

Abstract
The nuclear exit of messenger RNA (mRNA) molecules through the nuclear pore complex (NPC) is an essential step in the translation process of all proteins. The current limitations of conventional fluorescence and electron microscopy have prevented elucidation of how mRNA exports through the NPCs of live cells. In the recent years, various single-molecule fluorescence (SMF) microscopy techniques have been developed to improve the temporal and spatial resolutions of live-cell imaging allowing a more comprehensive understanding of the dynamics of mRNA export through native NPCs. In this review, we firstly evaluate the necessity of single-molecule live-cell microscopy in the study of mRNA nuclear export. Then, we highlight the application of single-point edge-excitation sub-diffraction (SPEED) microscopy that combines high-speed SMF microscopy and a 2D-to-3D transformation algorithm in the studies of nuclear transport kinetics and route for mRNAs. Finally, we summarize the new features of mRNA nuclear export found with SPEED microscopy as well as the reliability and accuracy of SPEED microscopy in mapping the 3D spatial locations of transport routes adopted by proteins and mRNAs through the NPCs.
Keywords: Nucleocytoplasmic transport, Single-particle tracking, Super-resolution microscopy, 3D live-cell imaging microscopy
1. Introduction
1.1. Overview of the NPC and RNA export
The nuclear pore complex (NPC) is a large protein structure embedded in the nuclear envelope (NE), a double bilayer membrane that separates the nucleus from the cytoplasm in eukaryotic cells. The NPC is the sole known gateway mediating nucleocytoplasmic transport of macromolecules [1,2]. The number of NPCs on the NE vary from hundreds (yeast) to thousands (human cells) [3–5]. With a molecular weight of ~ 60–120 MDa and dimensions of ~200 nm in length and ~50 nm diameter at the narrowest portion [6–9], the human NPC consists of approximately 30 different nucleoporins (Nups) organized into three major regions along the nucleocytoplasmic transport axis: 1) the cytoplasmic ring moiety with the cytoplasmic fibrils, 2) the nuclear ring moiety with the nuclear basket, and 3) the central framework [10,11]. Each of the Nups is present as 8-fold rotationally symmetric rings respective to the cross section of the NPC [7]. Moreover, about one third of these Nups are rich in phenylalanine-glycine (FG) repeats and together create a permeability barrier which allows the free diffusion of small cargo molecules (< 40–60 kDa) through the central channel of the NPC while restricting the passive diffusion of larger cargo molecules (> 60 kDa) into or out of the nucleus. However, when these larger cargos carry nuclear localization signals (NLSs) or nuclear export signals (NESs), transport receptors (TR) will bind to these signals and facilitate their efficient entry to or exit from the nucleus through direct interactions with FG-Nups in the NPC. [6,12–14].
Ribonucleic acid (RNA) is an essential bio-macromolecule to the central dogma of current biology [15]. In regards to their cellular function, RNA molecules may be coding RNA (e.g. mRNA) that are synthesized in the nucleus during transcription of genes, non-coding RNAs that are involved in RNA processing [e.g. small nuclear RNAs (snRNAs)], as well as components of protein translation [e.g. ribosomal RNA (rRNA), transfer RNA (tRNA), and microRNA(miRNA)] [16]. These RNAs: mRNA, tRNA, rRNA, miRNA and snRNA typically require the signal-dependent facilitated export from the nucleus into the cytoplasm [17,18]. In particular, mRNAs mainly use a transport receptor heterodimer (NXF1/Nxf1, also known as TAP/p15 in mammalian cells) to facilitate translocation through the NPC. This process involves direct interactions between the TAP/p15 heterodimer and FG-Nups to facilitate transport of mRNPs through the NPC [19,20]. Meanwhile, a certain subset of mRNAs were found to transport through the NPC using another major transport receptor, CRM1 (Chromosomal Maintenance 1, also known as Exportin 1) [21]. Additionally, some non-coding RNAs, such as rRNAs and snRNAs, can also use Crm1-dependent pathways for their nuclear export, while miRNAs predominantly use Exportin-5 for nuclear export [22–28].
1.2. Limitations of conventional light microscopy and electron microscopy in studies of mRNA nuclear export
Undoubtedly, it is critical to understand the basic mechanism of mRNA nuclear export, which forms a cornerstone in cancer therapy, prophylactic vaccines, gene therapy, and protein replacement therapies [29–31]. However, there are still many unknowns in the basic nuclear export process of mRNAs. To further study this critical process, it is necessary to complement conventional fluorescence microscopy and electron microscopy (EM) mRNA studies by employing techniques that expand into previously unfilled technical niches. Conventional fluorescence microscopy has been widely used to visualize molecular dynamics in live cells, taking advantage of the capability to tag molecules of interest through fusion with fluorescent proteins or dyes. However, the Abbe diffraction limit for light microscopy restricts the spatial resolution to approximately half of the wavelength of the emission light (~ 250 nm in the x and y dimensions and about ~ 750 nm in the z dimension if a 500-nm emission light is collected) [32–38]. Considering the dimensions of the NPC and the sizes of various large and small passage macromolecules, such as GFP (a cylinder with a length of ~ 4 nm and diameter of ~ 2–3 nm) and RNP complexes (up to ~ 25 nm when assuming a spherical shape) [39,40], it is impossible to distinguish individual macromolecules within the NPC by conventional light microscopy [41]. Although electron microscopy can achieve high resolution (up to < 1 nm), preparation of the sample via chemical fixation or freezing prevents EM from visualizing the real-time dynamics in live cells [42–45]. To break these limitations, single-molecule fluorescence microscopy has been developed and successfully applied to track single mRNPs exporting through the NE or the NPC in live cells, and achieved the refined features of mRNAs exiting the nucleus [39,46,47].
2. Description of method
2.1. Single-molecule fluorescence microscopy techniques applied to study mRNA transit across the NE
As shown in Table 1, wide-field epi-fluorescence single-molecule microscopy was firstly applied to track mRNPs containing mRNA with various sizes of 14 kb, 8 kb, and 4.8 kb. Multiple mRNP particles were tracked in the nuclear and cytoplasmic regions simultaneously; the authors reported that mRNP particles may need 5–40 min to complete transcription and nucleocytoplasmic transport. Particularly, with the temporal resolution of 1 s (dictated by the frame rate of the CCD camera) the nuclear export of mRNAs across the NE was tracked and found to have facilitated diffusion that is 15-fold faster than simple diffusion in nucleoplasm [46]. Later, registration epi-fluorescence single-molecule microscopy was used to simultaneously image fluorescently labeled mRNPs at the NE. In the setup, the emission fluorescence from mRNPs and the NE were separated by a dichroic filter and directed into two different cameras with an additional registration precision of ~ 25 nm. With a 20 ms temporal resolution, they found mRNA export through the NE consisted of a docking stage (80 ms), a transport step (5–20 ms), and a release process (80 ms), totaling 180 ± 10 ms in export time [39]. Soon after, light-sheet microscopy was used to detect mRNA nuclear export at the single-molecule level as well. In light-sheet microscopy the illuminating light takes the form of a thin sheet at the optical plane that arrives at the sample perpendicular to the detection path. Reduction of photobleaching, phototoxic effects, out-of-focus fluorescence, and a higher signal-to-noise ratio (SNR) comprise the advantages of light-sheet microscopy. With a spatial resolution in x and y dimensions of 10 nm and a temporal resolution of 20 ms, the authors reported that export time ranged from 65 ms to several seconds while the success rate of mRNA export across the NE was ~25% [47].
Table 1.
Studies of mRNP export of the nucleus using a variety of microscopy techniques. Techniques are listed alphabetically.
| Microscopy Technique | Main Observations/Conclusions | References |
|---|---|---|
| Light Sheet Fluorescence Microscopy (LSFM) | The transport efficiency and time of mRNPs across the NE were determined by using LSFM setup. They found that ~ 25% of mRNPs successfully exit nucleus after interacting with NE/NPCs with an averaged nuclear export time ranging from 65 ms to several seconds. | [48] |
| Single-Point Edge-Excitation sub-Diffraction (SPEED) | SPEED microscopy revealed that ~36% of all the NPC-interacting mRNP molecules successfully complete their nuclear export by adopting a fast-slow-fast diffusion pattern through the NPC during an averaged nuclear export time of ~ 12 ms. Furthermore, the major selectivity barrier in the NPC that separates the successful and abortive mRNPs export events locates at the NPC’s nuclear basket and central scaffold. | [40] |
| Super-Registration Microscopy | The nuclear export of mRNA crossing the NE includes a three-step process - docking, transport, and release. These events occurred in durations of ~ 80, 5–20, ~ 80 ms respectively. | [39] |
| Wide-Field Epi-Fluorescence Microscopy | The authors found that the nuclear export of mRNP occurs faster than nucleoplasmic diffusion. They concluded that the transcription, transport, and export process ranges from 5 to 40 min. | [46] |
| Wide-Field Epi-Fluorescence Microscopy and Transmission Electron Microscopy (TEM) | The authors found an alternative route for nuclear export of very large RNP complexes, via budding through the NE. This unique pathway might arise from mutations in nuclear lamins and inner nuclear membrane proteins. | [49] |
2.2. SPEED microscopy illuminates individual mRNA molecules through single NPCs
The above studies have provided great insight into the dynamics of mRNA export by imaging single mRNP particles across the NE in live cells. However, we were further motivated to refine the imaging process of mRNP nuclear export by investigating more details inside the sub-micrometer NPCs. These details included the nuclear export time and efficiency for mRNP particles through the NPCs instead of the NE, the diffusion patterns of mRNPs through the NPC’s sub-regions including the cytoplasmic side, the central scaffold and the nuclear basket of the NPC, and the three-dimensional (3D) probability density map of spatial export routes of mRNPs and their transport receptors within the NPC [40]. To answer these questions, our lab has developed single-point edge-excitation sub-diffraction (SPEED) microscopy and applied it to conduct single particle tracking (SPT) of single mRNP translocation through single NPCs in live HeLa cells by collaborating with the laboratory of Dr. Nils Walter at the University of Michigan-Ann Arbor. SPEED microscopy involves new technical developments in both microscopy imaging and single-molecule data analyses [50,51].
2.2.1. Small illumination point spread function and high detection speed of SPEED microscopy
As shown in Fig. 1, the average distance of ~ 400–600 nm between the nearest neighboring NPCs causes an overlapping of fluorescence signal from the fluorescence-protein-labeled NPCs illuminated by wide- or narrow-field epi-fluorescence microscopy [52,53], which allows a determination of NE’s middle plane but prevents a precise localization of individual NPCs. As a result, detailed interaction information between existing mRNAs and individual NPCs remained difficult to obtain. By using an inclined diffraction-limit illumination point spread function (iPSF) and a high-speed CCD camera in SPEED microscopy, we have shaped the iPSF to illuminate single NPCs and track single mRNPs within NPC’s sub-regions with high temporal and spatial resolutions. Technical advances of SPEED microscopy are briefly summarized in the following [40,51]: (1) Compared to wide-field and narrow-field microscopy, SPEED microscopy generated a diffraction-limited iPSF (~ 320 nm in x, y, and z dimensions with a 488-nm illumination laser) at the NE of live cells, which is smaller than the average distance between NPCs on the NE, allowing the excitation of single GFP-labeled NPCs in each dimension of the 3D Cartesian coordinate system (Fig. 1A and B). (2) The small iPSF of SPEED microscopy allows the imaging of single molecules within a small pixel area of the CCD camera, resulting in a very fast detection speed (up to 0.2 ms per frame). (3) The high optical density (100–500 kW/cm2) of the small iPSF also produce a high number of photons in a short time period from a single mRNP complex tagged with approximately ten copies of mCherry [50]. Notably, we could obtain more than 5000 photons from a single mRNP particle within 2 ms detection time. (4) The inclined iPSF greatly avoids out-of-focus background fluorescence and auto-fluorescence of the objective, resulting in a much improved SNR [54]. (5) Pinpointed illumination in live cells causes less photo-induced toxicity. Altogether, these features enabled SPEED microscopy to image single fluorophores with a spatiotemporal resolution of < 1 ms and < 10 nm. Moreover, these experimentally obtained high-quality 2D sub-diffraction-limited data provide the input for the post-localization 2D to 3D transformation data process (Figs. 2 and 3).
Fig. 1.
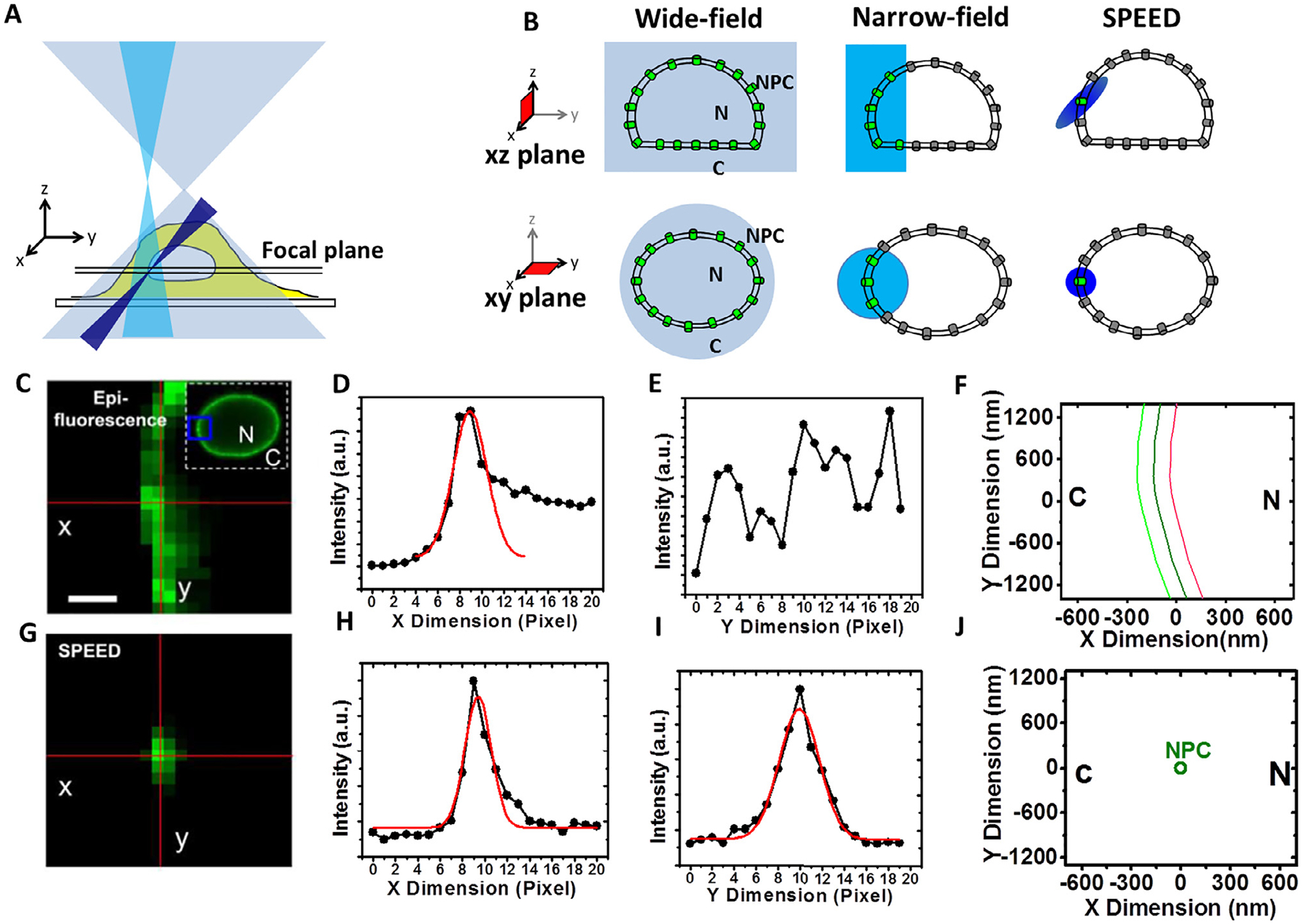
Illuminations of various single-molecule microscopy techniques. (A and B) Simplified optical diagrams illustrate the different excitation beam paths and the illumination volumes of the wide-field (light blue), narrow-field (cyan) epi-fluorescence microscopy and SPEED (blue) microscopy. GFP-labeled NPCs embedded in NE were highlighted inside (green) and outside (gray) the microscopy illumination volumes in the xy and xz planes [54]. (C) Overlapped fluorescence of GFP-NPCs on the NE under the illumination of wide-field or narrow field epi-fluorescence microscopy. The selected area is enclosed by a blue box in the image of the entire fluorescent NE. Scale bar, 1 μm. (D and E) The line-scan intensities for the pixels (red lines) in the epi-fluorescence image (C) are plotted in the x dimension (D) and y dimension (E). For the x dimension, a 2D line-scanning fitting is performed across the intensities (D). (F) The dark-green curve shows the determined position of the middle plane of the NE. The light-green and red curves are for reference at −100 nm and +100 nm from the middle plane of the NE, respectively. (G) Only a single GFP-NPC was excited in the illumination volume of the SPEED microscopy. (H and I) The peak position of the 2D line-scanning fitting for a GFP-NPC fluorescent spot in (G). (J) The 2D centroid of the single NPC determined by 2D Gaussian fitting. N, nucleus; C, cytoplasm. Figures were adapted from our previous publications with permissions [40].
Fig. 2.
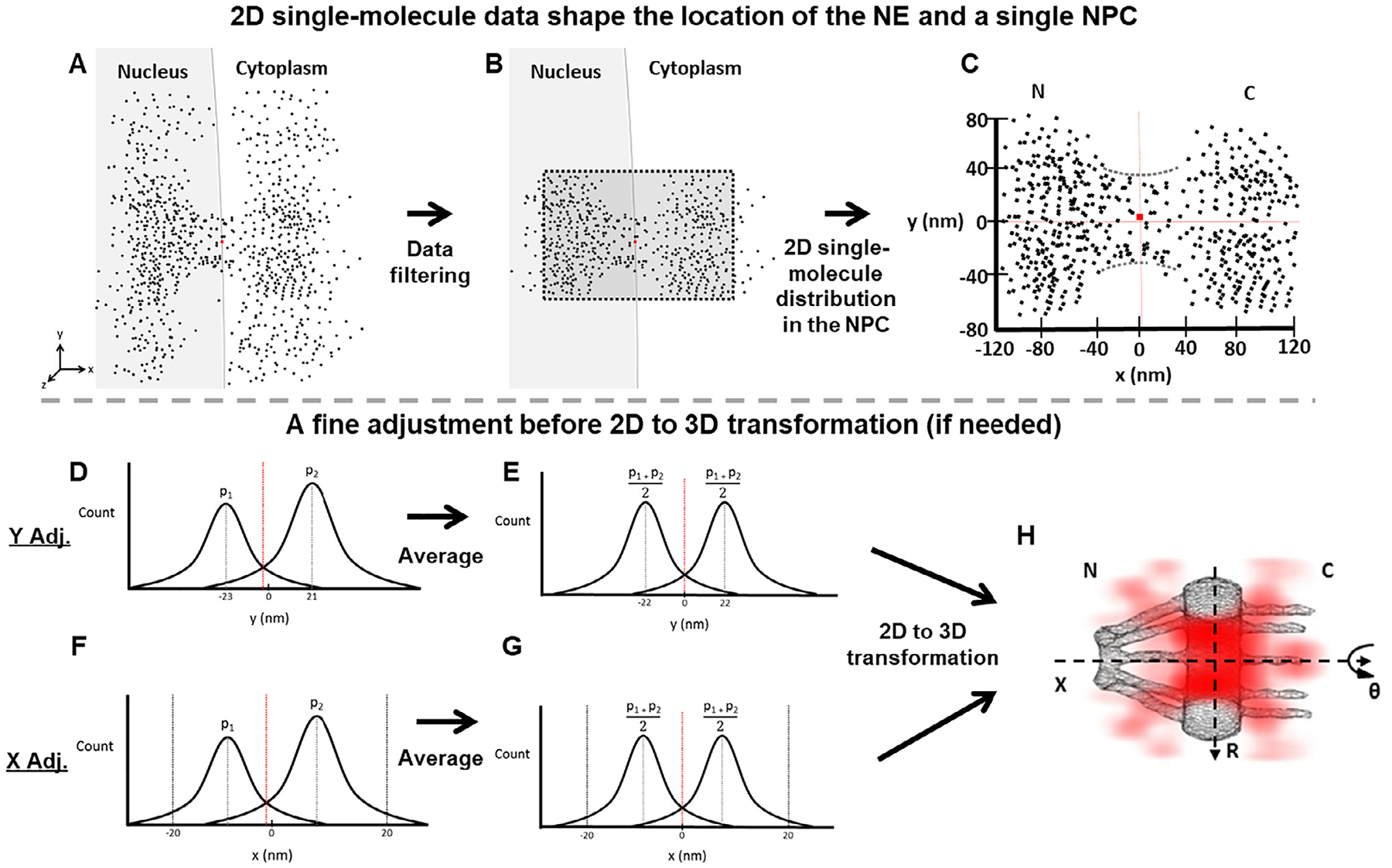
Determination of the central axis of the NPC illuminated by SPEED microscopy. Diagrams are used to demonstrate the detailed steps in our data analyses. (A) The initial single-molecule tracking data (black points) are plotted around the NPC’s marker (red dot) collected with SPEED. (B) The collected data after filtering by using single-molecule spatial localization precision. (C) The filtered 2D single-molecule data shape the spatial location of the NE and a single NPC. The red dot represents the location of the NPC’s marker. But sometimes the marker’s position is not perfectly overlapped with the averaged central positions in either x or y dimension suggested by the 2D single-molecule data. Also, the 2D single-molecule distribution also indicates the orientation of NPC. If the orientation of the NPC is within a free angle of 1.4° to the perpendicular direction to the NE [51], the 2D single-molecule data will be proceeded further. If it is bigger, the data will be dropped. (D) The plot of projected locations of these 2D single-molecule data in the y dimension will indicate if a fine adjustment for the central cytoplasmic transport axis is needed or not. Here we just show an example that peak 1 (p1) and peak 2 (p2) are not symmetrical locating at −23 nm and 21 nm respectively. If the peaks are symmetrical, the step E will be skipped to move onto the 2D to 3D transformation directly. (E) The two peaks are averaged as , resulting a fine adjustment in the NPC’s central cytoplasmic transport axis. The dotted red line has shifted to the corrected central position. (F and G) Similar to the y-dimensional data process, we next determine the precise location of the NPC’s central position along the x dimension by fitting the histogram of these 2D single-molecule locations within the NPC’s scaffold region projected into the x dimension (ranging from −20 nm to 20 nm, as clearly shown in the void region of the NE in (C) even if the NPC’s marker is a little bit off sometimes). (H) The 2D single-molecule data with the confirmed x and y axes will undergo the 2D to 3D transformation (Fig. 3) to produce the 3D density map of mRNP’s nuclear export routes (red clouds).
Fig. 3.
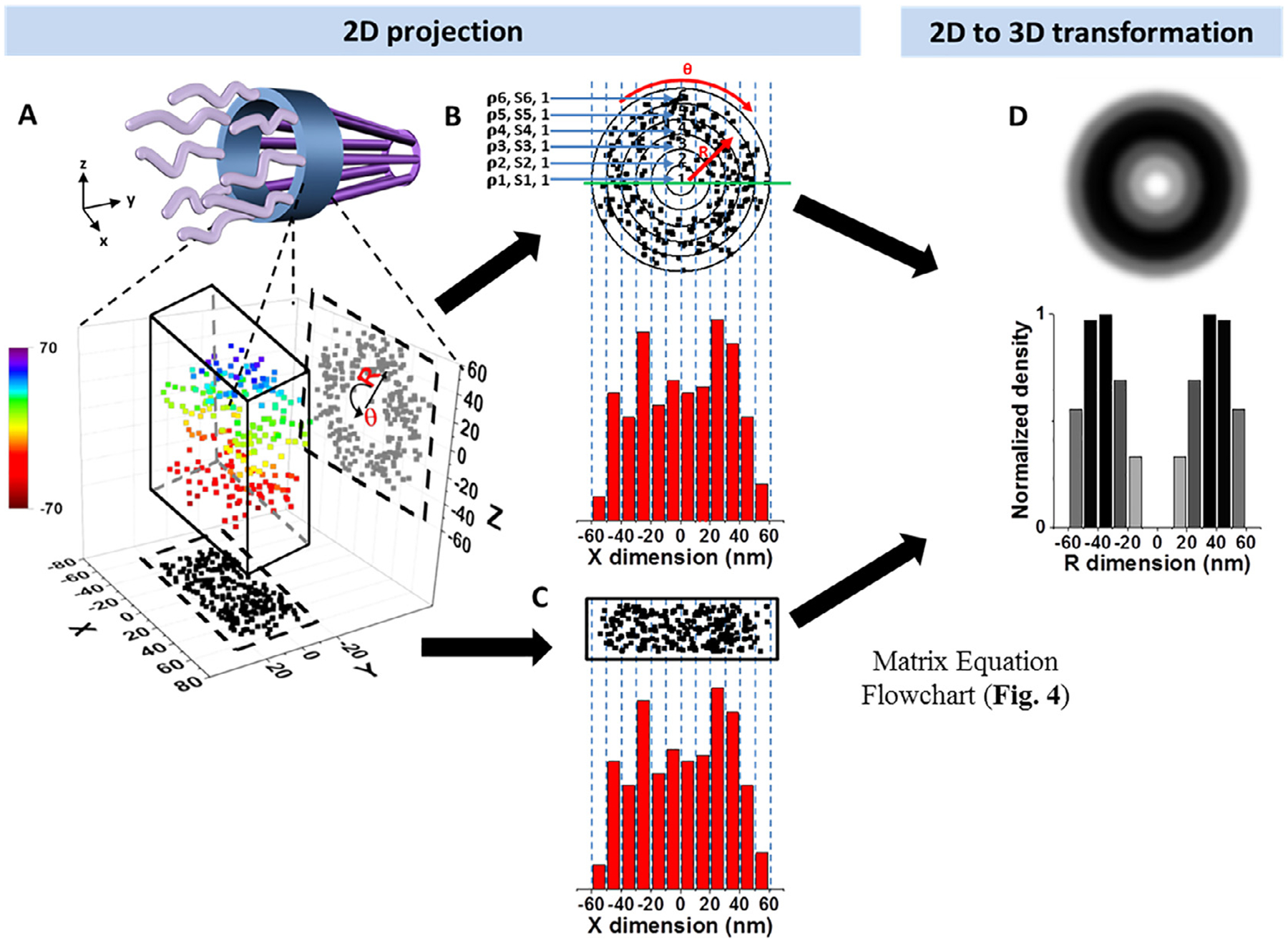
A schematic demonstration, with simulated data, of the 2D to 3D transformation algorithms for molecules that diffuse through the NPC. (A) 3D spatial locations of randomly diffusing molecules inside the NPC can be coordinated in a cylindrical coordination system (R, Ø, Y) due to the cylindrical rotational symmetry of the NPC. The 3D molecular locations in the NPC (rainbow colored for Z position) are projected onto a 2D plane in a Cartesian coordination system (X and Y, shown as black points) by microscopy imaging (X and Z shown as gray points). (B) The cross-sectional view of all the locations shown in Fig. 3A (same as the gray points from the X and Z dimension). These locations can be grouped into the sub-regions between concentric rings. Given the high number of randomly distributed molecules in the NPC the spatial density of locations (ρi) in each sub-region (s(i,j)) between two neighboring rings will be rotationally symmetrical and uniform. These locations can be further projected into 1D along the X dimension. The locations along X dimension can be clustered in a histogram with j columns. The total number of locations in each column (A(i,j)) is equal to , which is further explained in Fig. 4. (C) Histograms of 2D projected data from microscopy experiments are identical to B, thereby allowing us to use the aforementioned formula to determine the density of each concentric ring. (D) Using the algorithms, 2D projected data can be used to reconstruct the 3D spatial distribution of proteins traveling through the NPC.
2.2.2. Obtaining 3D transport routes for mRNAs and their transport receptors by using 2D to 3D transformation algorithms
Currently, it is still challenging to obtain 3D super-resolution information of structures and dynamics in live cells with a high spatiotemporal resolution. In SPEED microscopy, we have developed a new transformation algorithm, without using 3D super-resolution microscopy or real-time 3D particle tracking, to achieve 3D sub-diffraction-limited information. It is necessary to note that no z dimensional information is directly obtained for individual single molecule locations. Rather, this is a post-localization analysis that transforms 2D super-resolution images or 2D single-molecule localization distributions into their corresponding 3D spatial probability information. This method has been successfully applied to several sub-cellular organelles, such as the NPC and the primary cilia, because these sub-cellular structures have rotational symmetry [32,40,50,51,54–56].
As for nuclear export of mRNAs and their transport receptors, the 3D locations of their export routes have been revealed via the 2D-to-3D transformation algorithm by taking advantage of the structural rotational symmetry of the NPC and superimposing thousands of single-particle trajectories collected from multiple NPCs [5,32,57]. An assumption is made that single-molecule locations within the NPC are uniform in their rotational distribution for our 2D-to-3D transformations. This is based on the fact that an eightfold radial symmetrical structure has been revealed through EM studies [5], and additionally confirmed by single molecule study of the spatial distribution of single transiting molecules at the cross-section of NPC locating at the bottom of the NE [51]. Thus, the main advantage of this 2D-to-3D transformation algorithm is for elucidating the 3D density maps of dynamic transiting molecules through the NPC based on the fact that the NPC’s ultrastructure and rotational symmetry has been determined by other independent methods. Fig. 3 shows the mechanism of 2D-to-3D transformation. As molecules situate in or travel through the NPC, the obtained locations in the xy or the yz plane are in fact a projection of the actual 3D locations as indicated in cylindrical coordinates (Fig. 3A). In practice, it is much easier to obtain 2D super resolution images of these targeted molecules in the xy plane than the xz plane because of the higher resolution in the xy plane compared to the xz plane. Furthermore, projecting the 2D molecular spatial locations into the x dimension from either the xy or the xz plane, the obtained two x dimension histograms in principle are identical (Fig. 3B and C). Therefore, the same information can be obtained from projections in either the xy and xz plane. Then, based on the two identical x-dimension histograms, the value in each column in the x-dimension histogram projected from the xy plane will be equal to the areas times the densities for each radial bin of the xz plane (Fig. 3D). Then, we obtain the densities in the radial dimension by solving the matrix equations (Fig. 3D) [58]. Finally, a complete 3D spatial density map of mRNA molecules within the NPC was generated by combining all the concentric rings plotted from the densities in radial distributions from each y-dimension sliced region (Figs. 3D and 9).
Fig. 9.
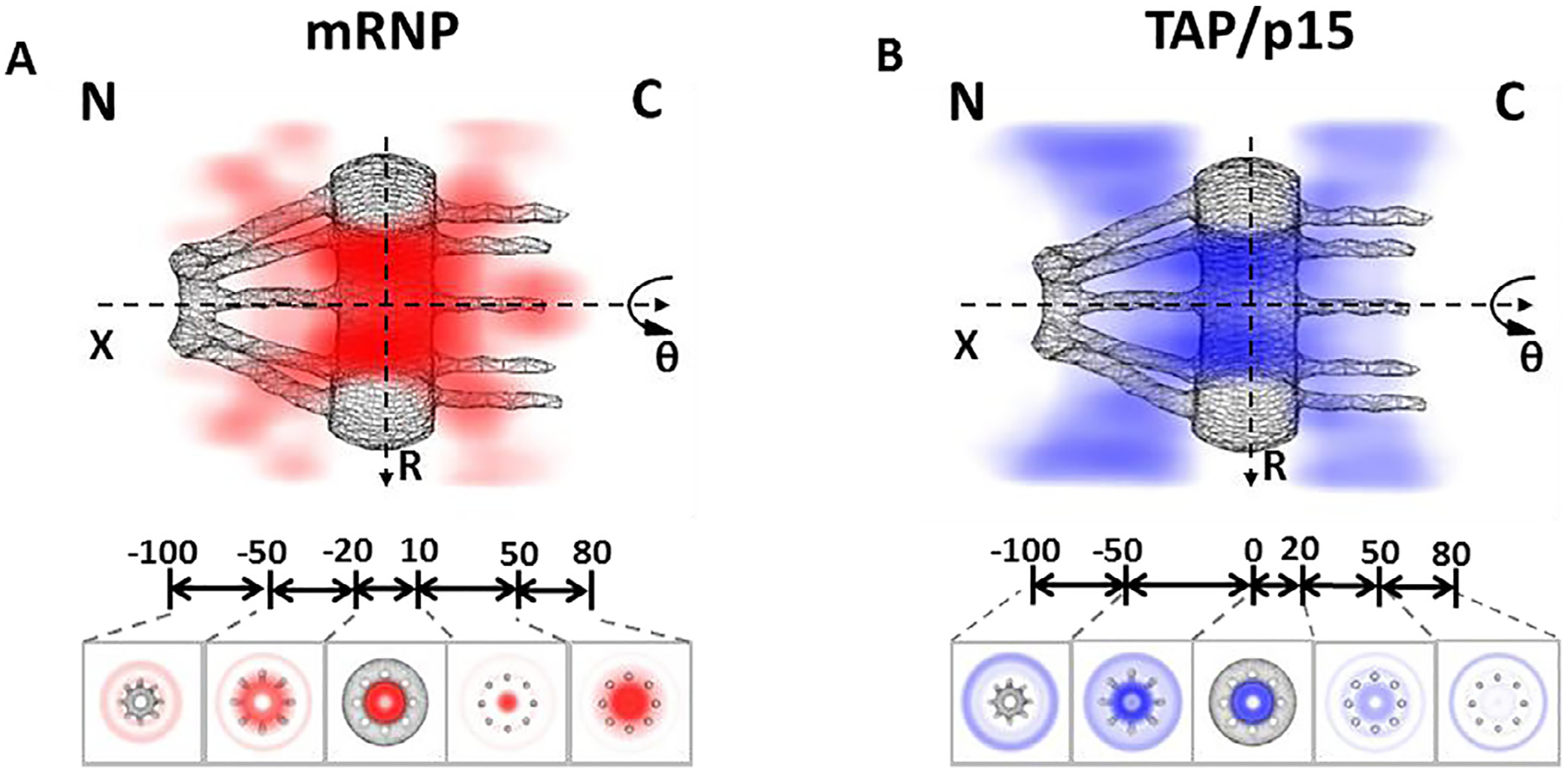
3D mapping of nuclear transport paths for mRNP and TAP/p15 through the NPC [40]. (A) 3D spatial probability density map of mRNPs (red, deeper shade indicates higher density), generated using a 2D-to-3D transformation algorithm, is shown in both a cut-away and a cross-section view superimposed on the NPC architecture (grey). Five regions with distinct spatial location distributions for mRNPs are marked with relative distances (in nm) from the centroid of the NPC. C, cytoplasmic side of the NPC; N, nucleoplasmic side of the NPC. (B) Cut-away and cross-section views of the 3D spatial probability density map for the TAP/p15 heterodimer (blue, deeper shade indicates higher density) superimposed on the NPC architecture (grey).
3. Description of the equipment
3.1. Microscopy
An Olympus IX81 microscope was used to perform our SMF experiments (Olympus). The microscope was equipped with 1.4 numerical aperture 100× oil-immersion apochromatic objective (UPLSAPO 100XO; Olympus). The stage clips (IX-SCL) were used to clamp the samples on Olympus IX-SVL2 stage. Optical axis plane movement was controlled by U-FH focus adjustment knob. Sample plane (x and y) movement was controlled by a mechanical IX-SVL2 stage. A 100 W halogen lamp was used for bright-field illumination. A 100 W mercury lamp was mounted to the epifluorescence excitation port. The entire SPEED microscope system was isolated from vibrations by mounting on a pneumatic isolator that was pre-mounted on a research grade optical table (Newport, Irvine, CA).
3.2. Laser illumination and filters setting
Three lasers were used in the above microscope system to provide the power necessary for rapid image acquisition of SMF signals. One set-up included a 35-mW 633-nm HeNe laser (Melles Griot). The other set-up included a 120-mWArKr tunable ion laser (561-nm) and a 50-mW argon-ion laser (488-nm) (both purchased from Melles Griot) running in single-line mode. Laser output was modulated with neutral density filters as necessary. Linearly polarized laser light was converted to circularly polarized light with a quarter-wave plate to allow homogeneous (polarization-independent) fluorophore excitation within the focal plane. An optical chopper (Newport) was used to generate an on-off mode of laser excitation. To minimize alignment problems in dual-color measurements, both green and red fluorescence emissions were collected by the same objective, filtered by a dichroic filter (Di01-R405/488/561/635–25 × 36; Semrock) and an emission filter (NF01–405/488/561/635–25 × 5.0; Semrock).
3.3. CCD cameras
On-chip electron multiplying charged-coupled device (EMCCD) cameras were used for signal detection. CCDs provide wide-field spatial information that cannot be obtained with photomultiplier tubes or avalanche photodiodes. Since frame rates are largely limited by frame-transfer speed and the number of pixels, we chose a 128 pixel × 128 pixel CCD camera (Cascade128 +; Roper Scientific) to monitor the fast nucleocytoplasmic transport of macromolecules. For this camera, full-frame, continuous image acquisition occurs at 500 frames per second (fps), with > 90% quantum efficiency. Faster frame rates can be obtained by limiting the acquisition area through software: e.g., for a 128 pixel × 20 pixel area, 2500 fps can be acquired. Dark current is a major factor limiting image quality for EMCCDs, especially at high frame rates. A solution is used to cool down the detection chip. The Cascade128+ camera is cooled to −30 °C, leading to a dark current of ≤1 e−/p/s. To minimize alignment problems in dual-color measurements, both green and red fluorescence emissions were imaged by an identical Cascade 128 + CCD camera.
3.4. Imaging software
Basic image acquisition software is typically obtained with the microscope from the manufacturer. It is desirable to obtain a software package that not only will run the image acquisition by the microscope, but also will control motorized microscope functions such as a z-stepper, objective changer, filter changer, and emission path changer (e.g., eye to camera, or camera to camera) as well as instrumentation add-ons (obtained initially or through expansion), such as (in our case) a laser shutter, and motorized stage. We use the Slidebook (Intelligent Imaging Innovations) software package for data acquisition, instrument control and most image processing. We use Glimpse, a Matlab-based program written by Jeff Gelles and freely available from his lab website, to fit fluorescent emission spots with two-dimensional Gaussian distributions for particle tracking purposes [59]. Specific parameters for Glimpse include: excluding single-molecule fittings with Gaussian widths less than half a pixel to avoid camera noise and greater than two pixels to avoid possible aggregate particles in the analysis.
4. Experimental procedures
4.1. Preparation of plasmid
4.1.1. Luciferase reporter plasmid bearing the MS2 stem loops
The ORF of IF2 (Initial Factor 2), PCR-amplified using a forward primer bearing SbfI and EcoRI restriction enzyme sites and a reverse primer bearing a NotI restriction enzyme site, was cloned into the corresponding SbfI and NotI sites in plasmid pmiR-GLO (Promega) to generate pmiR-GLO-IF2 (pmG-IF2).
Twenty-four copies of the MS2 stem loop from pSL-MS2_24x were cloned into the EcoRI-NotI restriction enzyme sites of the resultant plasmid, pmG-IF2, to generate pmG-MS2 (Fig. 5A, Hint 1) [39,60].
Fig. 5.
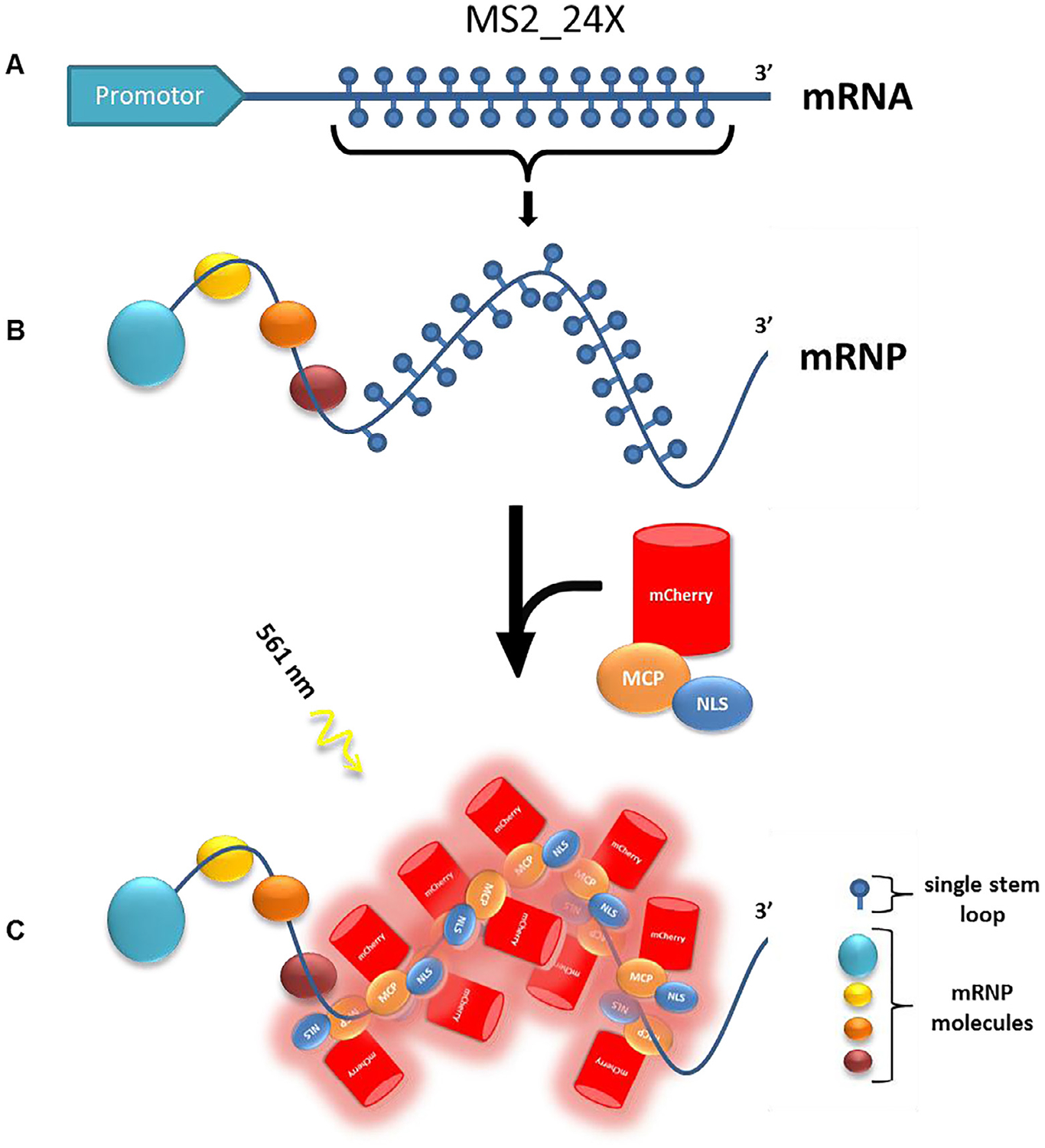
Plasmid cloning and labeling. (A) pmG-MS2 with 24 MS2 stem loops at the 3elings. B) mRNP with MS2_24X formed and incubated with mCherry-MCP-NLS. (C) The mCherry-MCP-NLS binds ~10 copies to the MS2_24X region of the mRNP. This complex can be excited by the 561-nm laser (Fig. 6).
4.1.2. MS2-coating protein (MCP) dimer fused to mCherry
pMCP-EGFP, a plasmid bearing the open reading frame (ORF) of the MCP fused to EGFP and an SV-40 NLS were received as generous gifts from Robert Singer (Albert Einstein College of Medicine) [39]. The GFP ORF in pMCP-GFP was replaced with PCR-amplified mCherry ORF bearing AgeI and BsrGI restriction enzyme sites, to result in pMCP-mCherry.
The NLS was added to MCP-mCherry to promote its import into the nucleus and enhance sufficient labelling of mRNAs even at the desired low MCP expression level (Fig. 5B).
4.2. Preparation of labelling proteins
By means of conventional heat-shock bacterial transformation, strains of E.coli (JM109 or BL21 (DE3)) are engineered in order to express TapΔNLS-p15 heterodimer proteins and are kept in stock at −80 °C. TapΔNLS-p15 plasmids containing the protein of interest contains a T7 promoter which can be activated by IPTG. 6x Histidine tags are present at either the N or C termini of the protein construct. Ampicillin resistance is additionally incorporated into the plasmid. A ~50 μL of frozen stock of transformed E.coli cells are transferred to 5 mL of LB media with addition of 5 μL of ampicillin (100 mg/mL) and grown aerobically with shaking for 12–14 h at 37 °C at 225 rpm to create a starter culture.
The 5 mL saturated starter culture is then transferred into 1 L of LB media (containing 1 mL ampicillin) and shaken in a 37 °C incubator for 5–6 h or until an OD of ~ 0.6 is reached. An addition of 1 mL of 1 M IPTG (Isopropyl-D-1-thiogalactopyranoside) is then sterilely placed into the mixture to activate protein production and incubation continues for another 5–6 h. (Hint 2)
The culture is then spun in the cooling high-speed centrifuge at 12,000 rpm for 15 min at 4 °C and the supernatant is discarded. Cells are then re-suspended in 40 mL of CelLytic B (Sigma-Aldritch). The volume should be based on 10 mL per 1 g of a pellet.
“protease inhibitor cocktail” is then added (400 μL of 0.2 mg/mL pepstatin A. 400 μL of 0.2 mg/mL leupeptin; 400 μL of 2 mg/mL trypsin inhibitor). This is to inhibit protein degradation from the E.coli lysate.
For histidine tagged plasmids: 1 mL of Ni-NTA resin is prepared by removing the ethanol, it is stored via centrifugation at 12000 rpm for 1 min followed by aspiration of the supernatant and re-suspension with CelLytic B. This process is repeated three times. Afterwards the Ni-NTA CelLytic B solution is added to the protein mixture.
Stir the mixture in dark on ice for 30 min to allow an adequate time for the binding reaction between the histidine tag and Ni-NTA beads, spin at 10000 rpm for 60 min in the cooling centrifuge at 4 °C.
Perform column chromatography as follows: the first fraction of 12 fractions to be collected is a flow through of the supernatant mixed with Ni-NTA from the previous step. The second is 10 mL of Lysis buffer (50 mM Na-phosphate, 300 mM NaCl, 20 mM imidazole, pH 8.0), the third is 10 mL of wash buffer (10 mM imidazole, 100 mM NaCl, pH 8.0), the fourth and the following fractions are 0.5 mL of elution buffer (250 mM imidazole, 100 mM NaCl, pH 8.0)
Prepare an SDS-PAGE polyacrylamide gel and prepare the samples by mixing 4 μL of a fraction and 1 μL of 5X SB (300 mM Tris 6.8, 25% BME, 10% SDS, 50% glycerol). Before loading boil samples for 5 min. After loading, run the gel at 120 V for 60 min. The fractions that contain the highest concentration of protein and yielding the purest product can be identified. Depending on the protein of interest, the bands should be located respectively to the size of a protein using a protein standard as a reference. This fraction should be selected for further protein purification with MonoQ and Superdex 200 (Amersham Pharmacia) to ensure purity above 90%. (Hint 3)
Concentration may be performed by spinning the most concentrated fractions through a Nanosep filter (with a pore size of 30 kDa at 14 rpm for 5 min, washing it with elution buffer every time).
Determine the final protein concentration utilizing the BSA assay for further protein labeling procedure.
For proteins containing surface cysteine residues labeling can be accomplished with an excess of a cysteine-reactive organic dye (Alexa flour 647 maleimide dye from Invitrogen) for 2 h at room temperature in 50 mM sodium phosphate, 150 mM NaCl after reducing with TCEP tris (2-carboxyethyl) for 20 min. Reactions can be quenched with 2-mercaptoethanol, and the protein solutions must be dialyzed to remove the free dye (Hint 4, 5).
Determine the concentration and labeling ratio of final labeled protein by Nanodrop spectrometry utilizing absorption at 280 nm and the corresponding absorbance for the dye used (Hint 6). The labeling ratio was about two Alexa Fluor 647 dye molecules per TapΔNLS/p15 heterodimer.
4.3. Preparation of cell system
4.3.1. Preparation of live cell system
HeLa cells stably expressing GFP fused to the NPC scaffold protein POM121 (HeLa POM121-GFP stable cell line) are used for our live cell system of single-molecule nuclear transport assays. Start a fresh culture of HeLa cell line from a stock by thawing at 37 °C and inoculating into a 25 cm2 culture flask with 5 mL DMEM (10% FBS, 10 mg/mL streptomycin, 100 U/mL penicillin). Incubate the cells at 37 °C within a 5% CO2 incubator for 24 h and split cells at least 3 times to ensure health.
Two days before the experiment, cells intended for a single molecule experiment should be spread on an autoclaved glass slide placed in a sterile Petri dish with 1–2 mL DMEM (10% FBS, 10 mg/mL streptomycin, 100 U/mL penicillin). These Petri dishes with the glass slides then are placed into the incubator and kept for 48 h prior to the experiment.
One day before the experiment, we transfected Firefly Luciferase mRNA plasmid bearing 24 MS2 stem loops and pMCP-mCherry plasmid into the HeLa POM121-GFP stable cell line with TransIT-LT1 transfection reagent (Mirus) following quick reference protocol of TransIT-LT1 transfection reagent.
After transfection, previous culture media should be replaced with 1–2 mL Opti-MEM and placed into the incubator and kept for 24 h prior to the experiment.
4.3.2. Preparation of the permeabilized cell system
Refer to Step 1 of preparation of live cell system.
One day before the experiment, cells intended for a single molecule experiment should be spread on an autoclaved glass slide placed in a sterile Petri dish containing DMEM (10% FBS, 10 mg/mL streptomycin, 100 U/mL penicillin). These Petri dishes with the glass slides then are placed into the incubator and kept for 24 h prior to the experiment.
Once the Petri dish is mounted on the microscope, the cells are washed two times with 25 μL Transport buffer (20 mM HEPES-KON, 110 mM KOAc, 5mM NaOAc, 2 mM MgOAc, 1 mM EGTA, pH 7.3) (Hint 7).
The cells are then permeabilized for ~2 min with 25 μL 40 μg/mL digitonin in transport buffer and washed 2 times again with 25 μL transport buffer supplemented with 1.5% polyvinylpyrrolidone (PVP; 360 kDa) (Hint 8).
4.4. SPEED microscopy tracking of single mCherry-labeled mRNPs through single GFP-NPCs in live cells, and separately tracking of individual Alexa Fluor 647-labeled Tap/p15 through single GFP-NPCs in digitonin-permeabilized cells
SPEED microscopy was used to conduct high-speed single molecule tracking of single mRNP and single TAP/p15 heterodimer complexes nuclear translocation through the NPC respectively in live HeLa cells and permeabilized HeLa cell [40,51].
As shown in Fig. 6, equivalent to the illumination pattern of a 488-nm laser, the 633-nm or 561-nm laser is shifted about 237 μm (d) by a micrometer stage off the center of the objective to generate an inclined illumination point spread function at 45° to the perpendicular direction. An optical chopper on the path of the laser beam provided an on–off laser mode (Hint 9). The 488-nm, 561-nm, and 633-nm lasers are used to respectively excite GFP-NPCs, mCherry-mRNPs and Alexa Fluor 647-Tap/p15.
For experiments of the nuclear export of mCherry labelled mRNPs in live cells, Cells must be incubated with transport buffer (20 mM HEPES-KOH, 110 mM KOAc, 5 mM NaOAc, 2 mM Mg (OAc)2 and 1 mM EGTA (pH 7.3)) for 30 min before the experiments (Hint 10).
For experiments of the nuclear export of Alexa Fluor 647-labeled TAP/p15 in permeabilized cell system, cells must be permeabilized (see 4.3.2 preparation of permeabilized cell system).
Focus the microscope to the equator of the GFP-fused NE. Target one GFP-labeled NPC at the edge of the NE.
First a single GFP-NPC was imaged with a 10-mW 488-nm laser for 1–2 s. The fluorescent spot of the NPC can then be fitted to a 2D elliptical Gaussian function to determine the centroid position of the NPC (Hint 11).
For a live cell system, the illumination area was photobleached by utilizing a 561-nm laser for 60 s until completely dark to improve signal to noise ratio for single-molecule tracking of mCherry-mRNPs.
If using a permeabilized cell system, add 0.1–1 nM Alexa 647-TAP/p15 into the transport buffer.
Set the 561-nm or the 633-nm laser to be 2–5 mW and the chopper with 5 Hz to generate 3:7 on-off ratio. A 561-nm laser was used to excite the mCherry-tagged mRNP in live cells, while a 633-nm laser was used to excite Alexa Fluor 647-labelled TAP/p15 in digitonin permeabilized cells (Hint 12).
Set the intensification and gain of the CCD to ~ 4000 and reduce the detection region to 128 pixels × 20 pixels. Use the “Stream” mode and set the exposure time to 0. With this setup, the CCD camera works on the speed of 400 μs/frame.
A series of videos of single-mRNP exports can be taken for 2 min per cell (Hint 13).
An example of typical trajectory is seen in Fig. 7A and B. The fluorescent spot was fitted to a 2D symmetrical Gaussian function and determined the positions (Fig. 7C and D).
Combine all collected trajectories and generate the 2D localization distributions via histograms (Fig. 7E and F; Hint 14)
Repeat step 3–11 for a different NPC in another cell until a suitable number of locations are obtained (Hint 15).
Fig. 6.
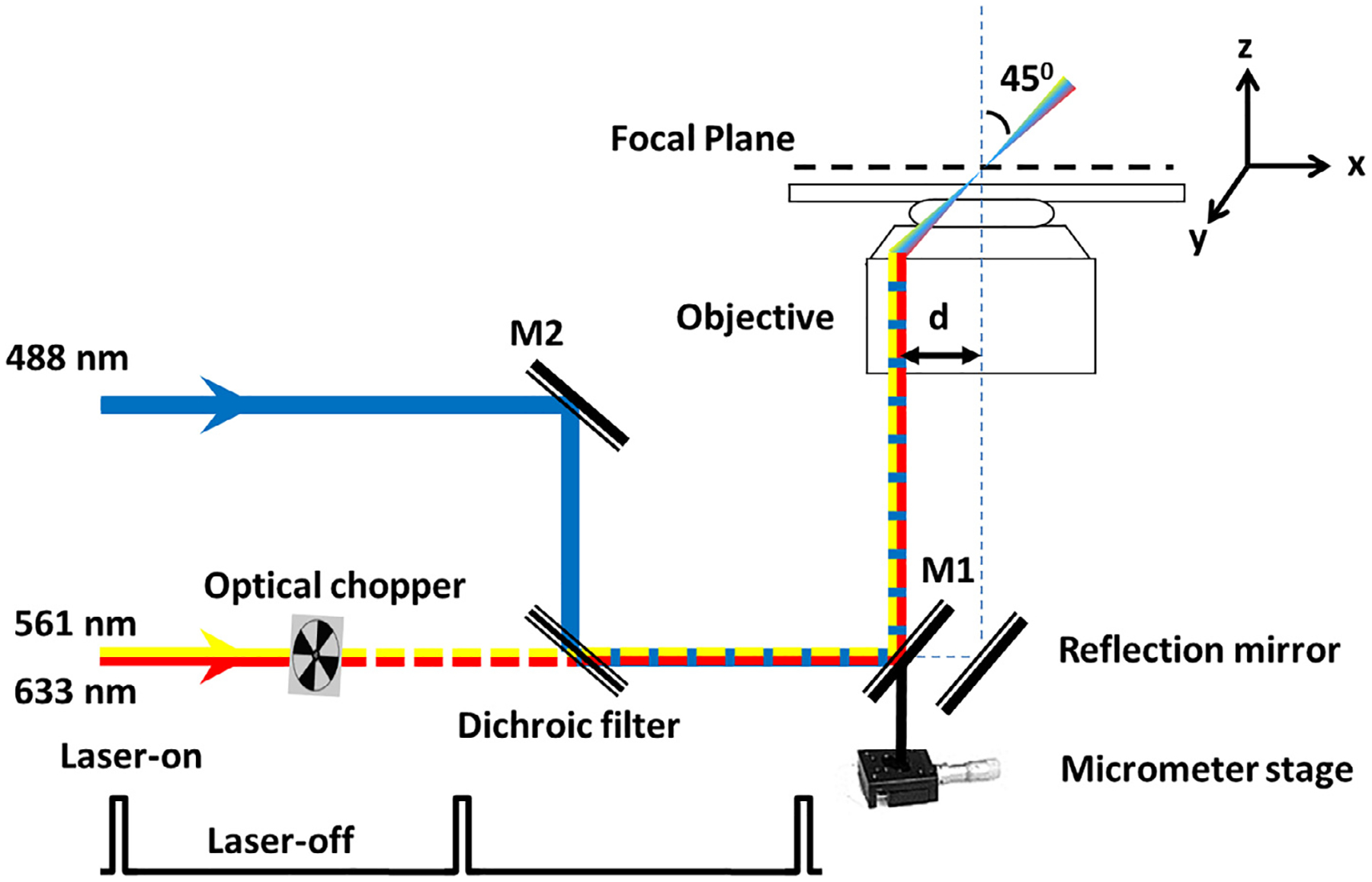
Optical schematic of the SPEED microscope setup [51]. A 488-nm, a 561-nm, and a 633-nm laser beams were co-aligned and then shifted together by ~237 μm (d) from the central optical axis of the objective to generate an inclined illumination volume at an angle of 45° to the perpendicular direction by using a micrometer stage. The 561-nm and 633-nm laser was chopped by an optical chopper to achieve an on-off laser mode with a laser-on time of 60 ms and a laser-off time of 140 ms. The longer laser-off time gives particles transiting the NPC sufficient time to escape from the illumination volume and for fresh fluorescent cargo to diffuse from the cytoplasm or the nucleus into the NPC.
Fig. 7.
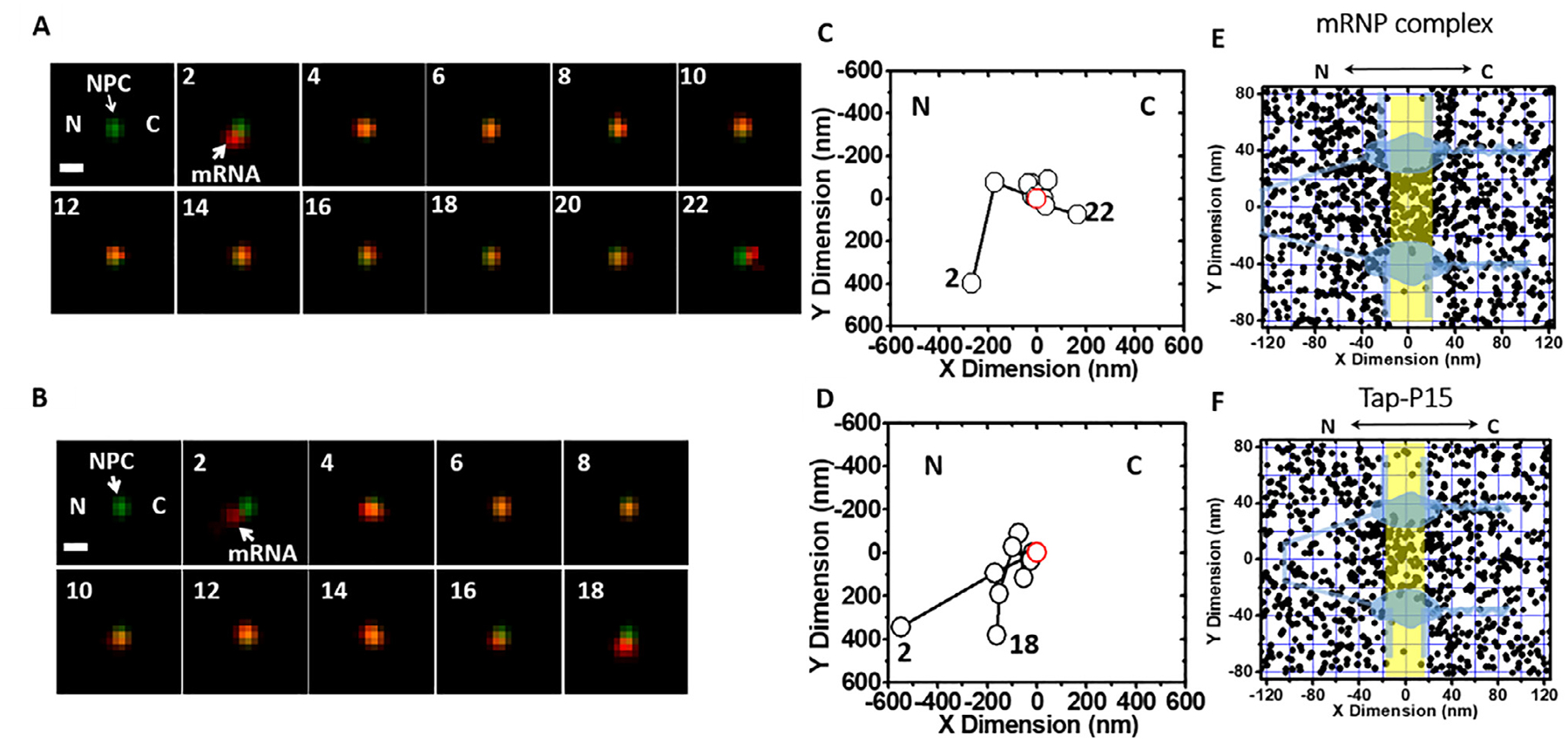
Single-molecule trajectories and 2D spatial locations of mRNPs in single NPCs [40]. (A) A typical successful single mRNP export event captured by SPEED microscopy. A single mCherry-tagged mRNP (red spot) started from the nucleus, interacted with a single GFP-tagged NPC (green spot) and arrived in the cytoplasm. Numbers denote time in milliseconds. (B) A typical abortive single mRNP export event. A single mCherry-tagged mRNP (red spot) started from nucleus, interacted with a single GFP-tagged NPC (green spot) and returned to the nucleus. (C and D) Single-particle tracks (black open dots) and the centroid of the NPC (red open dot) were acquired by 2D Gaussian fitting to point spread functions in a series of images for either the successful event in A or the abortive event in B. (E) Experimentally determined 2D spatial locations of mRNPs in the NPC. A schematic of the NPC (light blue) is superimposed, and the central region of the NPC (−20 nm to 20 nm) is highlighted in yellow. (F) Experimentally determined 2D spatial locations of Tap/p15 in the NPC. A schematic of the NPC (light blue) is superimposed, and the central region of the NPC (−20 nm to 20 nm) is highlighted in yellow. C, cytoplasmic side of the NPC; N, nucleoplasmic side of the NPC.
4.5. Copy number of GFPs per NPC and MCP-FPs per mRNP
Use SPEED microscopy to image single GFP and obtain the intensity of single GFP (Fig. 8).
Use the same setup to detect image of MCP-mCherry-mRNA (or MCP-GFP-mRNA) and GFP-NPC and determine their intensities (Fig. 8).
Our comparisons between the intensities of single GFP, GFP-NPC and mCherry-tagged mRNP revealed that there were ~ 8 copies of GFP in each NPC and ~ 10 copies of MCP-mCherry per mRNP (Fig. 8).
Fig. 8.
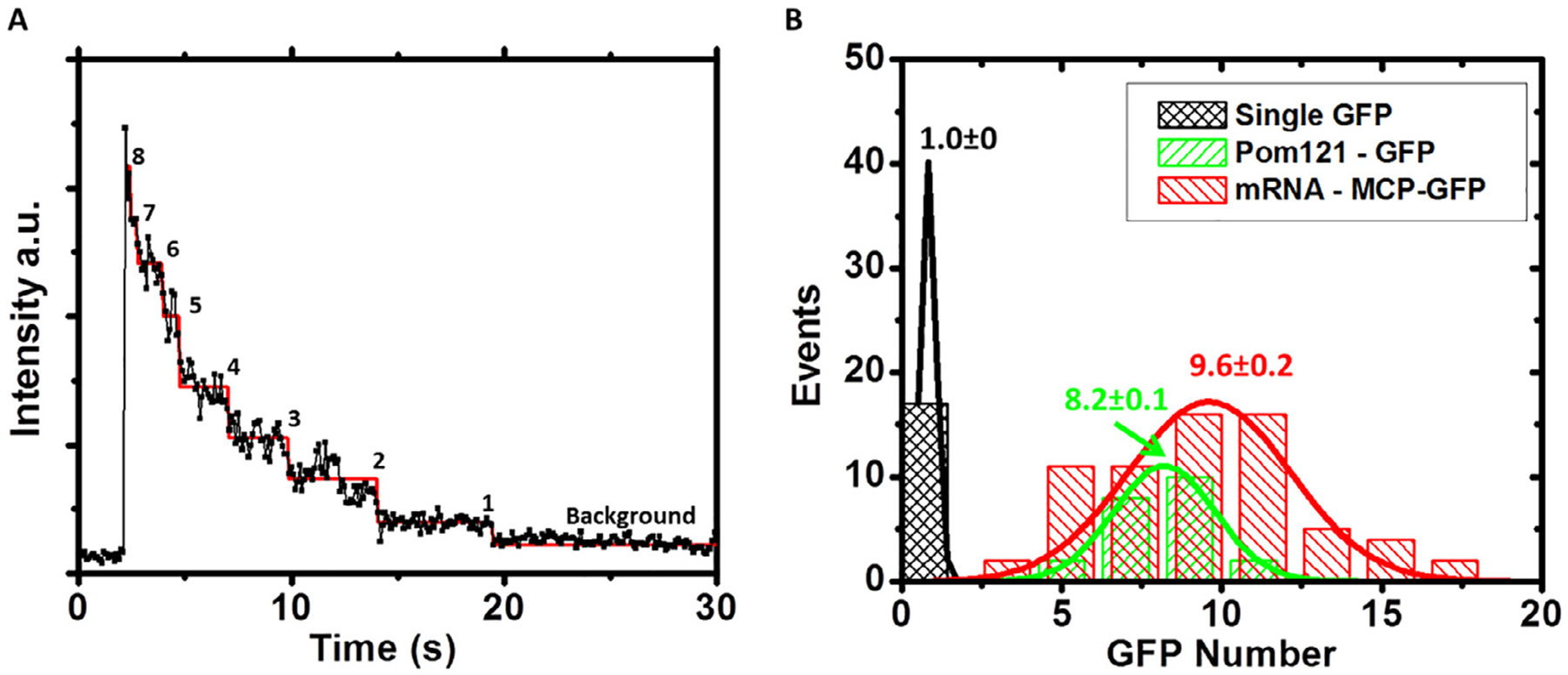
Determination of intensity of single GFPs, copy number of GFP-POM121 per NPC and copy number of MCP-GFP per mRNP in live cells [40]. (A) Photobleaching curve of eight copies of GFP-POM121 in a single NPC. The steps were determined by the maximum likelihood ratio method [61,62]. The intensity of single GFP was determined by averaging the step-intensity of GFP-POM121 in the NPC of live cells. (B) Compared to the fluorescence intensities of single GFP, ~8 copies of GFP-POM121 were found in each NPC and ~10 copies of MCP-GFP per mRNP. MCP-mCherry-mRNA was constructed with the exact same procedure as MCP-GFP-mRNA, and thus ~10 copies of MCP-mCherry per mRNA were concluded.
4.6. Localization of the NE and a single NPC on the NE
The GFP-NE can be excited using wide-field epi-fluorescence microscopy (Fig. 1C).
The pixel intensities within a row approximately perpendicular to the NE were fit with a Gaussian. The peak position of the Gaussian for a particular set of pixel intensities was considered the NE position for that row (Fig. 1D).
The peak positions of a series of such Gaussians were then fit with a second-degree polynomial, yielding the location of the NE within the entire image (Fig. 1F).
Individual GFP-NPC was excited using SPEED microscopy (Fig. 1G, Hint 16)
The fluorescent spot (GFP-NPC) was fitted by a 2D Gaussian function in both x and y directions (Fig. 1H–J).
4.7. Localization precision of isolated fluorescent spots
The localization precision for fluorescent NPCs, as well as immobile and moving fluorescent mRNPs, was defined as how precisely the central point of each detected fluorescent diffraction-limited spot was determined. For fluorescent NPCs and immobile mRNPs, the fluorescent spot was fitted with a 2D elliptical and symmetrical Gaussian, respectively, and the localization precision was determined by the standard deviation (s.d.) of multiple measurements of the central point. For moving mRNPs, the fluorescent spot was fitted with a 2D elliptical Gaussian function, and the localization precision (σ) was determined as:
| (1) |
where F is equal to 2, where N is the number of collected photons, a is the effective pixel size of the detector, b is the s.d. of the background in photons per pixel and
| (2) |
s0 is the standard deviation of the point spread function in the focal plane, D is the diffusion coefficient of substrate in the NPC and Δt is the image acquisition time [41,63–65].
In our measurements, typically 3000 signal photons versus 15 noise photons were collected from individual 10-mCherry-labelled mRNPs at a detection frame rate of 500 Hz in living cells. Correspondingly, the localization precision was ~8 nm for the immobile single mRNPs and ~10 nm for moving mRNPs. Because of the inevitable vibration of NPCs in the NE of living cells, the localization precision of the NPC centroid was 5–11 nm within our 2-minute detection time in total for one single-molecule experiment. The system error of aligned red and green fluorescence channels was determined to be 3.0 ± 0.1 nm. Therefore, the overall tracking precision for mCherry-fused mRNP export through the GFP-labelled NPC in living cells was estimated to be ~11–15 nm.
4.8. Calculation of transport time and transport efficiency
Overlay of all the trajectories of molecules and a NE or NPC identified the location of all functional NPCs within the imaging area.
We defined two classes of trajectories for cargos that approached within 100 nm of the NE from the nuclear side. The first class consists of trajectories for which the first and last points are at least 100 nm away from the NE and both are on the nuclear side of the NE. This class of trajectories identifies those molecules that did not successfully cross the NE or NPC (abortive export, Fig. 7B and D). The second class consists of trajectories for which the first and last points are at least 100 nm away from the NE, and the first is on the nuclear side of the NE while the last is on the cytoplasmic side. Therefore, this second class is considered that those molecules did successfully cross the NE or NPC (successful export, Fig. 7A and C).
Export efficiency is the number of class 2 trajectories divided by the number of class 1 and 2 trajectories combined and is reported as a percentage.
Transport times or nuclear export times can be obtained by multiplying the amount of frames obtained for each class 1 and class 2 trajectories in which the single molecule is localized within the single NPC by the frame rate utilized.
4.9. Diffusion coefficients of mRNPs inside and outside the NPC
Briefly, Imaris (Bitplane) was used for particle tracking [66]. Only particles that lasted for more than nine frames were used for further analysis. All tracks were visually inspected to ensure that they arose from well-isolate particles. In-house MATLAB routines were used to calculate the MSD and diffusion coefficients. Brownian diffusion coefficients were calculated by fitting the MSD for the first three time intervals to the equation:
| (3) |
where <r2> represents the MSD, D represents the diffusion coefficient, Δt represents time intervals, and the offset is noise correction. The MSD versus time plots were fitted using equations representing the appropriate diffusive motion as described [66].
Experimentally, mRNPs exhibited ~13% of their total spatial locations in the NPC center, ~ 46% on the nucleoplasmic side and ~41% on the cytoplasmic side during the total ~12 ms transport time. Correspondingly, they spent 1.6 ms in the center, ~5.5 ms on nuclear side and ~4.9 ms on cytoplasmic side. The volume (V) in the NPC center (rcen, 25 nm in radius; Lcen, 40 nm in length) is ~20 times smaller than that on either side of the NPC (rcyto = rnuc, 80 nm in radius; Lcyt = Lnuc, 80 nm in length). The volume of a central channel (rh, 10 nm in radius) barely occupied by mRNPs on the nucleoplasmic side and at the central region was subtracted from the calculation of diffusion volumes in these two areas.
According to the following equations:
| (4) |
| (5) |
| (6) |
The diffusion constants of mRNPs on the nucleoplasmic side, the center and the cytoplasmic side of the NPC are Dnuc = 0.16 mm2s−1, Dcen = 0.07 mm2s−1 and Dcyto = 0.19 mm2s−1.
4.10. 2D to 3D transformation process to obtain 3D spatial mapping of nuclear transport routes for mRNAs and their transport receptors through native NPCS
When thousands of 2D localizations are obtained, select one region along the x-dimension, for example from −20 nm to 20 nm relative to the obtained NPC centroid (Fig. 7E or F). The number of the locations obtained in an optimized bin-size (7 nm) (Fig. 11) increments along the y-dimension must be generated for the chosen x–dimensional region in Fig. 7E or F. This data is then placed into the matrix equations.
Use Matrix equations to calculate the relative spatial probability density distributions for the r-dimension (Fig. 4).
Normalize the relative density data.
Draw concentric rings to represent circular slices of the NPC along the axis and fill the rings with grayscale color based on the relative density for r-dimension by Photoshop, like Fig. 3D. The highest density area should be white (100%), while the areas lacking density will be completely black (0%).
Repeat step 1–4 for other x-dimensional regions (Hint 17).
Assemble the 3D structure by combining and stacking the generated ring picture (Fig. 9) obtained for each x-dimensional region with the program Amira.
Fig. 11.
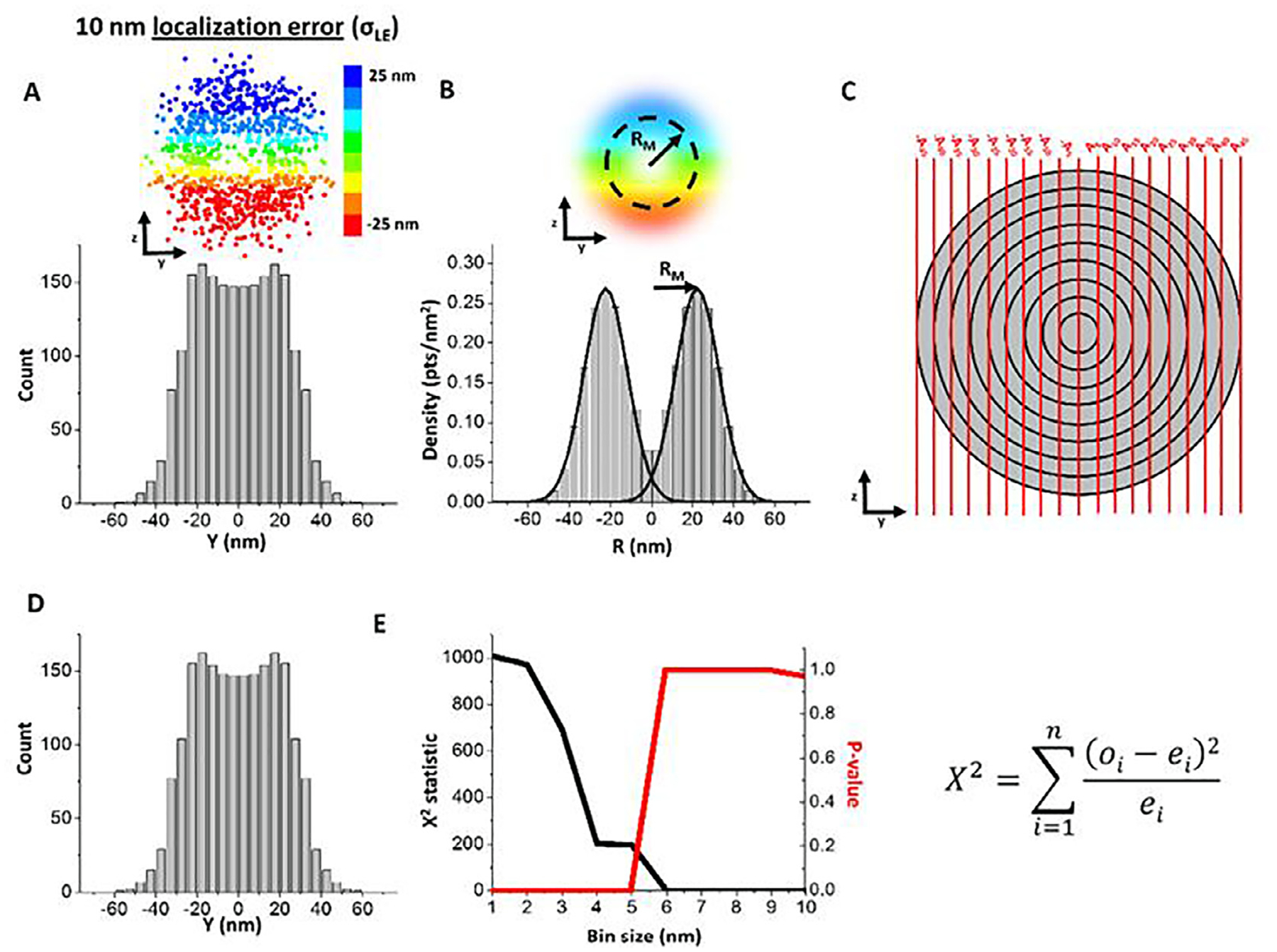
Optimal bin size determination using Chi-square error analysis. For any given set of simulated data, the bin size is varied from 1 nm to the precision that is 10 nm in this example. (A) Data sets were simulated in three dimensions. Color bar indicates z position of the simulated points. Each data set was simulated first with an ideal 25-nm radius (RI). Subsequently, a localization error (σLE) of 10 nm was added to each point. Using a 5-nm bin size for demonstration, the 2D histogram of a simulated data set with a 25-nm radius and 10-nm localization precision was determined. (B) The 3D density histogram was then obtained via the 2D to 3D transformation algorithm and the peaks were fit with Gaussian distributions. (C) The 5-nm bin size area matrix was calculated and multiplied by the 3D density distribution in (C) to reconstruct the 2D distribution. (D) The values of the reconstructed 2D distribution were then compared bin-by-bin to the original 2D distribution (as shown in A) using the Chi-square analysis equation where ‘o’ refers to the observed histogram values in (D), ‘e’ refers to the expected histogram values in (A), ‘i’ refers to the bin, and ‘n’ refers to the total number of bins with histogram values in them. (E) The Chi-square statistic and p-value were then plotted across the potential bin size values. A p-value ≤ 0.05 indicates that the 2D histograms in (A) and (D) are different from each other, suggesting the lack of enough data to allow sufficient sampling from each bin. A p-value > 0.05 indicates that the 2D histograms are not statistically different and likely have enough data points to accurately measure the value in each bin. Chi-square analysis was performed 10 times for each set of simulation parameters.
Fig. 4.
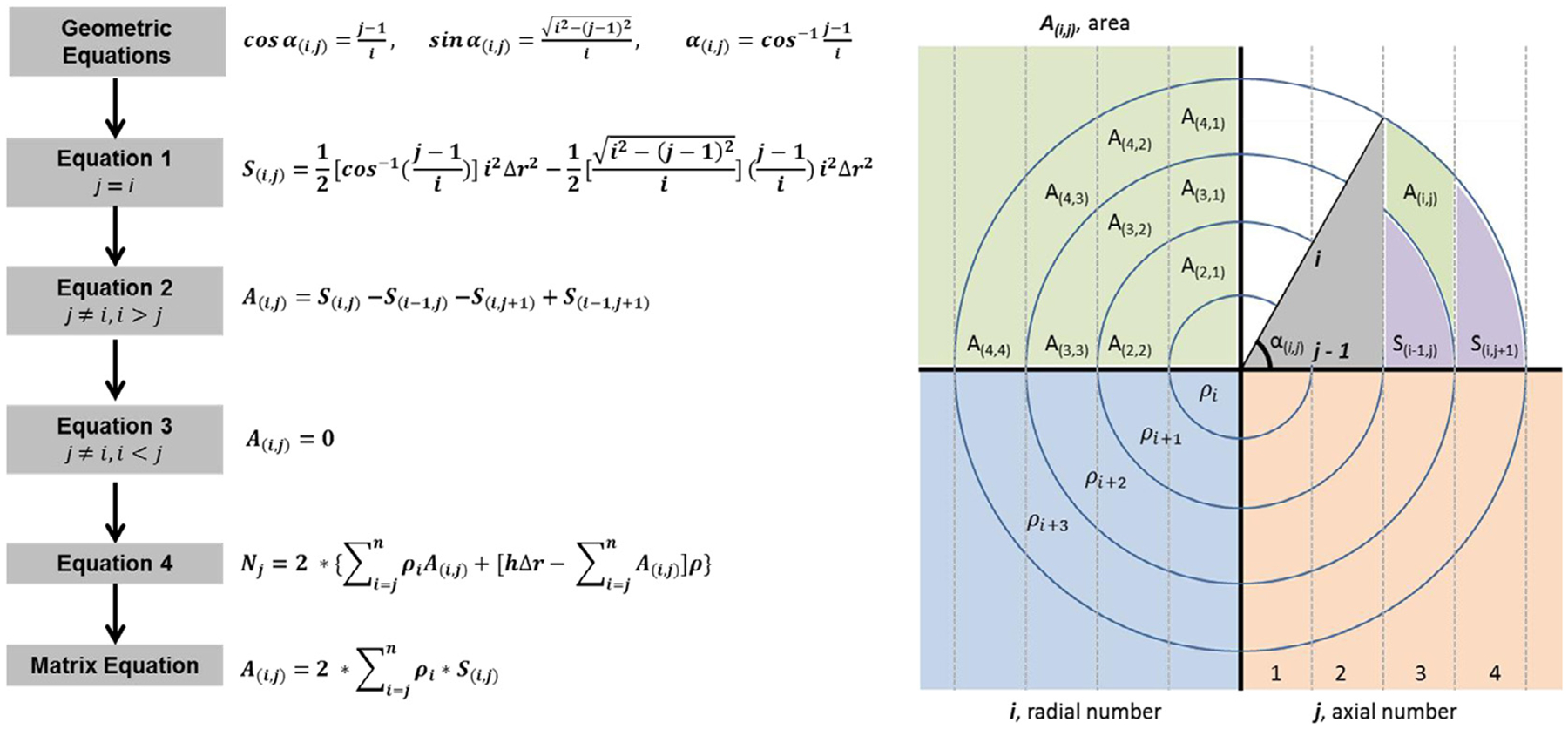
Matrix equation flowchart. (A(i,j)), area of the sub-region, (i), radial number, (j), axial number,(ρi), spatial probability density in each radial ring, (S(i,j)), area of the sub-region, (Nj), number of events, and (Δr), bin size. Eqs. (1)–(3) will determine the area of the sub-region given certain parameters (i.e. i = j). Equation (4) will determine the number of events in the given area. Once the sub-region area (A(i,j)) and events (Nj) are known, the spatial probability density in each radial ring (ρi) can be calculated.
4.11. The reproducibility of the 3D pathways of macromolecules through the NPC obtained by SPEED microscopy estimated by computational simulations [54]
We also determined the reproducibility of obtaining accurate 3D super-resolution information for mRNAs and TAP/p15 in the NPCs (Fig. 9 and Table 2) [67]. By using Monte Carlo simulations, we have demonstrated that two critical parameters, the single-molecule localization error of targeted molecules and the number of single-molecule locations, will determine the reproducibility of obtaining accurate 3D super-resolution information in the biological structures with rotational symmetry. Typically, the spatial localization of individual targeted molecules labeled with fluorophores was determined with non-zero localization error because of, primarily, background noise and limited photon collection in real experiments. To mimic typical transport routes of proteins in the NPC, our Monte Carlo simulations were performed where varying numbers of single-molecule locations were randomly simulated on an ideal radius (RI) (Fig. 10A and B). Then, single-molecule localization error (σLE) was added to RI by sampling an error value from a normal distribution with a standard deviation of σLE (Fig. 10C). Subsequently, the 2D-to-3D transformation algorithm was performed on only the y dimensional data of the simulated single-molecule localization distribution around RI to model the loss of z dimensional information during the 2D microscopy projection process. The peak position of the transformed 3D density histogram was then determined by Gaussian fitting to produce a measured mean radius (RM) which may deviate from the RI due to limited number of simulated locations and non-zero single-molecule localization error (Fig. 10D). We conducted 10,000 iterations of this process and obtained 10,000 RM values, in which the mean of the RM values converges on RI as expected (Fig. 10E). To quantify how reproducible a single experimental data set is, we set out to determine how many individual RM values from the whole distribution of RM fell within an acceptable range of RI. The acceptable range was defined as the RI ± σLE because, in principle, any single RM value can only be accurately localized within the range of approximately two standard deviations of its Gaussian fitting, similar to the concept of resolution stated by the Rayleigh Criterion (Fig. 10F). We expect that a high number of simulated single-molecule locations or low single-molecule localization errors would increase the number of iterations that fall within the acceptable range, thus resulting in a high reproducibility rate. To determine the reproducibility rate, we found that two critical steps should be correctly followed first in the process: first, optimize the bin size for each set of simulation parameters. This is accomplished by determining the smallest bin size that produces no statistical difference by Chi square analysis between the original 2D histogram and the back-calculated 2D histogram obtained by multiplying the 3D density histogram by the corresponding area matrix (Fig. 11). The second step is to account for the sensitivity of the inner bins of the area matrix when determining the accurate RM peak fitting (Fig. 12).
Table 2.
The reproducibility of 3D transport paths. Exp is the experimental parameters; Sim is the computational simulation.
| Axial Dimension (x, nm) | mRNP | TAP/p15 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| −100 to −50 | −50 to −20 | −20 to 10 | 10–50 | 50–80 | −100 to −50 | −50 to 0 | 0–20 | 20–50 | 50–80 | |
| Points (Exp.) | 191 | 161 | 99 | 207 | 89 | 158 | 189 | 43 | 134 | 103 |
| Radial Peak(s) (Exp., nm) | 56 | 36 | 22 | 0 | 0 | 75 | 0, 22 | 21 | 11 | 77 |
| Reproducibility (Exp., %) | 91 | 91 | 87 | 98 | 96 | 85 | 96, 91 | 75 | 99 | 76 |
| Points for ~90% Reproducibility (Sim.) | 165 | 140 | 130 | 12 | 12 | 225 | 12, 130 | 130 | 30 | 200 |
Fig. 10.
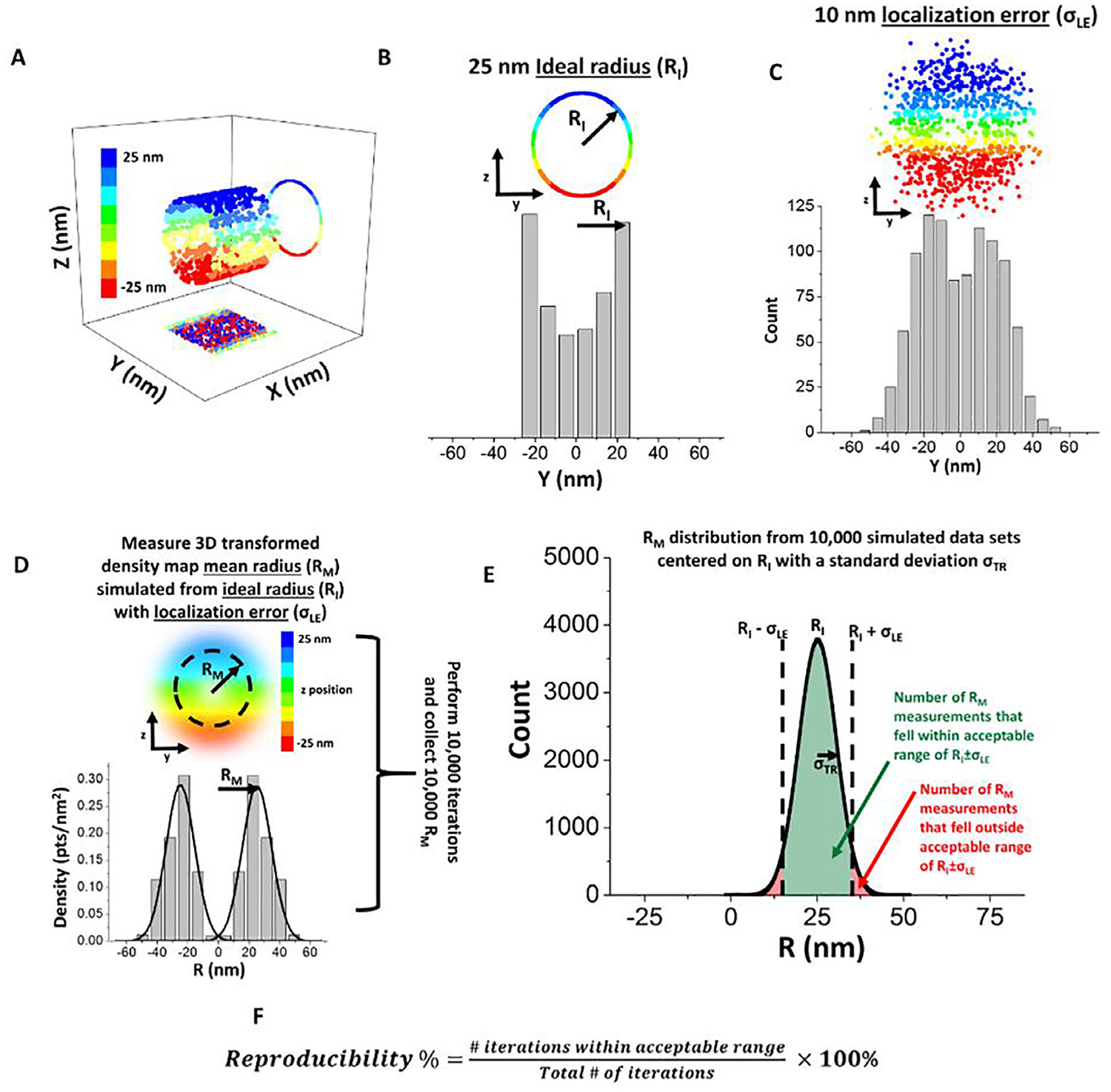
SPEED microscopy and 2D to 3D transformation reproducibility percentage using a simulation-based approach. For any given set of simulated data, the bin size is varied from 1 nm to the precision that is 10 nm in this example. (A) Data sets were simulated in three dimensions. Color bar indicates z position of the simulated points. (B) Each data set was simulated first with an ideal 25-nm radius (RI). (C) Subsequently, a localization error (σLE) of 10 nm was added to each point. (C) Using a 5-nm bin size for demonstration, the 2D histogram of the simulated data set with a 25-nm radius and 10-nm localization precision was determined. (D) 10,000 data sets were simulated with an ideal 25-nm radius (RI) and a localization error (σLE) of 10 nm. The resultant 3D histograms were then each fitted with a Gaussian function to localize the mean position of each peak, which is designated as the mean radius RM. (E) The histogram for all the RM values was determined and the number of simulated data sets that fell within the acceptable range of RI ± σLE were counted. The acceptable range of RI ± σLE was chosen because, in principle, the Rayleigh criterion limited the resolution of any single 3D histogram to the spread of that distribution, which was due to the simulated localization error (σLE). After 10,000 simulations, the histogram for RM values converges on the mean (RI) from which they were originally sampled, while the spread of the RM histogram (σTR) converges on a value that is due to the number of simulated points in each distribution and simulated localization error. (F) Reproducibility percentage was defined as the number of RM values that fell within the acceptable range of RI ± σLE divided by the total number of simulated data sets and multiplied by 100%.
Fig. 12.
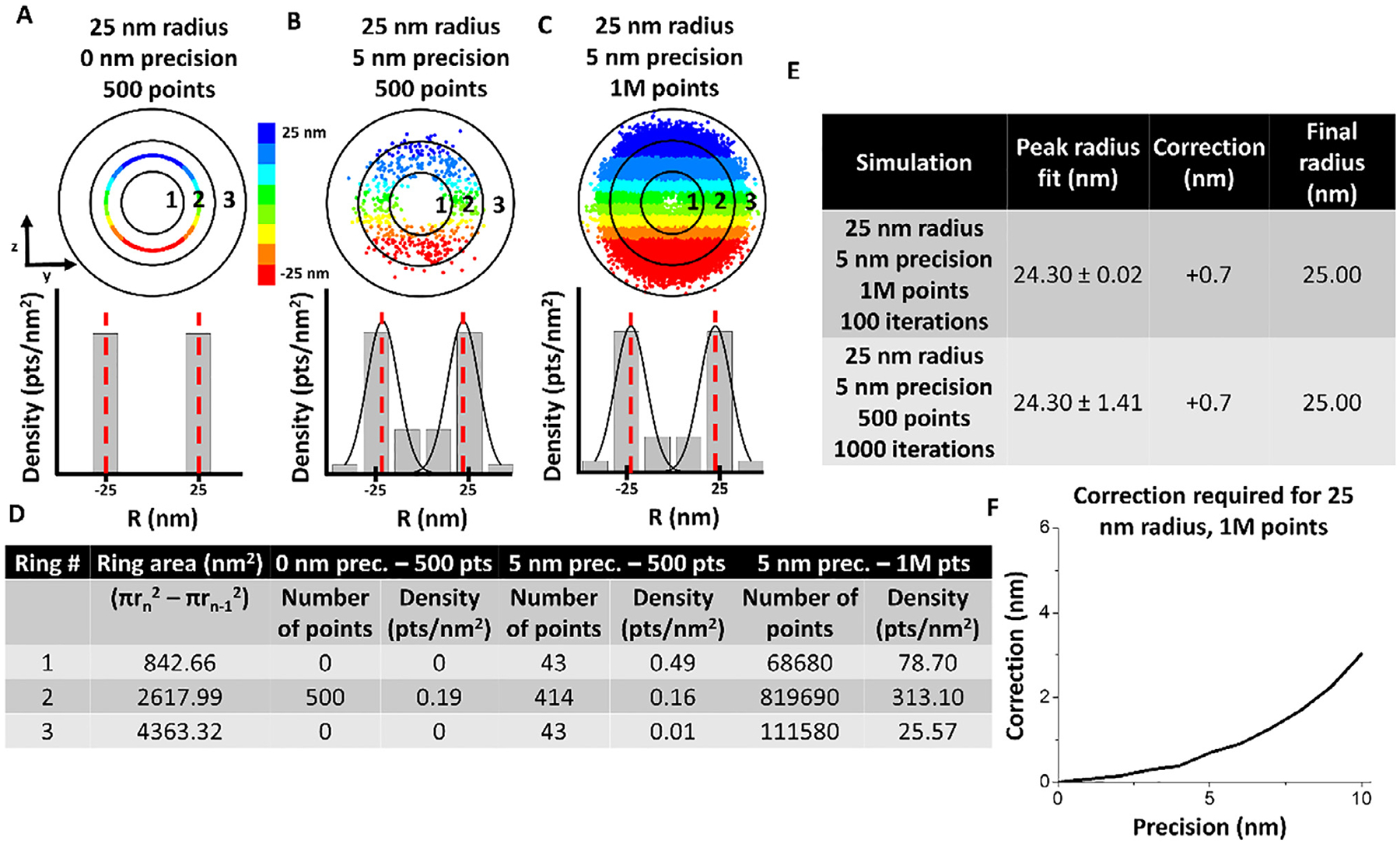
Sensitivity of inner bins necessitates slight correction of peak position during simulation. (A)–(C) Simulated single molecule data and corresponding 3D density histogram for simulation with 25 nm radius, 0 nm localization error, and 500 points. Red dashed lines indicate mean peak fitting. (B) Simulated single molecule data and corresponding 3D density histogram for simulation with 25 nm radius, 5 nm localization error, and 500 points. Red dashed lines indicate mean peak fitting. (C) Simulated single molecule data and corresponding 3D density histogram for simulation with 25 nm radius, 5 nm localization error, and 1 million points. Red dashed lines indicate mean peak fitting. (D) Table showing the calculation to obtain each bin of the 3D density histogram. (E) Table showing that even one million points does reconstruct a precise 25 nm peak fitting due to the fact that the inner radial bins have smaller area and are slightly more sensitive to changes in density. (F) Correction required for each precision up to 10 nm for a 25 nm radius. This correction process was performed before each simulation to accurately localize the RM density peak and correlate it to the ideal RI from which the data was simulated.
As a result, our simulation suggested that the high reproducibility is typically ensured by high number of points and high localization precision. For example, for the region of −100 to −50 along the axial dimension, simulations suggest that 165 points in this region are required to get 90% reproducibility of obtaining an accurate 3D path. Experimentally, we have collected 191 points and generated finally 91% reproducibility.
5. Hints
Clones containing the MS2 stem loops were created in SURE2 bacterial cells (Stratagene) to minimize recombination of the MS2 repeats with the bacterial genome. Still, we had to resort to a two-step cloning procedure, as linearized plasmids containing the MS2 stem loops often recombined with the genome of competent bacterial cells, thus resulting in clones bearing smaller plasmids.
IPTG is a lactose metabolite that triggers transcription of the lac operon, where the lacZ gene is replaced with the gene of interest and IPTG is then used to induce gene expression. This s used for proteins constructs under the power of T7 promoters.
The purification of the target protein should be more than 90% for single molecule-experiments.
Check the protein information to make sure there are surface cysteines available for labeling. For proteins without cysteines available, it could be labeled with another strategy.
The concentration of the free dye should be determined and be controlled to be less than 0.1% of the target dye.
If the labeling ratio > 1, the activity of the target molecule should be checked and the final concentration for the experiment should be calculate with labeled molecule. If the labeling ratio < 1, the final concentration for experiment should calculate by the real concentration of the labeled protein, which equal to the concentration of the protein
All the DMEM media should be removed prior to experimentation and replaced with transport buffer.
Monitor the permeabilization in real time using bright field imaging. This is accomplished by observing the intensity contrast between the nucleus and the other cellular components. Signal to noise ratio should improve significantly for fluorescence signal. The intact nucleus, together with a controlled cytoplasmic environment, provides a greatly simplified transport system that allows us to elucidate the complicated nuclear transport step by step.
Before single molecule experiments, the precision and photobleaching time of the single cargo molecule should be determined. Plate 10 pM cargo molecules on a glass cover slip to become immobile after 30 min due to non-specific absorption. The intensity and time course of the fluorescence can then be measured. The intensity could be transformed to determine the number of emitted photons to determine the precision. For example, the precision of four Alexa647 labeled Imp β1 could be 9 nm, which emit over 1100 photons per frame. The photobleaching time should be controlled as more than twice the transport time of the targeted cargo.
Fluorescent cargo concentrations were > 100 nM in bulk experiments, and ~100 pM for single molecule assays. At these concentrations, > 99% of the cargo was expected to form complexes with Importin α and Importin β1 and lone single-molecules are able to be imaged.
Examine the ratio of Gaussian widths in the long and short axis of the chosen GFP-NPC fluorescence spot. The ratio needs to fall in between 1.74 and 1.82. Within this range, an illuminated NPC only has a free angle of 1.4° to the perpendicular direction to the NE.
Be careful not to touch the sample. Take the image of the NE before and after experiment to help to determine if the cell was damaged.
The drift of the optical system should be determined before the experiment. The total detection time should be less than the time by which a large drift in precision can occur.
The position of the NPC could be rechecked with the NE and the symmetry of the y-dimension should also be monitored.
The single-molecule locations should be filtered based on localization precision and Gaussian width to remove imprecise points and positions that may not represent true single-molecules.
We focused on a GFP-NPC with ~eight copies of GFP-POM121, individual GFP-NPCs on the NE were selected when their fluorescence intensity was ~8-fold that of a single GFP. Second, we chose a fluorescent NPC on the equator of the nucleus such that the tangent of the NE at the location of this NPC was parallel to the y direction of the Cartesian coordinates (x, y) in the CCD camera.
The regions are determined by similar r-dimension distributions (peak positions) in each 5–10 nm (bin size may vary and an optimal bin size is chosen based on the optimization process in Fig. 11) binning of the data along the x-dimension.
6. Discussion
6.1. New features of mRNA export obtained by SPEED microscopy
First, the NPC was determined to operate as a major selectivity barrier for mRNPs. In our study, ~36% of all exporting events of mRNPs successfully export, crossing the barrier located in the nuclear basket and central scaffold region of the NPC, whereas the other 64% of exporting mRNPs failed their export mainly before they passed the central scaffold region. It is likely that FG-Nups at the central scaffold region form the dominant selective barrier, but the exact reason for the abortive export events of mRNA are still not fully understood. Second, a refined nuclear export time of ~12 ms for mRNPs was obtained. The export time of mRNPs we found is much shorter than previous studies possibly due to much faster detection speed and the high localization precision for single NPC [40]. Indeed, for comparison, we repeated the measurements with a slower detection speed for nuclear export of mRNPs, resulting in a longer nuclear export time for mRNPs [40]. Also, we found there are many slowly moving mRNPs at the periphery of NE, which could be mistakenly counted in nuclear export time of mRNPs if single NPCs could not be precisely localized [40]. Third, a new diffusion pattern of mRNPs in the NPC was revealed by this high-resolution 3D imaging approach. The diffusion coefficient of mRNPs in each sub-region of the NPC was directly determined as: ~0.16 μm2/s on the nucleoplasmic side, ~0.07 μm2/s in the central scaffold region, and ~0.19 μm2/s on the cytoplasmic side, revealing a fast-slow-fast diffusion pattern of mRNPs export through the NPC [40]. Fourth, a 3D export route revealed the actual paths of mRNPs through the NPC in live cells for the first time, the reliability and accuracy of which was supported by the reproducibility of obtaining 3D paths of mRNPs (Table. 2). Notably, mRNPs primarily interact with the periphery of, and seldom present in, the central axial channel that is reserved for small molecules to passively diffuse through the NPC within the NPC’s nuclear basket and central scaffold regions [40]. In summary, the great technical advances in SPEED microscopy brought more detailed dynamics of mRNP nuclear export into focus. In the near future, SPEED microscopy could be readily expanded for studying nucleocytoplasmic export of other types of RNAs, such as rRNA, microRNA and viral RNA.
6.2. Reliability of SPEED microscopy in distinguishing multiple transport pathways through the NPCs
In the past years, our applications of SPEED microscopy to study the nuclear transport kinetics and routes for proteins and mRNAs have distinguished two distinct pathways – the central passive diffusion path and the peripheral facilitated transport passageway in the NPC [2,32,40,50–51,58]. However, there might be additionally a combination of these two known pathways for macromolecules to transport through the NPC (one example is the routes of Tap/p15 at the NPC’s sub-region −50 nm to 0 nm as shown in Table 2). Therefore, we set out to test the reliability of SPEED microscopy in distinguishing three distinct distributions summarized in Fig. 13. We have simulated datasets from these three distinct distributions (peripheral, central and bimodal), and then compared the reconstructed distributions to the ground truth input distributions (obtained by simulating 1,000,000 points) via a categorization routine that compares the sum of the absolute valued residuals between the simulated data set and the ground truth data set (Fig. 13A–F). It is important to note that, prior to each simulation, the routine dynamically calculates an optimal categorization bin size for each set of parameters via a minimized Chi-square error algorithm (Fig. 11). When the simulation was applied to the 10-nm localization precision case, ~100–300 points were sufficient to distinguish the three distributions with > 90% success (Fig. 13G). Open source code for these simulations can be freely download [67]. Coupled with the simulations in this paper, we have shown that SPEED microscopy is able to collect sufficient quality data to characterize a reliable 3D spatial density distribution and localize the mean peak position for transport route through the NPC with high accuracy and reproducibility.
Fig. 13.
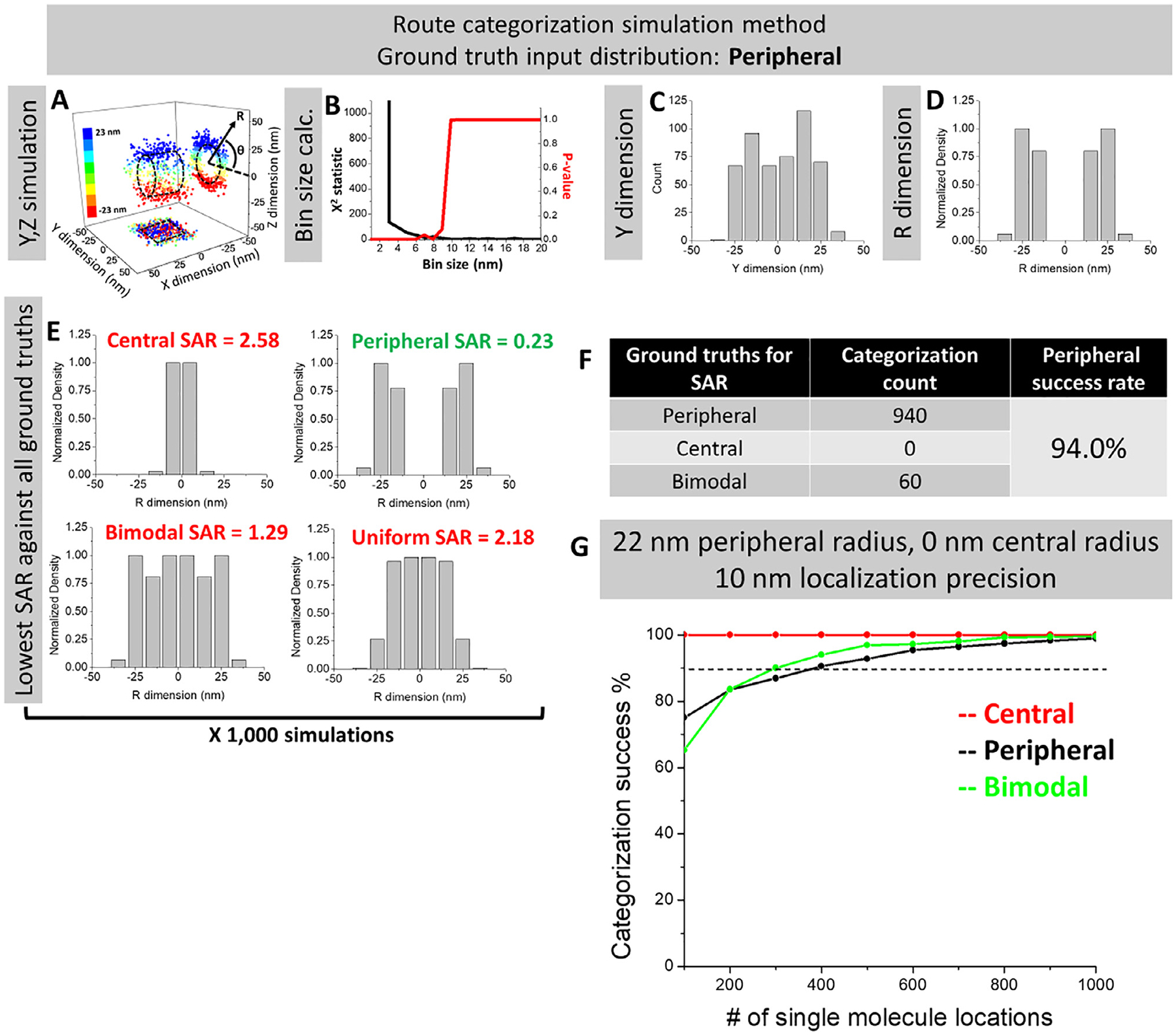
Route categorization simulations to confirm the high reliability of 3D mapping of transport routes for mRNAs and Tap/p15. (A) For each set of simulation parameters (distribution type, localization precision, peripheral radius, central radius, % transit in each route of a bimodal distribution, and number of single molecule locations), 1000 datasets were generated in the Y, Z dimensions. Displayed here is a representative dataset from a peripheral distribution, localization precision of 5 nm, peripheral radius of 23 nm, central radius of 0 nm, 50% central route localization and 500 single molecule locations to clearly visualize the distribution from the Y, Z scatter data. Y, Z coordinates can also be transformed to analogous R,θ coordinates. (B) The bin size optimization algorithm was performed according to in Fig. 11 using the given simulation parameters. (C) The Y-dimensional histogram was obtained with the optimized bin size for every dataset. (D) The 2D-to-3D transformation algorithm was performed on every dataset. (E) The resultant 3D density histograms were compared via SAR to the central, peripheral, and bimodal ground truth distributions, which were obtained by simulating a dataset with all the same parameters except for 1,000,000 single molecule locations. The lowest SAR indicates which distribution a given dataset is most similar to and, thus, which ground truth distribution it is classified as. (F) The count of all classifications for the ground truth peripheral parameters set of 5 nm localization precision, 23 nm peripheral radius, 0 nm central radius, and 100 single molecule locations. (G) Simulation results for the peripheral, central, and bimodal distributions with parameters of 10 nm localization precision, 22 nm peripheral radius, 0 nm central radius, 26% central route localization, and 10–20 nm bin sizes obtained from optimization algorithm.
Currently, it is highly desirable but still challenging to obtain 3D super-resolution information of structures in fixed specimens as well as dynamic processes in live cells with a high spatiotemporal resolution. Without using 3D super-resolution microscopy or real-time 3D particle tracking that typically involves complex optical implements, SPEED microscopy and its 2D-to-3D transformation algorithm provide an effective alternative approach to achieving 3D sub-diffraction-limited information in sub-cellular organelles that have rotational symmetry, such as NPC and primary cilia [51,56].
Acknowledgement
The project was supported by grants from the National Institutes of Health (NIH GM097037, GM116204 and GM122552 to W.Y.).
Footnotes
Conflicts of interest
The authors declare no conflict of interest.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.ymeth.2018.08.005.
References
- [1].Macara IG, Transport into and out of the nucleus, Microbiol. Mol. Biol. Rev 65 (4) (2001) 570–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Yang W, Distinct, but not completely separate spatial transport routes in the nuclear pore complex, Nucleus 4 (3) (2013) 166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rout MP, Blobel G, Isolation of the yeast nuclear pore complex, The Journal of cell biology 123 (4) (1993) 771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Görlich D, Kutay U, Transport between the cell nucleus and the cytoplasm, Annu. Rev. Cell Dev. Biol 15 (1) (1999) 607–660. [DOI] [PubMed] [Google Scholar]
- [5].Maimon T, Elad N, Dahan I, Medalia O, The human nuclear pore complex as revealed by cryo-electron tomography, Structure 20 (6) (2012) 998–1006. [DOI] [PubMed] [Google Scholar]
- [6].Wente SR, Rout MP, The nuclear pore complex and nuclear transport, Cold Spring Harbor Perspect. Biol 2 (10) (2010) a000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait BT, The molecular architecture of the nuclear pore complex, Nature 450 (7170) (2007) 695. [DOI] [PubMed] [Google Scholar]
- [8].Mattaj IW, Englmeier L, Nucleocytoplasmic transport: the soluble phase, Annual Reviews 4139 El Camino Way, PO Box 10139, Palo Alto, CA 94303–0139, USA, 1998. [DOI] [PubMed]
- [9].Vasu SK, Forbes DJ, Nuclear pores and nuclear assembly, Curr. Opin. Cell Biol 13 (3) (2001) 363–375. [DOI] [PubMed] [Google Scholar]
- [10].Schwartz TU, Modularity within the architecture of the nuclear pore complex, Curr. Opin. Struct. Biol 15 (2) (2005) 221–226. [DOI] [PubMed] [Google Scholar]
- [11].Fahrenkrog B, Köser J, Aebi U, The nuclear pore complex: a jack of all trades? Trends Biochem. Sci 29 (4) (2004) 175–182. [DOI] [PubMed] [Google Scholar]
- [12].Strawn LA, Shen T, Shulga N, Goldfarb DS, Wente SR, Minimal nuclear pore complexes define FG repeat domains essential for transport, Nat. Cell Biol 6 (3) (2004) 197. [DOI] [PubMed] [Google Scholar]
- [13].Milles S, Lemke EA, Single molecule study of the intrinsically disordered FG-repeat nucleoporin 153, Biophys. J 101 (7) (2011) 1710–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lim RY, Huang N-P, Köser J, Deng J, Lau KA, Schwarz-Herion K, Fahrenkrog B, Aebi U, Flexible phenylalanine-glycine nucleoporins as entropic barriers to nucleocytoplasmic transport, Proc. Natl. Acad. Sci 103 (25) (2006) 9512–9517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dieppois G, Stutz F, Connecting the transcription site to the nuclear pore: a multi-tether process that regulates gene expression, J. Cell Sci 123 (12) (2010) 1989–1999. [DOI] [PubMed] [Google Scholar]
- [16].Williams T, Ngo LH, Wickramasinghe VO, Nuclear export of RNA: Different sizes, shapes and functions, In: Seminars in cell & developmental biology, Elsevier, 2017. [DOI] [PubMed] [Google Scholar]
- [17].Zenklusen D, Stutz F, Nuclear export of mRNA, FEBS Lett. 498 (2–3) (2001) 150–156. [DOI] [PubMed] [Google Scholar]
- [18].Kloc M, Foreman V, Reddy SA, Binary function of mRNA, Biochimie 93 (11) (2011)1955–1961. [DOI] [PubMed] [Google Scholar]
- [19].Braun IC, Herold A, Rode M, Izaurralde E, Nuclear export of mRNA by TAP/NXF1 requires two nucleoporin-binding sites but not p15, Mol. Cell. Biol 22 (15) (2002) 5405–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Iglesias N, Stutz F, Regulation of mRNP dynamics along the export pathway, FEBS Lett. 582 (14) (2008) 1987–1996. [DOI] [PubMed] [Google Scholar]
- [21].Grosshans H, Deinert K, Hurt E, Simos G, Biogenesis of the signal recognition particle (SRP) involves import of SRP proteins into the nucleolus, assembly with the SRP-RNA, and Xpo1p-mediated export, J. Cell Biol 153 (4) (2001) 745–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Noh JH, Kim KM, Abdelmohsen K, Yoon J-H, Panda AC, Munk R, Kim J, Curtis J, Moad CA, Wohler CM, HuR and GRSF1 modulate the nuclear export and mitochondrial localization of the lncRNA RMRP, Genes Dev. 30 (10) (2016) 1224–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wild T, Horvath P, Wyler E, Widmann B, Badertscher L, Zemp I, Kozak K, Csucs G, Lund E, Kutay U, A protein inventory of human ribosome biogenesis reveals an essential function of exportin 5 in 60S subunit export, PLoS Biol. 8 (10) (2010) e1000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yao W, Roser D, Köhler A, Bradatsch B, Baßler J, Hurt E, Nuclear export of ribosomal 60S subunits by the general mRNA export receptor Mex67-Mtr2, Mol. Cell 26 (1) (2007) 51–62. [DOI] [PubMed] [Google Scholar]
- [25].Faza MB, Chang Y, Occhipinti L, Kemmler S, Panse VG, Role of Mex67-Mtr2 in the nuclear export of 40S pre-ribosomes, PLoS Genet. 8 (8) (2012) e1002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lund E, Güttinger S, Calado A, Dahlberg JE, Kutay U, Nuclear export of microRNA precursors, Science 303 (5654) (2004) 95–98. [DOI] [PubMed] [Google Scholar]
- [27].Yi R, Qin Y, Macara IG, Cullen BR, Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs, Genes Dev. 17 (24) (2003) 3011–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bohnsack MT, Czaplinski K, Görlich D, Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs, RNA 10 (2) (2004) 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gilboa E, Vieweg J, Cancer immunotherapy with mRNA-transfected dendritic cells, Immunol. Rev 199 (1) (2004) 251–263. [DOI] [PubMed] [Google Scholar]
- [30].Schlake T, Thess A, Fotin-Mleczek M, Kallen K-J, Developing mRNA-vaccine technologies, RNA Biol. 9 (11) (2012) 1319–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].McIvor RS, Therapeutic delivery of mRNA: the medium is the message, Mol. Ther 19 (5) (2011) 822–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Goryaynov A, Ma J, Yang W, Single-molecule studies of nucleocytoplasmic transport: from one dimension to three dimensions, Integr. Biol 4 (1) (2012) 10–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hell SW, Wichmann J, Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy, Opt. Lett 19 (11) (1994) 780–782. [DOI] [PubMed] [Google Scholar]
- [34].Huang B, Bates M, Zhuang X, Super-resolution fluorescence microscopy, Annu. Rev. Biochem 78 (2009) 993–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Schermelleh L, Heintzmann R, Leonhardt H, A guide to super-resolution fluorescence microscopy, J. Cell Biol 190 (2) (2010) 165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Van Oijen A, Köhler J, Schmidt J, Müller M, Brakenhoff G, Far-field fluorescence microscopy beyond the diffraction limit, JOSA A 16 (4) (1999) 909–915. [Google Scholar]
- [37].Klar TA, Engel E, Hell SW, Breaking Abbe’s diffraction resolution limit in fluorescence microscopy with stimulated emission depletion beams of various shapes, Phys. Rev. E 64 (6) (2001) 066613. [DOI] [PubMed] [Google Scholar]
- [38].Klar TA, Hell SW, Subdiffraction resolution in far-field fluorescence microscopy, Opt. Lett 24 (14) (1999) 954–956. [DOI] [PubMed] [Google Scholar]
- [39].Grünwald D, Singer RH, In vivo imaging of labelled endogenous β-actin mRNA during nucleocytoplasmic transport, Nature 467 (7315) (2010) 604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ma J, Liu Z, Michelotti N, Pitchiaya S, Veerapaneni R, Androsavich JR, Walter NG, Yang W, High-resolution three-dimensional mapping of mRNA export through the nuclear pore, Nat. Commun 4 (2013) 2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mortensen KI, Churchman LS, Spudich JA, Flyvbjerg H, Optimized localization analysis for single-molecule tracking and super-resolution microscopy, Nat. Methods 7 (5) (2010) 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Björk P, Wieslander L, Nucleocytoplasmic mRNP export is an integral part of mRNP biogenesis, Chromosoma 120 (1) (2011) 23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Erni R, Rossell MD, Kisielowski C, Dahmen U, Atomic-resolution imaging with a sub-50-pm electron probe, Phys. Rev. Lett 102 (9) (2009) 096101. [DOI] [PubMed] [Google Scholar]
- [44].Milazzo A-C, Cheng A, Moeller A, Lyumkis D, Jacovetty E, Polukas J, Ellisman MH, Xuong N-H, Carragher B, Potter CS, Initial evaluation of a direct detection device detector for single particle cryo-electron microscopy, J. Struct. Biol 176 (3) (2011) 404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bammes BE, Rochat RH, Jakana J, Chen D-H, Chiu W, Direct electron detection yields cryo-EM reconstructions at resolutions beyond 3/4 Nyquist frequency, J. Struct. Biol 177 (3) (2012) 589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mor A, Suliman S, Ben-Yishay R, Yunger S, Brody Y, Shav-Tal Y, Dynamics of single mRNP nucleocytoplasmic transport and export through the nuclear pore in living cells, Nat. Cell Biol 12 (6) (2010) 543. [DOI] [PubMed] [Google Scholar]
- [47].Siebrasse JP, Kaminski T, Kubitscheck U, Nuclear export of single native mRNA molecules observed by light sheet fluorescence microscopy, Proc. Natl. Acad. Sci 109 (24) (2012) 9426–9431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Jan TK, Peter Siebrasse, and Ulrich Kubitscheck, Nuclear export of single native mRNA molecules observed by light sheet fluorescence microscopy, Proc. Natl. Acad. Sci 109 (24) (2012) 9426–9431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Speese SD, Ashley J, Jokhi V, Nunnari J, Barria R, Li Y, Ataman B, Koon A, Chang YT, Li Q, Nuclear envelope budding enables large ribonucleoprotein particle export during synaptic Wnt signaling, Cell 149 (4) (2012) 832–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ma J, Goryaynov A, Sarma A, Yang W, Self-regulated viscous channel in the nuclear pore complex, Proc. Natl. Acad. Sci 109 (19) (2012) 7326–7331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ma J, Yang W, Three-dimensional distribution of transient interactions in the nuclear pore complex obtained from single-molecule snapshots, Proc. Natl. Acad. Sci 107 (16) (2010) 7305–7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Daigle N, Beaudouin J, Hartnell L, Imreh G, Hallberg E, Lippincott-Schwartz J, Ellenberg J, Nuclear pore complexes form immobile networks and have a very low turnover in live mammalian cells, J. Cell Biol 154 (1) (2001) 71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kubitscheck U, Wedekind P, Zeidler O, Grote M, Peters R, Single nuclear pores visualized by confocal microscopy and image processing, Biophys. J 70 (5) (1996) 2067–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Schnell SJ, Ma J, Yang W, Three-dimensional mapping of mRNA export through the nuclear pore complex, Genes 5 (4) (2014) 1032–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ruba A, Kelich Joseph, Luo Wangxi, Yang Weidong, Obtaining 3D Super-resolution Information from 2D Super-resolution Images through a 2D-to-3D Transformation Algorithm, bioRxiv (2017). [Google Scholar]
- [56].Luo W, Ruba Andrew, Takao Daisuke, Zweifel Ludovit P., Lim Roderick Y.H., Verhey Kristen J., Yang Weidong, Axonemal lumen dominates cytosolic protein diffusion inside the primary cilium, Sci. Rep 7 (15793) (2017) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Hiraoka Y, Sedat JW, Agard DA, Determination of three-dimensional imaging properties of a light microscope system Partial confocal behavior in epifluorescence microscopy, Biophys. J 57 (2) (1990) 325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Ma J, Kelich JM, Yang W, SPEED microscopy and its application in nucleocytoplasmic transport, Nucl. Envelope (2016) 503–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Gelles J, Gelles-brandeis, 2014. < https://github.com/gelles-brandeis/Glimpse >.
- [60].Evelina Tutucci MV, Biswas Jeetayu, Garcia Jennifer, Parker Roy, Singer Robert H, An improved MS2 system for accurate reporting of the mRNA life cycle, nature, Methods 15 (1) (2018) 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Boudjellaba BMPSH, On exact inference for change in a Poisson sequence, Commun. Stat. Theory Methods 30 (3) (2001) 407–434. [Google Scholar]
- [62].Lucas AHY, Watkins P, Detection of intensity change points in time-resolved single-molecule measurements, J Phys. Chem 109 (1) (2005) 617–628. [DOI] [PubMed] [Google Scholar]
- [63].Thompson RE, Larson DR, Webb WW, Precise nanometer localization analysis for individual fluorescent probes, Biophys. J 82 (5) (2002) 2775–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Quan T, Zeng S, Huang Z, Localization capability and limitation of electron-multiplying charge-coupled, scientific complementary metal-oxide semiconductor, and charge-coupled devices for superresolution imaging, J. Biomed. Opt 15 (6) (2010) 066005. [DOI] [PubMed] [Google Scholar]
- [65].Kelich JM, Ma J, Dong B, Wang Q, Chin M, Magura CM, Xiao W, Yang W, Super-resolution imaging of nuclear import of adeno-associated virus in live cells, Mol. Ther. Methods Clin. Dev 2 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Pitchiaya S, Androsavich JR, Walter NG, Intracellular single molecule microscopy reveals time and mRNA dependent microRNA assembly [DOI] [PMC free article] [PubMed]
- [67].Ruba A, YangLab, 2017. < https://github.com/andrewruba/YangLab >.