

Summary:
Photoinduced electron transfer (PET) is a phenomenon wherein the absorption of light by a chemical species provides an energetic driving force for an electron transfer reaction.1–4 This mechanism is relevant in many areas of chemistry, including the study of natural and artificial photosynthesis, photovoltaics, and photosensitive materials. In recent years, research in the area of photoredox catalysis has leveraged PET for the catalytic generation of both neutral and charged organic free radical species. These technologies have enabled a wide range of previously inaccessible chemical transformations and have seen widespread utilization in both academic and industrial settings. These reactions are often catalyzed by visible-light absorbing organic molecules or transition-metal complexes of ruthenium, iridium, chromium, or copper.5,6 While a wide variety of closed shell organic molecules have been shown to behave as competent electron transfer catalysts in photoredox reactions, there are only limited reports of PET reactions involving neutral organic radicals as an excited state donor or acceptor. This is perhaps somewhat unsurprising in light of previously reported doublet excited state lifetimes for neutral organic radicals, which are typically several orders of magnitude shorter than singlet lifetimes for known transition metal photoredox catalysts.7–11 Herein we document the discovery, characterization, and reactivity of a neutral acridine radical with a maximum excited state oxidation potential of −3.36 V vs. SCE: significantly more reducing than elemental lithium and marking it as one of the most potent chemical reductants reported.12 Spectroscopic, computational, and chemical studies indicate that the formation of a twisted intramolecular charge transfer species enables the population of higher energy doublet excited states, leading to the observed potent photoreductant behavior. We demonstrate that this catalytically-generated PET catalyst facilitates several chemical reactions that typically require alkali metal reductants and bodes well for the adoption of this system in additional organic transformations requiring dissolving metal reductants.
Our lab, as well as others, have published numerous examples highlighting the diverse reactivity of acridinium salts, such as Mes-Acr+BF4-, as photooxidation catalysts in the excited state (*Mes-Acr+; Figure 1A).13 Upon absorption of visible light, the corresponding excited state of the acridinium salt is populated and may be quenched via electron transfer from an electrochemically-matched substrate, resulting in the formation of an acridine radical (Mes-Acr•; Figure 1A). In past work utilizing acridinium photoredox catalysts, this radical is typically oxidized to regenerate the parent acridinium and close a catalytic cycle. During previous mechanistic studies conducted by our laboratory, it was noted that solutions of Mes-Acr• generated via reduction of Mes-Acr+BF4- with cobaltocene were indefinitely stable under oxygen-free conditions and possessed two major absorption features (350–400 nm and 450–550 nm; Figure 1B).14 These observations led us to explore the photophysical behavior of this radical, with a focus on identifying potential PET behavior. Previous studies have detailed the in situ generation and excitation of stable cation radical and anion radical species and their use in catalytic reactions,15–19 indicating the potential feasibility of this strategy and prompting our studies of the photophysical behavior of Mes-Acr•.
Figure 1: Mechanistic studies on Mes-Acr radical.
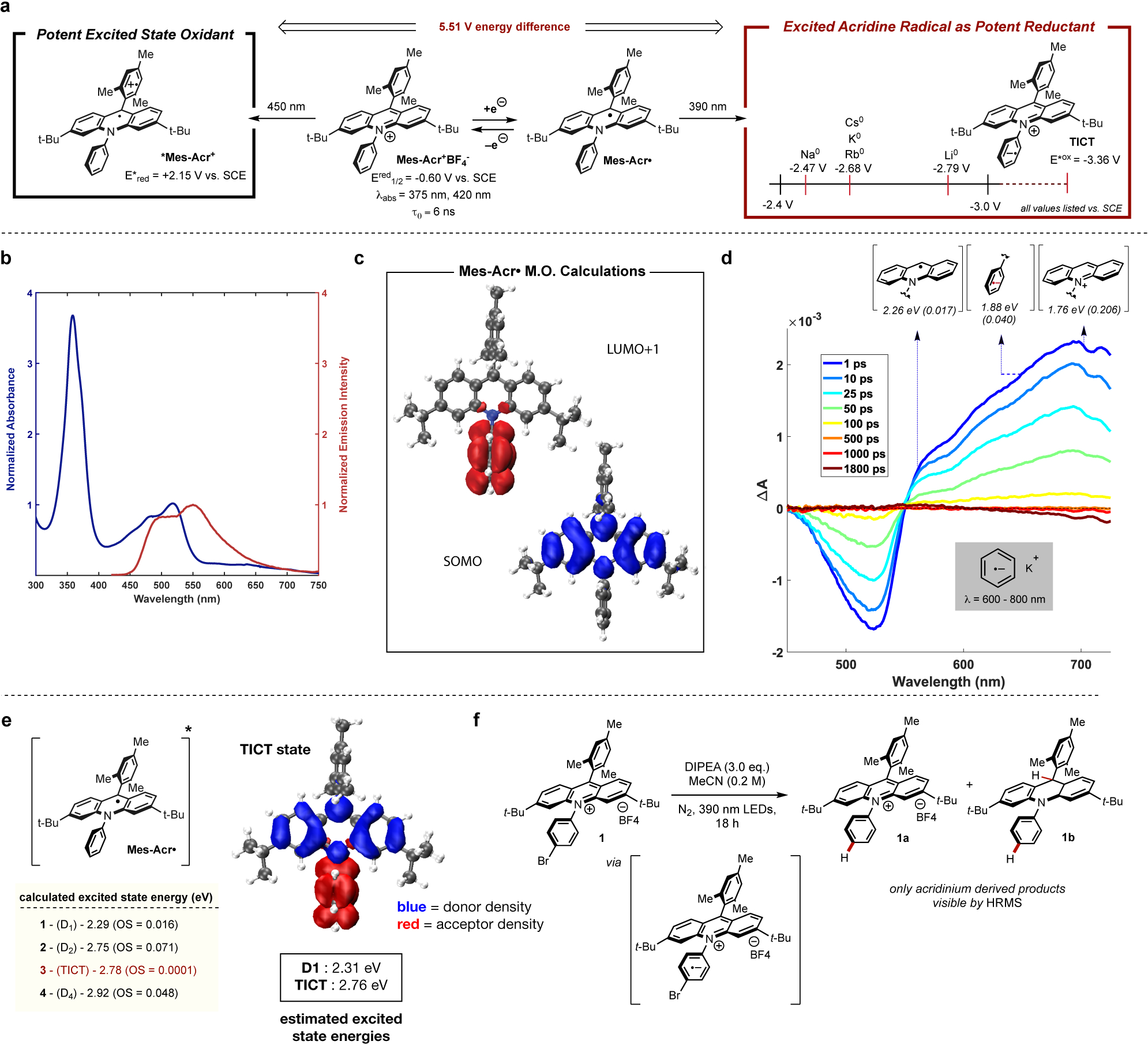
a, Reduction potential of various elemental alkali metals compared to peak reducing potential of Mes-Acr•, b, Absorbance and emission (excitation = 400 nm) profiles for Mes-Acr• in MeCN (5 mM, 1 mm path length) c, SOMO and LUMO +1 visualizations for Mes-Acr•. d, Transient absorption spectra of Mes-Acr• (2.5 mM, THF, 1 mm path length) collected with a 250 fs pump pulse centered at 400 nm e, Excited state energies calculated using SRSH-PCM/TD-DFT method for Mes-Acr• and frontier orbital plot showing donor and acceptor density for TICT excited state. f, Debromination reaction of brominated acridinium derivative giving circumstantial evidence for TICT state. Mes = mesityl; DIPEA = N,N-diisopropylethylamine; t-Bu = tert-butyl.
Upon investigation of the excited state dynamics of Mes-Acr•, we found that there are two main excited states, tentatively assigned as a lower energy doublet (D1) and higher energy twisted intramolecular charge transfer state (TICT). The excited state energy for the doublet excited state of Mes-Acr• is estimated by averaging the energies of the lowest energy absorption maximum and the highest energy emission observed upon 484 nm excitation. The energy of the proposed higher order excited state is estimated by averaging the energies of the emission maximum near 490 nm and the maximum of the corresponding excitation spectrum monitored at this wavelength (see SI for full details of excited state energy calculations). Estimation of excited state energies in this fashion gives values of 2.31 eV for the energy of the proposed D1 excited state and 2.76 eV for the corresponding higher energy excited state (Figure 1E). Using the known electrochemical potential of Mes-Acr•20 the excited state oxidation potentials of these states were estimated to be −2.91 V vs. SCE and −3.36 V vs. SCE, respectively. To our knowledge, these values represent some of the most negative excited state oxidation potentials reported for an organic molecule.
Before we proceed to discuss the calculated excited state energies, we consider the key orbitals involved in the low-lying excited states. We find that the singly occupied molecular orbital (SOMO) density is localized on acridine core and the lowest unoccupied MO (LUMO+1) is localized on the N-phenyl ring of Mes-Acr• (Figure 1C). Based on this observation of small spatial overlap between these two orbitals, we expect to find a relatively low lying excitation of an intramolecular CT (charge transfer) state. To further probe the excited state behavior of Mes-Acr•, transient absorption experiments were performed (Figure 1D). At early pump-probe delay times in THF, the ground state of Mes-Acr• is bleached (ΔA < 0) and excited state absorbance resonances (ΔA > 0) with maxima at 550 nm and ~650 nm are observed. Aromatic radical anions are known to exhibit broad absorbances in the 600–800 nm range as is aqueous solvated electron.21–23 The observed excited state absorbance signal at ~550 nm also matches the absorbance profile expected for a general acridine exitonic structure. Simple first-order decay to the ground state occurs after ~100 ps, matching well with previously reported values for excited state lifetimes of organic radicals. TD-DFT calculations indicate that other red-shifted absorptions present are well matched with energies calculated for a general acridinium structure. These spectral features support the formation of a charge transfer state possessing both aromatic radical anion and acridinium features, as expected for the proposed TICT state.
To better understand the effect of rotation of the N-phenyl ring on the excited state energetics of the acridine radical, we employ the recently reported polarization consistent TD-DFT based framework for obtaining excited state energies of solvated molecular systems. The approach addresses dielectric polarization consistently by invoking the same dielectric constant in the screened range separated hybrid (SRSH) functional parameters and in the polarizable continuum mode (PCM). SRSH-PCM was benchmarked well in calculating CT state energies of solvated donor-acceptor complex and in analyzing the spectral trends of several pigments with increased accuracy where conventional TD-DFT calculations fail to reproduce the observed trends (see SI for full computational details).24–27
The calculated doublet excited state energies for Mes-Acr• agree very well (within 0.1 eV) with values estimated via spectroscopic measurements for both the absorption and emission spectra (Figure 1E). The lowest energy calculated D1 state, possessing an excited state energy of 2.29 eV, agrees with the experimentally determined D1 value of 2.31 eV. Additionally, two excited states possessing significant charge transfer character were identified and the corresponding energies were calculated to be 2.75 and 2.78 eV, matching closely with estimated spectroscopic values for the proposed TICT state energy of 2.76 eV. As such, the identified D1 (2.29 eV) state is assigned as an untwisted exitonic state, while the calculated 2.78 eV state is assigned as a TICT state. These excited state energy values also correspond well with previously reported excited state energies for neutral radical species.9 Additionally, visualizations of the geometries of the corresponding TICT state indicate significant rotation of the N-phenyl ring (36 degrees) relative to the more planarized geometry of the D1 state, providing further evidence of the profound effects of N-phenyl rotation on excited state energy.
With the electronic and excited state behavior of Mes-Acr• elucidated, we sought to utilize this species as a catalytic reductant in a photoredox manifold. Previous work in reductive photoredox catalysis has established the reduction of aryl halides as a common benchmark reaction for these types of transformations.15,28–30 Furthermore, the extremely potent reducing behavior of the acridine radical should enable the reduction of a wide range of electronically diverse substrates. Diisopropylamine (DIPEA) was identified as a suitable single electron reductant for the generation of Mes-Acr• from Mes-Acr+BF4- in situ. Following excitation, Mes-Acr+BF4- undergoes single electron reduction via electron transfer from DIPEA, generating the desired radical Mes-Acr•. To chemically probe the possibility of charge transfer to the N-phenyl ring, brominated acridinium 1 was prepared. In the presence of 3 eq. DIPEA, 1 was completely converted to a mixture of debrominated acridinum 1a and hydroacridine 1b following irradiation at 390 nm for 18 hours (Figure 1F). As aryl halide radical anions are known to quickly fragment to yield the corresponding aryl radicals, this experiment is indicative of the formation of radical anion character localized on the N-phenyl ring during excitation.
To evaluate the competency of this radical species as a catalytic reductant, conditions for the reductive dehalogenation of aryl halides were developed (Figure 2A). A variety of both electron-rich (6–13) and electron-poor (14, 15) aryl bromides afforded the desired hydrodebrominated products in good to excellent yields (NMR yields of products taken using HMDSO as an internal standard). It is of note that reductively recalcitrant aryl chlorides also participated efficiently in this reaction, in contrast to previously reported methods which are only effective for electron-poor (under visible light irradiation) or moderately electron rich aryl chlorides (under UVA irradiation).31,32 A variety of both electron-donating (16–20) and electron-withdrawing (21–24) substituents were tolerated, with only slightly reduced yields in the case of electron-poor substrates. Substrates bearing ketone (30), carboxylic acid (31), and alcohol (28) functionalities all afforded the desired hydrodechlorinated products in good to excellent yield. Medicinally relevant pyridine (25, 26) and aryl carbamate (27) derivatives were also efficient substrates for this transformation. When substrate (23), which bears a trifluoromethyl substituent, was subjected to the reaction conditions, partial hydrodefluorination (5%) to yield the corresponding difluoromethyl derivative was observed in addition to hydrodechlorination. In all other examples, no Birch-type products resulting from overreduction are detected. The bis-reduction of polyhalogenated compounds (9a) and (9b) gave the corresponding bis-hydrodebromination (9) and bis-hydrodechlorination products in 58% yield and 46% yield, respectively. For compound 9b, 49% yield of the product resulting from mono-hydrodechlorination (9c) was observed in addition to the fully dechlorinated product.
Figure 2: Reductive dehalogenation of aryl halides enabled by acridine radical photoredox catalysis.
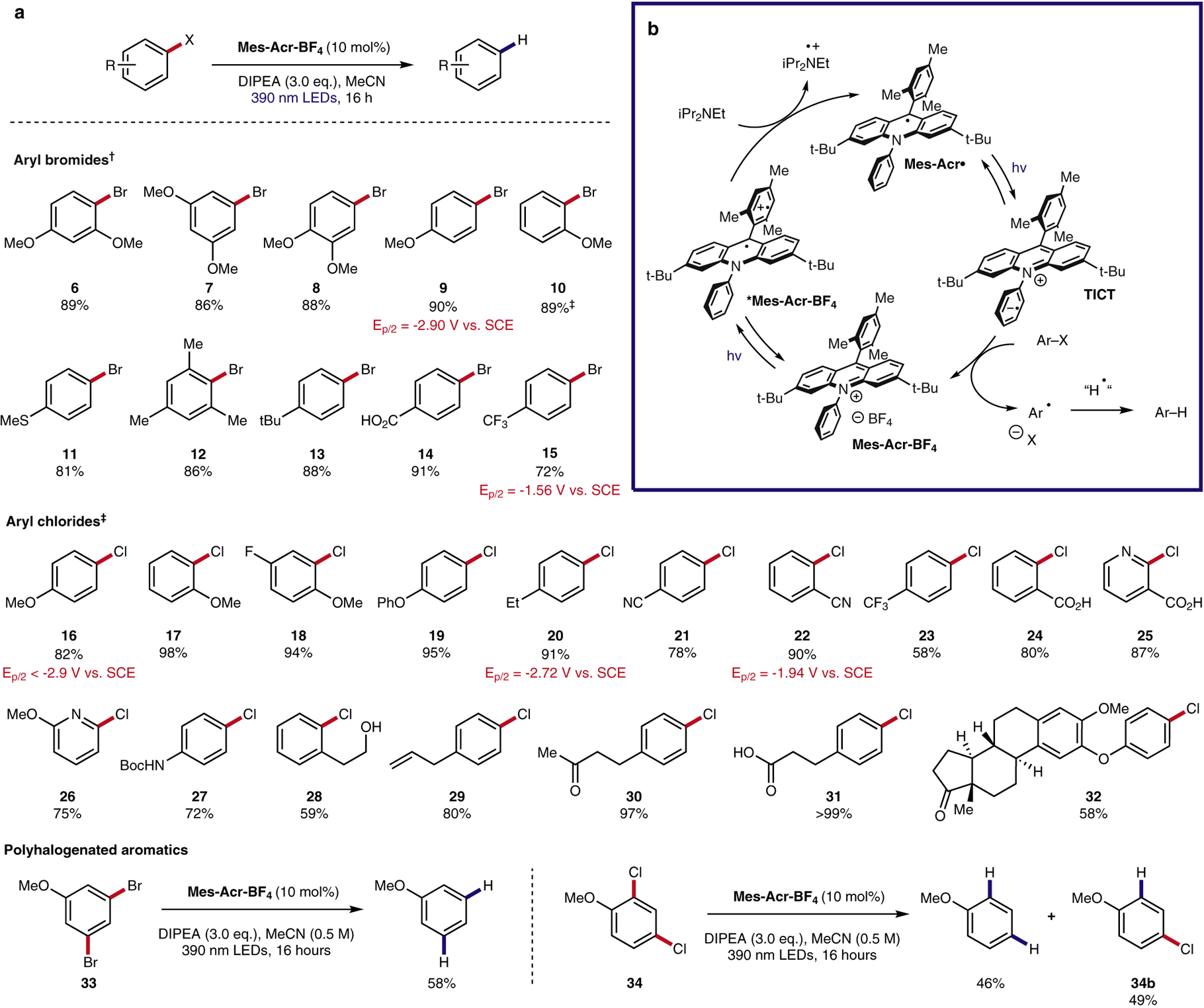
a, Reaction scope. b, proposed mechanism. †0.3 M reaction concentration. ‡0.5 M reaction concentration.
Based on prior work in reductive dehalogenation, the following mechanism is proposed (Figure 2B). Following excitation, Mes-Acr+BF4- engages in single electron transfer with the tertiary amine reductant (DIPEA), generating Mes-Acr• and the corresponding amine cation radical. Mes-Acr• is then excited by 390 nm light, populating a combination of highly reducing Dn/TICT excited states, and undergoes electron transfer with an electronically matched aryl halide generating an arene radical anion and reforming Mes-Acr+BF4-. The arene radical anion then fragments yielding an aryl radical. The resulting aryl radical abstracts a hydrogen atom from the amine cation radical, yielding the desired product as well as the corresponding iminium salt. Deuterium labeling studies confirmed the amine cation radical as the primary source of hydrogen atoms in this system (see SI – no deuterium incorporation in MeCN-d3).
The reductive detosylation of amines was identified as another possible transformation, which may be facilitated by Mes-Acr• (Figure 3). Typically, strong acid, dissolving metal (Li/Mg), or low-valent transition metal reductions are employed in detosylation reactions.33–35 A variety of electronically diverse tosylated aniline derivatives were smoothly converted to the desired free anilines in moderate to excellent yield. Interestingly, substrates containing aryl halides were tolerated under the reaction conditions. As this reaction is conducted at a significantly lower concentration of substrate compared to the reductive dehalogenation method (0.1 M vs. 0.5 M), the observed lack of aryl halide reduction may be a function of concentration. Esters (43, 73), free carboxylic acids (44), ketones (48), and free alcohols (58) were tolerated under the reaction conditions, showcasing the high functional group tolerance of this method relative to methods relying on harsh dissolving metal conditions. Benzylic (52) and secondary alkyl amines (45, 53, 65–68) were efficient substrates for this transformation as well. Medicinally-relevant heterocycles, including pyridines (59), indoles (58), pyrroles (62), pyrrolidines (67), indazoles (63), benzimidazoles (64), and morpholines (65) were deprotected in good to excellent yields, with no reduction of the aromatic system observed in all cases. Of note is the ability of this method to chemoselectively and efficiently deprotect tosyl-amines over mesyl protected-amines, as shown by the reaction of substrate 51, yielding the desired detosylation product in 61% yield with no observed cleavage of the mesyl-amine. Additionally, the reaction performed well on 1.28 g scale, with substrate 64 undergoing the desired detosylation in 92% yield when conducted in a standard round bottom flask irradiated with LED lamps (see SI for experimental details).
Figure 3: Scope of reductive detosylation catalyzed by Mes-Acr•.

1H NMR yields using DMSO or pyrazine as internal standard. †48 h reaction time. ‡0.5 M reaction concentration. Ts = p-toluenesulfonyl
In conclusion, an acridine radical generated in situ from single electron reduction of an acridinium derivative may act as potent single electron reductant upon excitation with 390 nm light. Spectroscopic and computational investigations indicate the formation of at least two distinct excited states, one of which may be characterized as a TICT state. The development of chemoselective dehalogenation and desulfonylation reactions utilizing Mes-Acr• compliment the well-known oxidative chemistry associated with acridinium salts and highlight the potential for the development of other types of reactions based on excitation of organic radicals.
Supplementary Material
Acknowledgements:
This work was supported in part by the National Institutes of Health (NIGMS) Award No. R01 GM120186 (D.A.N.). L.F.W. was supported by the National Natural Science Foundation of China (21801011) and the International Postdoctoral Exchange Fellowship Program (20180033). O.F.W. and A.M. were supported by the National Science Foundation under CHE-1763207. Photophysical measurements were performed in the AMPED EFRC Instrumentation Facility established by the Alliance for Molecular PhotoElectrode Design for Solar Fuels (AMPED), an Energy Frontier Research Center (EFRC) funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award DE-SC0001011. B.D.D acknowledges support for this project by the Department of Energy (DOE)- Basic Energy Sciences through the Chemical Sciences Geosciences and Biosciences Division, through grant no. DE- SC0016501.
Footnotes
Data Availability
The authors declare that the data supporting the findings of this study are available within the paper and its supplementary information.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Competing Interests: The authors declare no competing interests.
References:
- 1.Mattay J Photoinduced Electron Transfer in Organic Synthesis. Synthesis 1989, 233–252 (1989). [Google Scholar]
- 2.Bauer A, Westkämper F, Grimme S & Bach T Catalytic enantioselective reactions driven by photoinduced electron transfer. Nature 436, 1139–1140 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Fox MA Photoinduced Electron Transfer. Photochem. Photobiol 52, 617–627 (1990). [Google Scholar]
- 4.Fukuzumi S New development of photoinduced electron-transfer catalytic systems. Pure Appl. Chem 79, 981–991 (2007). [Google Scholar]
- 5.Romero NA & Nicewicz DA Organic Photoredox Catalysis. Chem. Rev 116, 10075–10166 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Prier CK, Rankic DA & MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 113, 5322–5363 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnston LJ Photochemistry of radicals and biradicals. Chem. Rev 93, 251–266 (1993). [Google Scholar]
- 8.Arnold BR, Scaiano JC & McGimpsey WG Electron-transfer quenching of excited diphenylmethyl radicals. J. Am. Chem. Soc 114, 9978–9982 (1992). [Google Scholar]
- 9.Scaiano JC, Tanner M & Weir D Exploratory study of the intermolecular reactivity of excited diphenylmethyl radicals. J. Am. Chem. Soc 107, 4396–4403 (1985). [Google Scholar]
- 10.Samanta A Bhattacharyya K, Das PK, Kamat PV, Weir D, Hug GL Quenching of excited doublet states of organic radicals by stable radicals. J. Phys. Chem 93, 3651–3656 (1989). [Google Scholar]
- 11.Weir D & Scaiano JC Substituent effects on the lifetime and fluorescence of excited diphenylmethyl radicals in solution. Chem. Phys. Lett 128, 156–159 (1986). [Google Scholar]
- 12.Scordilis-Kelley C Alkali Metal Reduction Potentials Measured in Chloroaluminate Ambient-Temperature Molten Salts. J. Electrochem. Soc 139, 694 (1992). [Google Scholar]
- 13.Margrey KA & Nicewicz DA A General Approach to Catalytic Alkene Anti-Markovnikov Hydrofunctionalization Reactions via Acridinium Photoredox Catalysis. Acc. Chem. Res 49, 1997–2006 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Romero NA & Nicewicz DA Mechanistic Insight into the Photoredox Catalysis of Anti-Markovnikov Alkene Hydrofunctionalization Reactions. J. Am. Chem. Soc 136, 17024–17035 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghosh I, Ghosh T, Bardagi JI & König B Reduction of aryl halides by consecutive visible light-induced electron transfer processes. Science 346, 725–728 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Connell TU, Fraser CL, Czyz ML, Smith ZM, Hayne DJ, Doeven EH, Agugiaro J, Wilson DJD, Adcock JL, Scully AD, Gómez DE, Barnett NW, Polyzos A & Francis PS The Tandem Photoredox Catalysis Mechanism of [Ir(ppy)2(dtb-bpy)]+ Enabling Access to Energy Demanding Organic Substrates. J. Am. Chem. Soc 141, 17646–17658 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Lu C, Fujitsuka M, Sugimoto A & Majima T Dual Character of Excited Radical Anions in Aromatic Diimide Bis(radical anion)s: Donor or Acceptor? J. Phys. Chem C 121, 4558–4563 (2017). [Google Scholar]
- 18.Christensen JA et al. Phenothiazine Radical Cation Excited States as Super-oxidants for Energy-Demanding Reactions. J. Am. Chem. Soc 140, 5290–5299 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Gumy J-C & Vauthey E Investigation of the Excited-State Dynamics of Radical Ions in the Condensed Phase Using the Picosecond Transient Grating Technique. J. Phys. Chem. A 101, 8575–8580 (1997). [Google Scholar]
- 20.Brancato G, Signore G, Neyroz P, Polli D, Cerullo G, Abbandonato G, Nucara L, Barone V, Beltram F & Bizzarri R Dual Fluorescence through Kasha’s Rule Breaking: An Unconventional Photomechanism for Intracellular Probe Design. J. Phys. Chem. B 119, 6144–6154 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Demchenko AP, Tomin VI & Chou P-T Breaking the Kasha Rule for More Efficient Photochemistry. Chem. Rev 117, 13353–13381 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Peng Z Wang Z, Huang Z, Liu S, Lu P & Wang Y Expression of anti-Kasha’s emission from amino benzothiadiazole and its utilization for fluorescent chemosensors and organic light emitting materials. J. Mater. Chem C 6, 7864–7873 (2018). [Google Scholar]
- 23.Scuppa S, Orian L, Donoli A, Santi S & Meneghetti M Anti-Kasha’s Rule Fluorescence Emission in (2-Ferrocenyl)indene Generated by a Twisted Intramolecular Charge-Transfer (TICT) Process. J. Phys. Chem. A 115, 8344–8349 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Romero NA, Margrey KA, Tay NE & Nicewicz DA Site-selective arene C-H amination via photoredox catalysis. Science 349, 1326–1330 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Bhandari S & Dunietz BD Quantitative Accuracy in Calculating Charge Transfer State Energies in Solvated Molecular Complexes Using a Screened Range Separated Hybrid Functional within a Polarized Continuum Model. J. Chem. Theory Comput 15, 4305–4311 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Song Y Schubert A, Maret E, Burdick RK, Dunietz BD, Geva E, Ogilvie JP Vibronic structure of photosynthetic pigments probed by polarized two-dimensional electronic spectroscopy and ab initio calculations. Chem. Sci 10, 8143–8153 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhandari S, Cheung MS, Geva E, Kronik L & Dunietz BD Fundamental Gaps of Condensed-Phase Organic Semiconductors from Single-Molecule Calculations using Polarization-Consistent Optimally Tuned Screened Range-Separated Hybrid Functionals. J. Chem. Theory Comput 14, 6287–6294 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Maiti B, Schubert A, Sarkar S, Bhandari S, Wang K, Li Z, Geva E, Twieg RJ & Dunietz BD, Enhancing charge mobilities in organic semiconductors by selective fluorination: a design approach based on a quantum mechanical perspective. Chem. Sci 8, 6947–6953 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shida T Electronic Absorption Spectra of Radical Ions. (Elsevier, 1988). [Google Scholar]
- 30.Kerzig C & Goez M Generating hydrated electrons through photoredox catalysis with 9-anthrolate. Phys. Chem. Chem. Phys 17, 13829–13836 (2015). [DOI] [PubMed] [Google Scholar]
- 31.Kerzig C, Guo X & Wenger OS Unexpected Hydrated Electron Source for Preparative Visible-Light Driven Photoredox Catalysis. J. Am. Chem. Soc 141, 2122–2127 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Poelma SO, Burnett GL, Discekici EH, Mattson KM, Treat NJ, Luo Y, Hudson ZM, Shankel SL, Clark PG, Kramer JK, Hawker CJ & Read de Alaniz J Chemoselective Radical Dehalogenation and C–C Bond Formation on Aryl Halide Substrates Using Organic Photoredox Catalysts. J. Org. Chem 81, 7155–7160 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Discekici EH Treat NJ, Poelma SO, Mattson KM, Hudson ZM, Luo Y, Hawker CJ & Read de Alaniz J A highly reducing metal-free photoredox catalyst: design and application in radical dehalogenations. Chem. Commun 51, 11705–11708 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Narayanam JMR, Tucker JW & Stephenson CRJ Electron-Transfer Photoredox Catalysis: Development of a Tin-Free Reductive Dehalogenation Reaction. J. Am. Chem. Soc 131, 8756–8757 (2009). [DOI] [PubMed] [Google Scholar]
- 35.Yin H, Jin Y, Hertzog JE, Mullane KC, Carroll PJ, Manor BC, Anna JM & Schelter EJ The Hexachlorocerate(III) Anion: A Potent, Benchtop Stable, and Readily Available Ultraviolet A Photosensitizer for Aryl Chlorides. J. Am. Chem. Soc 138, 16266–16273 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Poelma SO, Burnett GL, Discekici EH, Mattson KM, Treat NJ, Luo Y, Hudson ZM, Shankel SL, Clark PG, Kramer JK, Hawker CJ & Read de Alaniz J Chemoselective Radical Dehalogenation and C–C Bond Formation on Aryl Halide Substrates Using Organic Photoredox Catalysts. J. Org. Chem 81, 7155–7160 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Javorskis T & Orentas E Chemoselective Deprotection of Sulfonamides Under Acidic Conditions: Scope, Sulfonyl Group Migration, and Synthetic Applications. J. Org. Chem 82, 13423–13439 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Shohji N, Kawaji T & Okamoto S Ti(O-i-Pr)4/Me3SiCl/Mg-Mediated Reductive Cleavage of Sulfonamides and Sulfonates to Amines and Alcohols. Org. Lett 13, 2626–2629 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Alonso E, Ramón DJ & Yus M Reductive deprotection of allyl, benzyl and sulfonyl substituted alcohols, amines and amides using a naphthalene-catalysed lithiation. Tetrahedron 53, 14355–14368 (1997). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.