may have a relatively minor role at different steps and simultaneous loss of function of both genes causes a synthetic defect in autophagy. In other tissues of the double KO mice, no SQSTM1 aggregates are detected. Together, these results suggest that WDR45B/45 have a tissue-specific function in autophagy, or that their functions can be substituted in part by other WIPI proteins. WDR45B/45 may also have autophagy-independent functions which contribute to the death of double KO mice within 1 day of delivery.
Differential function of WIPI proteins in the autophagy pathway
The PtdIns3P effector protein Atg18 is essential for autophagosome formation in yeast [2]. In mammalian cells, WIPI2 directly interacts with ATG16 and mediates the formation of the ATG12–ATG5-ATG16L1 complex that functions as an E3 for LC3 lipidation, which is critical for the early step of autophagosome formation [24]. Previous studies revealed that WIPI1 and WIPI2 do not act redundantly in autophagy [10]. WIPI1 may negatively regulate autophagy [10].
PtdIns3P acts at multiple steps of the autophagy pathway [25]. The VPS34 PtdIns3-kinase complex generates PtdIns3P that is essential for autophagosome formation [26]. Temporal turnover of PtdIns3P from nearly closed phagophores/nascent autophagosomes triggers disassociation of PtdIns3P-binding proteins and possibly the disassembly of other ATG proteins, which is a pre-requisite for subsequent autophagosome maturation [27,28]. PtdIns3P also functions on autophagosomes, where it may be generated from re-targeting of the ATG14-VPS34 complex [8]. One function of PtdIns3P on autophagosomes appears to be to promote Mon1- and Ccz1-mediated recruitment of Ypt7/RAB7 [6–8]. In wdr45b KO mice, double membrane autophagosomes accumulated in affected axons, suggesting a role of WDR45B in the maturation step of autophagy. Thus, different WIPIs may be recruited to autophagic structures by PtdIns3P generated at different steps to fulfill their distinct functions in the autophagy pathway.
Impairment of autophagy and human diseases
Depletion of autophagy genes acting at different steps of autophagosome formation appears to cause distinct neurodegeneration phenotypes. Mice deficient in autophagy genes essential for autophagosome formation, such as Atg5, Atg7 and Ei24 (the mammalian epg-4 homolog), exhibit massive neuron loss and axonal degeneration, accompanied by dramatic accumulation of ubiquitinated protein aggregates [29–31]. These defects fail to recapitulate the key features of human neurodegenerative diseases, which include accumulation of non-degradative autophagosomes/autolysosomes, and also damage to a certain population of neurons [32].
Loss of function of EPG5, which acts as a tether protein mediating the fusion between autophagosomes and lysosomes for the formation of degradative autolysosomes [33], results in accumulation of many non-degradative autolysosomes [34]. Absence of Epg5 in mice causes vulnerability only in selective populations of neurons, including motor neurons and retinal photoreceptors [34,35]. In humans, mutations in EPG5 cause Vici syndrome, which is a multisystem disorder characterized by callosal agenesis, cataract, cardiomyopathy and immunodeficiency [36]. Mutations in WDR45 cause the neurodegenerative disease BPAN, which displays accumulation of axonal spheroids and intellectual deterioration [14–16]. Multiple recent studies show that mutations in WDR45B are associated with recessive intellectual disability [17–19]. The distinct neuropathological deficits could because these autophagy genes are differentially required in different types of neural cells and their depletion causes a different degree of autophagy defect. Different types of neurons also exhibit different vulnerability to autophagy impairment. The differential autophagy-independent functions may also contribute to selective neural damage. Further mechanistic study of the function of WDR45B andWDR45 in the autophagy pathway will help us to understand the role of autophagy in the pathogenesis of intellectual disability and BPAN, respectively.
Materials and methods
Generation of wdr45b KO mice using CRISPR-Cas9
DNA vectors expressing target sgRNAs driven by a T7 promotor were constructed. Oligos for sgRNA expression were denatured at 95°C for 5 min, annealed at room temperature and then cloned between two BsaI sites of a linearized PUC57 sgRNA expression vector (Addgene, 51132). The vectors for sgRNA expression were linearized by DraIII (NEB, R3510) and transcribed in vitro with a MEGAshortscript T7 kit (Life technologies, AM1354). DNA vector expressing Cas9 mRNA was linearized by Agel or XmaI. Cas9 mRNA was obtained with a mMESSAGE mMACHINE T7 kit (Life Technologies, AM1344). After purification in vitro, sgRNA (100 ng/µl) and Cas9 mRNA (150 ng/µl) were mixed and microinjected into the cytoplasm of fertilized eggs collected from oviducts of female C57BL/6N mice. About 200 injected zygotes were transferred to the oviducts of pseudo-pregnant ICR females. Finally, viable founder mice were obtained. The sgRNA sequence for Wdr45b was:
Wdr45b: 5ʹ-CGTGTAACCCTCACGGCAAC-3ʹ.
The wdr45b heterozygous mice were outcrossed with WT C57BL/6N mice and then intercrossed to generate wdr45b homozygous KO mice. The genotypes of the offspring were identified by PCR and sequencing using the following primers:
Forward, 5ʹ-ACCTACTTCCCTGCAGTGG-3ʹ,
Reverse, 5ʹ-AGTCTCCGGTTAGGGTCC-3ʹ.
Constitutive wdr45 KO mice were generated by crossing wdr45fl/Y mice with Zp3-Cre mice (Jackson Laboratory). wdr45fl/Y mice were obtained as described previously [11]. All the mice were C57BL/6N in background. The WT C57BL/6N mice were bought from Beijing Vital River Laboratory Animal Technology Co., Ltd. All mice were maintained with free access to food and water under specific pathogen-free conditions in the animal facility at the Institute of Biophysics, Chinese Academy of Sciences, Beijing. All animal experiments were approved by the institutional committee of the institute.
RNAscope ISH assay
This assay was performed with 20ZZ probes targeting mouse Wdr45b (NM-025793.3) and mouse Wdr45 (NM-001290792.1) mRNA using an RNAscope 2.5 HD Duplex Detection Kit (Advanced Cell Diagnostics, 322436) according to the user manual provided by the manufacturer. Mice were deeply anesthetized and transcardially perfused with fresh 10% neutral buffered formalin (Sigma-Aldrich, HT501128), then post-fixed, dehydrated, embedded in paraffin and sectioned. FFPE sections were deparaffinized, followed by tissue pretreatment, probe hybridization and signal amplification. Positive fluorescence ISH signals were identified as punctate dots in the nucleus using a Vectra® 3 automated quantitative pathology imaging system.
Antibodies
The following antibodies were used: rabbit anti-SQSTM1 (MBL, PM045), mouse anti-SQSTM1 (Abcam, ab56416), mouse anti-ubiquitin (Cell Signaling Technology, 3936), rabbit anti-GFAP (Abcam, ab48050), rabbit anti-LC3 (Cell Signaling Technology, 2775S), anti-ACTB (Proteintech, 60008-I-Ig), mouse anti-RBFOX3 (Millipore Sigma, MAB377), rabbit anti-MBP (Abcam, ab40390) and mouse anti-CALB1 (Sigma-Aldrich, C9848).
Histology and immunohistochemistry
Mice were anesthetized, then transcardially perfused with 10% neutral buffered formalin. After post-fixation, tissues were dehydrated, embedded in paraffin and then sectioned at 5 μm with a histotome. Samples were stained with hematoxylin and eosin and examined by light microscopy (Imager. A1; Carl Zeiss) with a 40×/0.75 objective lens (Plan-NeoFluar; Carl Zeiss) and a camera (AxioCam MRc 5; Carl Zeiss) used at room temperature. Images were captured with AxioVision 40 (v4.6.3.0; Carl Zeiss).
For immunostaining, sections were deparaffinized with xylene and rehydrated with a graded alcohol series as described previously [11]. After heat-induced antigen retrieval with 0.1 M citrate buffer, sections were blocked, and then incubated with diluted primary antibodies overnight at 4°C, followed by incubation with fluorescently-labeled secondary antibodies for 1 h at room temperature. Sections were counterstained with DAPI, and detected under a confocal microscope (LSM 710 Meta plus Zeiss Axiovert zoom; Carl Zeiss) with a 63X/1.40 oil-immersion objective lens (Plan-Apochromatlan; Carl Zeiss) and a camera (AxiocamHRm; Carl Zeiss).
Rotarod test
Motor coordination was measured as described previously [11]. The rotarod test was performed with 10 pairs of mice. Mice were trained on the constant rolling rod for two days before the test. On training days, the rolling speed was 5 rpm, and the mice were trained twice a day with 5 min each time. On the testing day, mice were tested for their ability to stay on the rotarod. The time that mice stayed on the rotarod at different rolling speeds (5, 10, 20 and 40 rpm) was recorded. The interval between two trials was 30 min. The maximum observation time was 5 min for each speed.
Grip strength test
A Grip Strength Meter (Bioseb, BIO-GS3) was used to measure the grip strength of the four limbs of the mice. Mice were placed in the center of the metal grid, and then pulled backwards by the tail when they grasped the grid by four paws. The maximum grip force was recorded in newtons (N). The test was carried out three times and the inter-trial interval was 1 min.
Morris water maze test
The Morris water maze test was done as described previously [11]. To create the maze, a 12-cm diameter circular platform was located in a fixed place in the center of one specific quadrant in a pool with a diameter of 1.2 m and a depth of 45 cm, and the pool was divided into four quadrants. The pool was filled with water until it was 1 cm above the platform. Visual cues (different shapes in colored paper sticking to the curtain surrounding the maze) were used as spatial cues. The water was equilibrated to room temperature (21°C). Since C57BL/6N mice are black, non-toxic white powdered food additives was added into the water and mixed thoroughly. Prior to behavioral testing, the mice were transferred to the behavior room and allowed an adaptation period of 30 min in the testing room. Then each mouse was placed in the maze from one of four evenly distributed spots in sequence, facing the tank wall. The mice were allowed to swim for up to 1 min to locate the fixed platform. For the first day of the learning phase, the mice were guided by the experimenter to the platform if they did not find it within 1 min, and then they were allowed to stay on the platform for 15 s. If they could find the platform, they were allowed to stay on the platform for 5 s. After a trial, the mice were dried off and returned to their home cage. The distance traveled and the escape latency of four trials from each of the four starting positions to the platform were recorded by a Smart 2.0 video tracking system (Panlab, Harvard Apparatus, Cornellà [Barcelona], Spain). After learning for five consecutive days, the probe trial was performed on the 6th day. The platform was removed and the mice were allowed to search for 1 min. The time that the mice stayed in the target quadrant and the number of times the mice swam across the platform site was recorded.
Fear conditioning test
In this test, a Startle and Fear Combined System (Panlab, Harvard Apparatus, Cornellà [Barcelona], Spain) was used. Mice were transferred to the testing room 30 min before each test for adaption. The day before training, mice were put into the conditioning chamber for 10 min to adapt to the environment. On the conditioning day, following a 2 min acclimation period to the conditioning chamber, mice received six conditioning trials consisting of a 70 dB tone lasting for 30 s as a conditioned stimulus. During the last 2 s, a 0.3 mA foot shock was delivered through a shock generator, co-terminating with the tone. Each trial was separated by a 15 s interval. Freezing, which is a defensive reaction of mice, was characterized as a lack of movement besides respiration. The fine movement of the mice was recorded by PACKWIN software through a weight transducer situated on the floor. For the contextual test on the second day, mice were placed into the same conditioning chamber, but without the tone or electric foot shock. The time of freezing behavior was measured for 5 min. For the cued test, which took place on the next day after the contextual test, the mice were placed into the same chamber with some decoration to modify its appearance. Mice were given the same sound without the electric foot shock, and the freezing time was recorded. The percentage of freezing time to the total observation time was used as an indicator of learning and memory ability.
Electrophysiology
Electrophysiology was measured as previously described with some modifications [11]. Mice were sacrificed, and hippocampi were transversely sliced (300 μm) using a vibratome (Leica, VT1200S, Wetzlar, Germany) in oxygenated (95% O2 and 5% CO2) ice-cold artificial cerebrospinal fluid (ACSF, 126 mM NaCl, 3 mM KCl, 26 mM NaHCO3, 1.2 mM NaH2PO4, 10 mM D-glucose, 2.4 mM CaCl2 and 1.3 mM MgCl2). The slices were placed in an incubating chamber filled with oxygenated ACSF at 28 to 30°C for a recovery period of at least 80 min. An individual slice was transferred to a recording chamber and was continuously superfused with oxygenated ACSF (5 mL/min) at 30°C. ACSF-filled glass pipettes (1 to 5 MΩ) were placed at the stratum radiatum of area CA1 to record fEPSPs. A concentric bipolar electrode (FHC, CBBEB75) was used to stimulate the Schaffer collateral fibers with a brief current pulse (50 ms), and the current pulse was delivered by a stimulus isolation unit (ISO-Flex; A.M.P.I., Jerusalem, Israel) every 30 s. The distance between the stimulating electrode and recording pipette was 200 to 300 μm. An input-output curve was made to calculate the stimulating strength to 30–50% of the maximal slope. After acquiring baseline for 30 min, LTP was induced by θ-burst stimulation (each burst contained 5 pulses at 100 Hz repeated at 5 Hz and 3 10-burst trains separated by 20 s). Evoked fEPSPs were recorded for 1 h after tetanization. Signals were filtered at 2 kHz and digitized at 100 kHz using Digidata 1440A (Molecular Devices). Data acquisition and slope measurement were performed using pClamp 10.2 (Molecular Devices). Pulse generation was achieved using a Master 8 stimulator (A.M.P.I., Jerusalem, Israel).
Immunoblotting
Total protein was extracted from tissues with RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.1% SDS [SERVA, 20765.03], 1% NP40 [Amresco, E109], protease inhibitor cocktail [Biotool, B14001]). Protein concentration was detected by Bradford protein assay (Genstar, E161–01). Then samples with equal amounts of protein were separated by SDS-PAGE electrophoresis. Proteins were transferred to a PVDF membrane (Millipore, IPVH00010), and blocking was performed with 5% non-fat milk (Inner Mongolia YILI Industrial Group Co., Ltd., Q/NYLB 0039S) for 1 h at room temperature. The membrane was incubated with the indicated primary antibodies and HRP-labeled secondary antibodies: peroxidase-AffiniPure goat anti-Mouse IgG (H + L) (Jackson Immunoresearch, 115-035-003); peroxidase-AffiniPure goat anti-Rabbit IgG (H + L) (Jackson Immunoresearch, 111-035-003). Immunoreactivity was examined with an Enhanced Chemiluminescent kit (Sigma-Aldrich, RPN2232).
Proteasome activity assay
Mouse brains were homogenized with lysis buffer containing 50 mM HEPES, pH 7.5, 5 mM EDTA, 150 mM NaCl, 1% Triton X-100 (Amresco, 0694) and 2 mM ATP. After centrifugation for 20 min at 15,871 x g, the supernatant was collected and protein concentrations were measured. 250-μl samples containing equal amounts of total protein (2 to 4 μg) were mixed with 2.5 μl of each substrate (final concentration, 50 nM), and incubated for 30 min at 37°C protected from light. The reaction was terminated by adding 252.5 μl ice-cold 96% ethanol. Finally, the fluorescence from aminomethylcoumarin (AMC) hydrolysis was measured (380 nm excitation and 460 nm emission). In this assay, the following AMC-linked synthetic peptide substrates were used: Ac-Gly-Pro-Leu-Asp-AMC for caspase-like activity, Suc-Leu-Val-Tyr-AMC for chymotrypsin-like activity, and Ac-Arg-Leu-Arg-AMC for trypsin-like activity (Enzo Life Sciences, PW9905-0001).
Quantitative RT-PCR
Total RNA was prepared with TRIzol (ThermoFisher Scientific, 15596018). cDNA was reverse transcribed using a RevertAid First Strand cDNA Synthesis Kit (ThermoFisher Scientific, K1622). Quantitative PCR was performed on a QuantStudio 7 Flex PCR System (Applied Biosystems, CA, USA) with UltraSYBR Mixture (CWBIO, CW2602M).
The following primers were used:
F-Wdr45b, 5ʹ-CACAGAGGTCAAGGCTGTCA-3ʹ;
R-Wdr45b, 5ʹ-ACTGTTGGGACACAGGACACAG-3ʹ;
F-Wdr45, 5ʹ-CACCTTCACCAAGCCAGTG-3ʹ;
R-Wdr45, 5ʹ-AGCTGCTTCTCCAGGCTTG-3ʹ;
F-Sqstm1, 5ʹ-GCTGCCCTATACCCACATCT-3ʹ;
R-Sqstm1, 5ʹ-CGCCTTCATCCGAGAAAC-3ʹ;
F-Actb, 5ʹ-CTGGCTCCTAGCACCATGAAGAT-3ʹ;
R-Actb, 5ʹ-GGTGGACAGTGAGGCCAGGAT-3ʹ.
Transmission electron microscopy
Mice were anaesthetized and transcardially perfused, then the DCN region was isolated from cerebellar tissues and post-fixed with 2.5% glutaraldehyde overnight at 4°C. After post-fixation in 1% osmium tetroxide and 0.05% potassium ferrocyanide for 60 min, samples were placed successively in thiocarbohydrazide solution for 30 min at room temperature, 1% osmium tetroxide in ddH2O for 60 min at room temperature, and 1% uranyl acetate (aqueous) at 4°C overnight. After dehydration with a graded series of ethanol, the tissues were embedded in epoxy resin using EMBed-812 Embeding Kit (Electron Microscopy Sciences, 14120). Ultrathin sections were stained with uranyl acetate and lead citrate, then observed under a SPIRIT electron microscope (FEI, Hillsboro, USA).
Statistical analysis
The statistical parameters, including n, SD and SEM, are described in the figure legends. Statistical significance was calculated by one-way ANOVA analysis. A P value less than 0.05 was considered significant.
Funding Statement
This work was supported by the National Natural Science Foundation of China [31630048]; National Natural Science Foundation of China [31671430]; National Natural Science Foundation of China [31561143001]; the Orphan Disease Center’s Million Dollar Bike Ride pilot grant program [MDBR–18–104–BPAN]
Acknowledgments
We are grateful to Dr. Isabel Hanson for editing work. This work was supported by the National Natural Science Foundation of China (NSFC) (31671430 to Y.G.Z. and 31561143001, 31630048 to H.Z.) and the Orphan Disease Center’s Million Dollar Bike Ride pilot grant program (MDBR–18–104–BPAN, MDBR-19-101-BPAN to H.Z.).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary materials:
The supplementary material for this article can be accessed here.
References
- [1].Xie Z, Klionsky DJ.. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–1109. [DOI] [PubMed] [Google Scholar]
- [2].Nakatogawa H, Suzuki K, Kamada Y, et al. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009;10:458–467. [DOI] [PubMed] [Google Scholar]
- [3].Kihara A, Noda T, Ishihara N, et al. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001;152:519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Reggiori F, Tucker KA, Stromhaug PE, et al. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev Cell. 2004;6:79–90. [DOI] [PubMed] [Google Scholar]
- [5].Dove SK, Piper RC, McEwen RK, et al. Svp1p defines a family of phosphatidylinositol 3,5-bisphosphate effectors. Embo J. 2004;23:1922–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hegedűs K, Takáts S, Boda A, et al. The Ccz1-Mon1-Rab7 module and Rab5 control distinct steps of autophagy. Mol Biol Cell. 2016;27:3132–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gao J, Langemeyer L, Kümmel D, et al. Molecular mechanism to target the endosomal Mon1-Ccz1 GEF complex to the pre-autophagosomal structure. Elife. 2018;7:pii: e31145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bas L, Papinski D, Licheva M, et al. Reconstitution reveals Ykt6 as the autophagosomal SNARE in autophagosome-vacuole fusion. J Cell Biol. 2018;217:3656–3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lu Q, Yang P, Huang X, et al. The WD40 repeat PtdIns(3)P-binding protein EPG-6 regulates progression of omegasomes to autophagosomes. Dev Cell. 2011;21:343–357. [DOI] [PubMed] [Google Scholar]
- [10].Polson HE, de Lartigue J, Rigden DJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates lc3 lipidation. Autophagy. 2010;6:506–522. [DOI] [PubMed] [Google Scholar]
- [11].Zhao YG, Sun L, Miao G, et al. The autophagy gene Wipi4 regulates learning and memory function and axonal homeostasis. Autophagy. 2015;11:881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nikoletopoulou V, Papandreou ME, Tavernarakis N. Autophagy in the physiology and pathology of the central nervous system. Cell Death Differ. 2015;22:398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Haack TB, Hogarth P, Kruer MC, et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am J Hum Genet. 2012;91:1144–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hayflick SJ, Kruer MC, Gregory A, et al. β-Propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. Brain. 2013;136:1708–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Saitsu H, Nishimura T, Muramatsu K, et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat Genet. 2013;45:445–449. [DOI] [PubMed] [Google Scholar]
- [17].Anazi S, Maddirevula S, Faqeih E, et al. Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Mol Psychiatry. 2017;22:615–624. [DOI] [PubMed] [Google Scholar]
- [18].Najmabadi H, Hu H, Garshasbi M, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63. [DOI] [PubMed] [Google Scholar]
- [19].Suleiman J, Allingham-Hawkins D, Hashem M, et al. WDR45B-related intellectual disability, spastic quadriplegia, epilepsy, and cerebral hypoplasia: A consistent neurodevelopmental syndrome. Clin Genet. 2018;93:360–364. [DOI] [PubMed] [Google Scholar]
- [20].Malenka RC. The long-term potential of LTP. Nat Rev Neurosci. 2003;4:923–926. [DOI] [PubMed] [Google Scholar]
- [21].Packard MG, Cahill L. Affective modulation of multiple memory systems. Curr Opin Neurobiol. 2001;11:752–756. [DOI] [PubMed] [Google Scholar]
- [22].Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. [DOI] [PubMed] [Google Scholar]
- [23].Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Dooley HC, Razi M, Polson HE, et al. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12–5–16L1. Mol Cell. 2014;55:238–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhao YG, Zhang H. Autophagosome maturation: an epic journey from the ER to lysosomes. J Cell Biol. 2018;218:757–770. pii: jcb.201810099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Feng YC, He D, Yao ZY, et al. The machinery of macroautophagy. Cell Res. 2014;24:24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cebollero E, van der Vaart A, Zhao M, et al. Phosphatidylinositol-3-phosphate clearance plays a key role in autophagosome completion. Curr Biol. 2012;22:1545–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wu Y, Cheng S, Zhao H, et al. PI3P phosphatase activity is required for autophagosome maturation and autolysosome formation. EMBO Rep. 2014;15:973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. [DOI] [PubMed] [Google Scholar]
- [30].Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. [DOI] [PubMed] [Google Scholar]
- [31].Zhao YG, Zhao H, Miao L, et al. The p53-induced gene Ei24 is an essential component of the basal autophagy pathway. J Biol Chem. 2012;287:42053–42063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Saxena S, Caroni P. Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron. 2011;71:35–48. [DOI] [PubMed] [Google Scholar]
- [33].Wang Z, Miao G, Xue X, et al. The vici syndrome protein EPG5 is a Rab7 effector that determines the fusion specificity of autophagosomes with late endosomes/lysosomes. Mol Cell. 2016;63:781–795. [DOI] [PubMed] [Google Scholar]
- [34].Zhao H, Zhao YG, Wang X, et al. Mice deficient in Epg5 exhibit selective neuronal vulnerability to degeneration. J Cell Biol. 2013;200:731–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Miao G, Zhao YG, Zhao H, et al. Mice deficient in the vici syndrome gene Epg5 exhibit features of retinitis pigmentosa. Autophagy. 2016;12:2263–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cullup T, Kho AL, Dionisi-Vici C, et al. Recessive mutations in EPG5 cause vici syndrome, a multisystem disorder with defective autophagy. Nat Genet. 2013;45:83–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.