

Abstract
NEK2 is associated with drug resistance in multiple cancers. Our previous studies indicated that high NEK2 confers inferior survival in multiple myeloma (MM); thus, a better understanding of the mechanisms by which NEK2 induces drug resistance in MM is required. In this study, we discovered that NEK2 enhances MM cell autophagy, and a combination of autophagy inhibitor chloroquine (CQ) and chemotherapeutic bortezomib (BTZ) significantly prevents NEK2‐induced drug resistance in MM cells. Interestingly, NEK2 was found to bind and stabilize Beclin‐1 protein but did not affect its mRNA expression and phosphorylation. Moreover, autophagy enhanced by NEK2 was significantly prevented by knockdown of Beclin‐1 in MM cells, suggesting that Beclin‐1 mediates NEK2‐induced autophagy. Further studies demonstrated that Beclin‐1 ubiquitination is decreased through NEK2 interaction with USP7. Importantly, knockdown of Beclin‐1 sensitized NEK2‐overexpressing MM cells to BTZ in vitro and in vivo. In conclusion, we identify a novel mechanism whereby autophagy is activated by the complex of NEK2/USP7/Beclin‐1 in MM cells. Targeting the autophagy signaling pathway may provide a promising therapeutic strategy to overcome NEK2‐induced drug resistance in MM.
Keywords: autophagy, Beclin‐1, multiple myeloma, NEK2, ubiquitination
Autophagy is associated with bortezomib resistance in multiple myeloma (MM). Here, we show that NEK2 stabilizes Beclin‐1 through USP7‐mediated deubiquitination, thereby enhancing autophagy and eventually inducing bortezomib resistance in MM cell. Moreover, knockdown of Beclin‐1 profoundly inhibits NEK2‐mediated bortezomib resistance. These findings indicate that NEK2 induces autophagy‐mediated bortezomib resistance through stabilizing Beclin‐1 in MM cell.
Abbreviations
- BTZ
bortezomib
- CQ
chloroquine
- MM
multiple myeloma
- MS
mass spectrometry
- TEM
transmission electron microscopy
1. Introduction
Multiple myeloma (MM) is the second most frequent hematological malignancy characterized by transformed clonal plasma cells in the bone marrow (BM) microenvironment, monoclonal immunoglobulin in the blood or urine, and associated organ dysfunctions (Palumbo and Anderson, 2011). The combination of autologous stem cell transplantation (ASCT) and multiple chemotherapeutic drugs such as proteasome inhibitor [bortezomib (BTZ), carfilzomib, ixazomib], immunomodulatory drugs (thalidomide, lenalidomide, pomalidomide), alkylating agent (melphalan), and corticosteroid (dexamethasone) have significantly extended patient overall survival in MM (Palumbo and Anderson, 2011). However, MM is still a difficult‐to‐cure disease. A main reason for the treatment failure and disease relapse is the appearance of drug‐resistant subclones during therapy. A better understanding of the mechanisms by which drug resistance is caused is urgently required.
Never in mitosis‐related kinase 2 (NEK2) is a serine/threonine kinase that promotes centrosome splitting and ensures correct chromosome segregation during the G2/M phase of the cell cycle (Xia et al., 2015). Previous data from our group and others have indicated that NEK2 is overexpressed in various cancers, including MM (Zhou et al., 2013), cholangiocarcinoma (Kokuryo et al., 2007), testicular seminomas (Barbagallo et al., 2009), human breast cancer (Tsunoda et al., 2009; Wang et al., 2012), cervical cancer (Wang et al., 2012), prostate cancer (Wang et al., 2012), and colorectal cancer (Neal et al., 2014; Suzuki et al., 2010). Our previous studies have identified that NEK2 was the gene most strongly associated with inferior survival in MM and other cancers, suggesting that NEK2 is a potential therapeutic target in MM (Zhou et al., 2013). NEK2 was found to promote cancer cell proliferation and drug resistance in MM. Subsequent mechanistic studies showed that NEK2 induces drug resistance through up‐regulation of efflux drug pumps in MM cells. NEK2 was also found to bind to USP7, a deubiquitinase that contributes to malignancy and BTZ resistance, and was stabilized by USP7 (Franqui‐Machin et al., 2018). Destabilization of NEK2 by USP7 inhibitor was able to overcome resistance to BTZ in MM cells.
Autophagy is an evolutionarily conserved cellular process. During autophagy, unwanted cellular components, including proteins, lipids, entire organelles, and invading pathogens, are delivered by double‐membrane vesicles called autophagosomes to the lysosome, where they are degraded (Galluzzi and Green, 2019; Mizushima and Komatsu, 2011). This degradative process is critical for cellular homeostasis, and defects in autophagy lead to various diseases. In cancer progression, autophagy plays dual roles, either as a tumor suppressor or as a tumor progression promoter (Fu et al., 2017; Guo et al., 2013; Kocaturk et al., 2019). Therefore, it is difficult to develop efficient treatment strategies targeting autophagy in cancer. Recent studies indicated that inhibition of autophagy enhances MM cell death induced by proteasome inhibitors (Hoang et al., 2009). Intriguingly, a phase I clinical trial demonstrated that the combination of autophagy inhibitor, hydroxychloroquine, and BTZ is a useful therapeutic approach for patients with relapsed/refractory myeloma (Vogl et al., 2014). Subsequently, several groups further determined that inhibition of autophagy by other mechanisms significantly increases cell death in MM cell in response to BTZ or carfilzomib, which is a second generation of approved proteasome inhibitor (Jarauta et al., 2016; Lu et al., 2018; Roy et al., 2016; Zhang et al., 2015, 2014, 2018). In view of former studies, we hypothesized that inhibition of autophagy overcomes NEK2‐mediated BTZ resistance.
In this study, we explored whether NEK2 promotes autophagy in MM cells and tested whether inhibition of autophagy overcomes NEK2‐induced BTZ resistance. Moreover, we also uncovered the underlying mechanism by which NEK2 enhances autophagy in MM cells.
2. Materials and methods
2.1. Clinical samples
Bone marrow specimens derived from healthy donors (HD; n = 6), and MM patients who were newly diagnosed (n = 9) and relapsed (n = 7) were obtained from Xiangya Hospital, the Second Xiangya Hospital, the Third Xiangya Hospital of Central South University during the period of 2017 to 2019. Firstly, monocytes were isolated from freshly received BM specimen (1 mL) by lymphocyte separation medium (TBD, Tianjin, China), and then, CD138+ cells were isolated by using CD138 antibody‐conjugated magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany). All CD138+ cells were fixed in methanol for immunofluorescence until use. The Ethics Committee of Cancer Research Institute at the Central South University approved this study according to the Declaration of Helsinki. Informed written consent was obtained from all individual participants included in the study. The information of specimens is described in Table S1.
2.2. Reagents and antibodies
General laboratory reagents were obtained from Thermo Fisher Scientific (Waltham, MA, USA) and Sigma‐Aldrich (Saint Louis, MO, USA). Antibodies against LC3B (#2775S), PARP (#9542S), cleaved caspase‐3 (#9661S), USP7 (#4833S), ubiquitin (#3933S), HA‐tag (#3724S), Vps15 (#14580S), and Vps34 (#3811S) were obtained from Cell Signaling Technology (Danvers, MA, USA). Antibodies against NEK2 (sc‐55601), Beclin‐1 (sc‐48381), and phosphorylated mTOR (Ser 2448) (sc‐293133) were obtained from Santa Cruz Biotechnology, Inc (St Louis, MO, USA). β‐Actin antibody (66009‐1‐1g) and Flag antibody (20543‐1‐AP) were purchased from Proteintech Group (Rosemont, IL, USA). Phosphorylated Beclin‐1 (S90/93/96) antibody (#13232) and HRP‐conjugated secondary goat anti‐rabbit (#L3012) and goat anti‐mouse (#L3032) antibodies were purchased from Signalway Antibody (College Park, MD, USA). Alexa Fluor 488‐conjugated donkey anti‐rabbit second antibody (#A21202), Alexa Fluor 594‐conjugated donkey anti‐mouse second antibody (#A21207), and UltraPure™ LMP agarose were purchased from Thermo Fisher Scientific. IPKine HRP goat anti‐mouse IgG LCS (#A250112) was obtained from Abbkine Scientific Co (Wuhan, China).
2.3. Cell culture
Human MM cell lines, including KMS11, RPMI 8226, ARP1, KMS28‐PE, U266, MM1.S, and XG1, were cultured at 37 °C and 5% CO2 in RPMI 1640 medium (Gibco, Thermo Fisher Scientific) supplemented with 10% heat‐inactivated FBS (Gibco, Thermo Fisher Scientific) and 1% penicillin and streptomycin (Gibco, Thermo Fisher Scientific).
2.4. Plasmids and virus production
Human NEK2 cDNA sequence was amplified and then cloned into the pCDH‐CMV‐MCS‐EF1‐copRFP lentiviral vector. Short hairpin RNA sequences targeting human NEK2 or BECN1 were obtained from the RNAi consortium collection (MISSION® shRNA; Sigma, http://www.sigmaaldrich.com). shRNAs were annealed and ligated into pLKO‐tet‐on lentiviral vector. Recombinant lentivirus was produced by transient transfection of 293T cells. After lentivirus transduction, NEK2‐overexpressing (NEK2‐OE) MM cells were purified by flow cytometry sorting, and MM cells expressing NEK2‐shRNA RNA or BECN1‐shRNA were selected with puromycin (1 μg·L−1). All primer sequences are listed in Table S2.
2.5. Western blotting
Western blot analysis was performed as described previously (Gu et al., 2015). Briefly, total proteins were extracted using RAPI strong buffer (Auragene Bioscience, Changsha, China) with freshly added proteinase inhibitor. Proteins were then separated by 10–12% SDS/PAGE and transferred to polyvinylidene fluoride (Millipore‐Sigma). The membranes were blocked with 5% nonfat dry milk in Tris‐buffered saline (TBS) containing 0.05% Tween‐20 (TBST) prior to incubation with primary antibodies overnight at 4 °C. Respective HRP‐conjugated secondary antibodies were added, and protein signals were developed with SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific). The developed images were obtained and analyzed using ChemiDox XRS Chemiluminescence imaging system (Bio‐Rad, Hercules, CA, USA).
2.6. Immunofluorescence analysis
4 × 104 CD138+ cells were spun down on glass slides and then fixed with methanol for 15 min at −20 °C. NEK2 antibodies (1 : 100 final dilution) and LC3B antibodies (1 : 200 final dilution) were diluted in antibody dilution buffer (TBS, 0.1% Triton X‐100, 1% BSA). The diluted antibodies were dripped on glass slides and incubated overnight at 4 °C. Then, slides were washed with TBST for three times followed by incubation of secondary antibodies conjugated with Alexa Fluor 488 donkey anti‐rabbit or Alexa Fluor 594 donkey anti‐mouse for 1 h at room temperature (protect slides from light). The nuclei were labeled with DAPI (Solarbio, Beijing, China). Fluorescence was observed under fluorescence microscope.
2.7. Co‐immunoprecipitation
Co‐immunoprecipitation (Co‐IP) was performed based on previous publication (Gu et al., 2017). Briefly, total proteins were extracted with IP lysis buffer (Thermo Fisher Scientific). NEK2 antibodies (1 : 50 final dilution) or Beclin‐1 antibodies (1 : 50 final dilution) were added and incubated with cell lysate overnight at 4 °C. The same cell lysate incubated with normal mouse IgG was set as control, then followed by protein A Dynabeads (Thermo Fisher Scientific) incubation for 2 h at 4 °C. The beads were washed three times with PBST. The pulled‐down proteins were obtained and examined by western blotting as described above.
2.8. Soft agar colony formation assay
Soft agar colony formation assay was performed based on previous report (Yang et al., 2014). Cells were planted in 12‐well plate (2000 cells/well) and fed with RPMI 1640 complete medium in the presence or absence of drugs twice every week. One colony was defined if more than 40 cells were observed. Plates were imaged, and colonies were enumerated using imagej software (NIH, Bethesda, MD, USA). Each sample was repeated three times.
2.9. Cell viability
Cell counts and viable cell number were determined using trypan blue staining as previously described (Xia et al., 2017). Cell viability was calculated by dividing viable cells by total cell number, each sample was done in triplicate.
2.10. Flow cytometry
Apoptotic cells were labeled by FITC‐conjugated Annexin V (US Everbright Inc, San Francisco, CA, USA). Dead cells were labeled by PI (US Everbright Inc). Cell staining was performed according to the manufacturer’s protocol. Cells were then analyzed on LSR II cytometer (BD Biosciences, San Jose, CA, USA), and the results were analyzed by using flowjo software (BD Biosciences).
2.11. Autophagy detection with DALGreen staining
DALGreen staining was performed based on previous publication with some modifications (Iwashita et al., 2018). A total of 200 000 cells were planted in 24‐well plate and cultured at 37 °C with 1 mL of 0.5 μm DALGreen (Dojindo Laboratories, Minato‐ku, Tokyo, Japan) working solution for 30 min. After the cells were washed twice with RPMI 1640 medium, they were observed under fluorescence microscope.
2.12. Tumor xenografts in mice
Mouse experiment was performed under the protocol approved by the Institutional Animal Care and Local Veterinary Office and Ethics Committee of the Central South University, China (animal experimental license number: 2019SYDW0115). NEK2‐OE KMS11 MM cell line transduced with BECN1‐shRNA or scramble‐shRNA (1 × 106 cells in 200 μL RPMI 1640 medium) was injected subcutaneously into the left abdomen of immunocompromised B‐NDG (NOD‐Prkdcscid IL2rgtm1/Bcgen) mouse (Biocytogen, Beijing, China), which lacks T cell, B cell, and NK cell. Tumor burden was monitored by tumor volume. shRNA expression was induced by the addition of doxycycline into the drinking water following the previous publication (Zhou et al., 2013). The mice were euthanized when a humane endpoint was reached.
2.13. Statistical analysis
All data were shown as means ± standard error. Student’s t‐test was used to compare two experimental groups. A value of P < 0.05 was considered to be significant.
3. Results
3.1. Inhibition of autophagy sensitizes high NEK2 myeloma cells to bortezomib
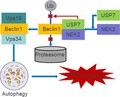
Several groups have reported that increased autophagy contributes to BTZ resistance, and targeting autophagy is a useful strategy to overcome BTZ resistance in MM (Vogl et al., 2014; Zhang et al., 2015, 2014, 2018). Our previous studies demonstrated high NEK2 is associated with poor prognosis in MM, and overexpression of NEK2 promotes BTZ resistance (Zhou et al., 2013). We thus speculated that autophagy might be involved in NEK2‐mediated BTZ resistance. To explore the correlation between NEK2 and autophagy during MM progression, immunofluorescence was performed to detect the expression of NEK2 and LC3B in CD138+ cells derived from HD (n = 6), newly diagnosed MM (NDMM) patients (n = 9), and relapsed MM (RMM) patients (n = 7). It has been widely accepted that the presence of LC3B in autophagosomes and the conversion of LC3B‐Ⅰ to the lower migrating form, LC3B‐Ⅱ, are indicators of autophagy (Kabeya et al., 2004). As shown in Fig. 1A, NEK2 is profoundly elevated along with the disease progression in MM; simultaneously, the number of autophagosomes labeled by LC3B is also increased.
Figure 1.
Inhibition of autophagy sensitizes NEK2‐OE MM cells to BTZ. (A) Representative images of immunofluorescence analysis for NEK2 (green) and LC3B (red) protein expression in CD138+ cells derived from HD (n = 6), NDMM patients (n = 9), and RMM patients (n = 7). (B) KMS11 EV, KMS11 NEK2‐OE, RPMI 8226 EV, and RPMI 8226 NEK2‐OE MM cells were treated with different doses of CQ (0, 10 μm, 20 μm, 40 μm) in combination or not with BTZ (10 nm), and cell viability was examined 48 h later. (C) Clonogenic analysis of KMS11 EV, KMS11 NEK2‐OE, RPMI 8226 EV, and RPMI 8226 NEK2‐OE MM cells after treatment with CQ (0, 10 μm, 20 μm) in combination or not with BTZ (1 nm), respectively (4×). (D) Western blots of full‐length PARP, cleaved PARP, cleaved caspase‐3, NEK2, and β‐actin in KMS11 EV, KMS11 NEK2‐OE, RPMI 8226 EV, and RPMI 8226 NEK2‐OE MM cells after treatment with CQ (0, 10 μm, 20 μm) in combination with or not with BTZ (10 nm). The ratio of integrated density between cleaved PARP and full‐length PARP was shown under the band of PARP. Scale bar: 5 μm. ns P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Significance was determined by Student’s t‐test. Error bars indicate SD.
We subsequently asked whether inhibition of autophagy overcomes NEK2‐mediated BTZ resistance in MM cells. To answer this question, we overexpressed NEK2 by lentivirus‐mediated NEK2‐cDNA transfection in both KMS11 and RPMI 8226 MM cell lines. Those cells transduced with empty vector were used as controls. KMS11 or RPMI 8226 MM cells overexpressing NEK2 were treated with BTZ in combination or not with different doses of a specific inhibitor of autophagy, chloroquine (CQ), for 2 days. Consistent with our previous report (Zhou et al., 2013), overexpression of NEK2 promoted BTZ resistance in both KMS11 and RPMI 8226; however, a combination of BTZ and CQ profoundly increased cell death compared with the drug BTZ or CQ alone in NEK2‐OE MM cell lines (Fig. 1B). The clonogenic soft agar assay was performed to confirm the synergistic effect between BTZ and CQ. The colonies were dramatically decreased in NEK2‐OE KMS11 or RPMI 8226 MM cell line treated with the combination of BTZ plus CQ, but showed only a slight decrease in those MM cell lines treated with BTZ or CQ alone (Fig. 1C). The synergistic effect between BTZ and CQ was further evidenced by increased cleavage of caspase‐3 and PARP in NEK2‐OE KMS11 or RPMI 8226 MM cell line in response to combination of BTZ and CQ compared with those cells treated with BTZ alone (Fig. 1D).
3.2. NEK2 enhances autophagy in myeloma cells
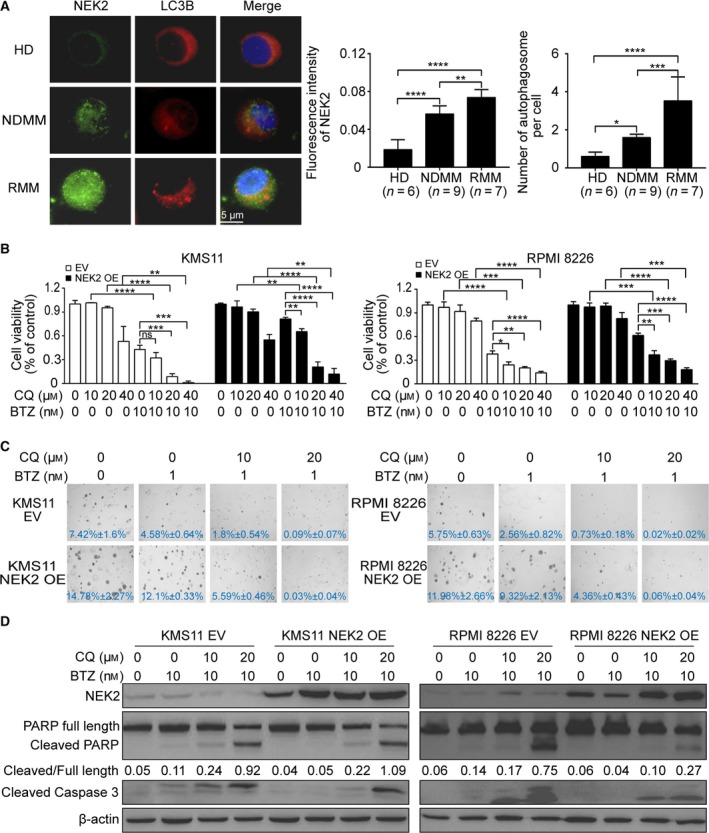
The data presented in Fig. 1 suggest that elevation of NEK2 is associated with autophagy activity in MM drug resistance. The conversion of LC3B‐Ⅰ to LC3B‐Ⅱ has been widely accepted as a marker for autophagy. As shown in Fig. 2A,B, overexpression of NEK2 increases LC3B‐Ⅱ in both KMS11 and RPMI 8226 MM cell lines by western blotting, while knockdown of NEK2 by a doxycycline‐inducible lentiviral expression system containing NEK2‐shRNA reduces LC3B‐Ⅱ in the same MM cell lines. To examine autophagy level, DALGreen, a small green hydrophobic molecule utilized for the imaging of autolysosomes, was introduced into MM cells. The DALGreen‐positive cells were significantly increased in NEK2‐OE KMS11 and RPMI 8226 MM cell lines but reduced in NEK2‐silenced KMS11 and RPMI 8226 MM cells (Fig. 2C,D). Transmission electron microscopy (TEM) was further performed to detect autophagosomes in NEK2‐overexpressed or knocked‐down KMS11 MM cells. Consistently, autophagosomes were increased in NEK2‐OE KMS11 and decreased in NEK2‐silenced KMS11 (Fig. 2E,F).
Figure 2.
NEK2 modulates autophagy in MM cells. (A) Western blots of NEK2, LC3B, and β‐actin in KMS11 EV, KMS11 NEK2‐OE, RPMI 8226 EV, and RPMI 8226 NEK2‐OE MM cells. (B) Western blots of NEK2, LC3B, and β‐actin in KMS11 Scr, KMS11 NEK2‐shRNA, RPMI 8226 Scr, and RPMI 8226 NEK2‐shRNA MM cells. (C) KMS11 EV, KMS11 NEK2‐OE, RPMI 8226 EV, and RPMI 8226 NEK2‐OE MM cells imaging with 0.5 μm DALGreen staining. (D) KMS11 Scr, KMS11 NEK2‐shRNA, RPMI 8226 Scr, and RPMI 8226 NEK2‐shRNA imaging with 0.5 μm DALGreen staining. (E) TEM analysis of autophagosomes in KMS11 EV and KMS11 NEK2‐OE MM cells. (F) TEM analysis of autophagosomes in KMS11 Sr and KMS11 NEK2‐shRNA MM cells. Scale bar: 5 or 2 μm. **P < 0.01, ****P < 0.0001. Significance was determined by Student’s t‐test. Error bars indicate SD.
3.3. NEK2 activates autophagy through up‐regulating Beclin‐1 at protein level
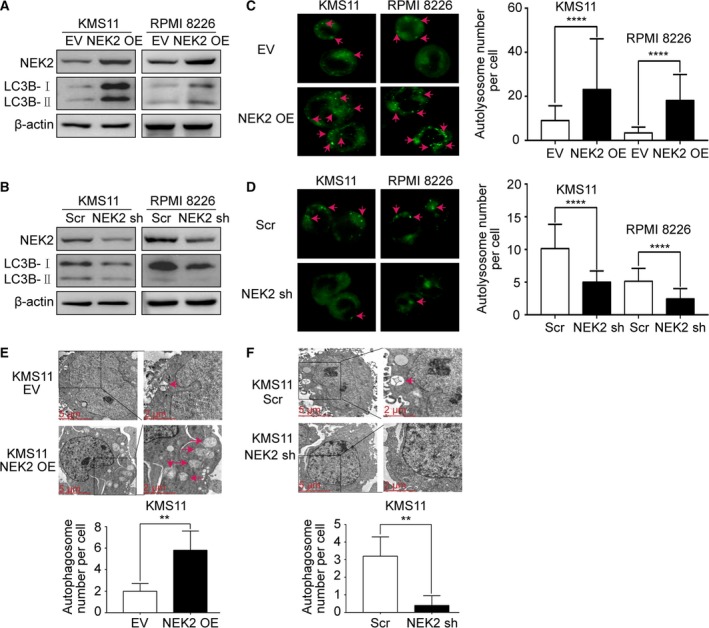
To determine the underlying mechanisms by which NEK2 promotes autophagy in MM cells, mass spectrometry (MS) was performed to identify NEK2‐interacting proteins in NEK2‐OE KMS11 MM cell line using NEK2 antibodies (Fig. S1A). Proteins pulled down by normal mouse IgG were identified as nonspecific binding. Western blots confirmed NEK2 was effectively pulled down by NEK2 antibodies (Fig. 3A). We obtained 156 proteins bound to NEK2, which were not observed in proteins pulled down by normal mouse IgG (data not shown). Interestingly, Beclin‐1, which interacts with Vps15‐Vps34 complex to play a crucial role in the initiation of autophagy, was found to bind to NEK2. The interaction between NEK2 and Beclin‐1 was confirmed by Co‐IP in NEK2‐OE KMS11 MM cell line (Fig. 3B). To further confirm the interaction between NEK2 and Beclin‐1, Co‐IPs of endogenous NEK2 in KMS11 and RPMI 8226 MM cell lines were performed using NEK2 antibodies. Western blots were applied to detect NEK2 and Beclin‐1. As shown in Fig. 3C, both NEK2 and Beclin‐1 are detected in NEK2 antibody‐immunoprecipitated proteins but not in IgG control. As the sizes of NEK2 protein band and Beclin‐1 protein band in SDS/PAGE are similar to IgG heavy chain, we used HRP‐conjugated second antibody only interacting with IgG light chain but not with heavy chain in Co‐IP experiments. In addition, we found an interesting phenomenon that the protein level of NEK2 was positively related to that of Beclin‐1 in seven MM cell lines (Figs 3D and S1B). We thus examined whether Beclin‐1 expression is regulated by NEK2. Western blots showed Beclin‐1 was significantly increased in NEK2‐OE KMS11 and RPMI 8226 MM cell lines compared with the controls, while silence of NEK2 decreased Beclin‐1 (Fig. 3E). We did not observe significant change in Beclin‐1 at mRNA level in the same MM cell lines (Fig. S1C). In addition, the expression of Beclin‐1‐interacting proteins Vps15 and Vps34 was not affected by NEK2 (Fig. 3E). Several studies have documented that autophagy is enhanced by phosphorylation of Beclin‐1 at Ser90, Ser93, and Ser96 (Menon and Dhamija, 2018). NEK2 is a serine/threonine kinase that was found to phosphorylate and then activate several oncogenes. The changes in Beclin‐1 phosphorylation were not observed in NEK2‐OE KMS11 MM cell line (Fig. S1D). Furthermore, the phosphorylation of mTOR, the upstream and negative regulator of Beclin‐1‐Vps15‐Vps34 complex (Navé et al., 1999), was not affected by NEK2 at ser‐2448, which is the active site of mTOR (Fig. S1D).
Figure 3.
NEK2 enhances autophagy through elevating Beclin‐1 at protein level. (A) NEK2 antibody was used to pull down NEK2 in KMS11 NEK2‐OE MM cells, and the efficiency of immunoprecipitation was confirmed by western blotting before MS. The lysates before IP were used as a positive control, and normal mouse IgG pulled‐down proteins, as a negative control. (B) NEK2 antibody was used to pull down NEK2 in KMS11 NEK2‐OE MM cells, and its interacting proteins were analyzed by western blotting. (C) NEK2 antibody was used to pull down NEK2 in KMS11 (left) and RPMI 8226 (right) MM cells, and its interacting proteins were analyzed by western blotting. (D) Western blots of NEK2, Beclin‐1, and β‐actin in RPMI 8226, KMS11, XG1, MM1.S, U266, KMS28‐PE, ARP1 MM cells. (E) Western blots of NEK2, Beclin‐1, Vps15, Vps34, and β‐actin in KMS11 EV, KMS11 NEK2‐OE, RPMI 8226 EV, RPMI 8226 NEK2‐OE, KMS11 Scr, KMS11 NEK2‐shRNA, RPMI 8226 Scr, RPMI 8226 NEK2‐shRNA MM cells. (F) Western blots of NEK2, Beclin‐1, LC3B, and β‐actin in KMS11 EV + Scr, KMS11 EV + BECN1‐shRNA, KMS11 NEK2‐OE + Scr, KMS11 NEK2‐OE + BECN1‐shRNA MM cells. (G) Western blots of NEK2, Beclin‐1, LC3B, and β‐actin in RPMI 8226 EV + Scr, RPMI 8226 EV + BECN1‐shRNA, RPMI 8226 NEK2‐OE + Scr, RPMI 8226 NEK2‐OE + BECN1‐shRNA MM cells. (H) KMS11 EV + Scr, KMS11 EV + BECN1‐shRNA, KMS11 NEK2‐OE + Scr, KMS11 NEK2‐OE + BECN1‐shRNA MM cells imaging with 0.5 μm DALGreen staining. (I) RPMI 8226 EV + Scr, RPMI 8226 EV + BECN1‐shRNA, RPMI 8226 NEK2‐OE + Scr, RPMI 8226 NEK2‐OE + BECN1‐shRNA MM cells imaging with 0.5 μm DALGreen staining. ****P < 0.0001. Significance was determined by Student’s t‐test. Error bars indicate SD.
To determine whether Beclin‐1 mediates NEK2‐enhanced autophagy, we knocked down Beclin‐1 in NEK2‐OE MM cell lines by a doxycycline‐inducible lentiviral expression system containing BECN1‐shRNA. These cell lines were exposed to doxycycline for 2 days, and then, western blots were performed to detect the expression of NEK2, Beclin‐1, and LC3B‐Ⅰ/Ⅱ. Beclin‐1 protein was clearly decreased in MM cell lines expressing BECN1‐shRNA (Fig. 3F,G). As expected, LC3B‐Ⅱ was also decreased in NEK2‐OE KMS11 and RPMI 8226 MM cell lines following the silence of Beclin‐1, indicating that NEK2 enhanced autophagy was attenuated by knockdown of Beclin‐1 (Fig. 3F,G). In addition, the number of autolysosomes labeled by DALGreen was significantly decreased in response to down‐regulation of Beclin‐1 in NEK2‐OE KMS11 and RPMI 8226 MM cell lines (Fig. 3H,I). Therefore, we conclude that NEK2 enhances autophagy through up‐regulating Beclin‐1 at protein level.
3.4. NEK2 stabilizes Beclin‐1 by USP7‐mediated deubiquitination
Beclin‐1 can be stabilized by several deubiquitinases, including USP19, USP13, and USP10 (Jin et al., 2016; Liu et al., 2011). In this study, we showed proteasome inhibitor, BTZ, elevated Beclin‐1 at protein level in MM cells (Fig. S2A). Addition of proteasome inhibitor MG132 to NEK2‐silenced MM cells profoundly increased Beclin‐1 protein (Fig. S2B). These data suggested that NEK2 might elevate Beclin‐1 through blocking proteasomal degradation. We have reported that USP7 binds and stabilizes NEK2 protein (Franqui‐Machin et al., 2018). NEK2 interacts with both USP7 and Beclin‐1 in MM cells, and we thus hypothesized that NEK2 stabilizes Beclin‐1 via interacting with USP7. To address this hypothesis, NEK2‐OE KMS11 and RPMI 8226 MM cell lines were treated with P005091, a USP7 inhibitor that selectively binds the USP7 active site and inhibits its activity, overnight at 10 μm. As shown in Fig. 4A, treatment with P005091 decreases Beclin‐1 in both NEK2‐OE MM cells and controls, suggesting that Beclin‐1 is stabilized by USP7. Since USP7 stabilizes its targets by preventing proteasomal degradation, we tested whether Beclin‐1 depletion by P005091 was dependent on proteasome activity. KMS11 and RPMI 8226 MM cell lines were treated with MG132 (10 μm) alone for 1 h or in a combination with P005091 (10 μm) for additional 9 h. Western blots showed P005091 alone depleted Beclin‐1; however, addition of MG132 blocked this depletion, indicating that USP7 stabilizes Beclin‐1 through preventing ubiquitination–proteasomal degradation of Beclin‐1 (Fig. 4B).
Figure 4.
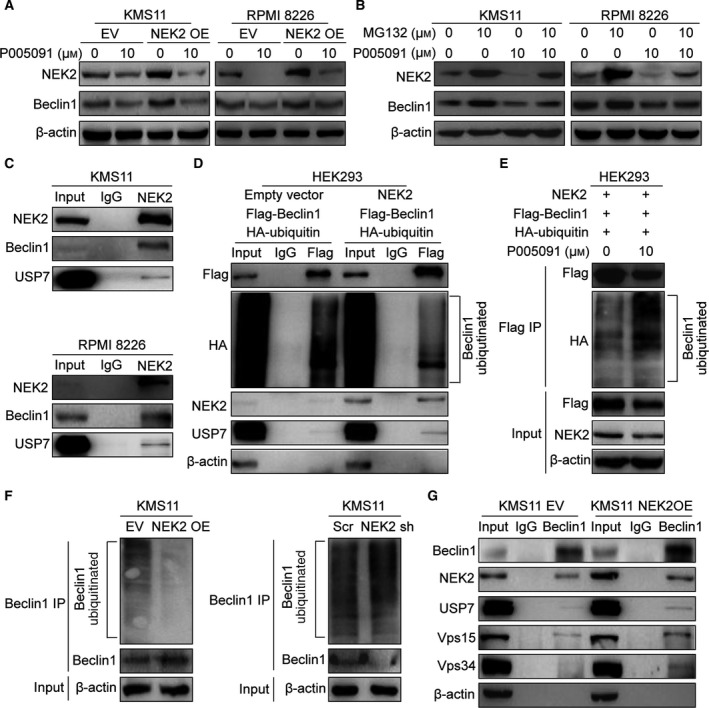
NEK2 stabilizes Beclin‐1 through USP7‐mediated deubiquitination. (A) KMS11 EV, KMS11 NEK2‐OE, RPMI 8226 EV, and RPMI 8226 NEK2‐OE MM cells were treated with 10 μm P005091 for 24 h, and the levels of NEK2 and Beclin‐1 were analyzed by western blotting. (B) KMS11 and RPMI 8226 MM cells were treated with MG132 (10 μm) alone for 1 h, or in combination with P005091 (10 μm) for an additional 9 h. Cells were lysed, and the levels of NEK2 and Beclin‐1 were analyzed by western blotting. (C) Endogenous NEK2 was pulled down by NEK2 antibodies in KMS11 (up) and RPMI 8226 (bottom) MM cells, NEK2‐interacting proteins were analyzed by western blotting. The lysates before IP were used as a positive control, and normal mouse IgG pulled‐down proteins, as a negative control. (D) HEK293 cells were transfected with HA‐ubiquitin vector, Beclin‐1‐Flag vector, and NEK2 overexpression vector or empty vector. After 48 h, cells were lysed, Beclin‐1‐flag fusion protein and its interacting proteins were pulled down by flag antibody‐conjugated beads, and the ubiquitination level of Beclin‐1 and its interacting proteins were analyzed by western blotting. (E) HEK293 cells were transfected with Beclin‐1‐Flag vector, HA‐ubiquitin vector, NEK2 overexpression vector; after 48 h, cells were treated with USP7 inhibitor P005091 (10 μm) for 10 h; cells were lysed; Beclin‐1‐flag fusion protein and its interacting proteins were pulled down by flag antibody‐conjugated beads; and the ubiquitination level of Beclin‐1 was analyzed by western blotting. (F) Endogenous Beclin‐1 was pulled down by Beclin‐1 antibodies in KMS11 EV, KMS11 NEK2‐OE, KMS11 Scr, and KMS11 NEK2‐shRNA MM cells, and Beclin‐1, ubiquitinated Beclin‐1, and β‐actin were analyzed by western blotting. (G) Beclin‐1 antibody was used to pull down Beclin‐1 in KMS11 EV and KMS11 NEK2‐OE MM cells, and western blots were performed to analyze the levels of Beclin‐1, NEK2, Vps15, Vps34, USP7, and β‐actin.
We subsequently examined the interaction between NEK2, Beclin‐1, and USP7 in MM cells. Endogenous NEK2 was pulled down by NEK2 antibodies in both KMS11 and RPMI 8226 MM cell lines. Western blots showed NEK2 was bound to Beclin‐1 and USP7, suggesting that a complex containing NEK2, Beclin‐1, and USP7 was persistent in KMS11 and RPMI 8226 MM cell lines (Fig. 4C). To determine whether increased deubiquitination of Beclin‐1 is induced by NEK2, HEK293 cells were transfected with HA‐ubiquitin vector, Beclin‐1‐Flag vector, and NEK2 overexpression vector or empty vector. Beclin‐1 was immunoprecipitated with flag antibody‐conjugated beads. Less ubiquitinated Beclin‐1 was observed in NEK2‐OE HEK293 cells compared with those cells transfected with empty vector. Furthermore, western blots detected more USP7 bound to Beclin‐1 in NEK2‐OE HEK293 cells (Fig. 4D). HEK293 cells transfected with HA‐ubiquitin vector, Beclin‐1‐Flag vector, and NEK2 overexpression vector were treated with P005091 and then pulled down Beclin‐1 by flag antibody‐conjugated beads. Western blots showed ubiquitinated Beclin‐1 was increased in response to P005091 treatment (Fig. 4E). We also examined the ubiquitination level of endogenous Beclin‐1 in NEK2‐OE and NEK2 knocked‐down KMS11 MM cell lines by Co‐IP. Endogenous Beclin‐1 was pulled down by Beclin‐1 antibodies. Figure 4F shows overexpression of NEK2 decreases the ubiquitination level of Beclin‐1, while silence of NEK2 enhances the ubiquitination level of Beclin‐1. To explore whether NEK2 promotes the formation of Beclin‐1‐Vps15‐Vps34 complex in MM cells, we pulled down Beclin‐1 using Beclin‐1 antibodies in both NEK2‐OE KMS11 MM cells and controls. As shown in Fig. 4G, increased Vps15 and Vps34 are obtained in Beclin‐1 antibody‐immunoprecipitated proteins derived from NEK2‐OE KMS11 MM cells compared with controls, indicating that overexpression of NEK2 promotes the formation of Beclin‐1‐Vps15‐Vps34 complex. Taken together, our data demonstrated that NEK2 up‐regulates Beclin‐1 through USP7‐mediated deubiquitination, thereby promoting autophagy.
3.5. Knockdown of Beclin‐1 prevents NEK2‐mediated bortezomib resistance in myeloma cells
Beclin‐1 was knocked down in NEK2‐OE KMS11 MM cells and controls by doxycycline‐induced expression of BECN1‐shRNA followed by BTZ treatment for 2 days. The viability of control cells showed significant reduction after BTZ treatment, while NEK2‐OE KMS11 MM cells were highly resistant to BTZ. However, knockdown of Beclin‐1 in NEK2‐OE KMS11 MM cells significantly reduced viability after treatment with BTZ, similar to control cells treated with BTZ (Fig. 5A). A similar result was observed in the RPMI 8226 MM cell line (Fig. 5B). Subsequently, clonogenic soft agar assay was performed with above MM cell lines with or without BTZ treatment. As shown in Fig. 5C,D, compared to nontreated controls, NEK2‐OE KMS11 or RPMI 8226 MM cell lines showed only a slight decrease in their capacity to form colonies, while those cells expressing BECN1‐shRNA showed a significant decrease in colony formation when treated with the same dose of BTZ, indicating that silence of Beclin‐1 prevents BTZ resistance in NEK2‐OE MM cell lines. We next examined cell apoptosis in NEK2‐OE/BECN1‐shRNA MM cell lines when treated with BTZ. Apoptotic cells were detected by flow cytometry using FITC‐conjugated Annexin V staining. Increased apoptotic cells were observed in Beclin‐1 knocked‐down MM cells compared with those cells expressing scramble‐shRNA after BTZ treatment (Fig. 5E). Western blots also showed knockdown of Beclin‐1 resulted in increase in cleaved caspase‐3 and PARP in NEK2‐OE MM cells when treated with BTZ (Fig. 5F).
Figure 5.
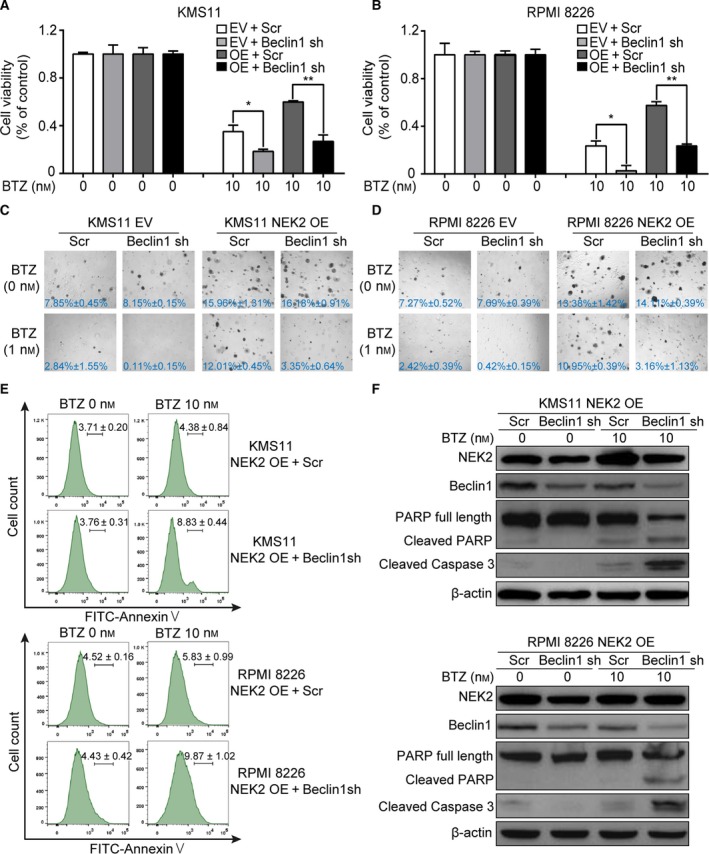
Knockdown of Beclin‐1 blocks NEK2‐mediated BTZ resistance in MM cells. (A) KMS11 EV + Scr, KMS11 EV + BECN1‐shRNA, KMS11 NEK2‐OE + Scr, KMS11 NEK2‐OE + BECN1‐shRNA MM cells were treated with BTZ (10 nm), cell viability was examined 48 h later. (B) RPMI 8226 EV + Scr, RPMI 8226 EV + BECN1‐shRNA, RPMI 8226 NEK2‐OE + Scr, RPMI 8226 NEK2‐OE + BECN1‐shRNA MM cells were treated with BTZ (10 nm), cell viability was examined 48 h later. (C) Clonogenic analysis of KMS11 EV + Scr, KMS11 EV + BECN1‐shRNA, KMS11 NEK2‐OE + Scr, KMS11 NEK2‐OE + BECN1‐shRNA MM cells after treatment with BTZ (1 nm), respectively (4×). (D) Clonogenic analysis of RPMI 8226 EV + Scr, RPMI 8226 EV + BECN1‐shRNA, RPMI 8226 NEK2‐OE + Scr, RPMI 8226 NEK2‐OE + BECN1‐shRNA MM cells after treatment with BTZ (1 nm), respectively (4×). (E) Assessment of apoptosis by using FITC‐Annexin V/PI staining in KMS11 NEK2‐OE + Scr, KMS11 NEK2‐OE + BECN1‐shRNA, RPMI 8226 NEK2‐OE + Scr, RPMI 8226 NEK2‐OE + BECN1‐shRNA MM cells after treatment with BTZ (10 nm). (F) Western blots of full‐length PARP, cleaved PARP, cleaved caspase‐3, NEK2, and β‐actin in KMS11 NEK2‐OE + Scr, KMS11 NEK2‐OE + BECN1‐shRNA, RPMI 8226 NEK2‐OE + Scr, RPMI 8226 NEK2‐OE + BECN1‐shRNA MM cells after treatment with BTZ (10 nm). *P < 0.05, **P < 0.01. Significance was determined by Student’s t‐test. Error bars indicate SD.
3.6. Knockdown of Beclin‐1 sensitizes NEK2‐OE KMS11 myeloma cells to bortezomib in vivo
NEK2‐OE KMS11 MM cells transduced with BECN1‐shRNA were injected subcutaneously into the left abdomen of 12 immunocompromised B‐NDG mice. The other 12 B‐NDG mice were injected with NEK2‐OE KMS11 MM cells transduced with scramble‐shRNA. Ten days after engraftment of the tumor cells, shRNA expression was induced by the addition of doxycycline (2 mg·mL−1) into the drinking water. Two days later, six mice selected randomly from the BECN1‐shRNA group or scramble‐shRNA group were treated with BTZ by intraperitoneal injection (1 mg·kg−1). As shown in Fig. 6A, similar tumor sizes are obtained in tumors expressing BECN1‐shRNA or scramble‐shRNA developed in nontreated mice; however, tumors expressing BECN1‐shRNA in BTZ‐treated mice are significantly smaller than those expressing scramble‐shRNA. Western blots confirmed that Beclin‐1 was indeed knocked down by addition of doxycycline in NEK2‐OE KMS11 MM cells (Fig. 6B). The volumes of tumors derived from each group were measured every 3 days after BTZ treatment. We found that knockdown of Beclin‐1 did not inhibit tumor growth, but significantly sensitized NEK2‐OE KMS11 MM cells to BTZ (Fig. 6C).
Figure 6.
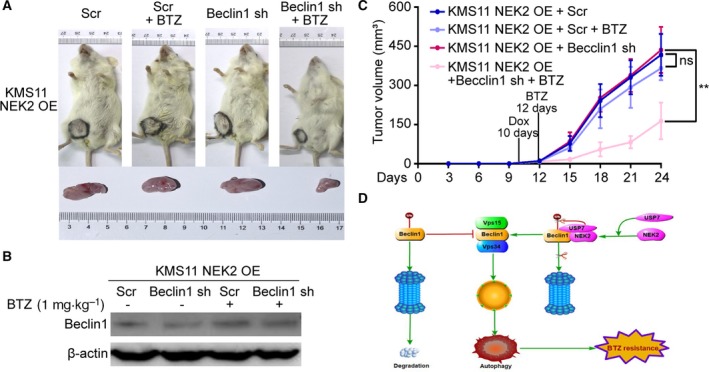
Knockdown of Beclin‐1 sensitizes NEK2‐OE KMS11 MM cells to BTZ in vivo. (A) KMS11 NEK2‐OE + Scr and KMS11 NEK2‐OE + BECN1‐shRNA MM cells were injected subcutaneously into the left abdomen of B‐NDG mice, and shRNA expression was induced by the addition of doxycycline to the drinking water about 10 days after injection of tumor cells. 2 days after addition of doxycycline, half mice of both groups were treated with BTZ (1 mg·kg−1). Differences in tumor size are shown between tumors transplanted with KMS11 NEK2‐OE + Scr or KMS11 NEK2‐OE + BECN1‐shRNA MM cells treated with or without BTZ. (B) Western blots showed Beclin‐1 is indeed knocked down in KMS11 NEK2‐OE + BECN1‐shRNA MM cells after BECN1‐shRNA expression. (C) Tumor volume assessments showed smaller tumor sizes in KMS11 NEK2‐OE + BECN1‐shRNA mice treated with BTZ than in other groups. (D) The model of our working hypothesis. ns P> 0.05, **P < 0.01. Significance was determined by Student’s t‐test. Error bars indicate SD.
4. Discussion
Multiple myeloma is an incurable hematological malignancy. Relapse resulted from drug resistance is the most important cause for the death of MM patients. The mechanisms of drug resistance have been explored for several years. In this study, we confirmed NEK2 promotes BTZ resistance in MM, which is consistent with our previous data (Zhou et al., 2013). Intriguingly, we also found NEK2 enhances autophagy through stabilizing Beclin‐1 by USP7‐mediated deubiquitination, which contributes to BTZ resistance in MM (Fig. 6D).
Multiple myeloma is a malignant plasma cell disease, which produces a mass of useless monoclonal immunoglobulin (Sirohi and Ray, 2004). Protein degradation systems that include ubiquitin–proteasome system and autophagy–lysosome system are essential for MM cells to maintain homeostasis. Thus, either ubiquitin–proteasome system or autophagy–lysosome system is a potential target for MM treatment. BTZ as the first proteasome inhibitor has achieved exciting treatment effect in MM therapy (Kane et al., 2003). However, the development of BTZ resistance limits its long‐term utility. A previous clinical trial indicated that the combination of BTZ and autophagy inhibitor is a promising therapeutic approach for patients with RMM (Vogl et al., 2014). In the present study, we found that NEK2 promotes autophagy in MM cells. Our data also showed that inhibition of autophagy by CQ sensitizes NEK2‐OE MM cell lines to BTZ. NEK2 is strongly associated with drug resistance and relapse in MM. At present, it is difficult to develop therapeutic approach through targeting NEK2 in MM because of a lack of specific NEK2 inhibitors. Thus, our study provides a potential novel treatment strategy in aggressive MM patients who express high NEK2 in tumor cells.
Autophagy is a multistep pathway requiring the interplay of numerous scaffolding and signaling molecules (Behrends et al., 2010; Green and Levine, 2014).The kinase mTOR is a critical regulator of autophagy induction, with activated mTOR suppressing autophagy, and vice versa (Jung et al., 2010). Beclin‐1‐Vps15‐Vps34 complex, the downstream target of mTOR, plays a central role in the initiation of autophagy (Ohashi et al., 2019). The expression of Beclin‐1 is regulated at the level of transcription, translation, and post‐translational modifications (Kang et al., 2011). Beclin‐1 serves not only as a key autophagic regulator with its specific interactors, but also as a potential therapeutic target in cancer (Fu et al., 2013; Toton et al., 2014). In this study, our MS and Co‐IP data showed NEK2 is bound to Beclin‐1. We also showed clearly that overexpression of NEK2 up‐regulates Beclin‐1 at protein level but not at mRNA expression. The phosphorylation of Beclin‐1 at Ser90, Ser93, and Ser96 was found to enhance autophagy (Menon and Dhamija, 2018). In our study, the change in phosphorylated Beclin‐1 at ser‐90, ser‐93, and ser‐96 was not observed in both NEK2‐overexpressed or down‐regulated MM cell lines. In addition, the expression of Beclin‐1‐interacting proteins Vps15 and Vps34 and the phosphorylation level of mTOR at ser‐2248, an indicator of mTOR intrinsic catalytic activity, were not regulated by NEK2 in MM cells. Importantly, knockdown of Beclin‐1 significantly inhibits NEK2‐induced autophagy in MM cells. Therefore, we conclude that NEK2 induces autophagy through up‐regulation of Beclin‐1 in MM cells.
Several deubiquitinases that include USP10, USP13, and USP19 were found to stabilize Beclin‐1 protein through deubiquitination (Boutouja et al., 2017), indicating that Beclin‐1 is regulated by ubiquitin–proteasome system. In this study, the USP7 inhibitor significantly decreases Beclin‐1 in NEK2‐OE MM cells, and the decrease in Beclin‐1 mediated by USP7 inhibitor can be blocked by MG132, suggesting that USP7 is a novel Beclin‐1 regulator, which stabilizes Beclin‐1 through deubiquitination. USP7 is known to regulate drug resistance by stabilizing its targets, which include a range of oncogenes and tumor suppressors (Chauhan et al., 2012; Song et al., 2008), NEK2 is one of the USP7 targets. Our data indicated that NEK2 binds to both Beclin‐1 and USP7, and we thus sought to determine whether NEK2 stabilizes Beclin‐1 through USP7‐mediated deubiquitination. The Co‐IPs showed overexpression of NEK2 decreases the ubiquitination of exogenous Beclin‐1, which is dramatically increased by USP7 inhibitor in the same cells. The ubiquitination of endogenous Beclin‐1 is also modulated by NEK2 in MM cells. These findings demonstrate NEK2 stabilizes Beclin‐1 through removing ubiquitins from Beclin‐1 by USP7. Interestingly, we also found knockdown of Beclin‐1 decreases NEK2 in both NEK2‐OE MM cells and controls. Liu et al have reported that Beclin‐1 is not only regulated by USP10 and USP13, but also affects the deubiquitination activity of USP10 and USP13 (Liu et al., 2011). In view of these data, we conclude that a regulatory network persists between NEK2, Beclin‐1, and USP7. Further studies are therefore necessary to uncover the underlying mechanisms by which NEK2, Beclin‐1, and USP7 regulate each other.
Previous studies have demonstrated inhibition of USP7 overcomes BTZ resistance in MM (Chauhan et al., 2012). Destabilizing NEK2 by USP7 inhibitor overcomes drug resistance to proteasome inhibitor in MM. USP7 binds to NEK2 and prevents NEK2 ubiquitination resulting in NEK2 stabilization. Increased NEK2 activates the canonical NF‐κB signaling pathway through the PP1α/AKT axis, which is the downstream targets of USP7‐NEK2 axis (Franqui‐Machin et al., 2018). In this study, we showed knockdown of Beclin‐1 prevents NEK2‐mediated BTZ resistance in MM both in vitro and in vivo. Thus, enhanced autophagy by up‐regulation of Beclin‐1 could be a novel mechanism by which the USP7‐NEK2 interaction induces BTZ resistance.
5. Conclusion
In summary, our findings demonstrate the interaction of NEK2 with USP7 enhances autophagy by stabilizing Beclin‐1 protein. Inhibition of autophagy significantly sensitizes NEK2‐OE MM cells to BTZ. Therefore, this study offers a promising novel therapeutic strategy to overcome NEK2‐induced drug resistance in MM.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
WZ and JX designed the research. JX, YH, BM, SC, YZ, and YW performed the experiments and analyzed the data. JZ, XW, QL, CK, and JG collected clinical samples. YS, XF, YG, LQ, GL, and GA provided technical assistance. JX wrote the manuscript. WZ and FZ critically revised the manuscript. All authors read and approved the final manuscript.
Supporting information
Fig. S1. NEK2 regulates Beclin‐1 at protein level but not affects its mRNA expression and phosphorylation.
Fig. S2. Beclin‐1 is regulated by proteasome inhibitors.
Table S1. Clinical characteristics of healthy donors and MM patients.
Table S2. The list of primer sequences.
Acknowledgements
The authors thank Professor Tiebang Kang (Collaborative Innovation Center for Cancer Medicine, Sun Yat‐sen University Cancer Center, Guangzhou, China) for providing Beclin‐1‐Flag vector and HA‐ubiquitin vector. We thank Professor Jiaxi Zhou for providing pLKO‐tet‐on vector (Institute of Hematology and Blood Diseases Hospital, China Academy of Medical Science, Tianjin, China). We thankfully acknowledge the Advanced Research Center at Central South University for technical support with TEM and analysis. This work was supported by grants from National Natural Science Foundation of China (81800209, 81570205, 81630007, and 81974010), China Postdoctoral Science Foundation (2018M640762), Postdoctoral Science Foundation of Central South University (198465), Hunan Province Natural Science Foundation of China (2019JJ50838), Ministry of Science and Technology of China (2018YFA0107800), Strategic Priority Research Program of Central South University (ZLXD2017004), and Open Sharing Fund for the Large‐scale Instruments and Equipments of Central South University (CSUZC201948, CSUZC201949).
Jiliang Xia and Yanjuan He contributed equally to this work
References
- Barbagallo F, Paronetto MP, Franco R, Chieffi P, Dolci S, Fry AM, Geremia R and Sette C (2009) Increased expression and nuclear localization of the centrosomal kinase Nek2 in human testicular seminomas. J Pathol 217, 431–441. [DOI] [PubMed] [Google Scholar]
- Behrends C, Sowa ME, Gygi SP and Harper JW (2010) Network organization of the human autophagy system. Nature 466, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutouja F, Brinkmeier R, Mastalski T, El‐Magraoui F and Platta HW (2017) Regulation of the tumor‐suppressor BECLIN 1 by distinct ubiquitination cascades. Int J Mol Sci 18, 2541–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan D, Tian Z, Nicholson B, Kumar KG, Zhou B, Carrasco R, McDermott JL, Leach CA, Fulcinniti M, Kodrasov MP et al (2012) A small molecule inhibitor of ubiquitin‐specific protease‐7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 22, 345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franqui‐Machin R, Hao M, Bai H, Gu Z, Zhan X, Habelhah H, Jethava Y, Qiu L, Frech I, Tricot G et al (2018) Destabilizing NEK2 overcomes resistance to proteasome inhibition in multiple myeloma. J Clin Invest 128, 2877–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu LL, Cheng Y and Liu B (2013) Beclin‐1: autophagic regulator and therapeutic target in cancer. Int J Biochem Cell Biol 45, 921–924. [DOI] [PubMed] [Google Scholar]
- Fu YF, Liu X, Gao M, Zhang YN and Liu J (2017) Endoplasmic reticulum stress induces autophagy and apoptosis while inhibiting proliferation and drug resistance in multiple myeloma through the PI3K/Akt/mTOR signaling pathway. Oncotarget 8, 61093–61106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L and Green DR (2019) Autophagy‐independent functions of the autophagy machinery. Cell 177, 1682–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR and Levine B (2014) To be or not to be? How selective autophagy and cell death govern cell fate. Cell 157, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Wang H, Xia J, Yang Y, Jin Z, Xu H, Shi J, De‐Domenico I, Tricot G and Zhan F (2015) Decreased ferroportin promotes myeloma cell growth and osteoclast differentiation. Cancer Res 75, 2211–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Xia J, Xu H, Frech I, Tricot G and Zhan F (2017) NEK2 promotes aerobic glycolysis in multiple myeloma through regulating splicing of pyruvate Kinase. J Hematol Oncol 10, 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Xia B and White E (2013) Autophagy‐mediated tumor promotion. Cell 155, 1216–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang B, Benavides A, Shi Y, Frost P and Lichtenstein A (2009) Effect of autophagy on multiple myeloma cell viability. Mol Cancer Ther 8, 1974–1984. [DOI] [PubMed] [Google Scholar]
- Iwashita H, Sakurai HT, Nagahora N, Ishiyama M, Shioji K, Sasamoto K, Okuma K, Shimizu S and Ueno Y (2018) Small fluorescent molecules for monitoring autophagic flux. FEBS Lett 592, 559–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarauta V, Jaime P, Gonzalo O, De Miguel D, Ramírez‐Labrada A, Martínez‐Lostao L, Anel A, Pardo J, Marzo I and Naval J (2016) Inhibition of autophagy with chloroquine potentiates carfilzomib‐induced apoptosis in myeloma cells in vitro and in vivo . Cancer Lett 382, 1–10. [DOI] [PubMed] [Google Scholar]
- Jin S, Tian S, Chen Y, Zhang C, Xie W, Xia X, Cui J and Wang RF (2016) USP19 modulates autophagy and antiviral immune responses by deubiquitinating Beclin‐1. EMBO J 35, 866–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung CH, Ro SH, Cao J, Otto NM and Kim DH (2010) mTOR regulation of autophagy. FEBS Lett 584, 1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Yamamoto A, Oshitani‐Okamoto S, Ohsumi Y and Yoshimori T (2004) LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form‐II formation. J Cell Sci 117, 2805–2812. [DOI] [PubMed] [Google Scholar]
- Kane RC, Bross PF, Farrell AT and Pazdur R (2003) Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 8, 508–513. [DOI] [PubMed] [Google Scholar]
- Kang R, Zeh HJ, Lotze MT and Tang D (2011) The Beclin1 network regulates autophagy and apoptosis. Cell Death Differ 18, 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocaturk NM, Akkoc Y, Kig C, Bayraktar O, Gozuacik D and Kutlu O (2019) Autophagy as a molecular target for cancer treatment. Eur J Pharm Sci 134, 116–137. [DOI] [PubMed] [Google Scholar]
- Kokuryo T, Senga T, Yokoyama Y, Nagino M, Nimura Y and Hamaguchi M (2007) Nek2 as an effective target for inhibition of tumorigenic growth and peritoneal dissemination of cholangiocarcinoma. Cancer Res 67, 9637–9642. [DOI] [PubMed] [Google Scholar]
- Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, Cai Y, Norberg HV, Zhang T, Furuya T et al (2011) Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 147, 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Wang Y, Xu H, Shi C, Jin F and Li W (2018) Profilin 1 induces drug resistance through Beclin1 complex‐mediated autophagy in multiple myeloma. Cancer Sci 109, 2706–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon MB and Dhamija S (2018) Beclin 1 phosphorylation at the center of autophagy regulation. Front Cell Dev Biol 6, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N and Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147, 728–741. [DOI] [PubMed] [Google Scholar]
- Navé BT, Ouwens M, Withers DJ, Alessi DR and Shepherd PR (1999) Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino‐acid deficiency on protein translation. Biochem J 344, 427–431. [PMC free article] [PubMed] [Google Scholar]
- Neal CP, Fry AM, Moreman C, McGregor A, Garcea G, Berry DP and Manson MM (2014) Overexpression of the Nek2 kinase in colorectal cancer correlates with beta‐catenin relocalization and shortened cancer‐specific survival. J Surg Oncol 110, 828–838. [DOI] [PubMed] [Google Scholar]
- Ohashi Y, Tremel S and Williams RL (2019) VPS34 complexes from a structural perspective. J Lipid Res 60, 229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palumbo A and Anderson K (2011) Multiple myeloma. N Engl J Med 364, 1046–1060. [DOI] [PubMed] [Google Scholar]
- Roy M, Liang L, Xiao X, Peng Y, Luo Y, Zhou W, Zhang J, Qiu L, Zhang S, Liu F et al (2016) Lycorine downregulates HMGB1 to inhibit autophagy and enhances bortezomib activity in multiple myeloma. Theranostics. 6, 2209–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirohi B and Ray P (2004) Multiple myeloma. Lancet 363, 875–887. [DOI] [PubMed] [Google Scholar]
- Song MS, Salmena L, Carracedo A, Egia A, Lo‐Coco F, Teruya‐Feldstein J and Pandolfi PP (2008) The deubiquitinylation and localization of PTEN are regulated by a HAUSP‐PML network. Nature 455, 813–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Kokuryo T, Senga T, Yokoyama Y, Nagino M and Hamaguchi M (2010) Novel combination treatment for colorectal cancer using Nek2 siRNA and cisplatin. Cancer Sci 101, 1163–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toton E, Lisiak N, Sawicka P and Rybczynska M (2014) Beclin‐1 and its role as a target for anticancer therapy. J Physiol Pharmacol 65, 459–467. [PubMed] [Google Scholar]
- Tsunoda N, Kokuryo T, Oda K, Senga T, Yokoyama Y, Nagino M, Nimura Y and Hamaguchi M (2009) Nek2 as a novel molecular target for the treatment of breast carcinoma. Cancer Sci 100, 111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogl DT, Stadtmauer EA, Tan KS, Heitjan DF, Davis LE, Pontiggia L, Rangwala R, Piao S, Chang YC, Scott EC et al (2014) Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 10, 1380–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Li W, Liu N, Zhang F, Liu H, Liu F, Liu J, Zhang T and Niu Y (2012) Nek2A contributes to tumorigenic growth and possibly functions as potential therapeutic target for human breast cancer. J Cell Biochem 113, 1904–1914. [DOI] [PubMed] [Google Scholar]
- Xia J, Franqui‐Machin R, Gu Z and Zhan F (2015) Role of NEK2A in human cancer and its therapeutic potentials. Biomed Res Int 2015, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Xu H, Zhang X, Allamargot C, Coleman KL, Nessler R, Frech I, Tricot G and Zhan F (2017) Multiple myeloma tumor cells are selectively killed by pharmacologically‐ dosed ascorbic acid. EBioMedicine 18, 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Zhou W, Xia J, Gu Z, Wendlandt E, Zhan X, Janz S, Tricot G and Zhan F (2014) NEK2 mediates ALDH1A1‐dependent drug resistance in multiple myeloma. Oncotarget 5, 11986–11997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Pang Y, Ma C, Li J, Wang H and Shao Z (2018) ClC5 decreases the sensitivity of multiple myeloma cells to bortezomib via promoting prosurvival autophagy. Oncol Res 26, 421–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, He J, Liu Z, Lu Y, Zheng Y, Li H, Xu J, Liu H, Qian J, Orlowski RZ et al (2015) Anti‐β2‐microglobulin monoclonal antibodies overcome bortezomib resistance in multiple myeloma by inhibiting autophagy. Oncotarget 6, 8567–8578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Li W, Wang C, Leng X, Lian S, Feng J, Li J and Wang H (2014) Inhibition of autophagy enhances apoptosis induced by proteasome inhibitor bortezomib in human glioblastoma U87 and U251 cells. Cancer Lett 385, 265–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Yang Y, Xia J, Wang H, Salama ME, Xiong W, Xu H, Shetty S, Chen T, Zeng Z et al (2013) NEK2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell 23, 48–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. NEK2 regulates Beclin‐1 at protein level but not affects its mRNA expression and phosphorylation.
Fig. S2. Beclin‐1 is regulated by proteasome inhibitors.
Table S1. Clinical characteristics of healthy donors and MM patients.
Table S2. The list of primer sequences.