

Abstract
As genome sequencing methodologies become more sensitive to the detection of low frequency rare variant events, the link between post-zygotic mutagenesis and somatic mosaicism in the etiology of several human genetic conditions other than cancers has become more clear. Since current clinical genomics diagnostic methods have limited detection sensitivity for mosaic events, a CNV deletion inherited from a low-level (< 10%) mosaic parent can be erroneously interpreted in the proband to represent a de novo germline event. Here, we describe three sensitive, precise, and cost-efficient methods: droplet digital PCR (ddPCR), PCR amplicon-based next generation sequencing (NGS), and quantitative PCR that can quantitatively assess the potential degree of parental somatic mosaic levels for CNV deletions. ddPCR using the EvaGreen fluorescent dye protocol can specifically quantify the deleted or non-deleted alleles by analyzing the number of droplets positive for a fluorescent signal for each event. PCR amplicon-based NGS assesses the allele frequencies of a heterozygous SNP within a deletion region. The difference in number of reads between the two genotypes indicates the level of somatic mosaicism for the CNV deletion. Quantitative PCR can be applied where the relative quantity of the deletion-junction specific product represents the level of mosaicism. Clinical implementation of these quantitative variant detection methods enables potentially more accurate assessment of a disease recurrence risk in family-based genetic counseling, allowing couples more informed family planning.
Single nucleotide variants (SNVs) or copy-number variants (CNVs) can arise during the approximately 1016 mitotic post-zygotic cell divisions required for organismal development and can result in different cell populations with distinct genotypes (Iourov et al., 2010; Lupski, 2013). The subsequent generation of these mosaic populations can be exclusive to the germline, somatic cells, or a combination of the two, i.e. gonosomal (Biesecker and Spinner, 2013). Recently, a growing body of evidence implicates the importance of somatic mosaicism in the pathogenesis of several human genetic diseases (Erickson, 2013; Biesecker and Spinner, 2013; Lim et al., 2017). Carriers of pathogenic mosaic variants, whether they are clinically affected or not, can transmit it to their children if the variant is also present in their germline cells. In 2014, Campbell et al. reported identifying low-level (<10%) parental somatic mosaicism for CNV deletions in four of 100 unrelated families (Campbell et al., 2014a). Application of genome sequencing (GS) in families with multiple siblings revealed that in the parental germline, 3.8% of mutations were mosaic for SNVs, resulting in 1.3% of mutations being shared by siblings (Rahbari et al., 2016). The level of somatic mosaicism in the parents has also been shown to correlate with overall recurrence risk (Campbell et al., 2014b; Rahbari et al., 2016; Jonsson et al., 2018).
In a diagnostic setting, CNVs are routinely detected using chromosomal microarray analysis (CMA) with either single nucleotide polymorphism (SNP) arrays or array comparative genomic hybridization (aCGH). The minimum level to detect a mosaic CNV using one of these methods ranges from 5% to 30% (Notini et al., 2008). If the level of a somatic mosaic deletion CNV in a parent is lower than this limit, the variant in the proband may be erroneously interpreted to represent a de novo mutational event.
In an effort to better assess the level of CNV mosaicism in a clinical setting, we queried the diagnostic CMA database at Baylor Genetics (BG) laboratories to select unrelated family trios with CNV deletions reported as pathogenic, likely pathogenic, or of uncertain significance with the inheritance status determined by CMA as apparently de novo. Based on CMA results, we then designed CNV deletion-specific primers targeting the deletion junctions in 50 trios using long-range PCR. The deletion breakpoints were determined by Sanger sequencing. Using this method, we identified four parents with the same CNV deletion as their child, indicating that the CNV deletions were present in a mosaic state in the parents below the level of detection of the CMA. Herein, we describe three cost-efficient assay methods by which the level of mosaicism for CNV deletions in somatic cells can be potentially more accurately quantified.
BASIC PROTOCOL
DROPLET DIGITAL PCR (ddPCR)
Based on our experience, ddPCR using the EvaGreen fluorescent dye is a robust method to assess the precise level of somatic mosaicism for a specified locus variant. A DNA sample is randomly distributed into ~20,000 uniform nanoliter-sized droplets in which an independent PCR reaction occurs within each droplet for both the patient and control samples. After PCR amplification, each droplet is analyzed for a fluorescent signal, indicating whether the targeted DNA region is present or not. Positive and negative droplets are quantified, and the concentration of target DNA is calculated as copies per microliter (μl) using the Poisson statistical model, allowing for a determination of the relative level of a specified variant in each sample (Hindson et al., 2011; Pinheiro et al., 2012).
Materials
Genomic DNA derived from the proband, parent, and an unrelated human control
-
Custom forward and reverse DNA oligo PCR primers designed using the Primer3 (v.0.4.0) online tool (http://bioinfo.ut.ee/primer3-0.4.0/. Koressaar and Remm, 2007; Untergasser et al., 2012; Koressaar et al., 2018)
Note: Primer sizes range between 18–30 bp. Primer melting temperature (Tm) ranges from 57˚C to 63˚C and GC content is between 40% and 60%. Suggested amplicon length is 80–250 bp (see Figure 1 for the design details of the primers’ locations).
DX200™ ddPCR™ EvaGreen Supermix (Bio-Rad, cat. no. 1864034): containing all components required for ddPCR except primers and template
Restriction enzyme for the fragmentation of the template >66 ng per reaction: HaeIII (New England Biolabs, NEB, cat. no. R0108S), MseI (NEB, cat. no. R0525S), AluI (NEB, cat. no. R0137S), HindIII (NEB, cat. no. R3104S), and CviQI (NEB, cat. no. R0639S) are recommended by Bio-Rad.
Eppendorf™ 96-Well twin.tec™ PCR Plate, semi-skirt (Fisher Scientific, cat. no. E951020320)
Sartorius™ Low Retention SafetySpace™ Filter Tips (Fisher Scientific, cat. no. 14-557-695)
Automated Droplet Generation Oil for EvaGreen (Bio-Rad, cat. no. 1864112)
DG32™ Automated Droplet Generator Cartridges (Bio-Rad, cat. no. 1864109)
PCR Plate Heat Seal, foil, pierceable (Bio-Rad, cat. no. 1814040)
ddPCR™ Droplet Reader Oil (Bio-Rad, cat. no. 1863004)
Figure 1.
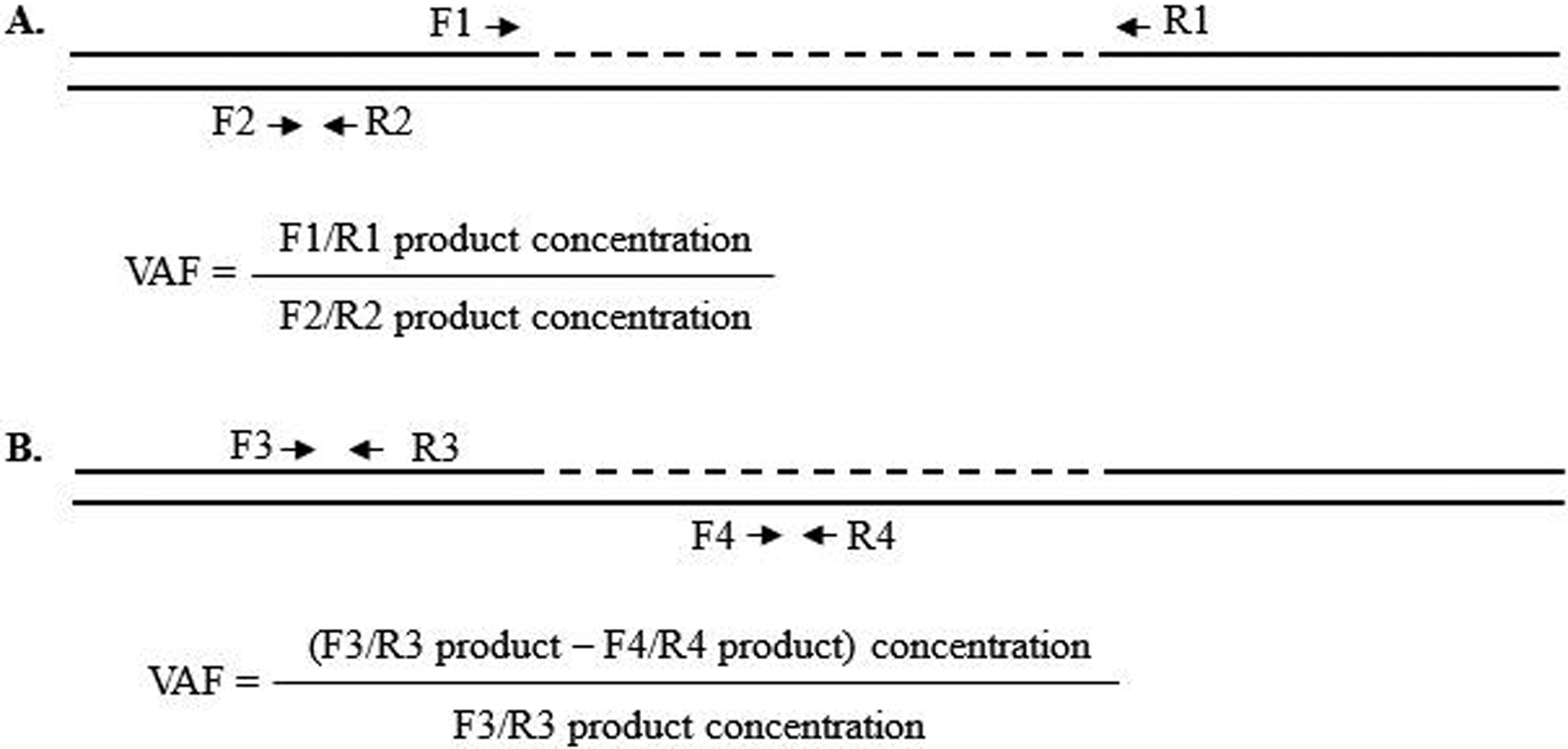
Primer design for ddPCR using EvaGreen fluorescent dye. Suggested amplicon length for each primer set is 80–250 bp. Dashed line indicates the deleted region. A. Preferred design: In one ddPCR reaction, deletion junction-specific primers F1 (forward 1) and R1 (reverse 1) are designed to amplify the allele with the deletion. In another reaction, primers (F2 and R2) are used to amplify both alleles. Variant allele frequency (VAF) is the fraction of the deleted allele when comparing both alleles. B. Often the breakpoint of a deletion locates within a repetitive element, which makes it hard to design deletion junction-specific primers with a product less than 250 bp. In these cases, primers F3 and R3 are designed to amplify both alleles in one reaction, while F4 and R4 are used to amplify the WT allele in another reaction. The difference between the two products is the amount of the variant allele, which is compared to the concentration of all alleles to calculate the VAF.
Equipment
Centrifuge for 96-well plate
PCR thermal cycler capable of adjusting ramp speed
QX200 AutoDG Droplet Digital PCR System, including Automated Droplet Generator (Bio-Rad, cat. no. 1864101), Droplet Reader (Bio-Rad, cat. no. 1864003), and QuantaSoft Analysis Pro Software (Bio-Rad)
PX1 PCR Plate Sealer (Bio-Rad, cat. no. 1814000)
Steps and annotations
- Choose a restriction enzyme for each ddPCR reaction:
- Go to NEBcutter V2.0 website (http://nc2.neb.com/NEBcutter2/, Vincze et al., 2003).
- Paste and submit the PCR product sequence.
- Click “0 cutters” (enzymes that do not cut) from the “List”.
- Choose one of the enzymes recommended by Bio-Rad.
- Prepare ddPCR reaction in an Eppendorf™ 96-Well twin.tec™ PCR Plate. Each 20 μl reaction contains:
- 10 μl of DX200™ ddPCR™ EvaGreen Supermix
- 1 μl of 5 μM forward primer
- 1 μl of 5 μM reverse primer
- 5 units of restriction enzyme
- 100 ng genomic DNA (may use as low as 25 ng)
- Add RNase-/DNase-free water to a total volume of 20 μl.
- Mix thoroughly by vortexing (in Eppendorf tube) or pipetting (in plate). Incubate at room temperature (RT) for 3 minutes, then apply 20 μl to each well in the PCR plate. Any unused wells in a column must be filled with 1x ddPCR buffer control. Centrifuge the plate at 1,000 g for 30 sec (aluminum foil may be used to temporarily cover the plate).
- Droplet generation is performed using the QX200 Automated Droplet Generator. To do so:
- Load the bottle of Automated Droplet Generation Oil for EvaGreen and press the “Oil Type” icon to select EvaGreen as the type of oil loaded.
- Configure sample plate by selecting columns.
- Set up cartridges, pipet tips and plates as the machine indicates (all indicator lights on the deck should turn green).
-
Press the “Start Droplet Generation” icon.Note: This step can also be performed by a manual droplet generator, such as QX200 Droplet Generator (Bio-Rad, cat. no. 1864002), which is cheaper than the automated machine. The rest of the protocol would be identical.
When the droplet generation is completed, carefully remove the droplet plate and transfer it to the PX1 PCR Plate Sealer. Place one piece of PCR Plate Heat Seal foil on top of the plate. Seal the plate at 180˚C for 5 seconds.
- Transfer the sealed plate to a thermal cycler. Use a heated lid set to 105˚C and set the sample volume to 40 μl. Perform PCR using the following amplification protocols:
- When using a newly designed primer set, the optimal annealing/extension temperature should be determined by performing a thermal PCR gradient and subsequent droplet analysis with the QuantaSoft software suite. The determination criteria are described in procedure 6. Using the corresponding proband’s sample as template, prepare a master mix including all components for 8 reactions. Load up to 8 wells in the same column. The PCR conditions to determine initial annealing/extension temperatures are:
- 1 cycle: 95˚C for 5 min (enzyme activation)
- 40 cycles: 95˚C for 30 sec (denaturation)
- 50–65˚C gradient for 1 min (annealing/extension)
- 1 cycle: 4˚C for 5 min (signal stabilization)
- 90˚C for 5 min (signal stabilization)
- Hold: 12˚C infinite
- Ramp rate at 2˚C/sec for all steps.
- After the optimal annealing/extension temperature is determined for a primer set, prepare a master mix including all components except for the template. In the same column, use the proband’s sample as template in 3 wells (triplicates), parental sample in 3 wells, and DNAs from an unrelated control and no template control each in single wells. Perform PCR using the same condition as described in 4.1 except that the optimal temperature (instead of gradient) is applied at the annealing/extension step.
- Transfer the sealed PCR plate to the DX200 Droplet Reader. Open the QuantaSoft software to set up a new plate:
- Double click on a well to open the well editor dialog box.
- Input the sample name (use the same name for duplicates or triplicates).
- Select “ddPCR EvaGreen supermix” as the supermix type.
- Select “ABS” for “Experiment”.
- Select “unknown” for both Channel 1 and Channel 2 “Target”.
- Apply to well and press “OK”.
- Complete this for all wells in which a reaction is to take place.
- Once the plate layout is complete, save the file to the designated folder.
- Select “Run” to begin the droplet reading.
- When the run is complete, data are saved as a QLP file. To analyze the data by QuantaSoft Analysis Pro Software:
- For gradient ddPCR, open the software, select a well, and examine Channel 1 of the 1-D fluorescence amplitude plot, which shows all accepted droplets for the selected well. Thresholds may be established automatically or manually if there are two distinguishable clusters of droplets. Negative droplets are displayed in grey (lower cluster), while positive droplets in Channel 1 are blue (upper cluster). Choose the well(s) that clearly define negative and positive droplets and find the corresponding annealing/extension temperature(s) on the thermal cycler as the optimal temperature(s). The well corresponding to the greatest separation (amplitude change) between negative and positive droplets is the ideal temperature and usually more than one temperature will work. Click on the “Concentration” tab to read the concentration (measured in copies/μl) the target sequence for each well. For each of the proband’s sample, calculate the variant allele frequency (VAF) as described in Figure 1. The VAF in each proband is expected to be close to 50% as would be expected for a typical heterozygous variant.
- For the ddPCR reactions performed with the optimal annealing temperatures, open the software and select one column that used the same primer set. Examine Channel 1 of the 1-D fluorescence amplitude plot. Set the threshold automatically or manually based on the wells that amplified the proband’s sample. The same threshold should be established uniformly for the remainder of the samples in the column. The average concentration for each duplicate or triplicate can be automatically counted by selecting “Merged Wells”. Identify the concentrations and calculate the VAFs for each sample. The VAF for each proband is expected to be close to 50%, while in the unrelated control should be close to 0%. The final VAF in a parental sample is the level of mosaicism for the specified CNV deletion being assessed by ddPCR.
ALTERNATE PROTOCOL 1
PCR AMPLICON-BASED NEXT GENERATION SEQUENCING
If a mosaic parental sample harbors a heterozygous SNP within a region of deletion, fractions of the two genotypes can be used to properly assess the level of mosaicism for the deleted region (Summerer et al., 2019). PCR amplicon-based next generation sequencing (NGS) can be applied to measure the two allele frequencies of the SNP. Using this method, individual template molecules are tagged and sequenced to enable detection of different variants (Metzker, 2010; van Dijk et al., 2014).
Materials
Genomic DNA derived from the parental samples
5 U/μl recombinant Taq DNA Polymerase, 10X Taq Buffer with (NH4)2SO4 and 25 mM MgCl2 (ThermoFisher Scientific, cat. no. EP0405)
10 mM dNTP Mix (ThermoFisher Scientific, cat. no. R0192)
Forward and reverse PCR primers designed by Primer3 (v.0.4.0) online tool using the same criteria as previously described in the ddPCR method. Amplicon sizes range from 150 bp to 225 bp. The SNP should map in the middle of the proposed amplified sequence.
UltraPure™ agarose (ThermoFisher Scientific, cat. no. 16500500)
10x TBE buffer, 0.2 μm filtered (ThermoFisher Scientific, cat. no. AM9863)
10 mg/ml UltraPure™ Ethidium Bromide (EtBr, ThermoFisher Scientific, cat. no. 15585011)
GeneRuler DNA Ladder Mix and 6x TriTrack DNA Loading Dye (ThermoFisher Scientific, cat. no. SM0331)
Exonuclease I (20 U/μl, ThermoFisher Scientific, cat. no. EN0582)
FastAP Thermosensitive Alkaline Phosphatase (1 U/μl, ThermoFisher Scientific, cat. no. EF0654)
PCR purification kit using columns, e.g., QIAquick PCR Purification Kit (Qiagen, cat. no. 28104)
Qubit™ dsDNA BR Assay Kit (ThermoFisher Scientific, cat. no. Q32850)
Qubit™ Assay Tubes (ThermoFisher Scientific, cat. no. Q32856)
Equipment:
PCR thermal cycler
Agarose gel electrophoresis system
Conventional microcentrifuge that accelerates to 14,000 rpm
Qubit™ 4 Fluorometer (ThermoFisher Scientific, cat. no. Q33226)
Steps and annotations
Using the UCSC Genome Browser (https://genome.ucsc.edu/), select 3–5 common SNPs within the deletion region, which each have a minimum allele frequency greater than 0.3.
- Prepare PCR reaction to amplify the region containing each SNP. Each 20 μl reaction contains:
- 2 μl of 10X Taq Buffer with (NH4)2SO4
- 1.2 μl of 25 mM MgCl2
- 2 μl of 2 mM dNTP mix
- 1 μl of 10 μM forward primer
- 1 μl of 10 μM reverse primer
- 0.1 μl of Taq DNA Polymerase
- 50 ng genomic DNA
- Add RNase-/DNase-free water to a total volume of 20 μl.
- Perform PCR using the following condition:
- 1 cycle: 95˚C for 3 min (initial denaturation)
- 35 cycles: 95˚C for 30 sec (denaturation)
- 5˚C lower than the primer Tm for 30 sec (annealing)
- 72˚C for 30 sec (extension)
- 1 cycle: 72˚C for 5 min (final extension)
- Hold: 4˚C infinite
Prepare a 1% agarose gel solution in 1x TBE buffer in a microwavable flask. Mix by swirling and heat in a microwave oven until the agarose is completely dissolved. Let the solution cool to room temperature for 5 min. Add 5 μl EtBr to every 100 ml agarose solution. Mix thoroughly by swirling. Pour the solution into a gel tray with a thin well comb. Wait until the gel is completely solidified and then remove the comb.
Add 1 μl of the 6x TriTrack DNA Loading Dye (three colors) to 5 μl of the PCR product. Load each of the samples and a GeneRuler DNA Ladder Mix into the wells. Run the gel in 1x TBE buffer until the lowest dye reaches the middle of the gel. Use UV light to visualize the DNA fragments. A specific PCR product should show a single band of the designed size corresponding to the amplified region.
To purify the amplicon for Sanger sequencing, add 0.5 μl of Exonuclease I and 1 μl of FastAP Thermosensitive Alkaline Phosphatase to 5 μl of PCR product in a PCR tube. Mix by vortexing and briefly centrifuge. Use a thermal cycler to incubate the mixture at 37˚C for 20 min to allow for primer degradation and dNTP dephosphorylation followed by 85˚C for 15 min to deactivate the enzymes.
Send the purified samples for Sanger sequencing using either the forward or reverse primer as the sequencing primer. Sequencher 5.4.6 software (Gene Codes Corporation) can be used to visually analyze the Sanger sequencing chromatogram. The guidance for Sanger DNA analysis using Sequencher software can be found at https://www.genecodes.com/training/tutorials. If a SNP is heterozygous, two peaks representing different nucleotides with comparable heights would present at the same position. When a SNP is determined to be heterozygous, proceed to the next step.
Amplify the heterozygous SNP in a 120–150 μl reaction using the same PCR gradients (proportionally adjust the amount) and condition as described previously in steps 2 and 3. Use separate PCR tubes if the reaction volume exceeds the thermal cycler capacity. Check the agarose gel to confirm the PCR amplification is specific for the proposed region.
- Purify the PCR product using a PCR purification kit with columns. Basic protocol includes:
- Add binding buffer to the PCR reaction.
- Load to the column to bind DNA.
- Centrifuge to remove non-DNA components.
- Wash the column with buffer containing ethanol.
- Centrifuge to remove residual non-DNA components and wash buffer.
- Elute DNA with elution buffer (30–50 μl).
Qubit™ dsDNA BR (broad range) Assay is specific for double-stranded DNA (dsDNA). This method can accurately measure initial sample concentration from 100 pg/μl to 1,000 ng/μl. First, dilute the Qubit™ reagent 1:200 in Qubit™ buffer to prepare the working solution sufficient to accommodate all standards and samples. Two DNA standards, Standard 1 and Standard 2 from the Qubit™ dsDNA BR Assay kit, are used to calibrate the assay. For each DNA standard, add 10 μl of the standard to 190 μl of working solution in a Qubit™ Assay Tube to a final volume of 200 μl. Add 1–2 μl purified PCR product to the working solution in a Qubit™ Assay Tube to a final volume of 200 μl. Label the top of the Qubit™ assay tubes with the corresponding sample name as labeling on the side could interfere with the sample read. Vortex tubes for 2–3 seconds, incubate at room temperature for 2 min and read standard 1, then standard 2 using the Qubit™ 4 fluorometer. Insert sample tubes one by one to read the sample concentrations. The amount of DNA required for PCR amplicon-based NGS differs among companies that provides the service, ranging from 0.5 μg to 3 μg with a minimum concentration requirement.
Send the purified PCR product for PCR amplicon-based NGS on a HiSeq or NovaSeq platform with 150 bp paired-end (PE) reads.
Standard bioinformatics can be provided by the same company and a BAM file (data file) generated for each sample. Integrative Genomics Viewer (IGV) software is used to visually interpret the data (available for free downloading at https://software.broadinstitute.org/software/igv/download. Robinson et al., 2011; Thorvaldsdottir et al., 2013). Open both the BAM file (.bam) and its index file (.bam.bai) to view data for a specified sample. Select the genome version and input the variant coordinate. The software will display the sequence flanking the variant. Read the result at the variant position, which shows the total counts and the counts of each genotype.
Read the counts of the two genotypes of the heterozygous SNP. The genotype with more counts represents the allele without deletion, while the genotype with less counts represents the allele with a mosaic deletion. The relative level of mosaicism for the original deletion for which the SNP is embedded may be estimated as the difference in number of reads between the two genotypes divided by 2 times the more frequent genotype reads.
ALTERNATE PROTOCOL 2
QUANTITATIVE REAL-TIME PCR (qPCR)
Similar to the EvaGreen ddPCR protocol, qPCR monitors incorporation of SYBR green dye into dsDNA. During the exponential amplification phase of the PCR reaction, the quantity of the target DNA product doubles for each PCR cycle, leading to a two-fold increase of fluorescence intensity (Wittwer et al., 2013). Relative quantification of the target DNA product (in our case, the deletion junction-specific DNA fragment) in each sample is calculated based on the internal reference genes. By comparing the fold-change differences between the parental and proband samples, the relative level of mosaicism in the parental sample can be determined.
Materials
Genomic DNA derived from samples from the proband, parent, and an unrelated human control.
PowerUp™ SYBR™ Green Master Mix (ThermoFisher Scientific, cat. no. A25779). SsoAdvanced™ Universal SYBR® Green Supermix (Bio-Rad, cat. no. 1725270) can be used instead according to manufacturer’s protocol.
Forward and reverse PCR primers designed by Primer3 online tool. Primer Tm ranges from 60˚C to 63˚C and GC content is between 40% and 60%. Optimal amplicon size is 70–200 bp. Longer product (but < 500bp) may work with appropriate reaction condition. Primers used for each family include CNV deletion junction-specific primers and primers amplifying 1–2 internal reference genes (e.g., GAPDH and ACTB).
Hard-Shell® 96-Well PCR Plates, thin wall (Bio-Rad, cat. no. HSP9601)
Microseal ‘B’ PCR Plate Sealing Film (Bio-Rad, cat. no. MSB1001)
Equipment:
CFX Connect Real-Time PCR Detection System (Bio-Rad, cat. no. 1855201). Real-time thermal cycler from other manufacturer may be used.
Steps and annotations
- Prepare qPCR reactions. Each 20 μl reaction contains:
- 10 μl of PowerUp™ SYBR™ Green Master Mix
- 1 μl of 5 μM forward primer
- 1 μl of 5 μM reverse primer
- 50 ng genomic DNA
-
Add RNase-/DNase-free water to a total volume of 20 μl.Note: Triplicate is recommended for each reaction.
Transfer the reactions to a Hard-Shell® 96-Well PCR Plate. Seal the plate with a Microseal ‘B’ PCR Plate Sealing Film. Place the reaction plate in the real-time thermal cycler.
- Input the sample information in Bio-Rad CFX Maestro software. Perform qPCR using the following condition:
- 1 cycle: 95˚C for 10 min (Taq Enzyme Activation)
- 40 cycles: 95˚C for 15 sec (denaturation)
- 60˚C for 1 min (annealing/extension)
- Calculate the relative quantity of the deletion junction-specific product in the parental sample as 50% (2−ΔΔCq). The step-by-step calculations are:
- Read the quantitation cycle (Cq) value for each reaction.
- Calculate the average Cq of each triplicate and use it for the subsequent calculation.
- Calculate the difference in Cq between the deletion junction-specific amplification and the reference gene amplification. ΔCq = (Cq of deletion junction-specific amplification) – (Cq of reference gene amplification).
- Calculate the difference in ΔCq between the parental and proband’s samples. ΔΔCq = (ΔCq in parental sample) – (ΔCq in proband’s sample).
-
Calculate the value of 2−ΔΔCq to get the amplification fold-change. The value in the proband’s sample would be 20 = 1.Note: Since the proband is heterozygous for the CNV deletion, the default quantity of the deleted allele should be approximately 50% in the proband’s sample. Calculate the relative quantity of the deleted allele in the parental sample by 50% times 2−ΔΔCq. This value represents the level of mosaicism.
(Optional) Check the specificity and density of the qPCR products by running on an agarose gel.
COMMENTARY
Background Information
Due to inherent detection limitations of many routine genetic tests performed in clinical diagnostic laboratories, a substantial fraction of families carrying pathogenic SNVs or CNVs may be erroneously determined to be non-mosaic. We have demonstrated that the true inheritance status of CNV deletions can be determined by long-range PCR (Campbell et al., 2014). Because the level of somatic mosaicism correlates with disease recurrence risk, it is important to assess the level of mosaicism for a specified variant using a more sensitive and quantitative test. These data can provide more information to parents relating to genetic counseling and recurrence risk information regarding choices for family planning purposes. At the same time, the test should be easy to design and perform, as well as time and cost efficient.
Critical Parameters
Herein we describe three quantitative methods to evaluate the levels of somatic mosaicism for CNV deletions in parental samples with our primary method being ddPCR. Though the experimental design is similar to traditional PCR, data analysis may require some training from the Bio-Rad representative or from an experienced researcher. Use of the ddPCR system and potential problems are described in Bio-Rad’s manual in great detail. At least 300 ng of proband DNA is required for the gradient PCR and as a positive control. To apply this method, special equipment including the ddPCR droplet generator, plate reader and PCR plate sealer are required. For testing each family, the consumable materials cost is approximately $160.
PCR amplicon-based NGS on the other hand does not require special equipment in the laboratory. The work primarily involves standard PCR amplification and DNA purification as well as concentration measurements. Although the deletion regions are often large and a great number of SNPs may be present within a deletion region, considering the cost and time, we only screen five SNPs for heterozygosity in each sample and find heterozygous SNPs in two out of four parental samples. To sequence one amplicon in each family, the total cost is approximately $150.
The qPCR experimental design is less strict than ddPCR in that the amplicon size can go up to 500 bp, which makes it more feasible to design deletion junction-specific primers. According to Bio-Rad, the EvaGreen dye has less background, is less inhibitory in the PCR reaction and more stable compared to the SYBR® Green dye. Furthermore, ddPCR uses absolute values, which is generally more precise compared to the relative value used in qPCR. The qPCR method will require a Real-Time PCR detection system. The cost for testing one family by qPCR is less generally less than $40.
These three methods represent robust protocols to successfully assess the levels of parental mosaicism for CNV deletions in most instances. However, because each CNV deletion is unique, there is a chance that none of these methods will work.
Troubleshooting
When preforming a ddPCR experiment, a common problem that can occur is when the positive and negative droplets are not clearly separated in the 1-D plot. To ensure that a ddPCR reaction generates true signals for the target amplicon, the efficiency and specificity of each primer set should be tested by standard PCR before using those primers in a ddPCR reaction. Performing a thermal gradient to determine the optimal annealing temperature when using newly designed primers is also crucial when starting a new experiment. The optimal annealing temperature may be different in a ddPCR versus traditional PCR reaction even when using the identical primer sets. Furthermore, loading an accurate total amount of DNA for each sample is critical for proper experimental interpretation.
For PCR amplicon-based NGS, specificity of the PCR product is critical for obtaining an accurate result, and must be checked by agarose gel electrophoresis. If a sufficient amount of DNA is not generated after PCR amplification, a second-round amplification can be performed using 1 μl of the purified PCR product as template with 25–30 cycles of denaturation, annealing and extension. The purified PCR products from both amplifications can then be combined and sent for PCR amplicon-based NGS. The final VAF calculation will not be affected by the re-amplification step.
For qPCR, it is also recommended to test the primer sets by standard PCR first to ensure efficiency and specificity. If using PowerUp SYBR Green Master Mix, general troubleshooting can be found on ThermoFisher website at https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0013511_PowerUp_mastermix_UG.pdf.
Understanding and Interpreting Results
Using ddPCR, we were able to detect varying levels of mosaicism in 3 out of 4 samples that were tested. Our experience indicates that this method is suitable for mosaicism levels above 5% though one may be able to visualize levels as low as 1%. To evaluate the samples better, the two primer sets for each sample should have equal efficiency which can be confirmed by obtaining a VAF close to 50% in the proband’s sample (which is presumed to be a normal heterozygous call).
In our PCR amplicon-based NGS, we examined three SNPs in each of the two samples. The average level of the somatic mosaicism in one sample was determined to be 15.7%, which is comparable to the ddPCR result. The results among the three SNPs in this sample were similar and the standard deviation was 1.7%. The other sample, in which the somatic mosaicism was undetectable by ddPCR, showed variable results (0.7%, 2.3% and 7.3%), indicating that PCR amplicon-based NGS may not be reliable for very low-level mosaicism.
We utilized qPCR to test parental samples with very low-level mosaicism using GAPDH as an internal control. Using this method, we determined the VAF as 0.4%. The gel image showed that the qPCR product was specific and weaker in the parental sample when compared to the proband’s sample. This result indicates that qPCR is useful for detecting very low-level mosaicism (<1%) if the deletion junction-specific primers are available.
Time Considerations
To perform ddPCR, 1–2 days are sufficient to finish all the procedures and obtain results. Preparing samples for PCR amplicon-based NGS takes 1 day, but screening the SNPs may take up to 3 days and NGS by the company of choice usually takes 3–6 weeks. The qPCR method of mosaic detection takes about 1 day.
Acknowledgements
This work was financially supported by the NIH grant R01 1HD087292 (P. S.). This work was also funded in part by the US National Human Genome Research Institute (NHGRI)/National Heart Lung and Blood Institute (NHLBI) grant UM1HG006542 to the Baylor-Hopkins Center for Mendelian Genomics (BHCMG), National Institute of Neurological Disorders and Stroke (NINDS) R35 NS105078, and National Institute of General Medical Sciences (NIGMS) GM106373 to J.R.L.
Footnotes
Conflict of Interests
JRL has stock ownership in 23andMe, is a paid consultant for Regeneron Pharmaceuticals and Novartis, and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from the chromosomal microarray analysis (CMA), clinical exome sequencing and GS offered in the Baylor Genetics (BG) Laboratory (https://www.baylorgenetics.com/). JRL serves on the SAB of BG. WB receives partial compensation from BG as part of a professional services agreement. The remaining authors declare that they have no conflict of interests.
Literature Cited
- Biesecker LG, and Spinner NB (2013). A genomic view of mosaicism and human disease. Nat. Rev. Genet, 14:307–320. doi: 10.1038/nrg3424. [DOI] [PubMed] [Google Scholar]
- Campbell IM, Yuan B, Robberecht C, Pfundt R, Szafranski P, McEntagart ME, Nagamani SCS, Erez A, Bartnik M, Wisniowiecka-Kowalnik B, Plunkett KS, Pursley AN, Kang SL, Bi W, Lalani SR, Bacino CA, Vast M, Marks K, Patton M, Olofsson P, Patel A, Veltman JA, Cheung SW, Shaw CA, Vissers LELM, Vermeesch JR, Lupski JR, and Stankiewicz P (2014a). Parental Somatic Mosaicism Is Underrecognizedand Influences Recurrence Risk of Genomic Disorders. Am. J. Hum. Genet, 95:173–182. doi: 10.1016/j.ajhg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell IM, Stewart JR, James RA, Lupski JR, Stankiewicz P, Olofsson P, and Shaw CA (2014b). Parent of origin, mosaicism, and recurrence risk: probabilistic modeling explains the broken symmetry of transmission genetics. Am. J. Hum. Genet, 95:345–359. doi: 10.1016/j.ajhg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson RP (2010). Somatic gene mutation and human disease other than cancer: an update. Mutat. Res, 705(2):96–106. doi: 10.1016/j.mrrev.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, Bright IJ, Lucero MY, Hiddessen AL, Legler TC, Kitano TK, Hodel MR, Petersen JF, Wyatt PW, Steenblock ER, Shah PH, Bousse LJ, Troup CB, Mellen JC, Wittmann DK, Erndt NG, Cauley TH, Koehler RT, So AP, Dube S, Rose KA, Montesclaros L, Wang S, Stumbo DP,, Hodges SP, Romine S, Milanovich FP, White HE, Regan JF, Karlin-Neumann GA, Hindson CM, Saxonov S, and Colston BW (2011). High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem, 83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes the mechanism and application of the basic protocol, ddPCR.
- Iourov IY, Vorsanova SG, and Yurov YB (2010). Somatic genome variations in health and disease. Curr. Genomics, 11:387–396. doi: 10.2174/138920210793176065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jónsson H, Sulem P, Arnadottir GA, Pálsson G, Eggertsson HP, Kristmundsdottir S, Zink F, Kehr B, Hjorleifsson KE, Jensson BÖ, Jonsdottir I, Marelsson SE, Gudjonsson SA, Gylfason A, Jonasdottir A, Jonasdottir A, Stacey SN, Magnusson OT, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, and Stefansson K (2018). Multiple transmissions of de novo mutations in families. Nat. Genet, 50:1674–1680. doi: 10.1038/s41588-018-0259-9. [DOI] [PubMed] [Google Scholar]
- Koressaar T, and Remm M (2007). Enhancements and modifications of primer design program Primer3. Bioinformatics, 23:1289–1291. [DOI] [PubMed] [Google Scholar]
- Koressaar T, Lepamets M, Kaplinski L, Raime K, Andreson R, and Remm M (2018). Primer3_masker: integrating masking of template sequence with primer design software. Bioinformatics, 34:1937–1938. [DOI] [PubMed] [Google Scholar]
- Lim ET, Uddin M, De Rubeis S, Chan Y, Kamumbu AS, Zhang X, D’Gama AM, Kim SN, Hill RS, Goldberg AP, Poultney C, Minshew NJ, Kushima I, Aleksic B, Ozaki N, Parellada M, Arango C, Penzol MJ, Carracedo A, Kolevzon A, Hultman CM, Weiss LA, Fromer M, Chiocchetti AG, Freitag CM, Autism Sequencing Consortium, Church GM, Scherer SW, Buxbaum JD, Walsh CA (2017). Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat Neurosci, 20:1217–1224. doi: 10.1038/nn.4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR (2013). Genetics. Genome mosaicism--one human, multiple genomes. Science, 341:358–359. doi: 10.1126/science.1239503. [DOI] [PubMed] [Google Scholar]
- Metzker ML (2010). Sequencing technologies – the next generation. Nat. Rev. Genet, 11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Notini AJ, Craig JM, and White SJ (2008). Copy number variation and mosaicism. Cytogenet. Genome. Res, 123:270–277. doi: 10.1159/000184717. [DOI] [PubMed] [Google Scholar]
- Pinheiro LB, Coleman VA, Hindson CM, Herrmann J, Hindson BJ, Bhat S, and Emslie KR (2012). Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal. Chem, 84:1003–1011. doi: 10.1021/ac202578x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR UK10K Consortium, and Hurles ME (2016). Timing, rates and spectra of human germline mutation. Nat. Genet, 48:126–133. doi: 10.1038/ng.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative Genomics Viewer. Nat. Biotechnol, 29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summerer A, Schäfer E, Mautner VF, Messiaen L, Cooper DN, and Kehrer-Sawatzki H (2019). Ultra-deep amplicon sequencing indicates absence of low-grade mosaicism with normal cells in patients with type-1 NF1 deletions. Hum. Genet, 138:73–81. doi: 10.1007/s00439-018-1961-5. [DOI] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, and Mesirov JP (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform, 14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, and Rozen SG (2012). Primer3--new capabilities and interfaces. Nucleic Acids Res, 40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk EL, Auger H, Jaszczyszyn Y, and Thermes C (2014). Ten years of next-generation sequencing technology. Trends Genet, 30:418–426. doi: 10.1016/j.tig. [DOI] [PubMed] [Google Scholar]
- Vincze T, Posfai J, and Roberts RJ (2003). NEBcutter: a program to cleave DNA with restriction enzymes. Nucleic Acids Res, 31:3688–3691. doi: 10.1093/nar/gkg526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittwer CT, Herrmann MG, Moss AA, and Rasmussen RP (2013). Continuous fluorescence monitoring of rapid cycle DNA amplification. 1997. Biotechniques, 54:314–320. [DOI] [PubMed] [Google Scholar]
Internet Resources with Annotation
- https://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6407.pdf.Droplet Digital PCR applications guide published by Bio-Rad.
- https://www.bio-rad.com/webroot/web/pdf/lsr/literature/10043138.pdf.Detailed QX200 automated droplet generator instruction manual published by Bio-Rad.
- https://www.bio-rad.com/webroot/web/pdf/lsr/literature/10031906.pdf.Detailed QX200 droplet reader and QuantaSoft software instruction manual published by Bio-Rad.
- https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0013511_PowerUp_mastermix_UG.pdf.Troubleshooting of qPCR using PowerUp SYBR Green Master Mix.