

Abstract
As the human population grows from 7.8 billion to 10 billion over the next 30 years, breeders must do everything possible to create crops that are highly productive and nutritious, while simultaneously having less of an environmental footprint. Rice will play a critical role in meeting this demand and thus, knowledge of the full repertoire of genetic diversity that exists in germplasm banks across the globe is required. To meet this demand, we describe the generation, validation and preliminary analyses of transposable element and long-range structural variation content of 12 near-gap-free reference genome sequences (RefSeqs) from representatives of 12 of 15 subpopulations of cultivated Asian rice. When combined with 4 existing RefSeqs, that represent the 3 remaining rice subpopulations and the largest admixed population, this collection of 16 Platinum Standard RefSeqs (PSRefSeq) can be used as a template to map resequencing data to detect virtually all standing natural variation that exists in the pan-genome of cultivated Asian rice.
Subject terms: Agriculture, Plant genetics, DNA sequencing, Structural variation, Evolutionary genetics
| Measurement(s) | genome • DNA • sequence_assembly • sequence feature annotation • physical map |
| Technology Type(s) | DNA sequencing • PacBio Sequel System • sequence assembly process • transposable elements annotation • Optical Mapping Illumina sequencing |
| Factor Type(s) | Oryza sativa cv. variety |
| Sample Characteristic - Organism | Oryza sativa |
Machine-accessible metadata file describing the reported data: 10.6084/m9.figshare.11950596
Background & Summary
Asian cultivated rice is a staple food for half of the world population. With the planet’s population expected to reach 10 billion by 2050, farmers must increase production by at least 100 million metric tons per year1,2. To address this need, future rice cultivars should provide higher yields, be more nutritious, be resilient to multiple abiotic and biotic stresses, and have less of an environmental footprint3,4. To achieve this goal, a comprehensive and more in-depth understanding of the full range of genetic diversity of the pan-cultivated rice genome and its wild relatives will be needed5.
With a genome size of ~390 Mb, rice has the smallest genome among the domesticated cereals, making it particularly amenable to genomic studies6 and the primary reason why it was the first crop genome to be sequenced 15 years ago6,7. To better understand the full-range of genetic diversity that is stored in rice germplasm banks around the world, several studies have been conducted using microarrays8,9 and low coverage skim sequencing10,11. In 2018, a detailed analysis of the Illumina resequencing of more than 3,000 diverse rice accessions (a.k.a. 3K-RG), aligned to the O. sativa v.g. japonica cv. Nipponbare reference genome sequence (a.k.a. IRGSP RefSeq), showed how the high genetic diversity present in domesticated rice populations provides a solid base for the improvement of rice cultivars12. One key finding from a population structure analysis of this dataset showed that the 3,000 accessions can be subdivided into nine subpopulations, where most accessions from close sub-groups could be associated to geographic origin12.
One critical piece of information missing from these analyses is the fact that single nucleotide polymorphisms (SNPs) and structural variations (SVs) present in subpopulation specific genomic regions have yet to be detected because the 3K-RG data set was only aligned to a single reference genome. Therefore, the next logical step, to capture and understand genetic variation pan-subpopulation-wide is to map the 3K-RG dataset to high-quality reference genomes that represent each of the subpopulations of cultivated Asian rice. At present, only a handful high-quality rice genomes for cultivated rice are publicly available5,6,13,14, thus, there is an immediate need for such a comprehensive resource to be created, which is the subject of this Data Descriptor.
Here we present a reanalysis of the population structure analysis discussed above12 and show that the 3K-RG dataset can be further subdivided into a total of 15 subpopulations. We then present the generation of 12 new and near-gap-free high-quality PacBio long-read reference genomes from representative accessions of the 12 subpopulations of cultivated Asian rice for which no high-quality reference genomes exist. All 12 genomes were assembled with more than 100x genome coverage PacBio long-read sequence data and then validated with Bionano optical maps15. The number of contigs covering each of the twelve assemblies, excluding unplaced contigs, ranged from 15 (GOBOL SAIL (BALAM)::IRGC 26624-2) to 104 (IR 64). The contig N50 value for the 12-genome dataset ranged from 7.35 Mb to 31.91 Mb. When combined with 4 previously published genomes (i.e. Minghui 63 (MH 63), Zhenshan 97 (ZS 97)13,14, N 225 and the IRGSP RefSeq.6), this 16-genome dataset can be used to represent the K = 15 population/admixture structure of cultivated Asian rice.
Methods
Ethics statement
This work was approved by the University of Arizona (UA), the King Abdullah University of Science and Technology (KAUST), Huazhong Agricultural University (HZAU), the International Rice Research Institute (IRRI) and the International Center for Tropical Agriculture (CIAT). All methods used in this study were carried out following approved guidelines.
Population structure
We extracted 30 subsets of 100,000 randomly chosen SNPs out of the 3K-RG Core SNP set v0.4 (996,009 SNPs, available at https://snp-seek.irri.org/_download.zul). For each subset, we ran ADMIXTURE16 with the number of ancestral groups K ranging from 5 to 15. We then aligned the resulting Q matrices using CLUMPP software17. Since different runs at a given value of K often give rise to different refinements (splits) of the lower level grouping, we first clustered the runs for each K according to similarity of Q matrices using hierarchical clustering, thus obtaining several clusters of runs for each K. We discarded one-element clusters (outlier runs), and averaged the Q matrices within each remaining cluster. Figure S1 shows the admixture proportions taken from the averaged Q matrices of the final clusters for K = 5 to 15. The columns of these averaged Q matrices, representing admixture proportions for groups discovered in different runs, were then used to define the “K15” grouping. At K = 9, 12, and 13, the Q matrices converged to two different modes according to whether XI-1A or GJ-trop is split (these are labeled as K = 9.1, 12.1 and 13.1).
Group membership for each sample was defined by applying a threshold of 0.65 to admixture components. Samples with no admixture components exceeding 0.65 were classified as follows. If the sum of components for subpopulations within the major groups cA (circum-Aus), XI (Xian-indica), and GJ (Geng-japonica) was ≥0.65, the samples were classified as cA-adm (admixed within cA), XI-adm (admixed within XI) or GJ-adm (admixed within GJ), respectively, and the remaining samples were deemed ‘fully’ admixed. The newly defined groups were mostly align with the previous K = 9 grouping, or were refined and named accordingly (e.g. XI-1B1 and XI-1B2 are two new subgroups within XI-1B).
The phenogram shown in Fig. 1 was constructed with DARwin v6 (http://darwin.cirad.fr/, unweighted Neighbor-joining) using the identity by state (IBS) distance matrix from Plink on the 4.8 M Filtered SNP set (available at https://snp-seek.irri.org/_download.zul). Colors were assigned to subpopulations based on K15 Admixture results. One entry, MH 63 (XI-adm) represents the admixed types among the XI group.
Fig. 1.

Phylogenetic tree with the accession selected for PSRefSeq sequencing for each of the K = 15 subpopulations and a single admixture group. Groups are colored according to the assignment from Admixture analysis. The subpopulation designation is in parentheses following the name.
Sample selection, collection and nucleic acid preparation
To select accessions to represent the 12 subpopulations of Asian rice that lack high-quality reference genome assemblies, the following strategy was employed. The IBS distance matrix was used for a principal component analysis (PCA) analysis in R to generate 5 component axes. Then, for each of the 12 subpopulations, i.e. circum-Aus2 = cA2, circum-Basmati = cB, Geng-japonica (GJ) subtropical (GJ-subtrp), tropical1 (GJ-trop1) and tropical2 (GJ-trop2), and Xian-indica (XI) subpopulations XI-1B1, XI-1B2, XI-2A, XI-2B, XI-3A, XI-3B1, XI-3B2, the centroid of each group in the space spanned by first 5 principal components was determined from the eigenvectors, and the entry closest to the centroid for which seed was available was chosen as the representative for that subpopulation (Table 1).
Table 1.
Sample collection information for the 12 Oryza sativa accessions.
| Variety Name | Genetic Stock ID | Country Origin | 15 subpops |
|---|---|---|---|
| CHAO MEO::IRGC 80273-1 | IRGC 132278 | Lao PDR | GJ-subtrp |
| Azucena | I1A41685 | Philippines | GJ-trop1 |
| KETAN NANGKA::IRGC 19961-2 | IRGC 128077 | Indonesia | GJ-trop2 |
| ARC 10497::IRGC 12485-1 | IRGC 117425 | India | cB |
| IR 64 | I1A42114 | Philippines | XI-1B1 |
| PR 106::IRGC 53418-1 | IRGC 127742 | India | XI-1B2 |
| LIMA::IRGC 81487-1 | IRGC 127564 | Indonesia | XI-3A |
| KHAO YAI GUANG::IRGC 65972-1 | IRGC 127518 | Thailand | XI-3B1 |
| GOBOL SAIL (BALAM)::IRGC 26624-2 | IRGC 132424 | Bangladesh | XI-2A |
| LIU XU::IRGC 109232-1 | IRGC 125827 | China | XI-3B2 |
| LARHA MUGAD::IRGC 52339-1 | IRGC 125619 | India | XI-2B |
| NATEL BORO::IRGC 34749-1 | IRGC 127652 | Bangladesh | cA2 |
Subpopulations: GJ = Geng-japonica where trop = tropical, subtrp = subtropical; cB = circum-Basmati; XI = Xian-indica; cA = circum-Aus.
Single seed decent (SSD) seed from IR 64 and Azucena were obtained from the Rice Genetics and Genomics Laboratory, CIAT, in Cali, Colombia, and SSD seed from the remaining 10 accessions (Table 1) were obtained from the International Rice Genebank, maintained by IRRI, Los Baños, Philippines. All seed were sown in potting soil and grown under standard greenhouse conditions at UA, Tucson, USA for 6 weeks at which point they were dark treated for 48-hours to reduce starch accumulation. Approximately 20–50 grams of young leaf tissue was then harvested from each accession and immediately flash frozen in liquid nitrogen before being stored at −80 °C prior to DNA extraction. High molecular weight genomic DNA was isolated using a modified CTAB procedure as previously described18. The quality of each extraction was checked by pulsed-field electrophoresis (CHEF) on 1% agarose gels for size and restriction enzyme digestibility, and quantified by Qubit fluorometry (Thermo Fisher Scientific, Waltham, MA).
Library construction and sequencing
Genomic DNA from all 12 accessions were sequenced using the PacBio single-molecule real-time (SMRT) platform, and the Illumina platform for genome size estimations and sequence polishing. High molecular weight (HMW) DNA from each accession was gently sheared into large fragments (i.e. 30 Kb–60 Kb) using 26-gauge needles and then end-repaired according to manufacturer’s instructions (Pacific Biosciences). Briefly, using a SMRTbell Express Template Prep Kit, blunt hairpins and sequencing adaptors were ligated to HMW DNA fragments, and DNA sequencing polymerases were bound to the SMRTbell templates. Size selection of large fragments (above 15 Kb) was performed using a BluePippin electrophoresis system (Sage Science). The libraries were quantified using a Qubit Fluorometer (Invitrogen, USA) and the insert mode size was determined using an Agilent fragment analyzer system with sizes ranging between 30 Kb–40 Kb. The libraries then were sequenced using SMRT Cell 1 M chemistry version 3.0 on a PacBio Sequel instrument. The number of long-reads generated per accession ranged from 2.01 million (LIMA::IRGC 81487-1) to 5.40 million (Azucena). The distribution of subreads is shown in Fig. S2 and the average lengths ranged from 10.58 Kb (Azucena) to 20.61 Kb (LIMA::IRGC 81487-1) (Table 2). According to the estimated genome size of the IRGSP RefSeq, the average PacBio sequence coverage for each accession varied from 103x (LIMA::IRGC 81487-1) to 149x (IR 64) (Table 2).
Table 2.
Sequencing platforms used and data statistics for the 12 Oryza sativa genomes.
| Variety Name | Sequencing platform | Raw data (Gb) | Depth | Number of subreads (M) | Mean subread length (Kb) |
|---|---|---|---|---|---|
| CHAO MEO::IRGC 80273-1 | PacBio Sequel | 49.1 | 123× | 4.26 | 11.526 |
| Azucena | PacBio Sequel | 57.1 | 143× | 5.40 | 10.581 |
| KETAN NANGKA::IRGC 19961-2 | PacBio Sequel | 49.8 | 125× | 2.78 | 17.876 |
| ARC 10497::IRGC 12485-1 | PacBio Sequel | 44.7 | 112× | 4.06 | 11.026 |
| IR 64 | PacBio Sequel | 59.7 | 149× | 5.24 | 11.393 |
| PR 106::IRGC 53418-1 | PacBio Sequel | 42.2 | 105× | 2.08 | 20.317 |
| LIMA::IRGC 81487-1 | PacBio Sequel | 41.4 | 103× | 2.01 | 20.612 |
| KHAO YAI GUANG::IRGC 65972-1 | PacBio Sequel | 42.5 | 106× | 2.37 | 17.954 |
| GOBOL SAIL (BALAM)::IRGC 26624-2 | PacBio Sequel | 42.2 | 105× | 2.13 | 19.777 |
| LIU XU::IRGC 109232-1 | PacBio Sequel | 55.3 | 138× | 3.66 | 15.109 |
| LARHA MUGAD::IRGC 52339-1 | PacBio Sequel | 45.1 | 113× | 3.22 | 14.011 |
| NATEL BORO::IRGC 34749-1 | PacBio Sequel | 44.4 | 111× | 2.74 | 16.2 |
For Illumina short-read sequencing, HMW DNA from each accession was sheared to between 250–1000 bp, followed by library construction targeting 350 bp inserts following standard Illumina protocols (San Diego, CA, USA). Each library was 2 × 150 bp paired-end sequenced using an Illumina X-ten platform. Low-quality bases and paired reads with Illumina adaptor sequences were removed using Trimmomatic19. Quality control for each library data set was carried out with FastQC20. Finally, between 36.52-Gb and 51.05-Gb of clean data for each accession was generated, and used for genome size estimation (Table S1) by Kmer analysis (Fig. S3) and the Genome Characteristics Estimation (GCE) program21.
Bionano optical genome maps
Bionano optical maps for each accession were generated as previously described22, except that ultra-HMW DNA isolation, from approximately 4 g of flash-frozen dark-treated (48 hour) leaf tissue per accession, was performed according to a modified version of the protocol described by Luo and Wing23. Prior to labeling, agarose plugs were digested with agarase and the starch and debris removed by short rounds of centrifugation at 13,000 × g. DNA samples were further purified and concentrated by drop dialysis against TE Buffer. Data processing, optical map assembly, hybrid scaffold construction and visualization were performed using the Bionano Solve (Version 3.4) and Bionano Access (v12.5.0) software packages (https://bionanogenomics.com/).
De novo genome assembly
Genome assembly for each of the 12 genomes followed a five-step procedure as shown in (Fig. 2):
Fig. 2.
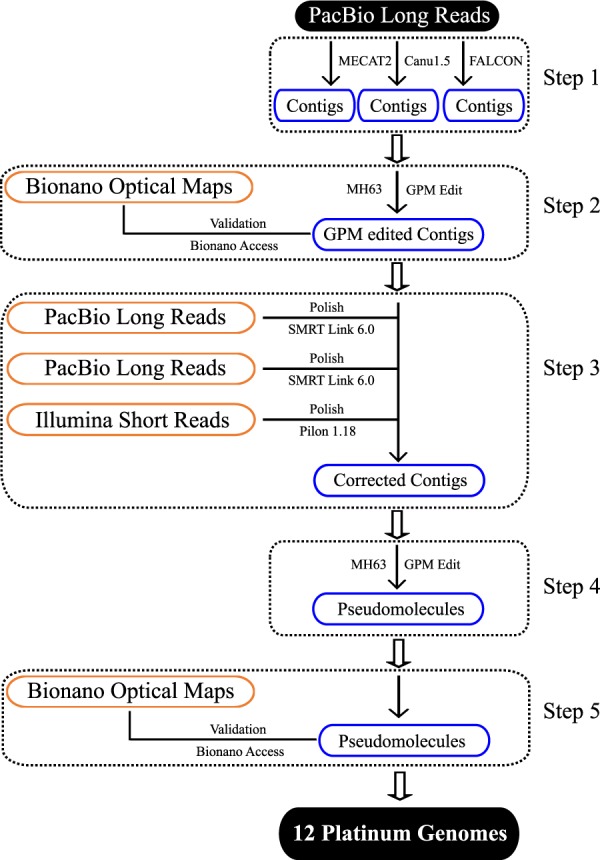
Genome assembly and validation pipeline.
Step 1: PacBio subreads were assembled de novo into contigs using three genome assembly programs: FALCON24, MECAT225 and Canu1.526. The number of de novo assembled contigs obtained varied from 51 (e.g. NATEL BORO::IRGC 34749-1 and KETAN NANGKA::IRGC 19961-2) to 1,473 (CHAO MEO::IRGC 80273-1) for the 12 genomes (Table S2).
Step 2: Genome Puzzle Master (GPM) software27 was used to merge the de novo assembled contigs from the three assemblers, using the high-quality O. sativa vg. indica cv. Minghui 63 reference genome sequence (MH63RS2)13,14 as a guide. GPM is a semi-automated pipeline that is used to integrate logical relationship data (i.e. contigs from three assemblers for each accession) based on a reference guide. Contigs were merged in the ‘assemblyRun’ step, with default parameters (minOverlapSeqToSeq was set at 1 Kb and identitySeqToSeq was set at 99%). Redundant overlapping sequences were also removed for each assembled contig. In addition, we gave contiguous contigs a higher priority than ones with gaps to be retained in each assembly. After manual checking, editing, and redundancy removal, the number of contigs in each assembly ranged from 26 (NATEL BORO::IRGC 34749-1) to 588 (LIU XU::IRGC 109232-1) (Table S2).
Step 3: The sequence quality of each contig was then improved by “sequence polishing”: twice with PacBio long reads and once with Illumina short reads. Briefly, PacBio subreads were aligned to GPM edited contigs using the software blasr28. All default parameters were used, except minimum align length, which was set to 500 bp. Secondly, the tool arrow as implemented in SMRTlink6.0 (Pacific Biosciences of California, Inc) was used for polishing the GPM edited contigs. The bwa-mem program29 was then used for mapping short Illumina reads onto assembled contigs, and the tool pilon30 was used for a final polishing step with default settings.
Step 4: The polished contigs for each accession were arranged into pseudomolecules using GPM, with MH63RS213,14 as the reference guide. The program blastn31 with a minimum alignment length of 1 Kb and an e-value < 1e−5 as the threshold was used to align the corrected contigs to the reference guide. In doing so, the corrected contigs were assigned to chromosomes, as well as ordered and orientated in the GPM assembly viewer function. The number of contigs after step 4 ranged from a minimum of 15 contigs (GOBOL SAIL (BALAM)::IRGC 26624-2) to a maximum of 104 contigs (IR 64) (Table 3). The assembly size for the 12 accessions ranged from 376.86 Mb (CHAO MEO::IRGC 80273-1) to 393.74 Mb (KHAO YAI GUANG::IRGC 65972-1) (Table 3) and the length of individual chromosome varied from 23.06 Mb (chromosome 9 of CHAO MEO::IRGC 80273-1) to 44.96 Mb (chromosome 1 of LIMA::IRGC 81487-1) (Table S4). The average N50 value was 23.10 Mb, with the highest and the lowest N50 values being 30.91 Mb (LIU XU::IRGC 109232-1) and 7.35 Mb (IR 64), respectively. The average number of gaps among the 12 new genome assemblies was 18, with 8 assemblies containing less than 10 gaps (Table 3).
Table 3.
De novo assembly, BUSCO evaluation and accession numbers in GenBank of the 12 Oryza sativa genomes.
| Variety Name | BioProject | BioSample | Genome size (bp) | #Contigs | Contig N50 (bp) | #Gaps | Scaffold N50 (bp) | BUSCO | Adjust BUSCO | Genome Accession | SRP | Supplementary Files (Bionano optical map) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CHAO MEO::IRGC 80273-1 | PRJNA565484 | SAMN12748601 | 376,856,903 | 55 | 11,024,768 | 43 | 30,350,168 | 97.60% | 98.49% | VYIH00000000 | SRP226088 | SUPPF_0000003210 |
| Azucena | PRJNA424001 | SAMN08217222 | 379,627,553 | 28 | 22,940,949 | 16 | 30,954,872 | 97.80% | 98.69% | PKQC000000000 | SRP227255 | SUPPF_0000003212 |
| KETAN NANGKA::IRGC 19961-2 | PRJNA564615 | SAMN12718029 | 380,759,091 | 21 | 22,679,302 | 9 | 30,696,581 | 98.00% | 98.89% | VYIC00000000 | SRP226080 | SUPPF_0000003204 |
| ARC 10497::IRGC 12485-1 | PRJNA565479 | SAMN12748569 | 378,463,869 | 40 | 17,921,520 | 28 | 30,566,713 | 98.40% | 99.30% | VYID00000000 | SRP226093 | SUPPF_0000003206 |
| IR 64 | PRJNA509165 | SAMN10564385 | 386,698,898 | 104 | 7,352,909 | 92 | 31,218,896 | 95.70% | 96.57% | RWKJ00000000 | SRP227298 | SUPPF_0000003213 |
| PR 106::IRGC 53418-1 | PRJNA563359 | SAMN12672924 | 391,176,105 | 16 | 27,051,416 | 4 | 32,028,703 | 96.60% | 97.48% | VYIB00000000 | SRP226078 | SUPPF_0000003202 |
| LIMA::IRGC 81487-1 | PRJNA564572 | SAMN12715984 | 392,625,308 | 17 | 27,369,091 | 5 | 32,421,942 | 98.50% | 99.40% | VXJH00000000 | SRP226079 | SUPPF_0000003203 |
| KHAO YAI GUANG::IRGC 65972-1 | PRJNA565481 | SAMN12748590 | 393,737,720 | 19 | 21,823,919 | 7 | 32,080,718 | 98.60% | 99.50% | VYIF00000000 | SRP226086 | SUPPF_0000003208 |
| GOBOL SAIL (BALAM)::IRGC 26624-2 | PRJNA564763 | SAMN12721963 | 391,772,995 | 15 | 29,604,901 | 3 | 31,753,752 | 97.90% | 98.79% | VXJI00000000 | SRP226082 | SUPPF_0000003205 |
| LIU XU::IRGC 109232-1 | PRJNA577228 | SAMN13021815 | 392,033,263 | 17 | 30,913,760 | 5 | 32,301,089 | 98.40% | 99.30% | WGGU00000000 | SRP226085 | SUPPF_0000003211 |
| LARHA MUGAD::IRGC 52339-1 | PRJNA565480 | SAMN12748589 | 390,195,943 | 16 | 30,747,645 | 4 | 32,107,744 | 98.60% | 99.50% | VYIE00000000 | SRP226084 | SUPPF_0000003207 |
| NATEL BORO::IRGC 34749-1 | PRJNA565483 | SAMN12748600 | 383,720,936 | 16 | 27,825,079 | 4 | 31,305,988 | 98.10% | 98.99% | VYIG00000000 | SRP226087 | SUPPF_0000003209 |
Step 5: To independently validate our assemblies, we generated and compared Bionano optical maps to each assembly. In total, 17 (Azucena) to 56 (LIU XU::IRGC 109232-1) Bionano optical contigs were constructed for all 12 rice accessions, which yielded contig N50 values of between 22.75 Mb (CHAO MEO::IRGC 80273-1) to 31.45 Mb (KHAO YAI GUANG::IRGC 65972-1) (Table S5). As shown in Figs. 3 and S4, the chromosomes and/or chromosome arms of all 12 de novo assemblies were highly supported by these ultra-long optical maps. Although rare, a few discrepancies between the optical maps and genome assemblies could be found and are likely due to small errors and chimeras that were produced through both the optical map and sequence assembly pipelines15.
Fig. 3.

Bionano optical map validation of chromosome 1 for 12 de novo assemblies.
Following these five steps, we were able to produce 12 near-gap-free Oryza sativa platinum standard reference genome sequences (PSRefSeqs) that represent 12 of 15 subpopulations of cultivated Asian rice.
BUSCO evaluation
The Benchmarking Universal Single-Copy Orthologs (BUSCO3.0) software package32 was employed to evaluate the gene space completeness of the 12 genome assemblies. These genomes captured, on average, 97.9% of the BUSCO reference gene set, with a minimum of 95.7% (IR64) and a maximum of 98.6% (LARHA MUGAD::IRGC 52339-1 and KHAO YAI GUANG::IRGC 65972-1) (Table 3).
Of note, when performing this analysis, we observed that on average 30 out of the 1,440 conserved BUSCO genes tested (https://www.orthodb.org/v9/index.html) were missing from each new assembly, 16 of which were not present in all 12, plus the IRGSP RefSeq-1.0, ZS 97, MH 63 and N 22 RefSeqs (Fig. S5). This result suggested that these 16 “conserved” genes may not exist in rice, or other cereal genomes, thereby artificially reducing the BUSCO gene space scores for our 12 assemblies. To test this hypothesis, we searched for all 16 genes missing in maize, which diverged from rice about 50 million years ago (MYA)33–35. We found that 13 of the 16 genes in question could not be found in 3 high-quality recently published maize genome assemblies (Fig. S5) and therefore, concluded that 13 of the 16 “conserved” genes in the BUSCO database are not present in cereals, and should be excluded from our gene space analysis. Taking this into account, we recalculated the BUSCO gene space content for each of 12 assemblies and found that 10 of 12 assemblies captured more than 98% of the BUSCO gene set (Table 3).
Transposable element (TE) prediction
To determine the pan-transposable element content of cultivated Asian rice, we analyzed the 12 new reference genomes, presented here, along with the MH 63, ZS 97, N 22 PacBio reference genomes. In addition, we also included a reanalysis of the IRGSP RefSeq-1.0, as it is conventionally considered the standard rice genome for which all comparisons are conducted.
A search for sequences similar to TEs was carried out using RepeatMasker36, run under default parameters with the exception of the option: -no_is –nolow, and that an updated in-house version of the publicly available MSU_6.9.5 library37, retrieved from https://github.com/oushujun/EDTA/blob/master/database/Rice_MSU7.fasta.std6.9.5.out, called “rice 7.0.0.liban” was used. The average TE content of this 16 genome data set was 47.66% with a minimum value of 46.07% in IRGSP RefSeq-1.0 and a maximum of 48.27% in KHAO YAI GUANG::IRGC 65972-1 (Table 4). The major contribution to this fraction was composed of long terminal repeat retrotransposons (LTR-RTs, min: 23.55%, max: 27.27% and average: 25.96%) followed by DNA-TEs (min:14.87%, max, 16.18% and average: 15.26%). Long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs) were identified as on average 1.43% and 0.39% of the 16 genomes, respectively.
Table 4.
Abundance of the major TE classes in the 16 Oryza sativa genomes.
| Variety Name | Total | LTR-RT | LINEs | SINEs | DNA_TEs | Unclassified |
|---|---|---|---|---|---|---|
| NIPPONBARE | 46.07 | 23.55 | 1.52 | 0.41 | 16.18 | 4.41 |
| CHAO MEO::IRGC 80273-1 | 46.25 | 24.00 | 1.46 | 0.40 | 15.59 | 4.80 |
| Azucena | 47.07 | 24.48 | 1.47 | 0.40 | 15.82 | 4.89 |
| KETAN NANGKA::IRGC 19961-2 | 46.99 | 24.87 | 1.47 | 0.40 | 15.72 | 4.53 |
| ARC 10497::IRGC 12485-1 | 46.95 | 24.74 | 1.48 | 0.40 | 15.68 | 4.65 |
| PR 106::IRGC 53418-1 | 47.95 | 26.82 | 1.41 | 0.39 | 15.05 | 4.28 |
| Minghui 63 | 47.97 | 26.61 | 1.44 | 0.4 | 15.3 | 4.22 |
| IR 64 | 47.87 | 26.82 | 1.42 | 0.40 | 14.97 | 4.26 |
| Zhenshan 97 | 47.95 | 26.79 | 1.42 | 0.39 | 15.19 | 4.16 |
| LIMA::IRGC 81487-1 | 48.04 | 26.87 | 1.40 | 0.39 | 15.01 | 4.37 |
| KHAO YAI GUANG::IRGC 65972-1 | 48.27 | 27.27 | 1.40 | 0.39 | 14.87 | 4.34 |
| GOBOL SAIL (BALAM)::IRGC 26624-2 | 48.15 | 26.99 | 1.40 | 0.39 | 14.99 | 4.38 |
| LIU XU::IRGC 109232-1 | 46.92 | 27.06 | 1.26 | 0.32 | 14.31 | 3.97 |
| LARHA MUGAD::IRGC 52339-1 | 48.05 | 26.74 | 1.41 | 0.39 | 15.09 | 4.42 |
| N 22::IRGC 19379-1 | 47.79 | 25.95 | 1.44 | 0.39 | 15.20 | 4.81 |
| NATEL BORO::IRGC 34749-1 | 47.33 | 25.75 | 1.42 | 0.40 | 15.12 | 4.64 |
Structural Variants
Each genome assembly, as described above, was fragmented using the EMBOSS tool splitter38 to create a 10x genome equivalent redundant set of 50 kb reads. These reads were then mapped onto every other genome assembly using the tool NGMLR39. Finally, the software SVIM40 was run under default parameters to parse the mapping output. Only insertions, deletions and tandem duplications up to a maximum length of 25 Kb were considered in this analysis.
The results of this analysis identified several thousand insertions and deletions whenever an assembly was compared to any other. Greater variability was found between varieties belonging to different major groups (e.g. Geng-japonica [GJ] vs. Xian-indica [XI] than occurred between those within these groups. The amount of genome sequences with structural variation between any two varieties ranged from 17.57 Mb to 41.54 Mb for those belonging to the indica (XI) varietal group (avg: 31.75 Mb) and from 18.55 Mb to 23.07 Mb (avg: 21.00 Mb) for those in the japonica (GJ) varietal group. When all 16 genomes are considered together, the range is between 17.57 Mb and 41.54 Mb, with an average value of 33.70 Mb (Table S6). The total unshared fraction collected out of all pairwise comparisons was composed for 89.89% by TE related sequences.
Data Records
Data for all 12 genome shotgun sequencing projects have been deposited in Genbank (https://www.ncbi.nlm.nih.gov/) including PacBio and Illumina raw data41–52, the twelve reference genome assemblies53–64 and Bionano optical maps. BioProjects, BioSamples, Genome assemblies, Sequence Read Archives (SRA) accession numbers and supplementary files (i.e. Bionano optical maps) of the 12 new assemblies are listed in Table 3. Transposable element, structural variation annotations and also the Bionano optical maps are available in the figshare link (https://figshare.com/s/dcdaea3adae5c44e2e31)65.
Technical Validation
DNA sample quality
DNA quality was checked by pulsed-field gel electrophoresis for size and restriction enzyme digestibility. Nucleic acid concentrations were quantified by Qubit fluorometry (Thermo Fisher Scientific, Waltham, MA).
Illumina libraries
Illumina libraries were quantified by qPCR using the KAPA Library Quantification Kit for Illumina Libraries (KapaBiosystems, Wilmington, MA, USA), and library profiles were evaluated with an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
Gene space completeness
Benchmarking Universal Single-Copy Orthologs (BUSCO3.0) was executed using the embryophyta_odb9.tar.gz database to assess the gene space of each genome, minus 13 genes that do not appear to exist in the cereal genomes tested (Fig. S5).
Assembly accuracy
Bionano optical maps were generated and used to validate all 12 genome assemblies.
Supplementary information
Acknowledgements
This research was supported by the AXA Research Fund (International Rice Research Institute), the King Abdullah University of Science & Technology, and the Bud Antle Endowed Chair for Excellence in Agriculture (University of Arizona) to R.A.W., the Start-up Fund of Huazhong Agricultural University to J.Z., and funding from the Taiwan Council of Agriculture to K.M.. The BUSCO analysis data for maize was kindly provided by Dr. Wu and Dr. Li from the Institute of Plant Physiology and Ecology, and Dr. Wang from Shanghai Jiao Tong University. One of two TE libraries used for repeat analysis was provided by Dr. Eric Laserre (University of Perpignan, France).
Author contributions
J.Z., K.M., D.C., M.L., N.A., N.R.S.H., H.L., R.M. and R.A.W. designed and conceived the research. D.C. and K.M. perform the population structure analysis. K.M., M.L., L.J.A. and N.L. generated and provided SSD seed 12 O. sativa accessions. D.K., S.L., S.R. and N.M. prepared DNA and performed PacBio and Illumina sequencing. C.S.-S. managed all PacBio and Illumina sequence data processing and transfer. P.P. and V.L. generated all Bionano optical maps. J.Z. and Y.Z. performed sequence assembly. Y.Z. carried out genome size estimation, GPM editing, assembly polishing and data submission. V.L. and Y.Z. analyzed the Bionano optical maps and the validation of 12 PSRefSeqs. A.Z. and Y.Z. carried out TE prediction and structural analysis. Y.Z., N.A., A.Z., J.Z., D.C., M.L., K.M., N.M. and R.A.W. wrote and edited the paper. All authors read and approved the final manuscript.
Code availability
The population re-analysis of 3K-RG dataset and 12 genome assemblies were obtained using several publicly available software packages. To allow researchers to precisely repeat any steps, the settings and the parameters used are provided below:
Population structure:
ADMIXTURE was run with default options. The R scripts for further population structure analysis, including setting up CLUMPP files, can be found in Github repository https://github.com/dchebotarov/Q-aggr.
Genome size estimation:
The K-mer and GCE program were employed for genome size estimation. Command line:
kmer_freq_hash -k (13-17) -l genome.list -a 10 -d 10 -t 8 -i 400000000 -o 0 -p genom_kmer(13-17) & > genome_kmer(13-17)_freq.log, and gce -f genom _kmer(13-17).freq.stat -c $peak -g #amount -m 1 -D 8 -b 1 -H 1 > genome.Table 2 > genom_kmer(13-17).log
Genome assembly:
(1) MECAT2: all parameters were set to the defaults. Command line:
mecat.pl config_file.txt, mecat.pl correct config_file.txt and mecat.pl assemble config_file.txt
(2) Canu1.5: all parameters were set to the defaults. Command line:
canu -d canu -p canu genomeSize = 400 m -pacbio-raw rawreads.fasta
(3) FALCON: all parameters were set to the defaults. Command line:
fc_run.py fc_run.cfg & > fc_run.out
(4) GPM: manual edit with merging de novo assemblies from MECAT2, Canu1.5, and FALCON
Polishing:
(1) arrow: all parameters were set to the defaults except alignment length = 500 bp. The arrow polish was carried out by the SMRT Link v6.0 webpage (https://www.pacb.com/support/software-downloads/).
(2) pilon1.18: all parameters were set to the defaults.
BUSCO:
The BUSCO3.0 version was employed in this study. Command line: run_BUSCO.py -i genome.fasta -o genome -l embryophyta_odb9 -m genome -c 16
RepeatMasker:
The repeat sequences were employed with the library rice7.0.0_liban in-house. Command line: RepeatMasker -pa 24 -x -no_is -nolow -cutoff 250 -lib rice7.0.0.liban.txt genome.fasta
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Yong Zhou, Dmytro Chebotarov.
Contributor Information
Andrea Zuccolo, Email: andrea.zuccolo@kaust.edu.sa.
Jianwei Zhang, Email: jzhang@mail.hzau.edu.cn.
Rod A. Wing, Email: rod.wing@kaust.edu.sa
Supplementary information
is available for this paper at 10.1038/s41597-020-0438-2.
References
- 1.Seck P-A, Diagne A, Mohanty S, Wopereis M-C. Crops that feed the world 7: Rice. Food security. 2012;4:7–24. doi: 10.1007/s12571-012-0168-1. [DOI] [Google Scholar]
- 2.Merrey, D.-J. et al. Agricultural Development and Sustainable Intensification. Routledge (2018).
- 3.Wing A-R, Michael D-P, Zhang Q-F. The rice genome revolution: from an ancient grain to Green Super Rice. Nature Reviews Genetics. 2018;19:505–517. doi: 10.1038/s41576-018-0024-z. [DOI] [PubMed] [Google Scholar]
- 4.3K RGP The 3,000 rice genomes project. GigaScience. 2014;3:2047–217X. doi: 10.1186/2047-217X-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stein J-C, et al. Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza. Nature genetics. 2018;50:285–296. doi: 10.1038/s41588-018-0040-0. [DOI] [PubMed] [Google Scholar]
- 6.Kawahara Y, et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice. 2013;6:4. doi: 10.1186/1939-8433-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.International Rice Genome Sequencing Project The map-based sequence of the rice genome. Nature. 2005;436:793–800. doi: 10.1038/nature03895. [DOI] [PubMed] [Google Scholar]
- 8.Thomson M-J, et al. Large-scale deployment of a rice 6 K SNP array for genetics and breeding applications. Rice. 2017;10:1–13. doi: 10.1186/s12284-017-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McNally K-L, et al. Genomewide SNP variation reveals relationships among landraces and modern varieties of rice. Proceedings of the National Academy of Sciences. 2009;106:12273–12278. doi: 10.1073/pnas.0900992106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang X-H, et al. A map of rice genome variation reveals the origin of cultivated rice. Nature. 2012;490:497–501. doi: 10.1038/nature11532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao Q, et al. Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nature genetics. 2018;50:278–284. doi: 10.1038/s41588-018-0041-z. [DOI] [PubMed] [Google Scholar]
- 12.Wang W, et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature. 2018;557:43–49. doi: 10.1038/s41586-018-0063-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang J, et al. Building two indica rice reference genomes with PacBio long-read and Illumina paired-end sequencing data. Scientific data. 2016;3:1–8. doi: 10.1038/sdata.2016.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J, et al. Extensive sequence divergence between the reference genomes of two elite indica rice varieties Zhenshan 97 and Minghui 63. Proc. Natl. Acad. Sci. 2016;113:E5163–E5171. doi: 10.1073/pnas.1611012113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Udall J-A, Kelly D. Is it ordered correctly? Validating genome assemblies by optical mapping. The Plant Cell. 2018;30:7–14. doi: 10.1105/tpc.17.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome research. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jakobsson M, Noah AR. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23:1801–1806. doi: 10.1093/bioinformatics/btm233. [DOI] [PubMed] [Google Scholar]
- 18.Porebski S, Bailey L-G, Baum B-R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant molecular biology reporter. 1997;15:8–15. doi: 10.1007/BF02772108. [DOI] [Google Scholar]
- 19.Bolger A-M, Marc L, Bjoern U. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown J, Meg P, Lee AM. FQC Dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics. 2017;33:3137–3139. doi: 10.1093/bioinformatics/btx373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu, B. et al. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. Preprint at, https://arxiv.org/abs/1308.2012 (2013).
- 22.Ou, S. et al. Effect of sequence depth and length in long-read assembly of the maize inbred nc358. Preprint at, 10.1101/858365v2.full (2019). [DOI] [PMC free article] [PubMed]
- 23.Luo M, Wing A-R. An improved method for plant BAC library construction. Plant functional genomics. Humana Press. 2003;236:3–19. doi: 10.1385/1-59259-413-1:3. [DOI] [PubMed] [Google Scholar]
- 24.Chin C, et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nature methods. 2016;13:1050. doi: 10.1038/nmeth.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao C, et al. MECAT: fast mapping, error correction, and de novo assembly for single-molecule sequencing reads. nature methods. 2017;14:1072. doi: 10.1038/nmeth.4432. [DOI] [PubMed] [Google Scholar]
- 26.Koren S, et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome research. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J, et al. Genome puzzle master (GPM): an integrated pipeline for building and editing pseudomolecules from fragmented sequences. Bioinformatics. 2016;32:3058–3064. doi: 10.1093/bioinformatics/btw370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chaisson M-J, Tesler G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC bioinformatics. 2012;13:238. doi: 10.1186/1471-2105-13-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at, https://arxiv.org/abs/1303.3997 (2013).
- 30.Walker B-J, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. Plos One. 2014;9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Altschul S-F, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic acids research. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simão F-A, et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 33.Wolfe K-H, et al. Date of the monocot-dicot divergence estimated from chloroplast DNA sequence data. Proceedings of the National Academy of Sciences. 1989;86:6201–6205. doi: 10.1073/pnas.86.16.6201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gale M-D, Katrien MD. Comparative genetics in the grasses. Proceedings of the National Academy of Sciences. 1998;95:1971–1974. doi: 10.1073/pnas.95.5.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo H, et al. Gene duplication and genetic innovation in cereal genomes. Genome research. 2019;29:261–269. doi: 10.1101/gr.237511.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maja T, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics. 2009;25:4–10. doi: 10.1002/0471250953.bi0410s05. [DOI] [PubMed] [Google Scholar]
- 37.Ou S-J, et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biology. 2019;20:1–18. doi: 10.1186/s13059-019-1905-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rice P, Ian L, Alan B. EMBOSS: the European molecular biology open software suite. Trends in Genetics. 2000;16:276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 39.Sedlazeck F-J, et al. Accurate detection of complex structural variations using single-molecule sequencing. Nature methods. 2018;15:461–468. doi: 10.1038/s41592-018-0001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heller D, Martin V. SVIM: structural variant identification using mapped long reads. Bioinformatics. 2019;35:2907–2915. doi: 10.1093/bioinformatics/btz041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.2019. NCBI Sequence Read Archive. SRP226085
- 42.2019. NCBI Sequence Read Archive. SRP226086
- 43.2019. NCBI Sequence Read Archive. SRP226088
- 44.2019. NCBI Sequence Read Archive. SRP227255
- 45.2019. NCBI Sequence Read Archive. SRP227298
- 46.2019. NCBI Sequence Read Archive. SRP226087
- 47.2019. NCBI Sequence Read Archive. SRP226084
- 48.2019. NCBI Sequence Read Archive. SRP226093
- 49.2019. NCBI Sequence Read Archive. SRP226080
- 50.2019. NCBI Sequence Read Archive. SRP226082
- 51.2019. NCBI Sequence Read Archive. SRP226079
- 52.2019. NCBI Sequence Read Archive. SRP226078
- 53.Zhang J, 2019. Whole genome shotgun (WGS) sequencing and assembly of the rice Azucena genome (Oryza sativa) with PacBio long-read technology. GenBank. PKQC00000000
- 54.Zhang J, 2019. IR64RS1 (Rice IR64 Reference Sequence Version 1) GenBank. RWKJ00000000
- 55.Zhou Y, 2019. Os125827RS1 (Rice IRGC 125827 Reference Sequence Version 1) GenBank. WGGU00000000
- 56.Zhou Y, 2019. Os127518RS1 (Rice IRGC 127518 Reference Sequence Version 1) GenBank. VYIF00000000
- 57.Zhou Y, 2019. Os132278RS1 (Rice IRGC 132278 Reference Sequence Version 1) GenBank. VYIH00000000
- 58.Zhou Y, 2019. Os127652RS1 (Rice IRGC 127652 Reference Sequence Version 1) GenBank. VYIG00000000
- 59.Zhou Y, 2019. Os125619RS1 (Rice IRGC 125619 Reference Sequence Version 1) GenBank. VYIE00000000
- 60.Zhou Y, 2019. Os117425RS1 (Rice IRGC 117425 Reference Sequence Version 1) GenBank. VYID00000000
- 61.Zhou Y, 2019. Os128077RS1 (Rice IRGC 128077 Reference Sequence Version 1) GenBank. VYIC00000000
- 62.Zhou Y, 2019. Os132424RS1 (Rice IRGC 132424 Reference Sequence Version 1) GenBank. VXJI00000000
- 63.Zhou Y, 2019. Os127564RS1 (Rice IRGC 127564 Reference Sequence Version 1) GenBank. VXJH00000000
- 64.Zhou Y, 2019. Os127742RS1 (Rice IRGC 127742 Reference Sequence Version 1) GenBank. VYIB00000000
- 65.Zhou Y, 2020. A platinum standard pan-genome resource that represents the population structure of Asian rice. figshare. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- 2019. NCBI Sequence Read Archive. SRP226085
- 2019. NCBI Sequence Read Archive. SRP226086
- 2019. NCBI Sequence Read Archive. SRP226088
- 2019. NCBI Sequence Read Archive. SRP227255
- 2019. NCBI Sequence Read Archive. SRP227298
- 2019. NCBI Sequence Read Archive. SRP226087
- 2019. NCBI Sequence Read Archive. SRP226084
- 2019. NCBI Sequence Read Archive. SRP226093
- 2019. NCBI Sequence Read Archive. SRP226080
- 2019. NCBI Sequence Read Archive. SRP226082
- 2019. NCBI Sequence Read Archive. SRP226079
- 2019. NCBI Sequence Read Archive. SRP226078
- Zhang J, 2019. Whole genome shotgun (WGS) sequencing and assembly of the rice Azucena genome (Oryza sativa) with PacBio long-read technology. GenBank. PKQC00000000
- Zhang J, 2019. IR64RS1 (Rice IR64 Reference Sequence Version 1) GenBank. RWKJ00000000
- Zhou Y, 2019. Os125827RS1 (Rice IRGC 125827 Reference Sequence Version 1) GenBank. WGGU00000000
- Zhou Y, 2019. Os127518RS1 (Rice IRGC 127518 Reference Sequence Version 1) GenBank. VYIF00000000
- Zhou Y, 2019. Os132278RS1 (Rice IRGC 132278 Reference Sequence Version 1) GenBank. VYIH00000000
- Zhou Y, 2019. Os127652RS1 (Rice IRGC 127652 Reference Sequence Version 1) GenBank. VYIG00000000
- Zhou Y, 2019. Os125619RS1 (Rice IRGC 125619 Reference Sequence Version 1) GenBank. VYIE00000000
- Zhou Y, 2019. Os117425RS1 (Rice IRGC 117425 Reference Sequence Version 1) GenBank. VYID00000000
- Zhou Y, 2019. Os128077RS1 (Rice IRGC 128077 Reference Sequence Version 1) GenBank. VYIC00000000
- Zhou Y, 2019. Os132424RS1 (Rice IRGC 132424 Reference Sequence Version 1) GenBank. VXJI00000000
- Zhou Y, 2019. Os127564RS1 (Rice IRGC 127564 Reference Sequence Version 1) GenBank. VXJH00000000
- Zhou Y, 2019. Os127742RS1 (Rice IRGC 127742 Reference Sequence Version 1) GenBank. VYIB00000000
- Zhou Y, 2020. A platinum standard pan-genome resource that represents the population structure of Asian rice. figshare. [DOI] [PMC free article] [PubMed]
Supplementary Materials
Data Availability Statement
The population re-analysis of 3K-RG dataset and 12 genome assemblies were obtained using several publicly available software packages. To allow researchers to precisely repeat any steps, the settings and the parameters used are provided below:
Population structure:
ADMIXTURE was run with default options. The R scripts for further population structure analysis, including setting up CLUMPP files, can be found in Github repository https://github.com/dchebotarov/Q-aggr.
Genome size estimation:
The K-mer and GCE program were employed for genome size estimation. Command line:
kmer_freq_hash -k (13-17) -l genome.list -a 10 -d 10 -t 8 -i 400000000 -o 0 -p genom_kmer(13-17) & > genome_kmer(13-17)_freq.log, and gce -f genom _kmer(13-17).freq.stat -c $peak -g #amount -m 1 -D 8 -b 1 -H 1 > genome.Table 2 > genom_kmer(13-17).log
Genome assembly:
(1) MECAT2: all parameters were set to the defaults. Command line:
mecat.pl config_file.txt, mecat.pl correct config_file.txt and mecat.pl assemble config_file.txt
(2) Canu1.5: all parameters were set to the defaults. Command line:
canu -d canu -p canu genomeSize = 400 m -pacbio-raw rawreads.fasta
(3) FALCON: all parameters were set to the defaults. Command line:
fc_run.py fc_run.cfg & > fc_run.out
(4) GPM: manual edit with merging de novo assemblies from MECAT2, Canu1.5, and FALCON
Polishing:
(1) arrow: all parameters were set to the defaults except alignment length = 500 bp. The arrow polish was carried out by the SMRT Link v6.0 webpage (https://www.pacb.com/support/software-downloads/).
(2) pilon1.18: all parameters were set to the defaults.
BUSCO:
The BUSCO3.0 version was employed in this study. Command line: run_BUSCO.py -i genome.fasta -o genome -l embryophyta_odb9 -m genome -c 16
RepeatMasker:
The repeat sequences were employed with the library rice7.0.0_liban in-house. Command line: RepeatMasker -pa 24 -x -no_is -nolow -cutoff 250 -lib rice7.0.0.liban.txt genome.fasta