

A multipartite DNA virus was isolated from a plant pathogenic fungus, which is infectious as cloned DNA and purified virions.
Abstract
Here, we describe a tripartite circular single-stranded (ss) DNA mycovirus, named Fusarium graminearum gemytripvirus 1 (FgGMTV1). The genome of FgGMTV1 comprises three circular ssDNA segments (DNA-A, DNA-B, and DNA-C). Sequence alignments and phylogenetic analyses showed that FgGMTV1 is nested within the family Genomoviridae. We also constructed the first infectious DNA clones of a DNA mycovirus. Our results show that DNA-A and DNA-B are mutually interdependent for their replication and are associated with severely reduced colony growth and hypovirulence. DNA-C relies on DNA-A and DNA-B for replication and is necessary for the recovery of abnormal fungal phenotypes. DNA-C also enhances the accumulation of viral DNA in infected fungi and permits stable colonization and easy transmission via conidia. This is the first multipartite DNA virus isolated from a fungus. Our phylogenetic analyses also suggest that the multipartite genome of FgGMTV1 may have evolved from a monopartite genome of an ancient genomovirus.
INTRODUCTION
Mycoviruses or fungal viruses are ubiquitous in all major taxa of fungi (1, 2). According to the latest list of fungal viruses approved by the International Committee on Taxonomy of Viruses (ICTV), mycoviruses have double-stranded (ds) RNA linear genomes, positive-sense single-stranded (+ss) RNA linear genomes including reverse-transcribing RNA linear genomes, negative-sense ssRNA (−ssRNA) linear genomes, or ssDNA circular genomes (3). The discovery of fungal viruses occurred relatively late compared to the identification and characterization of viruses of animals, plants, and prokaryotes, perhaps due primarily to the cryptic or asymptomatic nature of mycoviruses (4). However, the number of known mycoviruses has rapidly increased in the past few decades, and this trend is expected to continue, especially with the development and widespread use of next-generation sequencing techniques in the field of virology (3, 5). Studies based on this approach, along with conventional fungal virus hunting, have greatly promoted our understanding of mycovirus diversity and evolution (1).
Our knowledge of circular replication-associated protein-encoding single-stranded (CRESS) DNA virus diversity has increased markedly in recent years due to innovations in molecular techniques and sequencing technologies (6, 7). CRESS DNA viruses have been identified in environmental, fungal, plant, and animal samples. Because of their high diversity, variable genome organizations, and unknown hosts, a large number of the recently identified CRESS DNA viruses cannot be classified into any known viral families, which has resulted in large, novel CRESS DNA viral groups being proposed (8). One of the recently proposed groups is the family Genomoviridae (sigil: Ge- for geminivirus-like and nomo- for no movement protein) (9). Currently, there are nine approved genera in this family: Gemycircularvirus, Gemyduguivirus, Gemygorvirus, Gemykibivirus, Gemykolovirus, Gemykrogvirus, Gemykroznavirus, Gemytondvirus, and Gemyvongvirus (10). These viruses have small (~2 to 2.4 kb), circular ssDNA genomes, and they encode rolling-circle replication initiation proteins (Rep) that are mostly similar to those of geminiviruses, as well as unique capsid proteins (9, 10). Of all the identified genomoviruses, only Sclerotinia sclerotiorum hypovirulence-associated DNA virus 1 (SsHADV-1) (species, Sclerotinia gemycircularvirus 1; genus, Gemycircularvirus) has been associated with known hosts, S. sclerotiorum (11) and a mycophagous fly, Lycoriella ingenua (12).
The vast majority of the viruses and integrated virus-like sequences related to circoviruses, geminiviruses, nanoviruses, genomoviruses, and alphasatellites have been identified on the basis of recognizable DNA sequence of Rep involved in rolling circle replication (RCR) (13, 14). Compared to the capsid protein, Rep is better conserved across these viruses and satellites (14). Conserved motifs within the Rep are important for these viruses and satellites to replicate through RCR (14). These conserved motifs are divided into two main categories based on their roles: the RCR motifs I, II, and III in the N termini of Reps and the superfamily 3 helicase motifs in the C termini, known as Walker-A, Walker-B, and motif C (14). In addition, a conserved motif [geminivirus Rep sequence (GRS)] located between motifs II and III was initially only found in geminiviruses and later also identified in genomoviruses (10, 15). These CRESS DNA viruses replicate through the RCR mechanism, which is similar to that used by bacterial plasmids (14, 16, 17). The three conserved RCR motifs are well conserved across CRESS DNA viruses and plasmids (14, 18, 19).
Fusarium graminearum is a devastating fungal plant pathogen with worldwide distribution that causes Fusarium head blight (FHB) disease in wheat and barley (20). Many mycoviruses have been identified from F. graminearum, including dsRNA, (+)ssRNA, and (−)ssRNA viruses (21). In this study, we isolated a tripartite ssDNA virus, named Fusarium graminearum gemytripvirus 1 (FgGMTV1), from F. graminearum, with a unique genome organization. We constructed infectious clone vectors of these three DNA components and used them to identify essential and symptom-associated components, providing a good system for the study of virus-fungus interactions. We also propose that the multipartite genome of FgGMTV1 may represent a natural case of relatively recent multipartition.
RESULTS
Observation of viral particles and identification of viral genome sequences
To explore Fusarium mycovirus diversity, around 200 Fusarium strains, isolated from the diseased seeds or glumes of wheat of seven provinces in China, were conducted for high-throughput RNA sequencing. Unexpectedly, SsHADV-1–related sequences were identified in strain HB58 collected in the Hebei province of China. The strain HB58 was identified as F. graminearum based on sequencing of polymerase chain reaction (PCR) fragments and subsequent BLASTn searches of translation elongation factor 1α (EF-1α) (Fig. 1A and fig. S1) (22). Electron microscopy observation revealed that the negatively stained virus particles purified from strain HB58 were isometric with a diameter of ~19 to 21 nm (Fig. 1B), comparable to the 20 to 22 nm in diameter reported for SsHADV-1 (11). Nucleic acids extracted from the viral particles appeared as a band at ~1.3 kb in agarose gel electrophoresis (Fig. 1C). To confirm the properties of the extracted nucleic acids, different nucleases were used. The viral nucleic acids could be digested with both deoxyribonuclease I (DNase I) (active against ssDNA and dsDNA) and S1 nuclease (active against ssDNA or ssRNA), but not with exonuclease III (active against linear dsDNA) or exonuclease I (active against linear ssDNA) (Fig. 1D). This suggests that the viral nucleic acids are circular ssDNA molecules or linear ssDNA with modified 5′ and 3′ ends similar to that described by Yu et al. (11).
Fig. 1. Characterization of FgGMTV1 infecting F. graminearum strain HB58 and composition of the isolated virus particles.
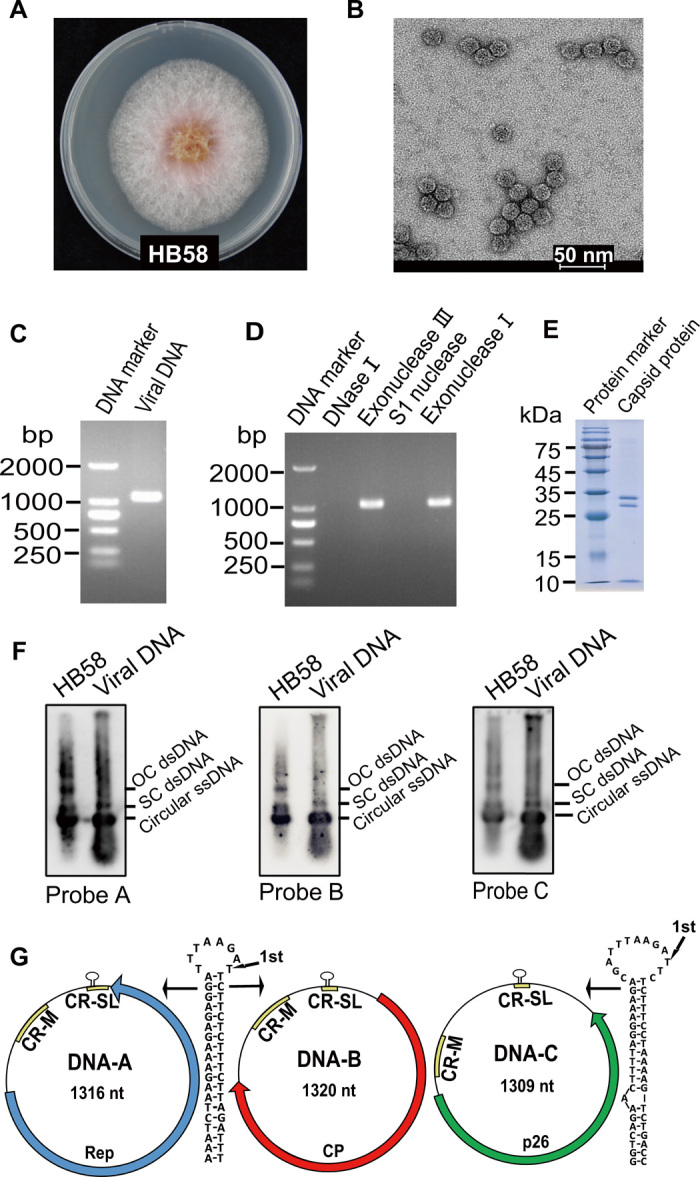
(A) Colony morphology of virus-carrying field strain HB58 of F. graminearum after 3 days of culture on potato dextrose agar (PDA) in the dark. (B) Viral particles observed under transmission electron microscopy. Scale bar, 50 nm. (C) One percent agarose gel electrophoresis of DNA extracted from virus particles. bp, base pair. (D) Electrophoresis analysis of enzyme-treated nucleic acid samples on 1% agarose gel. The samples were treated with DNase I, exonuclease III, S1 nuclease, and exonuclease I, respectively. (E) SDS–polyacrylamide gel electrophoresis analysis of purified virus particles from strain HB58 showing three protein bands. (F) Southern blot analyses of nucleic acids extracted from viral particles. Genomic DNA extracted from strain HB58 was used as a positive control. The forms of the viral DNA are indicated as OC dsDNA (open circular dsDNA), SC dsDNA (supercoiled dsDNA), and circular ssDNA. Three DNA fragments (probe A, probe B, and probe C, specific for DNA-A, DNA-B, and DNA-C, respectively) were PCR-amplified, labeled with DIG, and used as DNA probes. (G) Genome organization of the three components of FgGMTV1. The single ORF of each DNA component is displayed as a thick arrow. The positions of the potential stem-loop structure, common region major (CR-M) and common region stem-loop (CR-SL) are indicated. Photo credit: Pengfei Li, Institute of Plant Protection, Chinese Academy of Agricultural Sciences.
Nucleic acids from virion preparations purified from mycelia of strain HB58 were subjected to rolling circle amplification (RCA). Restriction digestion of the RCA products generated a unit-length DNA of ~1.3 kb using five tested restriction enzymes (Bam HI, Eco RI, Sal I, Bgl II, and Kpn I). In addition, using Sal I, a 0.9-kb DNA, and using Kpn I, a 1-kb DNA, were observed upon electrophoresis. The restricted and cloned RCA products were sequenced and analyzed. The full-length sequences were obtained by PCR using specific back-to-back primers designed based on the sequences obtained above. Last, a total of three circular ssDNA components (DNA-A, DNA-B, and DNA-C) associated with the nucleic acids from viral particles were identified, sequenced, and confirmed by Southern blot analyses (Fig. 1, F and G).
Molecular characterization of DNA-A, DNA-B, and DNA-C
DNA-A [1316 nucleotide (nt)] carried a large open reading frame (ORF) encoding a putative Rep with a calculated molecular mass of 34.4 kDa (Fig. 1G). BLASTp searches of the full-length amino acid sequence of the putative Rep showed a notable match to Gila monster–associated gemycircularvirus (GenBank accession number AWX63615.1; 1.0 × 10−63, 94% coverage and 39.72% identity), an unpublished virus in the family Genomoviridae. The putative Rep contained two conserved domains, namely, geminivirus Rep catalytic domain (Gemini_AL1) and geminivirus Rep protein central domain (Gemini_AL1_M) with conserved motifs for RCR. The Rep of many genomoviruses contains a putative intron (9); however, the Rep gene of DNA-A lacks an intron similarly to that of SsHADV-1 (11). DNA-B was 1320 nt in length and contained a large ORF with a coding potential for a protein of 215 amino acids (Fig. 1G). The deduced protein shared the highest similarity with the capsid protein (CP) of Finch-associated genomovirus 7 (GenBank accession number QCQ85257.1; 8.0 × 10−10, 77% coverage and 21.74% identity), an unpublished virus in the family Genomoviridae. Three bands could be observed in the SDS–polyacrylamide gel electrophoresis (SDS-PAGE) analysis of purified virus particles (Fig. 1E); unexpectedly, the relative molecular mass estimated by SDS-PAGE for the top band was similar to the deduced size (34.2 kDa) of the DNA-B encoding capsid protein, while the size of another two bands was smaller than the expected 34.2 kDa. The three bands were unambiguously identified by peptide mass fingerprinting (PMF) as the CP. This triple-band SDS-PAGE profile might be attributed to partial degradation of CP or to posttranslational modifications of the protein; further studies will be necessary to clarify this point. DNA-C (1309 nt) contained a large ORF potentially encoding a protein with a molecular mass of 25.8 kDa (Fig. 1G), hereafter named p26, which had no obvious similarities to other sequences in the National Center for Biotechnology Information (NCBI) protein database.
All three ssDNA segments had two common regions in the untranslated region. One was a 105- or 106-nt region that was 98% identical to regions in the three components, termed common region major (CR-M) (fig. S2). The other was a 77- or 82-nt region, termed common region stem loop (CR-SL), which was 89% identical to the other regions (fig. S2). The CR-SL contained a conserved unusual nonanucleotide (TTTAAGATT), which was not found in most reported genomoviruses (10). The potential stem-loop structures of DNA-A and DNA-B were the same, but different from that of DNA-C, apart from the aforementioned conserved nonanucleotide.
Sequence alignment and phylogenetic classification
With the exception of motif I, all conserved RCR motifs (motifs II and III) and three helicase motifs known as Walker-A, Walker-B, and motif C were found in the Rep encoded in DNA-A (table S1). A fourth conserved motif, GRS, was also found in this Rep (table S1). Sequence analyses of the full-length amino acid sequence of the putative Rep and CP of this ssDNA virus and selected genomoviruses showed that this novel virus shared higher Rep amino acid sequence identity (24.1 to 33.0%) than CP sequence identity (10.3 to 17.1%) with selected genomoviruses (table S2). Phylogenetic analysis of the Reps of this virus and selected ssDNA viruses indicated that this ssDNA virus was nested within the family Genomoviridae (Fig. 2). In many instances, the putative Rep encoded by divergent CRESS DNA viruses is the only protein that has any detectable sequence similarity to other known ssDNA viral Reps (10, 14). Compared to the Rep amino acid sequences, the CP sequences of this ssDNA virus and the members of nine genera in the family Genomoviridae were all found to be phylogenetically more divergent (fig. S3), possibly because they are involved in interactions with cell surface receptors in the hosts and/or vectors (10). In conclusion, we can verify that these three DNA segments are circular ssDNA molecules and tentatively assign this ssDNA virus the name “Fusarium graminearum gemytripvirus 1 (FgGMTV1).” We suggest that the mycovirus FgGMTV1 is a representative member of the new proposed genus Gemytripvirus (Gemini-like myco-infecting tripartite virus) in the family Genomoviridae. The GenBank accession numbers for the nucleotide sequence of DNA-A, DNA-B, and DNA-C of FgGMTV1 are MK430076, MK430077, and MK430078, respectively.
Fig. 2. Phylogenetic relationship of the Reps from FgGMTV1 and selected circular ssDNA viruses.
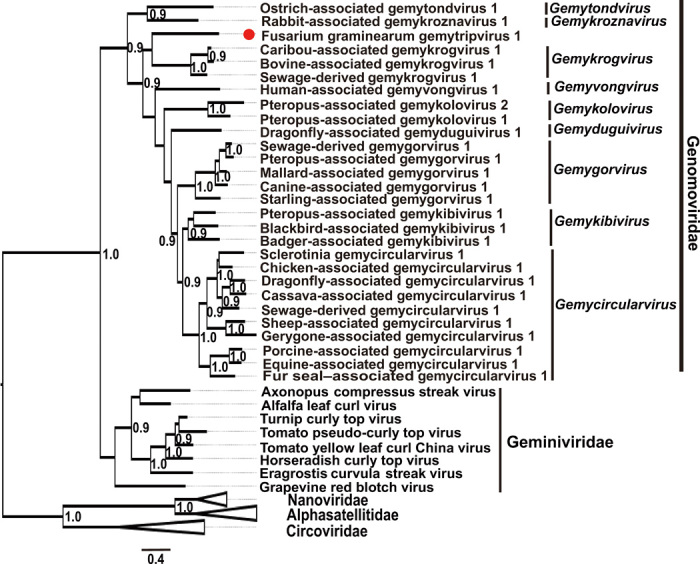
An unrooted phylogenetic tree was constructed by a maximum-likelihood method based on multiple amino acid sequence alignments of the Rep using PhyML 3.0. Ambiguously aligned regions were removed using the online software Gap Strip/Squeeze v2.1.0. The best-fit model “RtREV + G + I + F” was selected using the Smart Model Selection technique implemented in the PhyML 3.0. Numbers at the nodes represent approximate likelihood ratio test values derived using an SH-like calculation (only values greater than 0.9 are shown). The position of FgGMTV1 is indicated by a red dot.
Infectivity and symptoms induced by DNA-A, DNA-B, and DNA-C
To further test whether DNA-A, DNA-B, and DNA-C identified here represent the FgGMTV1 genome and assess their infectivity and effect on biological properties of the host, we constructed infectious clones of DNA-A (pSK-2A), DNA-B (pSK-2B), and DNA-C (pSK-1.6C), containing a dimer tandem repeat of DNA-A, a dimer tandem repeat of DNA-B, and a 1.6-mer tandem repeat of DNA-C, respectively, following a similar strategy to that used to construct infectious clones of geminiviruses (fig. S4) (23, 24). Eight transfectants were generated with different combinations of clones pSK-2A, pSK-2B, and pSK-1.6C and one transfectant using purified viral particles (table S3), among which strain S was used as a negative control. Southern blot analyses of viral DNA extracted from PH-1 and nine transfectants were conducted using probe A, probe B, and probe C (Fig. 3). When using probe A and probe B, viral DNA replicative forms were only present in strain A + B, strain A + B + C, and strain Virion, demonstrating a successful infection, but not in the other seven strains (Fig. 3). When using probe C, viral DNA replicative forms were only present in strain A + B + C and strain Virion, but not in the strain A + B or in the other seven strains (Fig. 3). Southern hybridization analyses showed that DNA-A, DNA-B, or DNA-C alone were not infectious; the combination of DNA-A and DNA-C or the combination of DNA-B and DNA-C were also shown not to be infectious, but the combination of DNA-A and DNA-B, the combination of DNA-A, DNA-B, and DNA-C, and viral particles were infectious. These results suggest that DNA-A and DNA-B are mutually interdependent and sufficient for their replication, while DNA-C relies on DNA-A and DNA-B. In addition, strain A + B + C and strain Virion accumulated more viral DNA than strain A + B, indicating that DNA-C enhances the accumulation level of viral DNA in infected fungi.
Fig. 3. Southern blot analysis of DNAs extracted from mycelia of PH-1 and nine transfectants.

Nucleic acids (~1 μg) were loaded in each lane. The blots were probed with probe A, probe B, or probe C, respectively. The stained gel below the blot indicates equal loading of DNA. The positions of OC dsDNA, SC dsDNA, and circular ssDNA forms are indicated.
To verify successful infection, viral particles, viral nucleic acids, and structural proteins were extracted from strains S, A + B, A + B + C, and HB58 (fig. S5, A to C). Viral particles, viral nucleic acids, and structural proteins, similar to those of strain HB58, were observed in strains A + B and A + B + C, not strain S (fig. S5, A to C), demonstrating a successful infection. Southern blot analyses of nucleic acids extracted from viral particles isolated from these strains were also conducted using probe A, probe B, and probe C (fig. S5D). Viral DNA was detected in strains A + B, A + B + C, and HB58 using probe A and probe B, while not present in strain A + B using probe C. The results demonstrate that the formation of viral particles does not correlate with the presence of DNA-C, and DNA-A, DNA-B, and DNA-C represent the FgGMTV1 genome.
We assessed the mycelial growth, conidiation, and virulence on wheat spikes of strains PH-1, S, A + B, A + B + C, Virion, and HB58. Compared to the virus-free strains PH-1 and S, strain A + B showed abnormal colony morphology (Fig. 4A). Strain A + B grew on potato dextrose agar (PDA) medium at a rate of 9.3 mm/day, whose growth was much slower than that of strain PH-1 (19.0 mm/day) and strain S (18.5 mm/day) (Fig. 4C). In addition, strain A + B had a significant reduction in colony diameter [45.5% reduction compared to strain PH-1 (P < 0.01) and 44.5% reduction compared to strain S (P < 0.01)] (Fig. 4B) and conidial production [55.1% reduction compared to strain PH-1 (P < 0.01) and 52.3% reduction compared to strain S (P < 0.01)] (Fig. 4D). Strains A + B + C, Virion, and HB58 showed similar characteristics to the virus-free strains PH-1 and S (Fig. 4). In the virulence assay, 10 μl of the conidial suspension was inoculated in the glume of a spikelet and cultured in the field for 15 days. Before the virulence assay, we tested the probability of the viral horizontal and vertical transmission. Southern blot analyses showed that the virus from strains A + B or A + B + C could be efficiently transmitted to the strain PH-1 incorporating a hygromycin-resistance gene (hph) (PH-1hph), but not from virus-carrying strain HB58 to the vegetative incompatible strain PH-1hph via hyphal anastomosis (fig. S6A). In addition, the virus in strain A + B + C was vertically transmitted through conidia at a 5 to 10% rate (figs. S6B and S7A), while no virus was detected from single ascospore isolates (0 infected of 150 tested) (fig. S6C). As for strain A + B, both the transmission rates through conidia (75 tested) and ascospores (150 tested) were 0% (figs. S6, B and C, and S7A). When inoculating a conidial suspension into the glume of a spikelet, there was no significant difference in virulence among these six strains (fig. S7B). However, when a small equisized mycelial plug was put into the glume of a spikelet, strain A + B spread more slowly from the inoculation sites to nearby spikelets than the other five strains (Fig. 4E). At 15 days after inoculation, there was a significant reduction [81.3% reduction compared to strain PH-1 (P < 0.01) and 79.1% reduction compared to strain S (P < 0.01)] in the number of diseased spikelets per invaded wheat head infected with strain A + B (Fig. 4E). The fungi were isolated from the glume of spikelets, and PCR was carried out using DNA-A–, DNA-B–, or DNA-C–specific degenerate primer pairs (table S4). It was worth mentioning that PCR products of the expected sizes were detected using primer pairs specific for DNA-A or DNA-B in the glumes of spikelets inoculated with mycelial plugs of strain A + B, but not the glumes of spikelets inoculated with a conidial suspension of strain A + B (fig. S8). Together, these results indicate that DNA-A and/or DNA-B might be related to hypovirulence of the fungal host, which could be countered by DNA-C; in addition, viral transmission through conidia requires DNA-C.
Fig. 4. Comparison of colony morphology, colonial diameter, growth rate, conidial production, and virulence among PH-1 and five different transfectants.
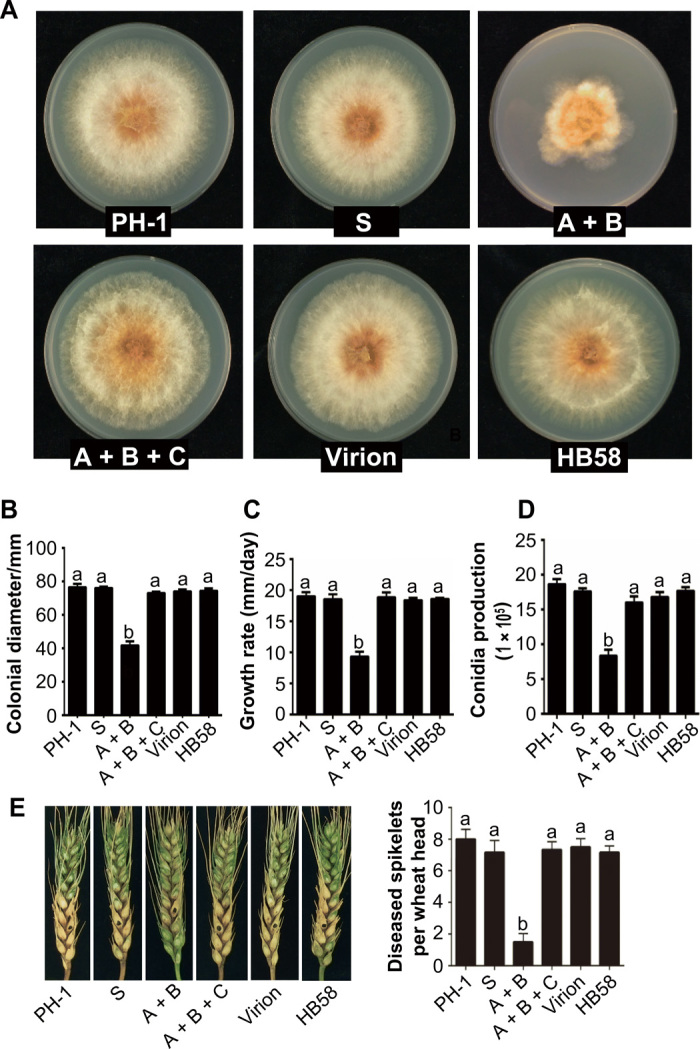
(A) Colony morphology of PH-1 and five different transfectants after 4 days of culture on PDA in the dark. (B) Comparison of the colonial diameter among PH-1 and five different transfectants (n = 3 replicates). (C) Growth rate after 4 days on PDA (n = 3 replicates). (D) Conidia production after 5 days in O-carboxymethylcellulose liquid medium (n = 3 replicates). (E) FHB symptoms and the numbers of diseased spikelets per invaded wheat head caused by putting small equisized mycelial plugs into the glume of spikelets (n = 6 replicates). At 15 days after inoculation, there was a significant reduction in the number of diseased spikelets per invaded wheat head infected with strain A + B. The black dots indicate the inoculation positions. Different letters (a and b) in (B to E) denote significant differences (P < 0.05 determined by Tukey’s post hoc test). Photo credit: Pengfei Li, Institute of Plant Protection, Chinese Academy of Agricultural Sciences.
DISCUSSION
Viruses of the family Genomoviridae are circular ssDNA viruses (~2 to 2.4 kb), encoding a Rep and a capsid protein in the same circular DNA molecule. Here, we report a novel ssDNA virus in the family Genomoviridae, and its genome comprises three circular ssDNA components of ~1.3 kb in size referred to as DNA-A (encoding the replication protein), DNA-B (encoding the capsid protein), and DNA-C (encoding an unknown protein). This research expands our knowledge of the diversity and evolution of viruses in the family Genomoviridae and CRESS DNA viruses.
Bacilladnaviridae, Circoviridae, Geminiviridae, Genomoviridae, Nanoviridae, and Smacoviridae are the only six eukaryotic CRESS DNA viral families currently recognized by the ICTV (7, 13, 25). Among those, members of the Circoviridae, Geminiviridae, Genomoviridae, and Smacoviridae families are circular ssDNA viruses (2 to 3 kb in size) that encode a Rep and a CP in the same circular DNA molecule (9, 13, 25). Viruses of the family Bacilladnaviridae have larger genome sizes than other eukaryotic CRESS DNA viruses (~4.5 to 6 kb) (7). Viruses of the genus Begomovirus in the family Geminiviridae have either one or two components (13). Members of two genera, Nanovirus and Babuvirus, in the family Nanoviridae encompass viruses with multicomponent genomes, where each component (~1 kb in size) encodes a single protein and is packaged into a separate particle (6, 13). Five homologous DNA components of these two genera (referred to as DNA-R, DNA-S, DNA-C, DNA-M, and DNA-N) encode a replication initiator protein (Rep), a capsid protein (CP), a cell cycle link protein, a movement protein, and a nuclear shuttle protein. Another DNA (DNA-U3) from babuviruses and three other DNAs (DNA-U1, DNA-U2, and DNA-U3) from nanoviruses encode potential proteins with presently unknown functions. All DNA components share two regions of sequence similarity known as the CR-SL and the CR-M (for babuviruses) or CR-II (for nanoviruses) (13). The DNA-R component encoding the master Rep is capable of self-replication and is required for replication of all other genome components (26, 27). Similarly, FgGMTV1 DNA-A, DNA-B, and DNA-C also have two common regions named CR-M and CR-SL, and DNA-A is required for replicating the two other genome components. However, in contrast to DNA-R, DNA-A alone could not be detected using Southern blotting analysis, suggesting that this molecule is not sufficient to sustain self-replication. A putative circovirus-related multicomponent virus, known as the Pacific flying fox feces–associated multicomponent virus 1 (PfffaMCV-1) that was sequenced from Pacific flying fox feces, harbored three ssDNA molecules of 1143 to 1163 nt that encoded a single ORF (6). In this viral genome, one ORF encoded a Rep, the second encoded a putative CP, and the third one showed no similarities with any sequences in GenBank. The PfffaMCV-1 Rep shared the highest sequence identity (48%) with the Rep of a circovirus and contained the conserved residues seen in the Rep motifs of eukaryotic CRESS DNA viruses. However, the hosts of this virus are not known. Another two putative multicomponent viruses, referred to as blackfly multicomponent virus (BfMCV) 1 and 2 that were recovered in the New Zealand blackflies, have Reps most similar to the multicomponent virus PfffaMCV-1, sharing 65 and 67% amino acid similarity, respectively (28). All these factors showed that, in addition to multicomponent viruses in the family Nanoviridae, other multipartite ssDNA viruses that encode a Rep and a CP in separate circular DNA molecule, like FgGMTV1, exist. To date, multiparticulate fungal viruses or mycoviruses have been classified into four families and one genus with linear dsRNA virus genomes (Chrysoviridae, Megabirnaviridae, Partitiviridae, Quadriviridae, and Botybirnavirus) (29). In addition, no mycoviruses with multipartite DNA genomes have previously been reported. FgGMTV1 is the first multipartite DNA virus infecting fungi.
Because of the RCR mechanism common to geminiviruses infecting plants and to plasmids replicating in bacteria, archaea, and algae, a scenario that geminiviruses, appearing relatively late in viral evolution, have originated from plasmids via the acquisition of capsid protein genes from ssRNA viruses has been proposed (19, 30–33). In this context, the existence of a pre-mastrevirus between plasmids and geminiviruses has been hypothesized (32). The family Genomoviridae is a geminivirus-like family, in which viruses encode a Rep with similarities to that of the geminiviruses (9). As Yu et al. (11) described, SsHADV-1 (from the family Genomoviridae) is likely to resemble a possible precursor of pre-mastreviruses. In this study, the phylogenetic analysis of the Reps and CPs of FgGMTV1 and selected ssDNA viruses indicated that FgGMTV1 was lodged within the family Genomoviridae (Fig. 2 and fig. S3). Furthermore, we suggest classifying the virus as a representative member of a new proposed genus Gemytripvirus within the family Genomoviridae. Our data suggest that the multipartite genome of FgGMTV1 may have evolved from a monopartite genome of an ancient genomovirus. Defective genomes, spontaneously generated upon replication of viral genomes, have been suggested as the important intermediate stages in the evolution of multipartitism and would essentially become components of multipartite genomes after being transreplicated and/or transencapsidated by the viable genomes (34–36). DNA-A and DNA-B of FgGMTV1, mutually interdependent and sufficient for their replication, may originally represent intermediate defective genomes of an ancient nonsegmented genomovirus. The subsequent bipartite viral form can encode a Rep and a CP in separate circular DNA molecules (Figs. 1G and 3, and fig. S5, A to D), which is different from an original genomovirus encoding a Rep and a CP in the same circular DNA molecule (9). Some in vitro studies have confirmed that two complementary defective molecules from a complete nonsegmental virus can evolve into two components of a bipartite virus (37–39). Therefore, the multipartite genome of FgGMTV1 may represent a natural case of relatively recent multipartition. Besides, defective genomic DNA, 236 to 284 nt in length, has been detected in SsHADV-1, which is the first member of the family Genomoviridae (9). However, the origin of the third segment DNA-C of FgGMTV1 and its function on the evolution of FgGMTV1 need to be further investigated.
The development of transformation and transfection techniques [e.g., transformation of fungal spheroplasts using complementary DNA (cDNA) infectious clones (40, 41), and transfection of fungal spheroplasts with dsRNA (42), in vitro–transcribed RNA transcripts (43–45), or purified virions (46, 47)] has facilitated the identification of viral and/or host factor(s) involved in symptom induction or virus replication for many mycovirus-host systems, especially in the hypovirus-host system (2). These techniques can also be used to expand the host ranges of some mycoviruses, without the obstacles of vegetative incompatibility (48). Among CRESS DNA viruses, the infectious DNA clones are mainly available for circoviruses and geminiviruses and are used to explore the properties of these ssDNA viruses and use ssDNA viruses as multifunctional vectors (23, 24, 49, 50). Previous to this work, only infectious cDNA clones of RNA mycoviruses have been constructed. In this study, the first infectious DNA clones of a DNA mycovirus were obtained. These clones will allow future further exploration of the interaction between FgGMTV1 and F. graminearum and could be also applicable to effectively extend the infection of FgGMTV1 to other hosts and enable manipulation of FgGMTV1 for both fundamental and practical applications.
Betasatellites associated with monopartite begomoviruses play important roles in symptom induction and suppression of both transcriptional and posttranscriptional gene silencing (51). FgGMTV1 DNA-A and/or DNA-B may cause hypovirulence, but DNA-C can counter this effect, indicating that DNA-A and/or DNA-B and DNA-C probably play important roles in changes of symptoms. FgGMTV1 cannot be transmitted through conidia in the absence of DNA-C, and DNA-C enhances the accumulation level of viral DNA in infected fungi; these observations suggest that DNA-C may be associated with viral movement and transmission and play important roles in the suppression of transcriptional or posttranscriptional gene silencing. However, it is unexpected that DNA-A and DNA-B cause more severe symptoms than the full set of the DNA components, which can replicate more efficiently (Figs. 3 and 4). DNA-C can enhance the ecological fitness potential of FgGMTV1 by retaining the growth and conidiation levels of infected strains in comparison with virus-free fungal strains. Collectively, the results obtained here indicate that the combination of F. graminearum–FgGMTV1 will provide an excellent system for examining fungal and viral factors involved in virus-host interactions, although the full characterization of this pathosystem will require further work.
MATERIALS AND METHODS
Fungal strains and culture conditions
F. graminearum strain HB58 was isolated from the diseased glumes of wheat infected with Fusarium spp., which were collected in Hebei province, China. The PH-1 strain of F. graminearum, a virulent virus-free strain, served as the wide-type and a receptor strain for infectious clones constructed in this study (52). PH-1hph was used for a viral transmission assay. All strains, including the transfectants generated here, and the strains arising from PH-1hph after dual culturing with strains A + B, A + B + C, and HB58, respectively, are listed in table S3 and were cultured at 25°C on PDA or in potato dextrose broth (PDB) in the dark and kept at 4°C until they were used.
Purification and protein components of virus particles
Strain HB58 of F. graminearum was grown in PDB and incubated on a shaking incubator at 25°C for 7 days. Mycelia (~3 g, wet weight) were harvested and ground to a fine powder in liquid nitrogen using a sterilized mortar and pestle. The powder was transferred to a plastic tube (50 ml) containing 15 ml of 0.1 M sodium phosphate (pH 7.4). The mixture was stirred on an orbital shaker for 2 hours at 4°C and then centrifuged at 5000 rpm for 20 min to remove the hyphal cell debris. The supernatant was carefully pipetted into another plastic tube containing the same volume of chloroform and stirred for 30 min at 4°C, followed by centrifugation at 12,000 rpm for 30 min to further remove the hyphal cell debris. The upper aqueous phase was subjected to a 3-hour ultracentrifugation at 35,000 rpm at 4°C to precipitate all of the particles. The resultant pellet was resuspended in 1 ml of sodium phosphate buffer (0.01 M, pH 7.4) overnight at 4°C. The suspension was clarified by centrifugation at 12,000 rpm for 20 min at 4°C. The supernatant was overlaid on a centrifuge tube containing sucrose solutions with a concentration gradient ranging from 10 to 40% (w/v) and centrifuged at 35,000 rpm for 3 hours at 4°C. Fractions in the middle portion of the tube were individually tested for the presence of the virus particles by detection of nucleic acids using 1% agarose gel electrophoresis. A virus-containing zone located at a sucrose concentration of ∼25 to 30% was further centrifuged to precipitate particles. Last, the pellets were carefully suspended in 1 ml of 0.01 M sodium phosphate buffer (pH 7.4). The virus particles were negatively stained with 1% uranyl acetate on carbon-coated 400-mesh copper grids and observed under transmission electron microscopy. Purified virus particles were electrophoresed through PAGE in 12% SDS-PAGE with 25 mM tris-glycine and 0.1% SDS. Following electrophoresis, the gel was stained using Coomassie brilliant blue R-250 [Sangon Biotech (Shanghai) Co. Ltd.]. PMF of three protein bands were performed by Shanghai Applied Protein Technology Co. Ltd., China.
Nucleic acid extraction from viral particles, molecular cloning, and sequencing
About 200 μl of purified virus particles was treated with 200 μl of extraction buffer [20 mM tris-HCl (pH 8.0), 1% SDS, 200 mM NaCl, and 5 mM EDTA] and 400 μl of phenol/chloroform/isoamyl alcohol (25:24:1) to remove viral proteins. The nucleic acids were precipitated with ethanol, dissolved in diethyl pyrocarbonate–treated water, and analyzed using 1% agarose gel electrophoresis. Last, the viral nucleic acids were treated with DNase I, exonuclease III, S1 nuclease, or exonuclease I to confirm the properties of the extracted nucleic acids.
RCA was conducted on nucleic acids extracted directly from virion samples using the Illustra TempliPhi Amplification Kit (GE Healthcare, USA). A volume of 1 μl of nucleic acids (~100 to 500 ng) was mixed with 5 μl of sample buffer, denatured at 95°C for 3 min, and then chilled on ice before adding 5 μl of reaction buffer and 0.2 μl of the Φ29 DNA polymerase, followed by incubation for 20 hours at 30°C and 10 min at 65°C. RCA products were digested with various restriction enzymes in appropriate buffers, and the fragments generated by Bam HI, Eco RI, Sal I, Bgl II, and Kpn I were resolved using 1% agarose gels, extracted, and inserted into the pET30a vector (Novagen) restricted with the appropriate restriction enzyme. The recombinant plasmids were then transformed into Trans5α chemically competent cells (TransGen) according to the manufacturer’s instructions. Candidate bacteria harboring recombinant plasmids were analyzed to measure the presence of the insert by colony PCRs, and every base was determined by sequencing in at least 10 independent overlapping clones. Positive clones were selected, sequenced, and analyzed via the DNAMAN software (v7) and the BLAST program on the NCBI website (http://blast.st-va.ncbi.nlm.nih.gov/Blast.cgi).
To obtain a complete DNA sequence representing a complete circular component, specific back-to-back primers were designed on the basis of the sequences obtained above and used in PCR on nucleic acid samples using 2× Phanta Max Master Mix (Vazyme Biotech Co. Ltd.). PCR primers (DNA-A–1F/1R for DNA-A, DNA-B–1F/1R for DNA-B, and DNA-C–1F/1R for DNA-C) are listed in table S5. All of the PCR products were purified, cloned, and sequenced according to the protocol described above.
DNA preparation and Southern blot analyses
Viral DNA was extracted from mycelia grown in PDB at 25°C for 7 days according to the manufacturer’s instructions of the Fungal DNA Kit (Omega Bio-tek Inc.). Approximately 1 μg of DNA was separated on 1% agarose gels in tris-acetate-EDTA buffer [40 mM tris-acetate and 1 mM EDTA (pH 8.0)], followed by alkali denaturation and neutralization, and then transferred onto Hybond-N+ membranes (Amersham Biosciences, Buckingham, England) by capillary blotting with 20× SSC [3 M NaCl and 0.3 M sodium citrate (pH 7.0)] overnight. The DNA was cross-linked to the membrane by ultraviolet (UV) irradiation for 10 min. Three DNA probes [359 base pair (bp) for probe A, 335 bp for probe B, and 326 bp for probe C, specific for DNA-A, DNA-B, and DNA-C, respectively] were produced by PCR amplification using specific primer pairs (table S6) and labeled with digoxigenin (DIG) using the DIG DNA Labeling and Detection Kit (Roche Diagnostics, Mannheim, Germany). After prehybridization and hybridization were complete, the blots were washed in 1× SSC (0.15 M sodium chloride and 0.015 M sodium citrate) and 0.1% SDS at 65°C for 30 min. Hybridization signals were detected on a Tanon 5200 imaging system (Tanon Science and Technology Co. Ltd.) using the DIG detection kit (Roche Diagnostics, Mannheim, Germany).
Sequence and phylogenetic analyses
The assembly and analysis of nucleotide sequences and translation of ORFs were performed using the DNAMAN version 7 software. The sequences of previously reported viruses referenced here were obtained from the NCBI GenBank database (www.ncbi.nlm.nih.gov/genome). Potential ORFs were deduced using DNAMAN and the ORF finder program in the NCBI website (http://ncbi.nlm.nih.gov/projects/gorf). Sequence similarity searches were performed using the NCBI BLAST program (http://blast.st-va.ncbi.nlm.nih.gov/Blast.cgi). Multiple alignments of nucleic and amino acid sequences were conducted using DNAMAN with default parameters (53). The phylogenetic trees were constructed using the maximum-likelihood method based on multiple amino acid sequence alignments in PhyML 3.0 (http://atgc-montpellier.fr/phyml/). See table S7 for all virus names and viral protein accession numbers used for phylogenetic analysis. The best-fit model was selected using the Smart Model Selection technique implemented in PhyML 3.0. Numbers at nodes in the phylogeny represent approximate likelihood ratio test values derived using an Shimodaira-Hasegawa (SH)–like calculation (only values greater than 0.9 are shown). Branch support was determined by bootstrapping (1000 replicates).
Construction of infectious clones of FgGMTV1 DNA-A, DNA-B, and DNA-C
For the construction of the infectious clone of FgGMTV1 DNA-A (fig. S4), RCA was conducted on viral DNA extracted from virion samples. RCA products were digested with Eco RI and Kpn I to obtain a full-length unit of DNA-A, and then the fragment obtained, of about 1.3 kb, was inserted into the Eco RI site of the vector pBluescript II SK(+) (pSK) (Stratagene) to produce the clone pSK-1A, containing a 1-mer of DNA-A. Candidate bacteria harboring the clone pSK-1A were Sanger-sequenced at Sangon Biotech Co. Ltd. (Shanghai, China) to confirm that no mutations had been introduced by RCA. The sequenced pSK-1A clone was digested with Eco RI and inserted into the Eco RI site of plasmid pSK to produce clone pSK-2A, which contained a tandem dimeric repeat of the DNA-A molecule. The positive recombinant plasmid was verified by restriction enzyme digestion and analyzed via 1% agarose gel electrophoresis. The infectious clone of FgGMTV1 DNA-B (pSK-2B) was constructed following a similar strategy but using different restriction enzymes Eco RI and Spe I to obtain a full-length unit of DNA-B (fig. S4).
Similarly, for the construction of the infectious clone of FgGMTV1 DNA-C (fig. S4), RCA products were digested with Xba I to obtain a full-length unit of DNA-C, and then the fragment obtained, of about 1.3 kb, was inserted into the Xba I site of the plasmid pSK to produce the clone pSK-1C, containing a 1-mer of DNA-C. The sequenced pSK-1C clone was digested with Xba I and Cla I and then inserted into the plasmid pSK to produce the clone pSK-0.6C. Clone pSK-1C was digested with Xba I to obtain a full-length unit of DNA-C and inserted into the unique Xba I site of pSK-0.6C to produce pSK-1.6C, containing a 1.6-mer tandem repeat of FgGMTV1 DNA-C. The positive recombinant plasmid was also verified through restriction enzyme digestion and analyzed by 1% agarose gel electrophoresis.
Protoplast preparation and transfection assays
Protoplast preparation and virus transfection were undertaken following a method described previously (54) with minor modifications. Briefly, ~1 × 107 conidia were germinated to produce hyphae overnight in liquid yeast extract peptone dextrose broth [yeast extract (5 g/liter), microbiological peptone (3 g/liter), and dextrose (10 g/liter)]. Germinated mycelia were harvested through one layer of Miracloth (Calbiochem) and washed three times with sterile distilled water and twice with 1 M sucrose. After being filtered on Miracloth, 0.5 g of mycelium was transferred to a 50-ml tube containing 40 ml of 1 M sucrose and 800 mg of cell wall–lysing enzymes (from Trichoderma harzianum; Sigma-Aldrich), followed by incubation for 3 hours at 28°C in a shaker at 90 rpm to release protoplasts. Protoplasts were filtered into a 50-ml tube and centrifuged at 5000 rpm for 10 min at 4°C. The protoplast pellet was washed three times with 10 ml of ice-cold sorbitol-tris-calcium chloride (STC) buffer (1 M sorbitol, 50 mM tris with a pH value of 8.0, and 50 mM CaCl2∙2H2O), centrifuged at 5000 rpm for 10 min, resuspended in 0.5 ml of STC, and kept on ice.
For transfection, 200 μl of protoplasts were mixed with 30 μl of purified virus particles or 30 μl (about 10 μg) of infectious clone plasmids and transferred to a 2-ml tube, followed by incubation on ice for 20 min. Then, 1.25 ml of STC buffer containing 40% polyethylene glycol 3350 was dropwise added into the above 2-ml tube and gently mixed for 15 s before incubation at room temperature for 20 min. Following incubation, the protoplast suspension was transferred to a 15-ml tube containing 5 ml of TB3 medium (0.3% yeast extract, 0.3% casamino acids, and 20% sucrose) and incubated for 16 hours at 25°C in a shaker at 90 rpm. A volume of 200 μl of aliquots of the mixture was transferred to 9-cm petri dishes, embedded in 20 ml of warm TB3 solid medium containing 0.7% low-melt agarose, and cultured for 2 to 3 days at 25°C in the dark. Mycelial plugs were randomly cut from the regenerated colonies, individually transferred to fresh PDA plates at one plug per plate, subcultured three times at weekly intervals, and then checked for the presence of the virus by Southern blotting.
Biological characterization
We assessed the mycelial growth, conidiation, and virulence on wheat spikes as previously described (55). After fungal isolates were grown on PDA plates for 5 days at 25°C, agar plugs with a diameter of 7 mm were cut from the actively growing margin and placed in the middle of a PDA plate. The growth rate of fungi was calculated by measuring colonial diameters daily after culturing for 4 days at 25°C. Three agar plugs were added to O-carboxymethylcellulose broth and cultured for 5 days, and conidial production was determined on blood count plates. For the virulence assay, 10 μl of conidial suspension (3 × 105 conidia/ml) or a small equisized mycelial plug was inoculated in the glume of a spikelet and cultured in the field for 15 days. At the end of the cultivation period, virulence was assessed by measuring the numbers of diseased spikelets per wheat head invaded. The infected glume was plucked from the spikelet, placed onto a PDA plate, and incubated for 3 to 5 days at 25°C. The generated fungi were transferred to PDA for subculture. Subsequently, the mycelial mass was collected for DNA extraction, followed by PCR using the indicated primer pairs (DNA-A–2F/2R for DNA-A, DNA-B–2F/2R for DNA-B, and DNA-C–2F/2R for DNA-C) (table S4).
Viral horizontal transmission via hyphal anastomosis and vertical transmission through conidia and ascospores
To assess the possibility of horizontal transmission of FgGMTV1 between strains of F. graminearum, fresh mycelial agar discs from strains A + B and PH-1hph, A + B + C and PH-1hph, or HB58 and PH-1hph were dual-cultured on PDA plates. In the contact culture in each dish (9 cm in diameter), strains A + B, A + B + C, and HB58 served as donors, whereas strain PH-1hph served as the recipient. After 4 days of dual culture, six mycelial derivative isolates were obtained from the growth side of six recipients and transferred to a fresh PDA plate containing hygromycin (200 μg/ml). New colonies were subcultured two times on hygromycin-containing PDA plates and then transferred onto fresh PDA plates overlaid with cellophane for further incubation for 3 days before viral detection. Subcultures of PH-1hph after dual culturing and contacting with strains A + B, A + B + C, and HB58, respectively, were analyzed for the presence of the mycovirus by performing total DNA extraction and Southern dot blot.
To test whether FgGMTV1 can be vertically transmitted through conidia, five aliquots (200 μl) of the conidial suspensions containing approximately 3 × 105 conidia/ml were inoculated in 50 ml of PDB broth and cultured for 5 days at 25°C on a rotary shaker. Germinated mycelia were harvested, dried, and ground to a fine powder. Viral DNA extraction and Southern blotting were conducted as described above. To assess the frequency of vertical transmission of FgGMTV1, 75 single conidia of strains A + B and A + B + C, respectively, were obtained, and the resultant monoconidial cultures were analyzed for the presence of the mycovirus by performing total DNA purification and Southern dot blot.
To test the possible transmission of FgGMTV1 through ascospores, mycelia plugs were inoculated onto carrot agar plates [carrots (400 g/liter) and agar (20 g/liter)] at 25°C for 5 days. Aerial hyphae were gently removed with a sterile toothpick and pressed down with 1 ml of sterile 0.2% Tween-20 solution to induce sexual development. The plates were continuously incubated under near-UV light at 23°C. After induction for 7 days, perithecia were observed under a dissecting microscope. At least 10 perithecia were collected with a needle and crushed on a microscope slide and examined for ascospores with a light microscope. The ascospores were washed off the slide with sterile water and quantified. Fifty single ascospores of strains A + B and A + B + C, respectively, were obtained, and the resultant single ascospore cultures were analyzed for the presence of the mycovirus by performing total DNA extraction and Southern dot blot. The experiment was repeated three times.
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 6.0 (GraphPad Software Inc., La Jolla, CA, USA). Significant differences between the control and treatment groups were analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test. Values are shown as means ± SD. Significant differences occurred when P < 0.05.
Supplementary Material
Acknowledgments
We thank N. Suzuki (Institute of Plant Science and Resources, Okayama University) for discussion and critical reading of the manuscript. Funding: This work was supported by the National Key R&D Program of China (2018YFD0200500). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. Author contributions: L.G. and P.L. designed the research. P.L., S.W., and L.Z. performed the research. L.G., P.L., D.Q., and X.Z. analyzed the data. P.L., L.G., and X.Z. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The GenBank accession numbers for the nucleotide sequence of DNA-A, DNA-B, and DNA-C of FgGMTV1 are MK430076, MK430077, and MK430078, respectively. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaay9634/DC1
REFERENCES AND NOTES
- 1.Hillman B. I., Annisa A., Suzuki N., Viruses of plant-interacting fungi. Adv. Virus Res. 100, 99–116 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Son M., Yu J., Kim K.-H., Five questions about mycoviruses. PLOS Pathog. 11, e1005172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kotta-Loizou I., Coutts R. H. A., Mycoviruses in Aspergilli: A comprehensive review. Front. Microbiol. 8, 1699 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hollings M., Mycoviruses: Viruses that infect fungi. Adv. Virus Res. 22, 1–53 (1978). [DOI] [PubMed] [Google Scholar]
- 5.Simmonds P., Adams M. J., Benkő M., Breitbart M., Brister J. R., Carstens E. B., Davison A. J., Delwart E., Gorbalenya A. E., Harrach B., Hull R., King A. M. Q., Koonin E. V., Krupovic M., Kuhn J. H., Lefkowitz E. J., Nibert M. L., Orton R., Roossinck M. J., Sabanadzovic S., Sullivan M. B., Suttle C. A., Tesh R. B., van der Vlugt R. A., Varsani A., Zerbini F. M., Consensus statement: Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 15, 161–168 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Male M. F., Kraberger S., Stainton D., Kami V., Varsani A., Cycloviruses, gemycircularviruses and other novel replication-associated protein encoding circular viruses in Pacific flying fox (Pteropus tonganus) faeces. Infect. Genet. Evol. 39, 279–292 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Zhao L., Rosario K., Breitbart M., Duffy S., Eukaryotic circular Rep-encoding single-stranded DNA (CRESS DNA) viruses: Ubiquitous viruses with small genomes and a diverse host range. Adv. Virus Res. 103, 71–133 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Kazlauskas D., Varsani A., Krupovic M., Pervasive chimerism in the replication-associated proteins of uncultured single-stranded DNA viruses. Viruses 10, E187 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krupovic M., Ghabrial S. A., Jiang D., Varsani A., Genomoviridae: A new family of widespread single-stranded DNA viruses. Arch. Virol. 161, 2633–2643 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Varsani A., Krupovic M., Sequence-based taxonomic framework for the classification of uncultured single-stranded DNA viruses of the family Genomoviridae. Virus Evol. 3, vew037 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu X., Li B., Fu Y., Jiang D., Ghabrial S. A., Li G., Peng Y., Xie J., Cheng J., Huang J., Yi X., A geminivirus-related DNA mycovirus that confers hypovirulence to a plant pathogenic fungus. Proc. Natl. Acad. Sci. U.S.A. 107, 8387–8392 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu S., Xie J., Cheng J., Li B., Chen T., Fu Y., Li G., Wang M., Jin H., Wan H., Jiang D., Fungal DNA virus infects a mycophagous insect and utilizes it as a transmission vector. Proc. Natl. Acad. Sci. U.S.A. 113, 12803–12808 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lefkowitz E. J., Dempsey D. M., Hendrickson R. C., Orton R. J., Siddell S. G., Smith D. B., Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 46, D708–D717 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosario K., Duffy S., Breitbart M., A field guide to eukaryotic circular single-stranded DNA viruses: Insights gained from metagenomics. Arch. Virol. 157, 1851–1871 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Nash T. E., Dallas M. B., Reyes M. I., Buhrman G. K., Ascencio-Ibañez J. T., Hanley-Bowdoin L., Functional analysis of a novel motif conserved across geminivirus Rep proteins. J. Virol. 85, 1182–1192 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chandler M., de la Cruz F., Dyda F., Hickman A. B., Moncalian G., Ton-Hoang B., Breaking and joining single-stranded DNA: The HUH endonuclease superfamily. Nat. Rev. Microbiol. 11, 525–538 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruiz-Masó J. A., MachóN C., Bordanaba-Ruiseco L., Espinosa M., Coll M., Del Solar G., Plasmid rolling-circle replication. Microbiol. Spectr. 3, PLAS-0035-2014 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Ilyina T. V., Koonin E. V., Conserved sequence motifs in the initiator proteins for rolling circle DNA replication encoded by diverse replicons from eubacteria, eucaryotes and archaebacteria. Nucleic Acids Res. 20, 3279–3285 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krupovic M., Networks of evolutionary interactions underlying the polyphyletic origin of ssDNA viruses. Curr. Opin. Virol. 3, 578–586 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Goswami R. S., Kistler H. C., Heading for disaster: Fusarium graminearum on cereal crops. Mol. Plant Pathol. 5, 515–525 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Cho W. K., Lee K.-M., Yu J., Son M., Kim K.-H., Insight into mycoviruses infecting Fusarium species. Adv. Virus Res. 86, 273–288 (2013). [DOI] [PubMed] [Google Scholar]
- 22.O'Donnell K., Kistler H. C., Tacke B. K., Casper H. H., Gene genealogies reveal global phylogeographic structure and reproductive isolation among lineages of Fusarium graminearum, the fungus causing wheat scab. Proc. Natl. Acad. Sci. U.S.A. 97, 7905–7910 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang X., Guo W., Ma X., An Q., Zhou X., Molecular characterization of Tomato leaf curl China virus, infecting tomato plants in China, and functional analyses of its associated betasatellite. Appl. Environ. Microbiol. 77, 3092–3101 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cui X., Tao X., Xie Y., Fauquet C. M., Zhou X., A DNA beta associated with Tomato yellow leaf curl China virus is required for symptom induction. J. Virol. 78, 13966–13974 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varsani A., Krupovic M., Smacoviridae: A new family of animal-associated single-stranded DNA viruses. Arch. Virol. 163, 2005–2015 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Timchenko T., de Kouchkovsky F., Katul L., David C., Vetten H. J., Gronenborn B., A single Rep protein initiates replication of multiple genome components of Faba bean necrotic yellows virus, a single-stranded DNA virus of plants. J. Virol. 73, 10173–10182 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horser C., Harding R. M., Dale J. L., Banana bunchy top nanovirus DNA-1 encodes the “master” replication initiation protein. J. Gen. Virol. 82, 459–464 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Kraberger S., Schmidlin K., Fontenele R. S., Walters M., Varsani A., Unravelling the single-stranded DNA virome of the New Zealand blackfly. Viruses 11, E532 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato Y., Castόn J. R., Suzuki N., The biological attributes, genome architecture and packaging of diverse multi-component fungal viruses. Curr. Opin. Virol. 33, 55–65 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Rojas M. R., Hagen C., Lucas W. J., Gilbertson R. L., Exploiting chinks in the plant's armor: Evolution and emergence of geminiviruses. Annu. Rev. Phytopathol. 43, 361–394 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Krupovic M., Ravantti J. J., Bamford D. H., Geminiviruses: A tale of a plasmid becoming a virus. BMC Evol. Biol. 9, 112 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nawaz-ul-Rehman M. S., Fauquet C. M., Evolution of geminiviruses and their satellites. FEBS Lett. 583, 1825–1832 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Stedman K., Mechanisms for RNA capture by ssDNA viruses: Grand theft RNA. J. Mol. Evol. 76, 359–364 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Lefeuvre P., Martin D. P., Elena S. F., Shepherd D. N., Roumagnac P., Varsani A., Evolution and ecology of plant viruses. Nat. Rev. Microbiol. 17, 632–644 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Lucía-Sanz A., Manrubia S., Multipartite viruses: Adaptive trick or evolutionary treat? Npj Syst. Biol. Appl. 3, 34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iranzo J., Manrubia S. C., Evolutionary dynamics of genome segmentation in multipartite viruses. Proc. Biol. Sci. 279, 3812–3819 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Neill F. J., Maryon E. B., Carroll D., Isolation and characterization of defective simian virus 40 genomes which complement for infectivity. J. Virol. 43, 18–25 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geigenmüller-Gnirke U., Weiss B., Wright R., Schlesinger S., Complementation between Sindbis viral RNAs produces infectious particles with a bipartite genome. Proc. Natl. Acad. Sci. U.S.A. 88, 3253–3257 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim K. H., Narayanan K., Makino S., Assembled coronavirus from complementation of two defective interfering RNAs. J. Virol. 71, 3922–3931 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Choi G. H., Nuss D. L., Hypovirulence of chestnut blight fungus conferred by an infectious viral cDNA. Science 257, 800–803 (1992). [DOI] [PubMed] [Google Scholar]
- 41.Zhang R., Hisano S., Tani A., Kondo H., Kanematsu S., Suzuki N., A capsidless ssRNA virus hosted by an unrelated dsRNA virus. Nat. Microbiol. 1, 15001 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Kanhayuwa L., Kotta-Loizou I., Özkan S., Gunning A. P., Coutts R. H. A., A novel mycovirus from Aspergillus fumigatus contains four unique dsRNAs as its genome and is infectious as dsRNA. Proc. Natl. Acad. Sci. U.S.A. 112, 9100–9105 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin H., Lan X., Liao H., Parsley T. B., Nuss D. L., Chen B., Genome sequence, full-length infectious cDNA clone, and mapping of viral double-stranded RNA accumulation determinant of hypovirus CHV1-EP721. J. Virol. 81, 1813–1820 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marzano S.-Y. L., Hobbs H. A., Nelson B. D., Hartman G. L., Eastburn D. M., McCoppin N. K., Domier L. L., Transfection of Sclerotinia sclerotiorum with in vitro transcripts of a naturally occurring interspecific recombinant of Sclerotinia sclerotiorum hypovirus 2 significantly reduces virulence of the fungus. J. Virol. 89, 5060–5071 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen B., Choi G. H., Nuss D. L., Attenuation of fungal virulence by synthetic infectious hypovirus transcripts. Science 264, 1762–1764 (1994). [DOI] [PubMed] [Google Scholar]
- 46.Hillman B. I., Supyani S., Kondo H., Suzuki N., A reovirus of the fungus Cryphonectria parasitica that is infectious as particles and related to the Coltivirus genus of animal pathogens. J. Virol. 78, 892–898 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sasaki A., Kanematsu S., Onoue M., Oikawa Y., Nakamura H., Yoshida K., Artificial infection of Rosellinia necatrix with purified viral particles of a member of the genus Mycoreovirus reveals its uneven distribution in single colonies. Phytopathology 97, 278–286 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Pearson M. N., Beever R. E., Boine B., Arthur K., Mycoviruses of filamentous fungi and their relevance to plant pathology. Mol. Plant Pathol. 10, 115–128 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fenaux M., Opriessnig T., Halbur P. G., Meng X. J., Immunogenicity and pathogenicity of chimeric infectious DNA clones of pathogenic porcine circovirus type 2 (PCV2) and nonpathogenic PCV1 in weanling pigs. J. Virol. 77, 11232–11243 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang L., Wu X., Shi J., Peng Z., Zheng S., Xu S., Han H., Xin C., Liu Y., Gao M., Yu J., Sun W., Cong X., Li J., Xu S., Wang J., The correlation between the mutual deletions of amino acids within porcine circovirus rep protein and the discrepancy of replication. Microb. Pathog. 117, 327–334 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Zhou X., Advances in understanding begomovirus satellites. Annu. Rev. Phytopathol. 51, 357–381 (2013). [DOI] [PubMed] [Google Scholar]
- 52.Cuomo C. A., Güldener U., Xu J.-R., Trail F., Turgeon B. G., Di Pietro A., Walton J. D., Ma L.-J., Baker S. E., Rep M., Adam G., Antoniw J., Baldwin T., Calvo S., Chang Y.-L., Decaprio D., Gale L. R., Gnerre S., Goswami R. S., Hammond-Kosack K., Harris L. J., Hilburn K., Kennell J. C., Kroken S., Magnuson J. K., Mannhaupt G., Mauceli E., Mewes H. W., Mitterbauer R., Muehlbauer G., Münsterkötter M., Nelson D., O'donnell K., Ouellet T., Qi W., Quesneville H., Roncero M. I. G., Seong K.-Y., Tetko I. V., Urban M., Waalwijk C., Ward T. J., Yao J., Birren B. W., Kistler H. C., The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science 317, 1400–1402 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Katoh K., Toh H., Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 9, 286–298 (2008). [DOI] [PubMed] [Google Scholar]
- 54.Ejmal M. A., Holland D. J., MacDiarmid R. M., Pearson M. N., The effect of Aspergillus thermomutatus chrysovirus 1 on the biology of three Aspergillus species. Viruses 10, 539 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li P., Lin Y., Zhang H., Wang S., Qiu D., Guo L., Molecular characterization of a novel mycovirus of the family Tymoviridae isolated from the plant pathogenic fungus Fusarium graminearum. Virology 489, 86–94 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/14/eaay9634/DC1