

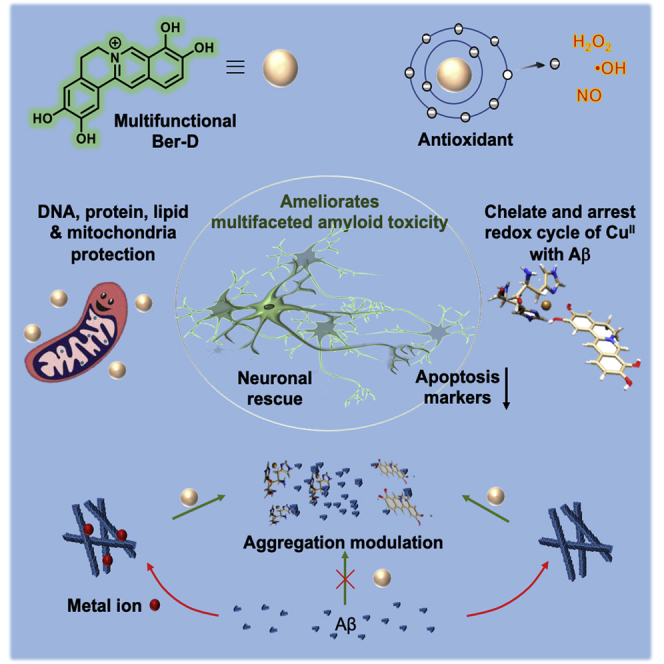
Summary
Multiple lines of evidence indicate that amyloid beta (Aβ) peptide is responsible for the pathological devastation caused in Alzheimer's disease (AD). Aβ aggregation species predominantly contribute to multifaceted toxicity observed in neuronal cells including generation of reactive oxygen species (ROS), mitochondrial dysfunction, interfering with synaptic signaling, and activation of premature apoptosis. Herein, we report a natural product berberine-derived (Ber-D) multifunctional inhibitor to ameliorate in cellulo multifaceted toxicity of AD. The structural attributes of polyphenolic Ber-D have contributed to its efficient Cu chelation and arresting the redox cycle to prevent the generation of ROS and rescue biomacromolecules from oxidative damage. Ber-D inhibits metal-dependent and -independent Aβ aggregation, which is supported by in silico studies. Ber-D treatment averts mitochondrial dysfunction and corresponding neuronal toxicity contributing to premature apoptosis. These key multifunctional attributes make Ber-D a potential therapeutic candidate to ameliorate multifaceted Aβ toxicity in AD.
Subject Areas: Drugs, Applied Chemistry, Molecular Biology
Graphical Abstract
Highlights
-
•
Multifaceted Aβ toxicity activates premature apoptosis and neuronal death in AD
-
•
Berberine is modified to nontoxic and polyphenolic multifunctional derivative (Ber-D)
-
•
Ber-D modulates Aβ aggregation, metal toxicity, ROS, oxidative and biomolecules damage
-
•
Study revealed mitochondrial protection and downregulation of apoptosis markers
Drugs; Applied Chemistry; Molecular Biology
Introduction
Neurodegenerative diseases are a major threat to global health and pose gigantic challenge for the scientific community to find effective treatments (Prince et al., 2013, Rajasekhar et al., 2015a). Alzheimer's disease (AD) is the most prevalent neurodegenerative disorder and accounts for more than 70% of all dementia (Knowles et al., 2014, Selkoe and Hardy, 2016). The multifactorial nature of the disease attributed to multifaceted toxicity has desisted researchers from developing effective medication for AD (Barnham and Bush, 2014, Huang and Mucke, 2012, Chiti and Dobson, 2017, Rajasekhar and Govindaraju, 2018). amyloid beta (Aβ) peptides with 37–43 amino acids generated by the proteolytic cleavage of a transmembrane human amyloid precursor protein (hAPP) are considered as the major culprits in causing multifaceted toxicity (Rauk, 2009, Nasica-Labouze et al., 2015, Wang et al., 2017). In particular, Aβ42 peptides misfold and undergo amyloidogenic self-assembly through hydrophobic interactions and β-sheet formation to form polymorphic species such as oligomers, protofibrils, and fibrillar aggregates, which deposit in the brain as Aβ plaques (Hamley, 2012, Rajasekhar et al., 2015a). The oligomeric forms of Aβ are considered to be highly lethal as they purge holes into the neuronal cell membrane and interact with synaptic cleft causing influx of excessive calcium into the cell disrupting the signaling pathways (Haass and Selkoe, 2007, Lee et al., 2017). Moreover, Aβ complexes with redox metal ions of Cu and Fe to generate reactive oxygen species (ROS) that cause DNA damage, protein oxidation, lipid peroxidation, and mitochondrial dysfunction eventually leading to neuronal toxicity and cell death (Bourassa and Miller, 2012, Savelieff et al., 2013, Rajasekhar et al., 2015a). In the past decades, various classes of molecules like peptides, peptidomimetics, polymers, and synthetic compounds have been extensively evaluated as aggregation modulators to prevent neurodegeneration, albeit with little or no success (Bulawa et al., 2012, Dwivedi and Iyer, 2014, Rajasekhar et al., 2015a, Rajasekhar et al., 2016, Rajasekhar et al., 2018, Rajasekhar et al., 2015b, Kaffy et al., 2016, Doig et al., 2017, Kumar et al., 2017, Samanta et al., 2019). The continuous failure of drug candidates at various stages of clinical trials has forced the scientific community to look for alternative design strategies to successfully develop therapeutic agents to target multiple toxicities. In fact, numerous approaches undertaken thus far to modulate individual toxicities (amyloid aggregation, metal inclusion, ROS generation, oxidative stress, biomolecular damage, or mitochondrial dysfunction) are ineffective and demand multifunctional inhibitor strategies to ameliorate multifaceted Aβ toxicity. Various attempts to use natural products or plant extracts to target AD have shown promising results. Natural products such as curcumin, resveratrol, and epigallocatechin-3-gallate (EGCG) have been shown to effectively decrease the Aβ toxicity in the mice brain attributed to their antioxidant and anti-aggregation properties. EGCG, a bioactive ingredient of green tea, exhibits aggregation modulation, anti-inflammatory, antioxidant, and neuroprotective properties (Ehrnhoefer et al., 2008). In recent times, several other natural products like brazilin, luteolin, tanshinone, and apigenin have been evaluated to assess the modulation of Aβ toxicity (Wang et al., 2013, Du et al., 2015). One of the major limitations of using natural products as anti-AD candidates is their slow relief or sluggish therapeutic action, which is ineffective for the treatment of moderate and advanced stages of the disease. Moreover, some of the drawbacks such as very low natural abundance, poor solubility, cellular toxicity, instability, and most importantly lack of multifunctional efficacy in targeting multifaceted Aβ toxicity limit the use of natural products as anti-AD agents.
Berberine is one of the isoquinoline alkaloids and a well-known Chinese medicine used to treat various disease conditions such as diarrhea, hypertension, inflammation, and tumors, among others (Yin et al., 2012, Ahmed et al., 2015, Zou et al., 2017). The pharmacological and bioactive properties of the natural product have also been extensively studied for diabetes, ischemia, cancer, arrhythmias, and neurodegenerative disorders such as Parkinson's disease and AD. Berberine has been shown to interfere with pathological pathways of AD by enforcing a reduction in the levels of Aβ generation by inhibiting secretase enzymes involved in APP processing, ameliorating gliosis, delaying oxidative stress, and preventing neuroinflammation (Cai et al., 2016). Several modifications and functional groups incorporated to the berberine core and their activity against acetylcholinesterase and other aspects have been evaluated (Huang et al., 2010a, Huang et al., 2016, Huang et al., 2010b, Tsai and Lee, 2010, Shan et al., 2011, Zou et al., 2017). The use of berberine as a therapeutic candidate for AD is hampered by its cytotoxic nature at relatively higher concentrations (Kysenius et al., 2014). The absence of a clear safety profile remains a major concern, and various reports have shown that berberine causes an increase in oxidative stress and mitochondrial fragmentation or swelling, decrease in mitochondrial membrane potential (MMP), and depleted ATP production, ultimately leading to neuronal death (Mahmoudi et al., 2016, Singh and Sharma, 2018). Herein, we report Ber-D multifunctional modulator to effectively target multifaceted Aβ toxicity of AD. The simple deprotection of berberine phenolic groups has yielded polyphenolic Ber-D with improved solubility and cell viability compared with the parent natural product. The polyphenolic Ber-D exhibits significantly improved antioxidant, redox metal chelation, and anti-Aβ aggregation properties. The detailed in vitro and in cellulo studies show that Ber-D actively modulates multifaceted Aβ toxicity.
Results
Design and Cytotoxicity
Berberine has been shown to have therapeutic value for several diseases including AD. In case of AD, berberine is reported to interfere with Aβ generation, acetylcholine esterase and monoamine oxidase activity, and cholesterol level maintenance (Cai et al., 2016). However, high cytotoxicity and low safety profiles have limited its further development as a potential therapeutic candidate. The role of polyphenolic moieties in natural and synthetic compounds to exhibit excellent antioxidant property has been well documented (Yan et al., 2017, Huyut et al., 2017). In fact, EGCG, resveratrol, brazilin, and tanshinone are some of the known examples that modulate Aβ aggregation in addition to antioxidant effects attributed to polyphenolic moieties in their molecular structures. Interestingly, berberine has a set of ortho-methoxy and methylenedioxy functionalities and their deprotection can generate four phenolic hydroxyl groups. The generation of multiple phenolic hydroxyl groups is anticipated to enhance the solubility, antioxidant property, and ability to chelate redox metal ions implicated in AD pathology. The starting material (berberine) is available abundantly and inexpensive compared with other natural products. The one-step modification procedure to obtain Ber-D from berberine (Transparent Methods, Data S1) makes it a cost-effective candidate to evaluate for the possible treatment of AD pathology. The aforementioned facts and design attributes encouraged us to undertake the detailed study to evaluate the ability of Ber-D as a multifunctional inhibitor of multifaceted Aβ toxicity.
Initially, we assessed the viability of PC12 cells through MTT assay in the presence of Ber-D or berberine in a concentration-dependent manner (Figure 1A). As expected berberine exhibited cytotoxicity to PC12 cells wherein the cell viability decreased with increasing concentration (95% cell viability at 2.5 μM reduces to 60% for 100 μM). Remarkably, Ber-D showed no observed adverse effects or minimal cytotoxicity to PC12 cells over the wide concentration range (0.5–100 μM) tested. As shown in Figure 1A, Ber-D is not cytotoxic up to 35 μM, whereas at higher concentrations (50 and 100 μM) very minimal cytotoxicity (cell viability of 97% and 93%, respectively) was observed. These data indicated significant improvement in the cell viability of Ber-D in comparison with the parent compound. For instance, Ber-D at 1 μM is nontoxic and at high concentration (100 μM) showed ∼33% improvement in the cell viability compared with berberine. The observed high cytotoxicity of berberine is attributed to its accumulation in the mitochondria causing cell-cycle arrest, decrease in MMP, mitochondrial fragmentation, oxidative stress, and depletion in ATP production (Yan et al., 2017). Next, we embarked on assessing the effect of Ber-D on the mitochondria of different cell lines (PC12, HeLa, and HEK cells) to gather a broader perspective. The cells were treated with 50 μM of Ber-D or berberine, and MMP was measured using an MMP-sensitive dye (Rhodamine 123) (Figure 1B). Berberine treatment has been shown to induce mitochondrial fragmentation in cells, which is a causative factor of mitochondrial damage and dysfunction. Berberine-treated cells exhibited MMP of 67% (PC12), 68% (HeLa), and 71% (HEK), respectively, in comparison with untreated cells. On the other hand, Ber-D showed MMP of 93%, 94%, and 94% in PC12, HeLa, and HEK, respectively, which show significant improvement in MMP compared with the parent compound and infer minimal interference of Ber-D on the mitochondrial functions. We visualized the effect of berberine and Ber-D on the mitochondrial morphology in HeLa cells under a fluorescence optical microscope (Figure 1C). HeLa cells were treated with 50 μM of berberine or Ber-D for 3 h, followed by staining with mitochondrial-specific dye (Mitotracker green FM), and cells were imaged. As anticipated, cells treated with berberine showed fragmented mitochondria, whereas Ber-D treatment showed normal morphology (elongated) of mitochondria similar to untreated cells (Figure 1C). These results clearly suggest that the Ber-D is nontoxic to cells and has minimal adverse effect on the mitochondrial morphology and function.
Figure 1.

Cell Viability Studies of Ber-D and Berberine
(A) Concentration-dependent (0.5, 1, 2.5, 5, 10, 25, 35, 50, and 100 μM) and comparative cell viability of Ber-D and berberine in PC12 cells.
(B) Quantification of Rho123 fluorescence at 534 nm (λex = 511 nm) corresponding to MMP of PC12, HeLa, and HEK cells treated independently with Ber-D (50 μM) and berberine (50 μM).
(C) Fluorescence microscopic images of Ber-D (50 mM) or berberine (50 mM) treated HeLa cells using Mitotracker green FM (mitochondria-specific dye) to study the mitochondrial morphology. Scale bar, 50 μM. Each experiment was repeated (n = 16, for cell viability; n = 4, for MMP quantification); error bars represent the standard deviation (SD). Ber, berberine.
Metal Chelation
The redox metal ion such as Cu plays a key role in the aggravation of Aβ toxicity (House et al., 2009). The presence of multiple phenolic hydroxyl groups in Ber-D encouraged us to study its metal-chelating property. The modification of berberine to Ber-D leads to a broad absorption band spanning from 200 to 800 nm with absorption maxima at 263, 367, and 648 nm (ε = 5230 M−1cm−1) (Figure 2A). Initially, we screened 20 μM of all the biologically relevant and key metal ions (CuII, FeII, FeIII, ZnII, CoII, NiII, MgII, AlIII, Na+, and K+) with Ber-D (20 μM) in PBS buffer (10 mM, pH = 7.4) (Figure S1A). Ber-D showed change in the absorption intensities (hypochromic or hyperchromic effect) or shifting of absorption maxima (bathochromic or hypsochromic shift) in the presence of metal ions tested. Interestingly, distinct absorption spectrum was observed for Ber-D in the presence of CuII with the appearance of new absorption bands at 400 and 505 nm. We performed competition study to evaluate the selective chelation by taking the advantage of distinctive absorption spectral change of Ber-D in the presence of CuII (Figure S1B). Ber-D+CuII (20 μM) in PBS buffer was treated with 20 μM of other metal ions (FeII, FeIII, ZnII, CoII, NiII, MgII, AlIII, Na+, and K+), and change in absorption spectra was monitored. The absorption spectra of Ber-D+CuII did not show significant change in the presence of other metals, which confirmed the preferential chelation of CuII by Ber-D. The selective chelation of CuII by Ber-D is a highly desirable property to target copper-dependent Aβ toxicity. Furthermore, we performed binding saturation study of Ber-D with increasing concentrations of CuII to determine the binding constant (kd) and was found to be 2.32 μM (Figures S1C and S1D).
Figure 2.

Metal Chelation and Anti-aggregation Properties of Ber-D
(A) Absorbance spectra of Ber-D, Ber-D+CuII, and Aβ42+CuII+Ber-D complex. Inset: proposed structure for Ber-D and two His units of Aβ chelation with CuII to form the Aβ42+CuII+Ber-D complex.
(B) Aβ42 (10 μM) was incubated with Ber-D (10, 20, 50, and 100 μM) or berberine (100 μM) in a concentration-dependent manner, and aggregation inhibition was quantified through fluorescence measurement of ThT at 485 nm (λex = 450 nm) after 48 h.
(C) Dot blot analysis of Aβ42 aggregates to study the anti-aggregation properties of Ber-D and berberine on the formation of Aβ42 fibrillar aggregates and Aβ42 oligomeric species. OC and A11 antibody were used for specific detection of Aβ42 fibrillar aggregates and Aβ42 oligomeric species, respectively.
(D) Aβ42 (10 μM) and CuII were incubated alone and independently with Ber-D (10, 20, 50, and 100 μM) or berberine (100 μM) in a concentration-dependent manner, and aggregation inhibition was quantified through fluorescence measurement of ThT at 485 nm (λex = 450 nm) after 24 h. Each experiment was repeated thrice (n = 3); error bars represent the standard deviation (SD) of the NFI. Ber, berberine.
See also Figures S1 and S4.
Metal chelators to sequestrate CuII from the metal-Aβ inclusion complex have been extensively studied as an effective strategy to ameliorate Cu-Aβ in AD pathology Atrián-Blasco et al., 2017. Interestingly, metal chelators such as clioquinol and PBT2 are some of the earliest drug candidates to reach the clinical trials (Crouch et al., 2011). Notwithstanding the failure of metal chelation-based drugs at various levels of clinical trials, several recent reports clearly emphasize the importance of metal-chelating ligands or structural elements in the drug design to tackle Cu-Aβ toxicity (Matlack et al., 2014). The redox-active Cu plays a predominant role in the acceleration of Aβ aggregation, stabilizing highly toxic oligomers and generation of excessive ROS when bound to Aβ, which is responsible for the additional trait of neuronal toxicity. We studied the effect of Ber-D on the Aβ42+CuII (20 μM) complex by monitoring the absorption spectra with increasing concentrations of Ber-D (2, 5, 10, 20, 40, 50, and 60 μM) in PBS buffer (Figure S1E). Remarkably, the absorption spectra of Aβ42+CuII complex transformed upon addition of Ber-D (2 μM), which is typical of Ber-D+CuII complex, and the spectral changes have become prominent with increase in concentration, a clear indication that Ber-D binds to CuII in the Aβ4+CuII complex. Notably, the binding affinity of Aβ42 for CuII is higher than that of the Ber-D and hence it cannot sequestrate the metal ion from Aβ4+CuII complex. However, Ber-D interacts with Aβ42+CuII and forms a cooperative complex of the type Aβ4+CuII+Ber-D, which arrests the redox cycle of CuII (Caballero et al., 2016). We performed a competitive assay to understand the binding strength of Ber-D toward CuII in the presence of Aβ42, where the absorption spectra of Ber-D+CuII was recorded with increasing concentrations of Aβ42 (1, 2, 5, 10, 15, 20, 40, 60, and 100 μM) (Figure S1F). Interestingly, the absorption spectra of Ber-D+CuII was unaffected by the addition of Aβ42 in the entire concentration range of 1–100 μM. The metal chelation or cooperative complexation of Ber-D with Aβ42+CuII to form Aβ42+CuII+Ber-D complex was studied by computational modeling using density functional theory. There are many hypotheses about the Cu-binding site in the amyloid fibril, and different proposals have been suggested regarding the position of His residues of Aβ42 involved in the coordination with CuII. Some experiments have suggested the involvement of His6 and His14 or His6 and His13 or His13 and His14 residues in the coordination (Shin and Saxena, 2011, Gunderson et al., 2012). A recent scanning tunneling microscopy, circular dichroism, and surface-enhanced Raman spectroscopy-based study proposes that CuII binds to two His residues (His13 and His14) of two adjacent beta strands (Atrián-Blasco et al., 2018). Analyses of His locations in Aβ fibril structure reveal the following possibilities for CuII coordination: (1) His6 and His13 of the same strand binding to CuII, (2) His6 of adjacent strands binding to CuII, (3) His13 of adjacent strands binding to CuII, and (4) His14 of adjacent residues binding to CuII (Figure 5C). The first three possibilities require the ligand (Ber-D) to diffuse into the core site, whereas the fourth involves the surface binding of the ligand and appears to be the potent site for Ber-D to form complex with Aβ bound CuII. We are not excluding the first three possibilities in this study despite the fact that the fourth possibility appears to be the most feasible mode for Ber-D binding to Aβ+CuII. Moreover, our modeling study revealed that CuII can bind to His14 of adjacent β-strands without any significant disruption to the fibril structure. The distance between the two α-carbons of the two His14 in adjacent strands after optimization with CuII is ∼5.4 Å, which suggests that CuII insertion does not alter the geometry of the amyloid fibril but rather stabilizes the inter-stand interaction through the electrostatic interaction and covalent bond formation. It is worth recalling that recently reported cryoelectron microscopy (cryo-EM)-based Aβ fibril structure showed the distance between two alpha-carbons of two His14 residues on the adjacent strands to be 4.78 Å. The interaction energies of CuII binding to two His14 residues on two β-strands are −393.25 and −379.54 kcal/mol, respectively, as predicted from the B3LYP and MO6-2X level of theory (these values are counter-poise corrected) (Figure S2). The comparable values for the complexation energy suggest that dispersion is not the dominant interaction rather electrostatic interactions. Such a larger magnitude of the energy suggests that the bonding is covalent in nature. Similarly, the calculated counterpoise corrected complexation energies of Ber-D and CuII complexation are −197.7 and −191.1 kcal/mol for B3LYP and MO6-2X levels of theory, respectively. This indicates that Ber-D binds to Aβ+CuII complex without sequestrating CuII from the metal-Aβ inclusion complex as the sequestrating process will require energy to break the covalent bond between the CuII and imidazole ring of His14 residues. These results are in good agreement with the fact that binding affinity of Aβ42 for CuII is higher than Ber-D and hence Ber-D cannot sequestrate the metal ion from Aβ+CuII complex (Figure S2). However, the complexation energies from the computational study suggest that Ber-D can bind to Aβ+CuII to form a tetrahedral cooperative complex where the two valences are occupied by two hydroxyl groups of Ber-D, whereas the other two valences are fulfilled by nitrogen atoms of the imidazole ring of His residues of Aβ (Figure 2A).
Figure 5.

Mechanism of Aβ Toxicity in PC12 Cells
(A) PC12 cell viability observed after independently incubated (24 h) with Aβ42 (10 μM) and Ber-D or berberine (Ber).
(B) Cell viability of PC12 cells after independently incubated (24 h) with Aβ42 (10 μM), CuII (10 μM), ascorbate (200 μm), and Ber-D or berberine.
(C) Fluorescence microscopic images of Rho123-treated PC12 cells after incubation (12 h) with Aβ42 (10 μM) and Ber-D or berberine (20 μM). Scale bar, 50 μM.
(D) Western blot analysis of the protein levels of cyt C in PC12 cells treated with Aβ42 (10 μM) and Ber-D or berberine (20 μM). Statistical analysis for the effect of Ber-D or berberine on the expression of Cyt C obtained from the western blot.
(E) Quantification of caspase 3 generations in PC12 cells treated with Aβ42 (10 μM) and Ber-D or berberine (20 μM). Ber, berberine.
Statistical analysis was performed through ordinary one-way ANOVA or Kruskal-Wallis one-way ANOVA test: ∗∗, p value 0.005; ∗, p value 0.05.
As discussed vide supra, the Aβ+CuII complex transforms into depot for ROS production through Fenton-type reaction and a major contributor to the oxidative stress and biomolecular damage (Smith et al., 2007). Thus, effective silencing of copper redox cycle in the Aβ+Cu complex and prevention of ROS generation is one of the essential criteria to qualify Ber-D as multifunctional inhibitor of Aβ toxicity. We performed ascorbate assay to evaluate the ability of Ber-D to redox-silence CuII and suppress generation of excessive ROS. The redox-active CuII in the reduced environment (ascorbate) generates ROS such as hydroxyl radicals that can be measured using coumarin-3-carboxylic acid (3-CCA), as the hydroxyl radicals transform 3-CCA (non-fluorescent) to 7-hydroxycoumarin-3-carboxylic acid (7-OH-CCA, fluorescent) (λex = 395 nm and λem = 452 nm). Fluorescence emission of 7-OH-CCA was monitored at 452 nm in the presence of CuII (5 μM), 3-CCA (50 μM), berberine (50 μM), or Ber-D (50 μM) and ascorbate (150 μM) for a period of 100 min at regular intervals of 4 min (Figure S3). Interestingly, the samples containing Ber-D showed negligible emission at 452 nm, which suggests that Ber-D binds to CuII and prevents its redox cycle, in spite of the highly reducing environment. On the other hand, berberine initially showed low fluorescence emission at 452 nm that increased with time and reached an emission intensity comparable with that of the control (Aβ+CuII). This clearly indicates the inability of berberine to maintain CuII in redox dormant state, which is attributed to the lack of coordination sites (hydroxyl groups) for effective metal chelation. Remarkably, Ber-D was found to be effective in maintaining CuII in redox dormant state at a concentration ratio of as low as 1:1 (Ber-D:Aβ+CuII) (Figure S3). The Cu chelation and redox silencing properties of Ber-D are expected to contribute cooperatively to ameliorate the multifaceted Aβ toxicity.
Modulation of Aβ Aggregation
The misfolding and assembly of Aβ42 to form soluble (oligomers) and insoluble (fibrils and plaques) polymorphic aggregation species is considered to be the hallmark of AD. Moreover, identifying and quantifying specific polymorphic species of Aβ has been an active area of research (Hatai et al., 2017, Rajasekhar et al., 2017). Inhibition of Aβ aggregation to form oligomers and fibrillar aggregates is considered as one of the prominent strategies to develop therapeutic agents (Frydman-Marom et al., 2011). In this context, propensity of Ber-D to inhibit the Aβ42 aggregation was evaluated by thioflavin (ThT) assay and dot blot analysis. The Aβ42 monomer (10 μM) was incubated with Ber-D (100 μM) and berberine (100 μM) independently at 37°C for 48 h. The extent of aggregation of Aβ42 was assessed by recording the fluorescence emission of added ThT (10 μM, λex = 450 nm, λem = 485 nm) (Figure 2B). The data in Figure 2B show that Ber-D inhibits the aggregation of Aβ42 by 60% and berberine by 20% compared with untreated control Aβ42 (100%). We performed dot blot analysis to further strengthen this finding, a reliable quantitative technique to study the effect of modulators of Aβ42 aggregation. In a set of independent experiments, Aβ42 (10 μM) in PBS (10 mM, pH = 7.4) was incubated with Ber-D (100 μM) and berberine (100 μM) at 37°C for 48 h. The samples were spotted on a polyvinylidene fluoride (PVDF) membrane and the membrane was treated with OC antibody (specific to Aβ fibrillar aggregates) followed by enhanced chemiluminescence (ECL) reagent, and the spot intensities were subject to quantification in comparison with the control (Aβ42) (Figure 2C). Dot blot analysis data showed 68% and 12% inhibition of aggregation for Ber-D and berberine, respectively. These data are in good agreement with ThT assay and clearly suggest the superior aggregation modulation by Ber-D compared with its parent compound. Inhibition of Aβ42 aggregation was found to be concentration dependent, as increase in concentration of Ber-D from 10 to 100 μM resulted in 20%–60% activity, respectively (Figure S4A).
The oligomeric form is considered the most toxic polymorphic species of Aβ42 and known to cause cell membrane disruption, mitochondrial dysfunction, generation of excessive ROS, and neuronal damage through synaptic dysfunction (Savelieff et al., 2013). Thus, effective modulator of Aβ aggregation must inhibit the formation of oligomeric species. To assess the inhibitory activity of Ber-D against oligomers, Aβ42 monomer (10 μM) was incubated independently with Ber-D (100 μM) and berberine (100 μM) for 24 h at 4°C. The samples were spotted on a PVDF membrane, treated with the A11 antibody (specific for Aβ oligomeric species), followed by ECL reagent, and the spot intensities were quantified to determine the extent of inhibition. The Ber-D-treated sample showed 85% inhibition of oligomer against 55% by berberine (Figure S4B). These results suggest that Ber-D is an effective modulator of polymorphic Aβ42 aggregation species.
Modulation of Metal-Dependent Aβ Aggregation
The redox-active Cu accelerates the Aβ aggregation process to generate stable oligomers and fibrillar aggregates, which in turn deteriorate the neuronal toxicity (Esmieu et al., 2019). This necessitates the need for an effective metal chelating-aggregation modulator to tackle metal-mediated Aβ toxicity, and this strategy has been extensively pursued by the research community (Robert et al., 2015). However, most of the reported metal chelating-aggregation modulators lack the multifunctional capability to target multifaceted Aβ toxicity. We anticipated that Ber-D with metal chelation and aggregation modulation ability can be a potential inhibitor of metal-mediated Aβ toxicity. The Aβ42 (10 μM) with CuII (10 μM) in the presence or absence of Ber-D (100 μM) or berberine (100 μM) was incubated at 37°C for 24 h, ThT was added, and fluorescence was recorded at 485 nm (Figure 2D). As anticipated, Ber-D showed 60% aggregation inhibition against 15% by berberine, which corroborates with the fact that Cu chelation and anti-aggregation properties of Ber-D work synergistically to prevent the metal-mediated Aβ aggregation.
Computational Study of Ber-D Interactions with Aβ Monomer and Fibrils
We performed computational modeling studies to investigate the molecular-level interactions that contributed to the enhanced anti-aggregation activity of Ber-D in comparison with its parent compound. Berberine and Ber-D were found to interact with the core and surface sites of the Aβ (17–42) protofibril (Figure 3B) (Murugan et al., 2016). Computational modeling studies using molecular docking and implicit solvent free energy calculation methods have shown that most of the Aβ aggregate-staining molecules bind preferentially to the core-2 site with high affinity (Kawai et al., 2018). In case of berberine and Ber-D ligands the entry cleft site was found to be associated with high binding affinity followed by the core-2 site (Figure 3A). The surface site and core-1 site were not associated with stronger binding interactions with the ligands. The binding affinity and inhibition constants for berberine and Ber-D with Aβ monomer and Aβ fibrils were derived from molecular docking study (Figure 3B). Berberine and Ber-D have low binding affinity (in the micromolar range) toward the monomer compared with the fibril (in the nanomolar range). Furthermore, Ber-D shows a 5- to 10-fold higher binding affinity (12 and 31.3 nM) toward fibril compared with that of berberine (131 and 160 nM) in the entry cleft and core-1 sites, respectively. The observed high binding affinity of Ber-D toward Aβ compared with berberine is in agreement with the experimental data. It can be proposed that the more stable complex formation of Ber-D with Aβ in the core sites will interfere with the adhesion of additional units of Aβ in the fibril growth direction, which is responsible for the observed aggregation inhibition. The most stable complex of berberine with Aβ monomer is stabilized by two hydrogen bonds formed between the ligand and residues His6 and His14, whereas in case of Ber-D additional hydrogen bonds are formed with the Asp7 residue. Using the LigPLOT program, the interaction of berberine and Ber-D with different amino acids of Aβ fibril and their nature was investigated (Figure 3C). The interaction of berberine with Aβ protofibril is mostly hydrophobic in nature (dominated by van der Waals than electrostatic interactions). Interestingly, the interaction between Ber-D and Aβ protofibril is mediated by the hydrogen bonding in addition to van der Waals-type interactions. This explains the stronger and specific interaction of Ber-D with Aβ protofibril compared with the parent compound. The binding affinity data discussed above were performed for the protofibril of Aβ peptide with amino acid residues 17–42 in each strand (based on 2BEG structure). Recently, the cryo-EM-based fibril structure of full-length Aβ42 has been reported (this full-length fibril is referred to as fibril-2) (Gremer et al., 2017). Next, we investigated the binding affinity of berberine and Ber-D to the newly reported Aβ42 fibrils using molecular docking and implicit solvent model-based free energy calculations, which is referred to as the MM-GBSA approach and discussed in the Transparent Methods section. Although molecular docking-based study or calculations are unable to reproduce the relative binding affinities of berberine and Ber-D with fibril-2, the MM-GBSA-based binding free energies correctly reproduce the trend observed in our experimental results. The binding sites for ligands berberine and Ber-D within fibril-2 are shown in Figure 3D. The binding free energies of Ber-D in site-1∗ and site-2∗ (−28.8 and −27.3 kcal/mol, respectively) are greater (in terms of magnitude) than that of berberine (−17.5 and −14.2, respectively), and the increased dispersion energy is responsible for the high binding affinity of the former toward fibril-2 (Table S1). The other striking feature is that berberine binds to only two sites of the Aβ42 fibril, whereas Ber-D binds to five different sites with significant affinities. These data suggest that the binding profiles of berberine and Ber-D and the number of binding sites for these compounds vary depending on the polymorphic nature of the fibrils (whether it is C-type structure as in fibril-1 or S-type structure as in fibril-2). Independent of the specific polymorphic form of Aβ fibrils, Ber-D has a greater binding affinity compared with berberine.
Figure 3.

Prediction of Binding Sites
(A) Aβ monomer (1–42) and protofibril (17–42).
(B) The binding constants for berberine (Ber) and Ber-D as predicted from molecular docking program for monomer and protofibril (Aβ17-42).
(C and D) (C) Protofibril (17–42)-ligand interaction diagram and (D) binding sites predicted for protofibril (Aβ1-42). The binding sites and binding constants for Ber and Ber-D compounds were predicted using autodock software. It can be seen, multiple binding sites were predicted for both protofibril (17–42) (based on 2BEG) and protofibril (1–42) (based on 5OQV).
As discussed in the previous section, one of the key mechanisms of Aβ oligomers-mediated toxicity is through cell membrane disruption (Quist et al., 2005). It is worth mentioning that the NMR study suggested a virus fusion domain-like structure for Aβ42 monomer in a non-polar environment (Crescenzi et al., 2002). Many experimental and computational studies have reported Aβ peptide and oligomer-induced membrane disruption and its contribution toward cell toxicity. In this context, we studied the membrane permeability of berberine and Ber-D. In addition, we addressed how the membrane permeability of Aβ peptide will be modified when bound to Ber-D. The polar surface area (PSA) for the isolated molecular systems, namely, berberine, Ber-D, and Aβ monomer, as in 1Z0Q and for Aβ in complexation with Ber-D was calculated. PSA has been considered as a suitable descriptor for estimating the cell permeability of compounds, and a larger value of PSA is related to lower value for the permeability. In order to understand the concentration-dependent change in membrane permeability, PSA calculations were performed for Aβ monomer complexed with one and two molecules of Ber-D (Table S2). From the data in Table S2 (Transparent Methods), Ber-D appears to have a lower PSA compared with berberine and easily passes through the cell membrane. The calculated PSA for Aβ monomer and its complex with Ber-D suggests a larger PSA in the bound state, which reduces the probability of membrane permeability and thereby reduces the cell toxicity. Furthermore, the increasing concentration of Ber-D increases the PSA of its complex with Aβ monomer, which indicates a concentration-dependent reduction in cell toxicity by the complex (Table S2).
Metal-Independent and -Dependent Antioxidant Assay
We investigated the ability of Ber-D to quench ROS and reactive nitrogen species (RNS) generated independent of redox metal using 2,2-diphenyl-1-picrylhydrazyl (DPPH), nitric oxide (NO), 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS), and oxygen radical absorbance capacity (ORAC) assays (Moon and Shibamoto, 2009). The stable free radical DPPH predominantly absorbs at 517 nm and the presence of a compound with radical quenching ability decreases the absorbance intensity, which is correlated to antioxidant property (or antioxidant capacity [AC]) of the compound. In this assay, DPPH (50 μM) was added to the methanol solutions of Ber-D (50 μM) or berberine (50 μM) or Asc (50 μM, positive control) and the absorption intensity at 517 nm was recorded for all the samples after incubation for 30 min (Figure 4A). Interestingly, Ber-D exhibited an AC of 97%, which is comparable with that of natural antioxidant ascorbic acid (Asc). Moreover, Ber-D was found to quench free radicals very effectively at lower concentrations (51%, 62%, and 81% AC for 5, 10, and 25 μM of Ber-D, respectively). In contrast, berberine showed an AC of 32% at 50 μM, and this poor activity is attributed to the lack of phenolic hydroxyl groups to quench the free radicals. Similar to DPPH, ABTS also generates relatively stable nitrogen radical with characteristic absorption at 734 nm on reaction with potassium persulfate. The extent of reduction in the absorption at 734 nm in the presence of radical quenchers corresponds to their AC. Interestingly, in ABTS assay Ber-D showed an AC of ∼100%, whereas the AC of berberine was 30% (Figure S5A). Overall, DPPH and ABTS assay clearly demonstrated the effective radical quenching ability of Ber-D compared with the parent natural product.
Figure 4.

In Vitro and In Cellulo Antioxidant Assay
(A and B) Radical scavenging property of Ber-D and berberine analyzed through DPPH (A) and nitric oxide (B) assay and plotted as their percentage of antioxidant capacity (% of AC) or nitric oxide percentage.
(C) Determination of oxygen radical quenching capacity (ORAC) value for Ber-D and berberine through ORAC assay with Trolox as internal standard.
(D) Resorufin fluorescence intensity (585 nm) measured in solutions of Aβ42 (5.1), CuII (5 μM), Asc (150 μM) HRP, and Amplex Red (10 μM) in presence of Ber-D or berberine with time for 100 min with incubation at 37°C.
(E) Agarose gel electrophoresis of plasmid DNA (pBR322, 2 μg/mL) treated with 20 μM of Ber-D or berberine and CuII (5 μM) in the presence of ascorbate (150 μM) incubated at 37°C. sc-DNA, supercoiled DNA; c-DNA, circular DNA.
(F) BSA (1 mg/mL), CuII (0.1 mM) and H2O2 (2.5 mM), Ber-D or berberine (0.1 mM) are added, and the percentage of BSA oxidation was assessed and quantified by measuring DNPH absorption at 370 nm.
(G) Linoleic acid (10 mM), CuII (0.1 mM), Ber-D or berberine (0.1 mM), and ascorbate (2 mM) are added, and the percentage of lipid peroxidation was assessed and quantified by measuring TBARS formation through absorbance change at 532 nm.
(H) Quantifying ROS generation in PC12 cells incubated with Aβ42 (10 μM), CuII (10 μM), Ber-D or berberine (10 μM), and ascorbate (200 μm) by measuring by DCF fluorescence intensity at 529. Each experiment was repeated three times (n = 3, n = 6 for DCFDA assay) and error bars represent the standard deviation (SD). Ber, berberine.
See also Figures S3 and S5.
Elevated levels of NO are implicated in the progression of AD pathology, and quenching NO is one of the viable therapeutic strategies (Togo et al., 2004). We evaluated the ability of Ber-D to quench NO through a simple NO assay. In this assay, NO generated from sodium nitroprusside is detected using Griess reagent by monitoring the change in absorbance intensity at 546 nm. Sodium nitroprusside (5 mM) was added to the ethanol solutions of Ber-D (50 μM) or berberine (50 μM) and incubated for 100 min at 25°C. Then the Griess reagent was added and absorbance at 546 nm was measured to determine the percentage of NO in each sample. Interestingly, Ber-D showed 60% reduction in NO generation, whereas berberine showed absorbance similar to the untreated control (NO generated from sodium nitroprusside in ethanol solution is considered 100%) indicating its inability to quench NO (Figure 4B). In agreement with other assays, Ber-D exhibited a concentration-dependent activity in quenching NO (5 μM: 96%, 10 μM: 67%, and 25 μM: 63% reduction, respectively). Next, we estimated the oxygen radical quenching ability of Ber-D in ORAC assay (Ou et al., 2002). Trolox, a water-soluble analogue of natural antioxidant vitamin E, was used as an internal standard and reference to determine the ORAC for Ber-D and berberine (Figure 4C). The ORAC number of a given antioxidant compound is measured with respect to Trolox concentration at which a similar level of AC is observed, and the higher the ORAC value the better the ability of the compound to quench oxygen radicals. Ber-D and berberine showed ORAC values of 4.33 and 0.34, respectively, which confirm that Ber-D is an efficient quencher of oxygen radicals (Figure 4C).
Aβ42+CuII complex under reducing conditions generate ROS such as hydrogen peroxide (H2O2). The generation of H2O2 by Aβ42+CuII was measured through hydrogen peroxide assay. The antioxidant property of Ber-D was evaluated to assess its ability to prevent metal-dependent ROS generation from Aβ42+CuII complex. In this assay, horseradish peroxidase (HRP) was used in situ to generate H2O2 (from Aβ42-CuII complex) as substrate to produce hydroxyl radicals. The generated H2O2 transforms non-fluorescent Amplex Red into fluorescent resorufin dye (λex = 571 nm; λem = 585 nm). The measure of resorufin fluorescence is directly correlated to H2O2 generation. To a PBS solution of Aβ42 (5.1 μM), CuII (5 μM), HRP, Amplex Red (10 μM), and Ber-D or berberine (50 μM), Asc (150 μM) was added and the fluorescence emission recorded at 585 nm (λex = 571 nm) as a function of time over a period of 100 min (Figure 4D). The maximum fluorescence recorded for the control sample (Aβ42+CuII) was considered 100%. The Ber-D-treated sample exhibited 85% reduction in the H2O2 generation, whereas berberine showed only 35% reduction confirming the superior radical quenching ability of the former (Figure S5B). Notably, Ber-D effectively prevented the generation of H2O2 by 70% and 80% at concentrations of 10 and 50 μM, respectively. These results clearly demonstrate that Ber-D binds to the Aβ42+CuII complex and silences the redox cycling activity of CuII to prevent the generation of ROS.
Ber-D Rescues Biomacromolecules from ROS
The ROS react with essential biomacromolecules such as DNA, protein, and lipid causing structural and functional damage (Schieber and Chandel, 2014). DNA damage, protein oxidation, and lipid peroxidation are common noxious events associated with AD pathology. The damaged biomolecules trigger a series of adverse biological responses ultimately leading to cellular apoptosis. A simple experiment was performed to determine the ability of Ber-D to protect DNA from ROS-induced damage wherein plasmid DNA (pDNA) was used to mimic the cellular DNA under in vitro conditions. The pDNA exists in supercoiled (sc-DNA) and relaxed forms (c-DNA), which can be analyzed by agarose gel electrophoresis (Figure 4E). To the PBS solution of CuII (5 μM), pDNA (pBR322) and Ber-D (20 μM) or berberine (20 μM) and Asc (150 μM) were added and incubated for 10 min at 37°C, and the samples were analyzed by agarose gel electrophoresis (Figure 4E). In case of the positive control, the hydroxyl radicals generated by the CuII-Asc system create breaks in the sc-DNA and relieve its supercoiled structure, which can be visualized by the disappearance of the scDNA band and appearance of a new band corresponding to c-DNA. Evidently, Ber-D-treated sample showed protection of DNA from ROS-induced cleavage, which is revealed by the retention of scDNA bands in gel electrophoresis similar to untreated pDNA. On the other hand, berberine treatment failed to protect pDNA from ROS damage, as the gel electrophoresis data showed a new band corresponding to c-DNA. Overall, the multifunctional nature of Ber-D including redox-metal chelation, antioxidant property and quenching of ROS resulted in effective protection of DNA.
Next, we assessed the protein oxidation by ROS through a simple in vitro assay. The destructive action of ROS on the proteins generate many oxidized products such as protein carbonyls. The measurement of protein carbonyls offers good approximation of the extent of protein oxidation (Mesquita et al., 2014). It is well known that 2,4-dinitrophenylhydrazine (DNPH) reacts with carbonyl compounds to form hydrazones, which can be spectroscopically detected by monitoring the absorbance intensity at 370 nm. To evaluate the protective effect of Ber-D, the oxidation of BSA was used to mimic cellular protein and CuII+H2O2 serves as redox system for in situ generation of ROS. To a solution of BSA (1 mg/mL) and Ber-D or berberine (100 μM) in PBS buffer, CuII (0.1 mM) and H2O2 (2.5 mM) were added, and the samples were incubated for 24 h at 37°C in the dark, followed by the addition of DNPH (5 mM). After incubation of the samples for 10 min, absorption was recorded at 370 nm and the percentage of protein oxidation was estimated. The control sample (BSA and CuII+H2O2) showed maximum absorption at 370 nm, which is considered as 100% protein oxidation (Figure 4F). Treatment with berberine (100 μM) reduced the protein oxidation by 16%, whereas Ber-D (100 μM) showed a maximum reduction of 84%. Ber-D was found to reduce the protein oxidation in a concentration-dependent manner (25, 50, and 100 μM of Ber-D treatment showed reduced protein oxidation levels of 30%, 18%, and 16%, respectively). Next, lipid peroxidation assay was performed using linoleic acid as a model lipid and CuII-Asc system to generate ROS (Choi et al., 2002). Malondialdehyde (MDA) is one of the most common products formed during lipid peroxidation, and estimation of the concentration of MDA can be directly correlated to the extent of lipid peroxidation. MDA was measured using thiobarbituric acid (TBA), where TBA reacts with MDA to form thiobarbituric acid reactive substances (TBARS) that exhibit characteristic absorbance at 532 nm. To a solution of CuII (0.1 mM), linoleic acid (10 mM), and Ber-D (100 μM) or berberine (100 μM), Asc (2 mM) was added, and the samples were incubated for 30 min at 37°C. These samples were treated with TBA, and absorbance at 532 nm was measured (Figure 4G). The estimated lipid peroxidation in the presence of Ber-D and berberine was found to be 60% and 86%, respectively, compared with 100% peroxidation in the case of the control sample (linoleic acid treated with CuII and Asc). A difference of 26% in the peroxidation of linoleic acid between Ber-D and berberine treatment demonstrated the better protective effect of the former against lipid oxidation. These results from three different assays to assess the biomolecular damage and protection clearly suggest that Ber-D with phenolic hydroxyl groups significantly prevents DNA damage, protein oxidation, and lipid peroxidation by effective metal chelation and arresting the redox cycle or quenching the ROS.
In Cellulo Antioxidant Assays
The excellent in vitro antioxidant property of Ber-D motivated us to evaluate its in cellulo antioxidant efficacy. The oxidative stress was induced in PC12 cells by exposing to high concentrations of H2O2, and the protective nature of Ber-D was assessed by estimating the cell viability. The cells were initially treated with Ber-D (50 μM) or berberine (50 μM) followed by the addition of H2O2 (200 μM) (Figure S5C), incubated for 24 h at 37°C, and the cell viability was assessed through 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Ber-D showed 80% cell viability, whereas the cell viability of berberine (51%) was comparable with that of H2O2 treated cells (∼50%). Furthermore, Ber-D showed a concentration-dependent improvement in the cell viability (60%, 65%, and 80% for 10, 25, and 50 μM, respectively). These results suggest that Ber-D effectively quenches ROS generated in both in vitro and in cellulo conditions. Next, we studied the effect of Ber-D on Aβ42+CuII-induced ROS generation in PC12 cells by (2′,7′-dichlorodihydrofluorescein diacetate) DCFDA assay. The cells were treated with DCFDA and independently incubated with Aβ42+CuII (10 μM), Ber-D or berberine (10 μM), and Asc (200 μM) (Figure 4H). After incubation for 6 h at 37°C, fluorescence was measured at 529 nm (DCFDA) to estimate the levels of ROS. The data in Figure 4 show 25% ROS in the presence of Ber-D when compared with Aβ42+CuII-treated cells (100%). In contrast, ROS generation in cells treated with berberine was found to be unaltered and comparable with that of Aβ42+CuII-treated cells (∼100%). Interestingly, Ber-D was found to be effective at a concentration as low as 5 μM, which showed 37% reduction in the ROS levels, whereas berberine showed moderate effect in reducing the ROS levels only at high concentrations >50 μM, which is five times the concentration of Ber-D (5 μM) with comparable activity. These in cellulo results are in good agreement with the in vitro antioxidant assays and confirm the excellent antioxidant activity of Ber-D.
In Cellulo Aβ Toxicity
The efficient in cellulo antioxidant property of Ber-D encouraged us to evaluate its ability to modulate Aβ toxicity. We used PC12 cells as neuronal cell mimic to assess the efficacy of Ber-D against Aβ42 and Aβ42+CuII toxicity. The Aβ42 (10 μM)-treated cells were incubated independently with Ber-D (100 μM) or berberine (100 μM) (Figure 5A). Aβ42-treated cells showed ∼50% cell viability in comparison with untreated cells (Ctrl, 100%). Remarkably, Ber-D-treated cells with Aβ42 toxicity showed 25% improvement in viability when compared with cells treated with Aβ42 alone. In contrast, berberine failed to show any significant improvement in the cell viability, which is possibly attributed to its cytotoxic nature. Furthermore, Ber-D showed concentration-dependent improvement in cell viability (60%, 62%, and 65% corresponds to 10, 20, and 50 μM, respectively) against Aβ cellular toxicity. Subsequently, we evaluated the protective effect of Ber-D against Aβ42+CuII-induced toxicity in PC12 cells. The cells were treated with Aβ42+CuII complex (10 μM) and incubated independently with Ber-D (100 μM) or berberine (100 μM) (Figure 5B). Aβ42+CuII-treated cells showed 45% cell viability when compared with untreated cells (Ctrl, 100%). Ber-D improved the viability of cells under Aβ42+CuII toxicity by 30% in comparison with the parent natural product, which showed a mere 10% improvement. The substantial improvement in the viability of cells affected by the Aβ42 or Aβ42+CuII toxicity upon treatment with Ber-D is attributed to its effective CuII chelation and redox cycle arresting, antioxidant, and anti-aggregation properties. Put together all the results and Ber-D emerged as an effective multifunctional candidate to modulate multifaceted Aβ42 toxicity.
Ber-D Prevents Aβ-Induced Apoptosis
Mitochondrial dysfunction induced by Aβ is one of the key contributors to multifaceted toxicity in AD. The mitochondrial impairment causes membrane disruption, oxidative phosphorylation, and disturbed ATP production, subsequently leading to neuronal toxicity (Knott et al., 2008, Guo et al., 2013). The preliminary symptom of mitochondrial dysfunction is evident in the form of reduction in MMP followed by the generation of hazardous ROS (Castellani et al., 2002). Aβ interacts with and disrupts the mitochondrial membrane causing a decrease in MMP. We studied the protective effect of Ber-D on the mitochondrial damage caused by Aβ in PC12 cells by monitoring the MMP using membrane potential dye (Rho123) (Baracca et al., 2003). PC12 cells treated with Aβ42 (10 μM) were incubated alone or independently with Ber-D or berberine (20 μM) followed by addition of Rho123 and monitoring of the fluorescence at 534 nm (Figure 5C). The Aβ42-treated cells showed a low MMP of 40% compared with untreated cells (100%) (Figure S5D). Remarkably, Ber-D treatment improved the MMP of Aβ42-treated cells to 75%, which is 35% higher than the cells treated with Aβ42 alone. In contrast, berberine-treated cells showed an MMP (45%) comparable with that of Aβ42-treated cells. In essence, significant improvement in the MMP implies that Ber-D prevents the interaction of toxic Aβ42 with mitochondrial membrane and protects mitochondria and cells from Aβ toxicity.
The mitochondrial damage leads to activation of apoptosis, a process of programmed cell death, in response to irreversible cell damage (Mattson, 2000, Taylor et al., 2008). Apoptosis is highly regulated in normal healthy cells and plays a crucial role in maintaining cell population in tissue. Because of mitochondrial damage, cytochrome c (Cyt C) is released into the cytoplasm, which triggers or activates the production of protease enzymes such as caspase 3 (Cai et al., 1998). The protease enzymes break down the essential proteins required for various cellular functions. Moreover, it has been shown that elevated levels of cytochrome c (Cyt C) and caspases are expressed in Aβ42-treated cells (Bobba et al., 2010). The prevention of mitochondrial damage by Aβ42 prompted us to study the effect of Ber-D on the downstream process of apoptosis. We measured the levels of Cyt C and caspase 3 in PC12 treated with Aβ42 (10 μM) and 20 μM of Ber-D or berberine (Figures 5D and 5E). The Cyt C extraction and quantification by western blot revealed high expression levels in Aβ42-treated cells (considered as 100%, control), whereas Ber-D treatment lowered its levels to 55% and the effect of berberine was not significant (Figure 5D). Similarly, overexpressed caspase 3 level was reduced by 47% upon treatment with Ber-D in comparison with Aβ42-treated cells (100%), whereas berberine showed only 24% reduction. These results are in good agreement with MMP measurements and emphasize the fact that Ber-D effectively prevents Aβ-induced mitochondrial dysfunction and averts cellular apoptosis.
Discussion
In this current study, we have developed berberine derived multifunctional inhibitor of multifaceted Aβ toxicity implicated in AD pathology (Figure 6). The therapeutic potential of berberine against AD condition has been studied in the literature. However, the high cytotoxic nature of berberine attributed to its mitochondrial accumulation, dysfunctional ATP production, and unregulated mitochondrial division has hampered its further evaluation and practical utility. Berberine has been subjected to numerous modifications to improve its therapeutic potential (Huang et al., 2010a, Huang et al., 2016, Huang et al., 2010b, Shan et al., 2011, Tsai and Lee, 2010, Zou et al., 2017). We performed a simple synthetic chemical modification on berberine to generate free phenolic hydroxyl groups, which resulted in multifunctional Ber-D with metal chelation and antioxidant property. Moreover, the synthetic modification to generate Ber-D has greatly improved the solubility and cell viability in comparison with the parent natural alkaloid (berberine). The mitochondrial accumulation leading to cellular damage was minimal in the case of Ber-D-treated cell lines (Figure 6). The presence of ortho phenolic groups have hugely contributed to the efficient metal-chelating property of Ber-D. Ber-D showed good selectivity toward CuII with a characteristic change in absorption spectrum in the bound state. According to metal ion hypothesis, CuII chelates with Aβ peptide and transforms into a depot for continuous generation of ROS. Development of metal chelators as a therapeutic candidate has met with partial success and is currently pursued as an active area of research. The metal-chelating property was completely absent in case of berberine as it lacked metal-chelating sites. In contrast, Ber-D could effectively chelate to redox metal (Cu) indicating its multifunctional nature and superiority over berberine. Ber-D exhibits a binding constant of 2.23 μM (kd) toward CuII, which is less than the affinity of Aβ42 for CuII, and therefore, Ber-D cannot sequestrate CuII from Aβ42+ CuII complex. However, the characteristic absorption spectrum corresponding to Ber-D+CuII upon addition of Ber-D to Aβ42+CuII complex revealed co-binding and the formation of Aβ42+CuII+Ber-D complex. Molecular modeling studies using density functional theory showed that CuII binds to Ber-D through its phenolic hydroxyl groups and Aβ through two His14 units (from two Aβ monomers) to attain a tetrahedral-like coordination complex structure. Notably, CuII bound to Ber-D and Aβ ceased to generate ROS as it is possibly maintained in the redox dormant state, which is an important and desired attribute of Ber-D to prevent ROS generation by Aβ42+CuII complex. The four hydroxyl groups of Ber-D efficiently quenched both ROS and RNS. ROS or RNS in the cellular context has been implicated to cause DNA damage, protein oxidation, and lipid peroxidation, which cause numerous adverse biochemical cascade reactions leading to neuronal death. We mimicked these cellular scenarios through various in vitro experiments to study the antioxidant property and protective nature of Ber-D (Figure 4). The metal chelation property or radical-quenching capacity or both together effectively rescued the biomacromolecules from reactive species in case of Ber-D; however, berberine was mostly ineffective in exhibiting antioxidant nature. These in vitro results were further validated trough in cellulo antioxidant assays. Our design strategy of synthetically transforming berberine to Ber-D as antioxidant has effectively worked in both in vitro and in cellulo conditions.
Figure 6.

Schematic Representation Shows the Inhibition of Multifaceted Aβ Toxicity by Ber-D
For a Figure 360 author presentation of Figure 6, see https://doi.org/10.1016/j.isci.2020.101005.
Ber-D inhibits the formation of toxic Aβ fibrillar aggregates and oligomeric species (Figure 4). Molecular docking studies reveled that Ber-D has a higher binding affinity or larger number of binding sites toward Aβ aggregates in comparison with berberine (Figure 4D). This could be attributed to the presence of free phenolic hydroxyl groups in Ber-D, which facilitates a larger number of molecular interactions, and hence strong binding affinity and aggregation inhibition. The metal-mediated Aβ aggregation was also significantly reduced owing to the combined effect of CuII chelation and anti-aggregation property of Ber-D. Similarly, the Aβ or Aβ+CuII complex-induced toxicity was significantly reduced under in cellulo conditions in the presence of Ber-D (Figure 5). Aβ interaction with mitochondria is one of the major pathways involved in causing neuronal toxicity (Robert et al., 2015). Mitochondrial fission is the early indication for cellular apoptosis; however, mitochondrial fusion indicates the healthy cell (Taylor et al., 2008). Aβ induces mitochondrial fragmentation and decreases its MMP. Treatment of cells with Ber-D effectively rescues mitochondria by averting the interaction of Aβ aggregates with the mitochondrial membrane, which results in restoration of the MMP (Figure 5). Another major consequence of mitochondrial damage is release (from mitochondria) and expression of excess Cyt C, which in turn activates a series of protease enzymes such as caspases. The data in Figure 5 show that Ber-D significantly lowers the levels of Cyt-C and caspase 3 and rescues the cells from apoptosis. However, berberine could partially prevent the expression of Cyt-C and caspase 3. These attributes have confirmed that Ber-D is a potential multifunctional inhibitor to target various aspects of Aβ toxicity as shown in multiple in vitro and in cellulo studies (Figure 6). In the current study we restricted our efforts to prove the multifunctional role of Ber-D in modulating Aβ-mediated toxicity in in vitro and cellular models. We estimated the partition coefficient (P) value of Ber-D, a simple and reasonable method to assess the possible blood-brain barrier (BBB) crossing ability. The calculated positive logP value of Ber-D indicates probable BBB permeability (Figure S6). However, effective BBB permeability, bioavailability, modulation of cognition, and improving life span and memory in an AD in vivo model have to be studied to assess the therapeutic efficacy of Ber-D. In addition to Aβ, tau has been shown to play a key role in AD progression and pathology (Wang and Mandelkow, 2016). Many aggregation inhibitors studied in the literature for Aβ are also explored for their effect on tau aggregation, because the aggregation in both cases is predominantly driven by the hydrophobic and hydrogen-bonding interactions. Therefore, Ber-D could be a good candidate to explore the modulation of tau aggregation and microtubule binding interactions.
Conclusion
We have developed a berberine-derived multifunctional inhibitor (Ber-D) of multifaceted Aβ toxicity implicated in AD pathology. A simple synthetic chemical modification was performed on berberine to generate free phenolic hydroxyl groups, which imparted multifunctional capability with metal chelation and antioxidant property. Moreover, the synthetic modification to generate Ber-D has greatly improved the solubility and cell viability in comparison with the parent natural alkaloid (berberine). Ber-D showed good binding affinity and selectivity toward CuII (kd, 2.23 μM) and effectively prevents its redox cycling. Moreover, Ber-D binds to Aβ42+CuII complex and prevents the generation of ROS. Molecular modeling studies using density functional theory showed that Ber-D cooperatively binds to Cu in complexation with two His14 units from two Aβ monomers through its phenolic hydroxyl groups and forms a tetrahedral-like coordination complex structure. The four hydroxyl groups of Ber-D efficiently quench both ROS and RNS and prevent DNA damage, protein oxidation, and lipid peroxidation, which cause numerous adverse biochemical cascade reactions leading to neuronal death. These in vitro results were further validated trough in cellulo antioxidant assays. Ber-D inhibits the formation of toxic Aβ fibrillar aggregates and oligomeric species. The Aβ42 aggregates-mediated mitochondrial toxicity was effectively inhibited by Ber-D, and it protects mitochondria from dysfunction, one of the major causes of neuronal death. Our design strategy of synthetically transforming berberine to Ber-D, a multifunctional antioxidant and aggregation modulator, effectively ameliorates multiple Aβ toxicity in both in vitro and in cellulo conditions. These multifunctional attributes make Ber-D a promising candidate for developing effective therapeutics to treat multifaceted toxicity of AD.
Limitation of the Study
Our study focusses on developing a natural product-derived multifunctional inhibitor (Ber-D) for modulating multifaceted Aβ toxicity of AD pathology. Our experimental methods have predominantly relied on using in vitro or cellular models to study the effectiveness of Ber-D as a multifunctional inhibitor of multifaceted Aβ toxicity. The determined positive LogP value predicts probable BBB crossing ability of Ber-D and warrants in vivo evaluation. In vivo modulation of amyloid burden and resulting improvements in cognitive functions and memory need to be assessed to ascertain the therapeutic potential of Ber-D. Neuroinflammation is a key contributor to multifaceted toxicity of AD, which necessitates in vitro and in vivo evaluation of anti-inflammatory activity. Evaluating the effect of Ber-D in modulating tau aggregation and microtubule interaction could be a relevant study.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Prof. C.N.R. Rao FRS for his constant support, JNCASR, Swarna Jayanti Fellowship, the Department of Science and Technology (DST) (Grant: DST/SJF/CSA-02/2015-2016), Department of Biotechnology, (DBT/VN-HB-NC-SB/4515), Govt. of India, and Sheikh Saqr Laboratory (SSL), ICMS- JNCASR, and SRL project for financial support. S.S. thanks CSIR, New Delhi for a research fellowship.
Author Contributions
K.R. and T.G. designed the project. K.R. undertook the in vitro and in cellulo experiments. S.S. performed in vitro ascorbate and hydrogen peroxide assay. V.B. synthesized Ber-D. N.A.M performed computational studies. K.R. and T.G. wrote the manuscript and all the authors read and commented.
Declaration of Interests
The authors declare no competing interests.
Published: April 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101005.
Supplemental Information
References
- Ahmed T., Gilani A.-H., Abdollahi M., Daglia M., Nabavi S.F., Nabavi S.M. Berberine and neurodegeneration: a review of literature. Pharmacol. Rep. 2015;67:970–979. doi: 10.1016/j.pharep.2015.03.002. [DOI] [PubMed] [Google Scholar]
- Atrián-Blasco E., Santoro A., Pountney D.L., Meloni G., Hureau C., Faller P. Chemistry of mammalian metallothioneins and their interaction with amyloidogenic peptides and proteins. Chem. Soc. Rev. 2017;46:7683–7693. doi: 10.1039/c7cs00448f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atrián-Blasco E., Gonzalez P., Santoro A., Alies B., Faller P., Hureau C. Cu and Zn coordination to amyloid peptides: from fascinating chemistry to debated pathological relevance. Coord. Chem. Rev. 2018;371:38–55. doi: 10.1016/j.ccr.2018.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baracca A., Sgarbi G., Solaini G., Lenaz G. Rhodamine 123 as a probe of mitochondrial membrane potential: evaluation of proton flux through F0 during ATP synthesis. Biochim. Biophys. Acta. 2003;1606:137–146. doi: 10.1016/s0005-2728(03)00110-5. [DOI] [PubMed] [Google Scholar]
- Barnham K.J., Bush A.I. Biological metals and metal-targeting compounds in major neurodegenerative diseases. Chem. Soc. Rev. 2014;43:6727–6749. doi: 10.1039/c4cs00138a. [DOI] [PubMed] [Google Scholar]
- Bobba A., Petragallo V.A., Marra E., Atlante A. Alzheimer’s proteins, oxidative stress, and mitochondrial dysfunction interplay in a neuronal model of Alzheimer’s disease. Int. J. Alzheimers Dis. 2010;2010:621870. doi: 10.4061/2010/621870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourassa M.W., Miller L.M. Metal imaging in neurodegenerative diseases. Metallomics. 2012;4:721–738. doi: 10.1039/c2mt20052j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulawa C.E., Connelly S., DeVit M., Wang L., Weigel C., Fleming J.A., Packman J., Powers E.T., Wiseman R.L., Foss T.R. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. U S A. 2012;109:9629–9634. doi: 10.1073/pnas.1121005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero A.B., Terol-Ordaz L., Espargaró A., Vázquez G., Nicolás E., Sabaté R., Gamez P. Histidine-rich oligopeptides to lessen copper-mediated amyloid-β toxicity. Chem. Eur. J. 2016;22:7268–7280. doi: 10.1002/chem.201600286. [DOI] [PubMed] [Google Scholar]
- Cai J., Yang J., Jones D. Mitochondrial control of apoptosis: the role of cytochrome c. Biochim. Biophys. Acta Bioenerg. 1998;1366:139–149. doi: 10.1016/s0005-2728(98)00109-1. [DOI] [PubMed] [Google Scholar]
- Cai Z., Wang C., Yang W. Role of berberine in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2016;12:2509–2520. doi: 10.2147/NDT.S114846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani R., Hirai K., Aliev G., Drew K.L., Nunomura A., Takeda A., Cash A.D., Obrenovich M.E., Perry G., Smith M.A. Role of mitochondrial dysfunction in Alzheimer’s disease. J. Neurosci. Res. 2002;70:357–360. doi: 10.1002/jnr.10389. [DOI] [PubMed] [Google Scholar]
- Chiti F., Dobson C.M. Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu. Rev. Biochem. 2017;86:27–68. doi: 10.1146/annurev-biochem-061516-045115. [DOI] [PubMed] [Google Scholar]
- Choi C.W., Kim S.C., Hwang S.S., Choi B.K., Ahn H.J., Lee M.Y., Park S.H., Kim S.K. Antioxidant activity and free radical scavenging capacity between Korean medicinal plants and flavonoids by assay-guided comparison. Plant Sci. 2002;163:1161–1168. [Google Scholar]
- Crescenzi O., Tomaselli S., Guerrini R., Salvadori S., D’Ursi A.M., Temussi P.A., Picone D. Solution structure of the Alzheimer amyloid β-peptide (1–42) in an apolar microenvironment. Eur. J. Biochem. 2002;269:5642–5648. doi: 10.1046/j.1432-1033.2002.03271.x. [DOI] [PubMed] [Google Scholar]
- Crouch P.J., Savva M.S., Hung L.W., Donnelly P.S., Mot A.I., Parker S.J., Greenough M.A., Volitakis I., Adlard P.A., Cherny R.A. The Alzheimer’s therapeutic PBT2 promotes amyloid-β degradation and GSK3 phosphorylation via a metal chaperone activity. J. Neurochem. 2011;119:220–230. doi: 10.1111/j.1471-4159.2011.07402.x. [DOI] [PubMed] [Google Scholar]
- Doig A.J., del Castillo-Frias M.P., Berthoumieu O., Tarus B., Nasica-Labouze J., Sterpone F., Nguyen P.H., Hooper N.M., Faller P., Derreumaux P. Why is research on amyloid-β failing to give new drugs for Alzheimer’s disease? ACS Chem. Neurosci. 2017;8:1435–1437. doi: 10.1021/acschemneuro.7b00188. [DOI] [PubMed] [Google Scholar]
- Du W.-J., Guo J.-J., Gao M.-T., Hu S.-Q., Dong X.-Y., Han Y.-F., Liu F.-F., Jiang S., Sun Y. Brazilin inhibits amyloid β-protein fibrillogenesis, remodels amyloid fibrils and reduces amyloid cytotoxicity. Sci. Rep. 2015;5:7992. doi: 10.1038/srep07992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi A.K., Iyer P.K. Therapeutic strategies to prevent Alzheimer’s disease pathogenesis using a fluorescent conjugated polyelectrolyte. Macromol. Biosci. 2014;14:508–514. doi: 10.1002/mabi.201300107. [DOI] [PubMed] [Google Scholar]
- Ehrnhoefer D.E., Bieschke J., Boeddrich A., Herbst M., Masino L., Lurz R., Engemann S., Pastore A., Wanker E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008;15:558. doi: 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- Esmieu C., Guettas D., Conte-Daban A., Sabater L., Faller P., Hureau C. Copper-targeting approaches in Alzheimer’s disease: how to improve the fallouts obtained from in vitro studies. Inorg. Chem. 2019;58:13509–13527. doi: 10.1021/acs.inorgchem.9b00995. [DOI] [PubMed] [Google Scholar]
- Frydman-Marom A., Shaltiel-Karyo R., Moshe S., Gazit E. The generic amyloid formation inhibition effect of a designed small aromatic β-breaking peptide. Amyloid. 2011;18:119–127. doi: 10.3109/13506129.2011.582902. [DOI] [PubMed] [Google Scholar]
- Gremer L., Schölzel D., Schenk C., Reinartz E., Labahn J., Ravelli R.B.G., Tusche M., Lopez-Iglesias C., Hoyer W., Heise H. Fibril structure of amyloid-β(1-42) by cryo-electron microscopy. Science. 2017;358:116–119. doi: 10.1126/science.aao2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunderson W.A., Hernández-Guzmán J., Karr J.W., Sun L., Szalai V.A., Warncke K. Local structure and global patterning of Cu(2+) binding in fibrillar amyloid-β [Aβ(1-40)] protein. J. Am. Chem. Soc. 2012;134:18330–18337. doi: 10.1021/ja306946q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C., Sun L., Chen X., Zhang D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013;8:2003–2014. doi: 10.3969/j.issn.1673-5374.2013.21.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C., Selkoe D.J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007;8:101. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hamley I.W. The amyloid beta peptide: a chemist’s perspective. role in Alzheimer’s and fibrillization. Chem. Rev. 2012;112:5147–5192. doi: 10.1021/cr3000994. [DOI] [PubMed] [Google Scholar]
- Hatai J., Motiei L., Margulies D. Analyzing amyloid beta aggregates with a combinatorial fluorescent molecular sensor. J. Am. Chem. Soc. 2017;139:2136–2139. doi: 10.1021/jacs.6b10809. [DOI] [PubMed] [Google Scholar]
- House E., Mold M., Collingwood J., Baldwin A., Goodwin S., Exley C. Copper abolishes the beta-sheet secondary structure of preformed amyloid fibrils of amyloid-beta(42) J. Alzheimers Dis. 2009;18:811–817. doi: 10.3233/JAD-2009-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L., Luo Z., He F., Lu J., Li X. Synthesis and biological evaluation of a new series of berberine derivatives as dual inhibitors of acetylcholinesterase and butyrylcholinesterase. Bioorg. Med. Chem. 2010;18:4475–4484. doi: 10.1016/j.bmc.2010.04.063. [DOI] [PubMed] [Google Scholar]
- Huang L., Luo Z., He F., Shi A., Qin F., Li X. Berberine derivatives, with substituted amino groups linked at the 9-position, as inhibitors of acetylcholinesterase/butyrylcholinesterase. Bioorg. Med. Chem. Lett. 2010;20:6649–6652. doi: 10.1016/j.bmcl.2010.09.013. [DOI] [PubMed] [Google Scholar]
- Huang M.Y., Lin J., Huang Z.J., Xu H.G., Hong J., Sun P.H., Guo J.L., Chen W.M. Design, synthesis and anti-inflammatory effects of novel 9-O-substituted-berberine derivatives (2016) Medchemcomm. 2016;7:730–731. [Google Scholar]
- Huyut Z., Beydemir Ş., Gülçin İ. Antioxidant and antiradical properties of selected flavonoids and phenolic compounds. Biochem. Res. Int. 2017;2017:7616791. doi: 10.1155/2017/7616791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaffy J., Brinet D., Soulier J.-L., Correia I., Tonali N., Fera K.F., Iacone Y., Hoffmann A.R.F., Khemtémourian L., Crousse B. Designed glycopeptidomimetics disrupt protein–protein interactions mediating amyloid β-peptide aggregation and restore neuroblastoma cell viability. J. Med. Chem. 2016;59:2025–2040. doi: 10.1021/acs.jmedchem.5b01629. [DOI] [PubMed] [Google Scholar]
- Kawai R., Araki M., Yoshimura M., Kamiya N., Ono M., Saji H., Okuno Y. Core binding site of a thioflavin-T-derived imaging probe on amyloid β fibrils predicted by computational methods. ACS Chem. Neurosci. 2018;9:957–966. doi: 10.1021/acschemneuro.7b00389. [DOI] [PubMed] [Google Scholar]
- Knott A.B., Perkins G., Schwarzenbacher R., Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 2008;9:505. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles T.P.J., Vendruscolo M., Dobson C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014;15:384. doi: 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- Kumar S., Henning-Knechtel A., Chehade I., Magzoub M., Hamilton A.D. Foldamer-mediated structural rearrangement attenuates Aβ oligomerization and cytotoxicity. J. Am. Chem. Soc. 2017;139:17098–17108. doi: 10.1021/jacs.7b08259. [DOI] [PubMed] [Google Scholar]
- Kysenius K., Brunello C.A., Huttunen H.J. Mitochondria and NMDA receptor-dependent toxicity of berberine sensitizes neurons to glutamate and rotenone injury. PLoS One. 2014;9:e107129. doi: 10.1371/journal.pone.0107129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.J.C., Nam E., Lee H.J., Savelieff M.G., Lim M.H. Towards an understanding of amyloid-β oligomers: characterization, toxicity mechanisms, and inhibitors. Chem. Soc. Rev. 2017;46:310–323. doi: 10.1039/c6cs00731g. [DOI] [PubMed] [Google Scholar]
- Mahmoudi M., Zamani Taghizadeh Rabe S., Balali-Mood M., Karimi G., Memar B., Rahnama M., Tabasi N., Khazaee M., Riahi-Zanjani B. Immunotoxicity induced in mice by subacute exposure to berberine. J. Immunotoxicol. 2016;13:255–262. doi: 10.3109/1547691X.2015.1058306. [DOI] [PubMed] [Google Scholar]
- Matlack K.E.S., Tardiff D.F., Narayan P., Hamamichi S., Caldwell K.A., Caldwell G.A., Lindquist S. Clioquinol promotes the degradation of metal-dependent amyloid-β (Aβ) oligomers to restore endocytosis and ameliorate Aβ toxicity. Proc. Natl. Acad. Sci. U S A. 2014;111:4013–4018. doi: 10.1073/pnas.1402228111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000;1:120. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- Mesquita C.S., Oliveira R., Bento F., Geraldo D., Rodrigues J.V., Marcos J.C. Simplified 2,4-dinitrophenylhydrazine spectrophotometric assay for quantification of carbonyls in oxidized proteins. Anal. Biochem. 2014;458:69–71. doi: 10.1016/j.ab.2014.04.034. [DOI] [PubMed] [Google Scholar]
- Moon J.-K., Shibamoto T. Antioxidant assays for plant and food components. J. Agric. Food Chem. 2009;57:1655–1666. doi: 10.1021/jf803537k. [DOI] [PubMed] [Google Scholar]
- Murugan N.A., Halldin C., Nordberg A., Långström B., Ågren H. The culprit is in the cave: the core sites explain the binding profiles of amyloid-specific tracers. J. Phys. Chem. Lett. 2016;7:3313–3321. doi: 10.1021/acs.jpclett.6b01586. [DOI] [PubMed] [Google Scholar]
- Nasica-Labouze J., Nguyen P.H., Sterpone F., Berthoumieu O., Buchete N.-V., Coté S., De Simone A., Doig A.J., Faller P., Garcia A. Amyloid β protein and Alzheimer’s disease: when computer simulations complement experimental studies. Chem. Rev. 2015;115:3518–3563. doi: 10.1021/cr500638n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou B., Huang D., Hampsch-Woodill M., Flanagan J.A., Deemer E.K. Analysis of antioxidant activities of common vegetables employing oxygen radical absorbance capacity (ORAC) and ferric reducing antioxidant power (FRAP) assays: a comparative study. J. Agric. Food Chem. 2002;50:3122–3128. doi: 10.1021/jf0116606. [DOI] [PubMed] [Google Scholar]
- Prince M., Bryce R., Albanese E., Wimo A., Ribeiro W., Ferri C.P. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 2013;9:63–75.e2. doi: 10.1016/j.jalz.2012.11.007. [DOI] [PubMed] [Google Scholar]
- Quist A., Doudevski I., Lin H., Azimova R., Ng D., Frangione B., Kagan B., Ghiso J., Lal R. Amyloid ion channels: a common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. U S A. 2005;102:10427–10432. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekhar K., Govindaraju T. Current progress, challenges and future prospects of diagnostic and therapeutic interventions in Alzheimer’s disease. RSC Adv. 2018;8:23780–23804. doi: 10.1039/c8ra03620a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekhar K., Chakrabarti M., Govindaraju T. Function and toxicity of amyloid beta and recent therapeutic interventions targeting amyloid beta in Alzheimer’s disease. Chem. Commun. 2015;51:13434–13450. doi: 10.1039/c5cc05264e. [DOI] [PubMed] [Google Scholar]
- Rajasekhar K., Suresh S.N., Manjithaya R., Govindaraju T. Rationally Designed peptidomimetic modulators of Aβ toxicity in Alzheimer’s disease. Sci. Rep. 2015;5:8139. doi: 10.1038/srep08139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekhar K., Madhu C., Govindaraju T. Natural tripeptide-based inhibitor of multifaceted amyloid β toxicity. ACS Chem.Neurosci. 2016;7:1300–1310. doi: 10.1021/acschemneuro.6b00175. [DOI] [PubMed] [Google Scholar]
- Rajasekhar K., Narayanaswamy N., Murugan N.A., Viccaro K., Lee H.-G., Shah K., Govindaraju T. Aβ plaque-selective NIR fluorescence probe to differentiate Alzheimer’s disease from tauopathies. Biosens. Bioelectron. 2017;98:54–61. doi: 10.1016/j.bios.2017.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekhar K., Mehta K., Govindaraju T. Hybrid multifunctional modulators inhibit multifaceted Aβ toxicity and prevent mitochondrial damage. ACS Chem.Neurosci. 2018;9:1432–1440. doi: 10.1021/acschemneuro.8b00033. [DOI] [PubMed] [Google Scholar]
- Rauk A. The chemistry of Alzheimer’s disease. Chem. Soc. Rev. 2009;38:2698–2715. doi: 10.1039/b807980n. [DOI] [PubMed] [Google Scholar]
- Robert A., Liu Y., Nguyen M., Meunier B. Regulation of copper and iron homeostasis by metal chelators: a possible chemotherapy for Alzheimer’s disease. Acc. Chem. Res. 2015;48:1332–1339. doi: 10.1021/acs.accounts.5b00119. [DOI] [PubMed] [Google Scholar]
- Samanta S., Rajasekhar K., Babagond V., Govindaraju T. Small molecule inhibits metal-dependent and -independent multifaceted toxicity of Alzheimer’s disease. ACS Chem. Neurosci. 2019;8:3611–3621. doi: 10.1021/acschemneuro.9b00216. [DOI] [PubMed] [Google Scholar]
- Savelieff M.G., Lee S., Liu Y., Lim M.H. Untangling amyloid-β, tau, and metals in Alzheimer’s disease. ACS Chem. Biol. 2013;8:856–865. doi: 10.1021/cb400080f. [DOI] [PubMed] [Google Scholar]
- Schieber M., Chandel N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014;24:R453–R462. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D.J., Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan W.J., Huang L., Zhou Q., Meng F.C., Li X.S. Synthesis, biological evaluation of 9-N-substituted berberine derivatives as multi-functional agents of antioxidant, inhibitors of acetylcholinesterase, butyrylcholinesterase and amyloid-beta aggregation. Eur. J. Med. Chem. 2011;46:5885–5893. doi: 10.1016/j.ejmech.2011.09.051. [DOI] [PubMed] [Google Scholar]
- Shin B., Saxena S. Substantial contribution of the two imidazole rings of the His13−His14 Dyad to Cu(II) binding in amyloid-β(1−16) at physiological pH and its significance. J. Phys. Chem. A. 2011;115:9590–9602. doi: 10.1021/jp200379m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N., Sharma B. Toxicological effects of berberine and sanguinarine. Front. Mol. Biosci. 2018;5:21. doi: 10.3389/fmolb.2018.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D.G., Cappai R., Barnham K.J. The redox chemistry of the Alzheimer’s disease amyloid β peptide. Biochim. Biophys. Acta Biomembr. 2007;1768:1976–1990. doi: 10.1016/j.bbamem.2007.02.002. [DOI] [PubMed] [Google Scholar]
- Taylor R.C., Cullen S.P., Martin S.J. Apoptosis: controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008;9:231. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- Togo T., Katsuse O., Iseki E. Nitric oxide pathways in Alzheimer’s disease and other neurodegenerative dementias. Neurol. Res. 2004;26:563–566. doi: 10.1179/016164104225016236. [DOI] [PubMed] [Google Scholar]
- Tsai S.F., Lee S.S. Characterization of acetylcholinesterase inhibitory constituents from annona glabra assisted by HPLC microfractionation. J. Nat. Prod. 2010;73:1632–1635. doi: 10.1021/np100247r. [DOI] [PubMed] [Google Scholar]
- Wang Y., Mandelkow E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016;17:5–21. doi: 10.1038/nrn.2015.1. [DOI] [PubMed] [Google Scholar]
- Wang Q., Yu X., Patal K., Hu R., Chuang S., Zhang G., Zheng J. Tanshinones inhibit amyloid aggregation by amyloid-β peptide, disaggregate amyloid fibrils, and protect cultured cells. ACS Chem. Neurosci. 2013;4:1004–1015. doi: 10.1021/cn400051e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Gu B.J., Masters C.L., Wang Y.-J. A systemic view of Alzheimer disease-insights from amyloid-β metabolism beyond the brain. Nat. Rev. Neurol. 2017;13:612. doi: 10.1038/nrneurol.2017.111. [DOI] [PubMed] [Google Scholar]
- Yan X.-J., Yu X., Wang X.-P., Jiang J.-F., Yuan Z.-Y., Lu X., Lei F., Xing D.-M. Mitochondria play an important role in the cell proliferation suppressing activity of berberine. Sci. Rep. 2017;7:41712. doi: 10.1038/srep41712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J., Ye J., Jia W. Effects and mechanisms of berberine in diabetes treatment. Acta Pharmacol. Sin. B. 2012;2:327–334. [Google Scholar]
- Zou K., Li Z., Zhang Y., Zhang H., Li B., Zhu W., Shi J., Jia Q., Li Y. Advances in the study of berberine and its derivatives: a focus on anti-inflammatory and anti-tumor effects in the digestive system. Acta Pharmacol. Sin. 2017;38:157–167. doi: 10.1038/aps.2016.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.