

Abstract
Genetic testing of probands in families with an initial diagnosis of autosomal dominant retinitis pigmentosa (adRP) usually confirms the diagnosis, but there are exceptions. We report results of genetic testing in a large cohort of adRP families with an emphasis on exceptional cases including X-linked RP with affected females; homozygous affected individuals in families with heterozygous, dominant disease; and independently segregating mutations in the same family. Genetic testing was conducted in more than 700 families with a provisional or probable diagnosis of adRP. Exceptions to the proposed mode of inheritance were extracted from our comprehensive patient and family database. In a subset of 300 well-characterized families with a probable diagnosis of adRP, 195 (70%) have dominant mutations in known adRP genes but 25 (8%) have X-linked mutations, 3 (1%) have multiple segregating mutations, and 3 (1%) have dominant-acting mutations in genes previously associated with recessive disease. It is currently possible to determine the underlying disease-causing gene and mutation in approximately 80% of families with an initial diagnosis of adRP, but 10% of “adRP” families have a variant mode of inheritance. Informed genetic diagnosis requires close collaboration between clinicians, genetic counselors, and laboratory scientists.
Keywords: Retinitis pigmentosa (RP), Autosomal dominant retinitis pigmentosa, Next-generation sequencing, Linkage mapping, X-linked retinitis pigmentosa, Semidominant inheritance, Prevalence of mutations
29.1. Introduction
Retinitis pigmentosa (RP) has a prevalence of approximately 1 in 3,100 and affects more than 1.5 million individuals worldwide (Haim 2002; Daiger et al. 2013). RP is extremely heterogeneous: mutations in more than 80 genes cause syndromic and non-syndromic forms of RP, more than 3,500 mutations have been described in these genes, and disease symptoms and progression are highly variable (Berger et al. 2010; Wright et al. 2010; Daiger et al. 2013; RetNet 2018). To date, mutations in 29 genes are known to cause autosomal dominant RP (adRP), and these genes are themselves highly heterogeneous (Daiger et al. 2014; RetNet 2018).
Our research focuses on genes and mutations causing adRP. Over the past 30 years, we have assembled a cohort of adRP families and applied a wide range of methods to detect the disease-causing mutation in each family, including linkage mapping and next-generation sequencing (NGS) (Sohocki et al. 2001; Sullivan et al. 2006; Bowne et al. 2011a; Koboldt et al. 2013; Wang et al. 2013). As more and more RP genes have been identified, the fraction of adRP families in which we can identify the underlying disease-causing gene and mutation has increased from a few percent to the majority of patients. However, a substantial fraction of families with a provisional diagnosis of adRP (5–10%) have an alternative or more complicated molecular diagnosis. To illustrate this problem, we provide several examples of “complicated” families selected from the larger set of adRP families.
29.2. Materials and Methods
29.2.1. Family Ascertainment and Clinical Characterization
Families in our studies are ascertained by clinical collaborators in Houston, the Retina Foundation of the Southwest, the Kellogg Eye Center, and other retinal genetics centers. Genetic testing is done in the Laboratory for Molecular Diagnosis of Inherited Eye Diseases, CLIA ID 45D0935007, Human Genetics Center, School of Public Health, the Univ. of Texas Health Science Center (UTHealth), Houston. Families are included in the AdRP Cohort if they have an initial diagnosis of adRP and three or more affected generations with affected females or two or more generations with male-to-male transmission.
The research adheres to tenets of the Declaration of Helsinki, and the studies were approved by the Committee for the Protection of Human Subjects at UTHealth and by human subjects review boards at participating institutions.
29.2.2. Next-Generation Sequencing (NGS)
Whole-exome and whole-genome NGS was done at The Genome Institute, Washington Univ., St. Louis (Bowne et al. 2011a). Retinal targeted-capture NGS was done in the DNA Diagnostic Laboratory, UTHealth, and in collaboration with Dr. Rui Chen, Dept. of Molecular and Human Genetics, Baylor College of Medicine, Houston (Wang et al. 2013; Bowne et al. 2015).
29.2.3. Linkage Mapping and Haplotype Analysis
Genotyping for linkage mapping was done at the Hussman Institute for Human Genomics, Univ. of Miami, using Affymetrix Genome-Wide Human SNP 6.0 array data, and at the UCLA Sequencing and Genotyping Center using an ABI High Density 5 cM STR marker set (Bowne et al. 2011b, 2016; Sullivan et al. 2014). Analysis was conducted using PLINK and Merlin. Haplotypes were determined by inspection and confirmed by segregation analysis.
29.3. Results
29.3.1. Background
The Laboratory for Molecular Diagnosis of Inherited Eye Diseases (LMDIED) in Houston is focused on finding genes and mutations causing autosomal dominant retinitis pigmentosa (adRP) and related diseases. Patients are enrolled and examined by clinical collaborators. Support is provided through several projects including FFB Research Centers, the NEI-NIH eyeGENE Network® (Sullivan et al. 2013), and the Texas 1000 Project (Daiger et al. 2016).
Currently, 2,300 families with inherited retinal disease are enrolled; of these, approximately 50% have a diagnosis of adRP. Samples are available from 4,600 members of these families; approximately 75% are affected, the rest are at-risk or unaffected family members for linkage testing. Among the families, a subset of 300 is designated as the AdRP Cohort and a further 400 are an adRP confirmation panel. The AdRP Cohort consists of families with a likely diagnosis of dominant disease based on clinical examination and pedigree. The confirmation panel has less stringent criteria. The following exceptional families are drawn from the AdRP Cohort.
29.3.2. X-Linked RP in Apparent adRP Families
Figure 29.1 is the pedigree of a family with a diagnosis of RP and apparent autosomal dominant inheritance (Mears et al. 2000). There are at least three generations of inheritance, and males and females are equally likely to be affected. However, there is no male-to-male transmission, which would exclude X-linked inheritance, and in retrospect, the affected females are less severely affected on average than the males. In fact, the family has X-linked RP with a mutation in RPGR. It is now clear that carrier females with X-linked RP mutations may be affected (Comander et al. 2015); however, the extent to which X-linked families may be misdiagnosed is striking. In the AdRP Cohort, at least 8% of families with a provisional diagnosis of adRP have X-linked RP (Churchill et al. 2013). Further, up to 15% of males with isolated (simplex) RP have RPGR or RP2 mutations (Branham et al. 2012). This may be the single most common diagnostic complication with RP.
Fig.29.1.
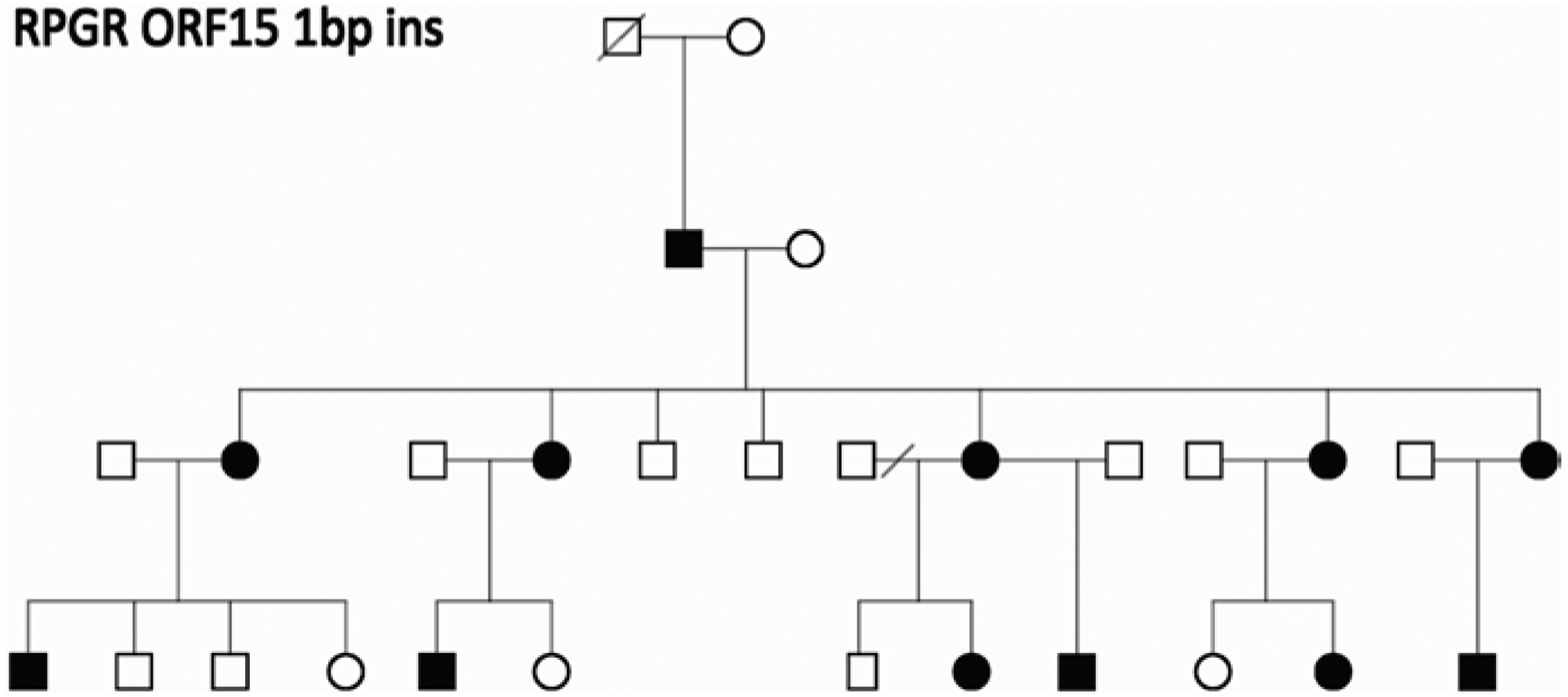
Pedigree of a family with a clinical diagnosis of RP and apparent autosomal dominant inheritance. The disease-causing mutation in the family is a 1 bp deletion in ORF15 of the X-linked RPGR gene (Mears et al. 2000)
29.3.3. Semidominant Inheritance
The expectation is that an autosomal mutation in a single family is either dominant acting or recessive acting, but not both. However, two families in the Cohort, Figs. 29.2 and 29.3, display both modes. Figure 29.2 is the pedigree of an adRP family with a Glu847Lys mutation in HK1 (Sullivan et al. 2014). Although consanguinity is not reported in the family, one individual, number 16, has two copies of the mutation and severe RP. Although he is homozygous for the mutation, the two associated haplotypes differ in flanking sequences, suggesting they descend from a distant ancestor. He has no other apparent clinical findings.
Fig.29.2.
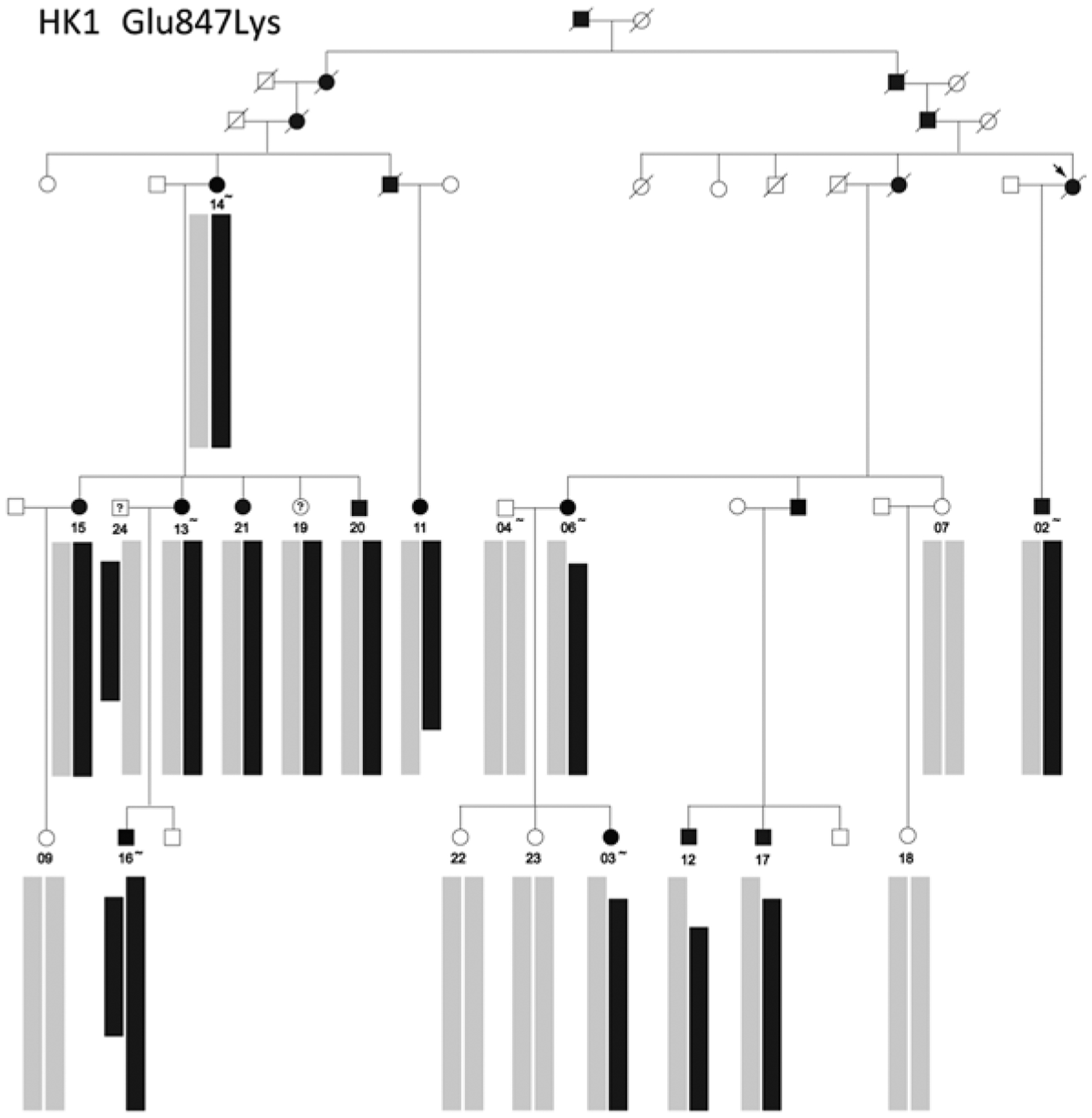
Pedigree and segregating haplotypes in a family with adRP caused by a Glu847Lys missense mutation in HK1 (Sullivan et al. 2014). Black bars show the shared haplotype segregating with the HK1 mutation on chromosome 10q22.1, and gray bars represent the unrelated haplotypes in trans. Individual 16 carries two copies of the mutation-associated haplotype but with different recombinant end points
Fig.29.3.
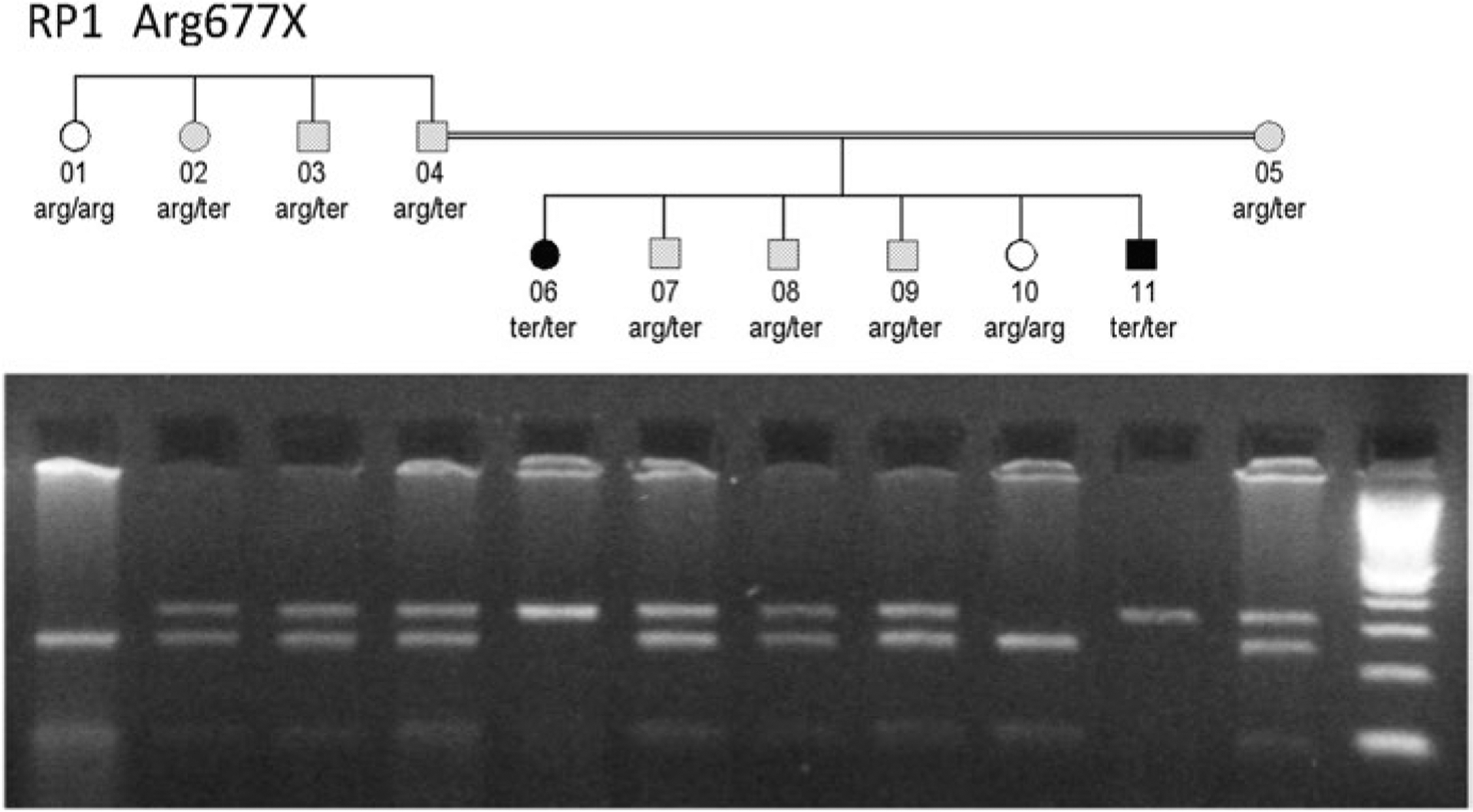
Pedigree of a nuclear family within a larger family with adRP caused by an RP1 Arg677X mutation (Sullivan et al. 1999). Parents 04 and 05 are related and both are heterozygous for the RP1 mutation. Individuals 07–09 inherited one copy of the mutation, and individuals 06 and 11 inherited two copies. Photograph below is a restriction digest showing segregation of the wild-type (lower band) and mutant (upper band) RP1 alleles of the individuals in the pedigree
Figure 29.3 is the pedigree of a nuclear family within a larger family with adRP caused by an RP1 Arg677X mutation (Sullivan et al. 1999). The parents, 04 and 05, are distantly related. Of the six children in the pedigree, three, 07–09, inherited one copy of the RP1 mutation; two, 06 and 07, inherited two copies; and 10 has wild-type RP1 only. Family members with one copy of the mutation have classical RP with onset in late adolescence, whereas individuals homozygous for the mutation have early-onset, rapidly progressing RP, consistent with Leber congenital amaurosis. Like the HK1 homozygote, the RP1 homozygotes did not show apparent non-ocular disease at the last examination.
This type of inheritance is semidominant, that is, a single allele causing both dominant and recessive disease but with more severe disease in the homozygote – in this case apparently limited to a single tissue.
29.3.4. Independently Segregating Mutations
Figure 29.4 is the pedigree of a family with apparent adRP but a possible non-penetrant individual (Wheaton et al. 2016). Targeted capture NGS, though, revealed mutations in two disease-causing genes, a dominant-acting RP1 Arg677X mutation, and compound heterozygous, recessive mutations in USH2A, Cys419Phe, and Glu767Serfs*21. This is notable for several reasons. First, if just the RP1 mutation had been reported, individual 3.3 would have been misdiagnosed. For this reason, it is wrong to assume that all affected individuals in a family share the same cause. Second, this is one example of many in which USH2A mutations are associated with RP alone, not Usher syndrome (Seyedahmadi et al. 2004). Finally, at least three families in our AdRP Cohort have multiple, independently segregating mutations, with additional families in the extended cohort: this is not a rare phenomenon.
Fig.29.4.
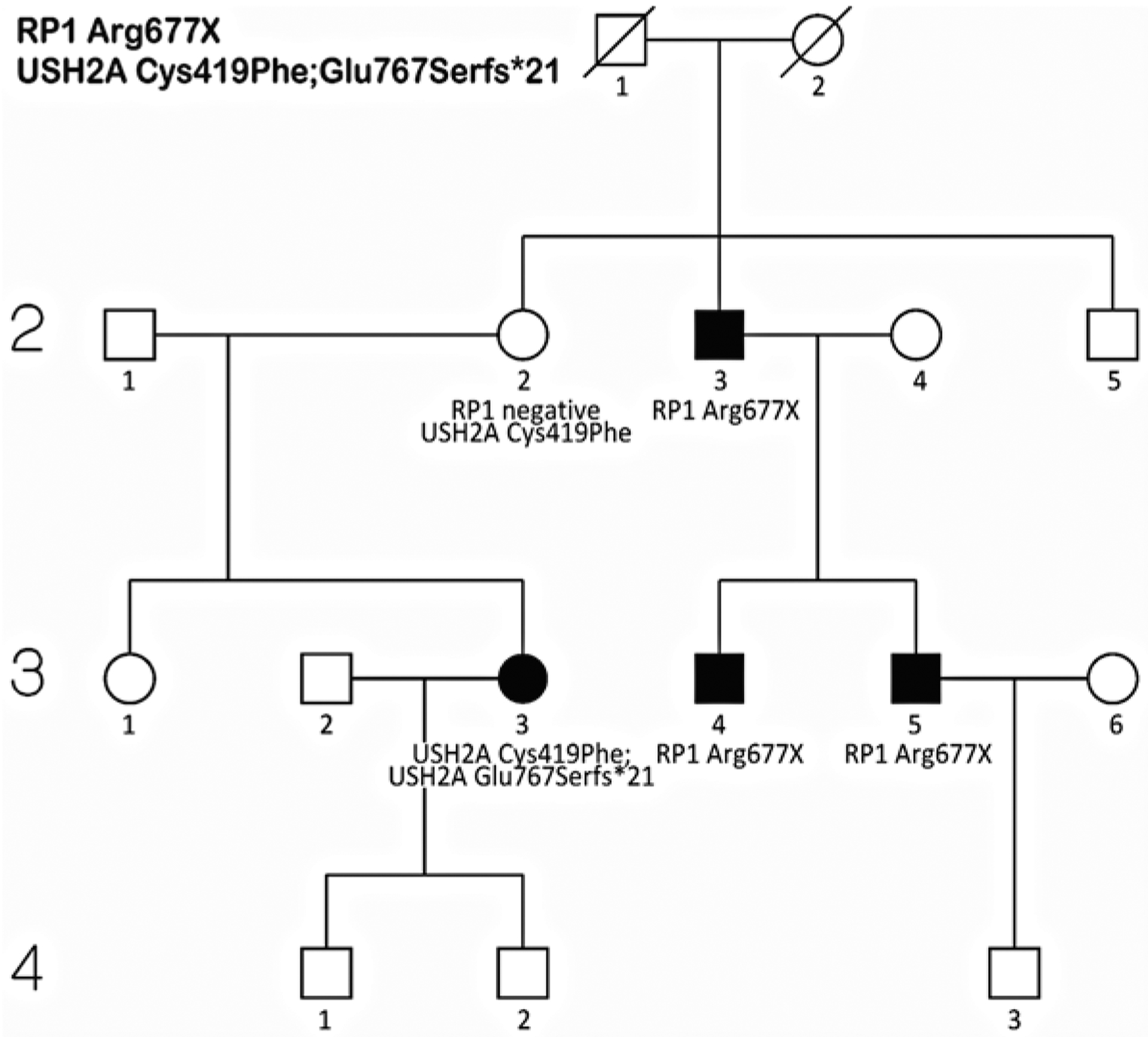
Pedigree of a family with a provisional diagnosis of adRP but an apparent non-penetrant individual, 2.2 (Wheaton et al. 2016). Molecular testing showed an autosomal dominant RP1 Arg677X mutation segregating in individuals 2.3, 3.4, and 3.5, whereas individual 3.3 is a compound heterozygote for USH2A [Cys419Phe];[Glu767Serfs*21] mutations. Individual 2.2 is a carrier of the USH2A Cys419Phe mutation but does not carry the RP1 mutation
29.4. Discussion and Conclusion
William Batson said “treasure your exceptions” (Bateson 1908). In genetics, and the biological sciences in general, exceptions provide insight into processes that would otherwise be inaccessible. For inherited retinal diseases, and RP in particular, families with exceptional modes of inheritance reveal new information about underlying mechanisms of disease. For example, the fact that specific mutations in RP1 and HK1 can be both dominant and recessive, with different degrees of retinal disease but no other obvious clinical consequences, shows that these mutations are not lethal and have dose-dependent effects but do not affect other tissues (Sullivan et al. 1999, 2014). From another perspective, though, the “treasure” is the affected families who participate in our research and expect reliable genetic information. From this perspective, the many exceptions to textbook models of inheritance for RP and related diseases mandate particular caution in diagnosis and interpretation of genetic findings.
In practice, evaluation of a patient or family with an inherited retinal disease can be divided into three overlapping stages: clinical characterization of the patient and available family members, family history including pedigree, and molecular testing. Because of the complexity of inherited retinal diseases, it is often necessary to reconcile clinical diagnosis, family diagnosis, and molecular diagnosis. This can best be done in centers that include retinal specialists, genetic counselors, molecular biologists, and other experts. Until we have a much better understanding of the complexity of RP and related diseases, diagnosis and treatment must be approached with caution and an open mind.
Acknowledgments
This work was supported by NIH grants EY007142 and EY09076, a Wynn-Gund TRAP Award, and grants from the Foundation Fighting Blindness, the William Stamps Farish Fund, and the Hermann Eye Fund.
Contributor Information
Stephen P. Daiger, Human Genetics Center, School of Public Health, The University of Texas Health Science Center (UTHealth), Houston, TX, USA Ruiz Department of Ophthalmology and Visual Science, UTHealth, Houston, TX, USA.
Kari Branham, Kellogg Eye Center, University of Michigan, Ann Arbor, MI, USA.
John R. Heckenlively, Kellogg Eye Center, University of Michigan, Ann Arbor, MI, USA
David G. Birch, The Retina Foundation of the Southwest, Dallas, TX, USA
References
- Bateson W (1908) The methods and scope of genetics: an inaugural lecture delivered 23 October 1908. Cambridge University Press, Fetter Lane, E.C, London, UK [Google Scholar]
- Berger W, Kloeckener-Gruissem B, Neidhardt J (2010) The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res 29:335–375 [DOI] [PubMed] [Google Scholar]
- Bowne SJ, Sullivan LS, Koboldt DC et al. (2011a) Identification of disease-causing mutations in autosomal dominant retinitis pigmentosa (adRP) using next-generation DNA sequencing. Invest Ophthalmol Vis Sci 52:494–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowne SJ, Humphries MM, Sullivan LS et al. (2011b) A dominant mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Euro. J Hum Genet 19:1074–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowne SJ, Sullivan LS, Wheaton DK et al. (2015) Retinal targeted-capture next generation sequencing and CLIA confirmation in a representative range of patients with inherited retinal degeneration. Invest Ophthalmol Vis Sci E-Abstr 56:1241 [Google Scholar]
- Bowne SJ, Sullivan LS, Wheaton DK et al. (2016) North Carolina macular dystrophy (MCDR1) caused by a novel tandem duplication of the PRDM13 gene. Mol Vis 22:1239–1247 [PMC free article] [PubMed] [Google Scholar]
- Branham K, Othman M, Brumm M et al. (2012) Mutations in RPGR and RP2 account for 15% of males with simplex retinal degenerative disease. Invest Ophthalmol Vis Sci 53:8232–8237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill JD, Bowne SJ, Sullivan LS et al. (2013) Mutations in the X-linked retinitis pigmentosa genes RPGR and RP2 found in 8.5% of families with a provisional diagnosis of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci 54:1411–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comander J, Weigel-DiFranco C, Sandberg MA et al. (2015) Visual function in carriers of X-linked retinitis pigmentosa. Ophthalmology 122:1899–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiger SP, Sullivan LS, Bowne SJ (2013) Genes and mutations causing retinitis pigmentosa. Clin Genet 84:132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiger SP, Bowne SJ, Sullivan LS (2014) Genes and mutations causing autosomal dominant retinitis pigmentosa. Cold Spring Harb Perspect Med October 2015;5:a017129 10.1101/cshperspect.a017129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiger SP, Bowne SJ, Sullivan LS et al. (2016) Retinal targeted-capture next generation sequencing and CLIA confirmation in patients with a range of inherited retinal degeneration. Invest Ophthalmol Vis Sci 57:e1417 [Google Scholar]
- Haim M (2002) Epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol Scand Suppl 233:1–34 [DOI] [PubMed] [Google Scholar]
- Koboldt DC, Larson DE, Sullivan LS et al. (2013) Linkage mapping using whole-exome next-generation sequencing in families with autosomal dominant retinitis pigmentosa Invest Ophthalmol Vis Sci E-Abstract, ARVO Annual Meeting [Google Scholar]
- Mears AJ, Hiriyanna S, Vervoort R et al. (2000) Remapping of the RP15 locus for X-linked cone-rod degeneration to Xp11.4-p21.1, and identification of a de novo insertion in the RPGR exon ORF15. Am J Hum Genet 67:1000–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- RetNet (2016) The Retinal Information Network, http://www.sph.uth.tmc.edu/RetNet/. In: Daiger, PhD Stephen P., Administrator, The Univ. of Texas Health Science Center at Houston [Google Scholar]
- Seyedahmadi BJ, Rivolta C, Keene JA et al. (2004) Comprehensive screening of the USH2A gene in Usher syndrome type II and non-syndromic recessive retinitis pigmentosa. Exp Eye Res 79:167–173 [DOI] [PubMed] [Google Scholar]
- Sohocki MM, Daiger SP, Bowne SJ et al. (2001) Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum Mutat 17:42–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LS, Heckenlively JR, Bowne SJ et al. (1999) Mutations in a novel retina-specific gene cause autosomal dominant retinitis pigmentosa. Nat Genet 22:255–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LS, Bowne SJ, Birch DG et al. (2006) Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: a screen of known genes in 200 families. Invest Ophthalmol Vis Sci 47:3052–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LS, Bowne SJ, Reeves MJ et al. (2013) Prevalence of mutations in eyeGENE probands with a diagnosis of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci 54:6255–6261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LS, Koboldt DC, Bowne SJ et al. (2014) A dominant mutation in hexokinase 1 (HK1) causes retinitis pigmentosa. Invest Ophthalmol Vis Sci 55:7147–7158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Wang H, Tuan HF et al. (2013) Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype-phenotype correlation and clinical refinements. Hum Genet 133:331–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheaton DK, Webb-Jones KD, Bowne SJ et al. (2016) Complex multi-allelic inherited retinal dystrophy: multiple genes contributing independently and concurrently in extended families. Invest Ophthalmol Vis Sci E-Abstr 57:3135 [Google Scholar]
- Wright AF, Chakarova CF, Abd El-Aziz MM et al. (2010) Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat Rev Genet 11:273–284 [DOI] [PubMed] [Google Scholar]