

Summary
Background
The cause of severe acute respiratory syndrome (SARS) has been identified as a new coronavirus. Whole genome sequence analysis of various isolates might provide an indication of potential strain differences of this new virus. Moreover, mutation analysis will help to develop effective vaccines.
Methods
We sequenced the entire SARS viral genome of cultured isolates from the index case (SIN2500) presenting in Singapore, from three primary contacts (SIN2774, SIN2748, and SIN2677), and one secondary contact (SIN2679). These sequences were compared with the isolates from Canada (TOR2), Hong Kong (CUHK-W1 and HKU39849), Hanoi (URBANI), Guangzhou (GZ01), and Beijing (BJ01, BJ02, BJ03, BJ04).
Findings
We identified 129 sequence variations among the 14 isolates, with 16 recurrent variant sequences. Common variant sequences at four loci define two distinct genotypes of the SARS virus. One genotype was linked with infections originating in Hotel M in Hong Kong, the second contained isolates from Hong Kong, Guangzhou, and Beijing with no association with Hotel M (p<0.0001). Moreover, other common sequence variants further distinguished the geographical origins of the isolates, especially between Singapore and Beijing.
Interpretation
Despite the recent onset of the SARS epidemic, genetic signatures are emerging that partition the worldwide SARS viral isolates into groups on the basis of contact source history and geography. These signatures can be used to trace sources of infection. In addition, a common variant associated with a non-conservative aminoacid change in the S1 region of the spike protein, suggests that immunological pressures might be starting to influence the evolution of the SARS virus in human populations.
Published online May 9, 2003 http://image.thelancet.com/extras/03art4454web.pdf
Introduction
The first cases of severe acute respiratory syndrome (SARS) were identified in November, 2002, in Guangdong Province, China. By April, 2003, the epidemic had spread worldwide, affecting 3547 individuals resulting in 182 deaths.1 In March, 2003, the putative cause of SARS was identified as a new coronavirus.2, 3 Oropharyngeal specimens from patients with SARS induced a cytopathic effect on Vero E6 tissue culture cells and revealed the presence of coronavirus-like particles. Reverse transcriptase-PCR analysis with random or broadly-specific coronavirus primers amplified a DNA fragment that resembled, but was distinct from, known coronavirus genomes. When tested, these diagnostic PCR methods detected the SARS virus in many clinical samples taken from affected patients. In addition, serological evidence has shown the presence of antibodies specific to the new coronavirus in the serum of patients with SARS.4 Collectively, these data strongly implicate the new coronavirus as the cause of SARS, which we designate as SARS-CoV.
SARS-CoV is a member of the Coronoviridae family of enveloped, positive-stranded rna viruses, which have a broad host range. Some coronavirus infections in people, cattle, and birds cause respiratory disease, whereas other coronavirus infections in rodents, cats, pigs, and cattle lead to enteric disease. The 27–32 kb genomes of coronaviruses, the largest of RNA viruses, encode 23 putative proteins, including four major structural proteins; nucleocapsid (N), spike (S), membrane (M), and small envelope (E). The spike protein, a glycoprotein projection on the viral surface, is crucial for viral attachment and entry into the host cell. In addition, variations of S protein among strains of coronavirus are responsible for host range and tissue tropism.5 Differences in virulence of mice coronaviruses have also been linked to genetic variance in the S protein.6, 7 and the serological response in the host is typically raised against the S protein.8 However, the S, M, and N mature proteins all contribute to generating the host immune response as seen in transmissible gastroenteritis coronavirus,9 infectious bronchitis virus,10, 11 pig respiratory coronavirus,12 and mouse hepatitis virus.13
A characteristic of RNA viruses is the high rate of genetic mutation, which leads to evolution of new viral strains and is a mechanism by which viruses escape host defenses. Therefore, from a public-health perspective, understanding the mutation rate of the SARS virus as it spreads through the population is important. Moreover, the genetic mutability of SARS-CoV, especially in the segments encoding the major antigenic proteins, would also have an effect on development of broadly effective vaccines.
We aimed to determine the complete genome sequence for five SARS-CoV related isolates from a single SARS index case, three associated primary contact cases, and one secondary contact and compare them with nine other SARS-CoV isolates available in public-domain databases.
Methods
Patients
We obtained five positive isolates for coronavirus from the index patient of the outbreak (SIN2500), three primary contacts of this patient (SIN2774, SIN2748, SIN2677), and one secondary contact (SIN2679) who was related to the index patient, but contracted SARS from another primary contact not included in this study. All patients fitted the WHO case definition for probable SARS14– fever of 38°C or higher, respiratory symptoms (eg, cough, shortness of breath, difficulty in breathing), hypoxia and chest radiograph changes suggestive of pneumonia, and history of close contact with another patient with SARS or travel to a region with documented community transmission of SARS within 10 days of onset of symptoms. The index patient had a history of travel to Hong Kong and had stayed in hotel M.15 The viral sources were all from respiratory samples: two endotracheal tube aspirates, two nasopharyngeal aspirates, and one throat swab obtained from the patients between 0 and 11 days after of onset of symptoms.
Procedures
The virus-containing samples were inoculated into various cell lines including Vero cells, which showed a cytopathic effect characterised by generalised rounding against a granular background seen 5–11 days after inoculation. We maintained the cells at 33°C and repassaged them after 7 days of incubation.
We spotted washed cell pellets from harvested cells showing cytopathic effects onto glass slides and overlaid them with acute and convalescent serum samples from the patients from whom we obtained the respiratory samples. All five cell lines showed reactivity by immunofluorescence (methods available from author) with convalescent serum samples from the respective patients. We confirmed the presence of the SARS coronavirus by PCR on the cell supernatants using SARS specific primers.24 When tested, two of the infected Vero cell lines also had coronavirus-like particles on electron microscopy. We isolated the viral RNA template from the supernatants of Vero cells that showed cytopathic effects by centrifugation at 23 000 relative centrifugal force for 2·5 h to pellet the viral particles and extracted RNA with the QiAmp viral RNA mini kit (Qiagen, Valencia, CA, USA).
To sequence the viral genome we used both shot-gun and specific priming approaches. From the RNA templates, we synthesised double strand cDNA with the SuperScript cDNA system (Invitrogen, Carlsbad, CA, USA). The cDNA were PCR amplified (20 cycles) with random 16-mer by Platinum Taq polymerase. The amplicons were then cloned into pCR2.1-TOPO vector (Invitrogen). We selected random clones for single pass sequencing analysis on ABI3730 sequencers (Applied Biosystems, Foster City, CA, USA). We compared sequence data generated from the library with human, mouse, and viral genome databases managed at the US National Center for Biotechnology Information (NCBI) site (www.ncbi.nlm.nih.gov). Since previous sequences from reverse transcriptase-PCR of SARS patient samples are most closely matched to the mouse hepatitis virus at protein level, we used the mouse hepatitis virus genome sequence (NC_001846) as a backbone to align the viral cDNA sequence fragments for positions on the viral genome. For sequence positioning we also used limited unpublished SARS viral sequences (EMC1 to EMC8) posted April 5, 2003 on a secure website at WHO.16 We designed sequence-specific primers on the basis of cDNA sequence data to fill the gaps of 1–2 kb distance. After we completed the first rough genome sequence for the sample SIN2500, we designed 30 primer pairs incorporating sequence information from the newly available TOR2 SARS virus17 to cover the whole viral genome with each primer pair amplifying about 1200 base pairs. We then used this sequence-specific priming approach to generate reverse transcriptase-PCR fragments for the five whole viral genomes. Each PCR fragment was directly sequenced from both directions inward and outward, in duplicate. Therefore, for any region of the genome there was six to eight-fold coverage. We used the phred/phrap/consed package (University of Washington, Seattle, WA, USA; www.phred.org) to process all the raw sequence reads for base calling, assembly, and editing. Nucleotide differences in the assembled genome sequences were also checked manually for accuracy. Sequence regions that were poor quality were resequenced either from purified PCR fragments or cloned plasmid. All genetic variations of Singapore isolates identified when compared with available SARS-CoV genome sequences were further confirmed by primer extension genotyping technology (Sequenom, San Diego, CA, USA). The panel shows the SARS-CoV isolates we accessed from GenBank. We determined the locations of point mutations by aligning the 14 SARS sequences with clustalw.18 We calculated putative coding sequences by completing the multialignment of the 14 SARS sequences with the TOR2 nucleotide sequence annotation as the reference. The annotations of the proteins are taken from the corresponding entries in the NCBI Entrez site.
Panel.
| Genome sequences | Accession number |
|---|---|
| SARS-CoV isolates | |
| Complete | |
| SIN2500 | AY283794 |
| SIN2677 | AY283795 |
| SIN2679 | AY283796 |
| SIN2748 | AY283797 |
| SIN2774 | AY283798 |
| TOR2 | NC_004718 |
| URBANI | AY278741 |
| CUHKU-W1 | AY278554 |
| HKU-39849 | AY278491 |
| Partial | |
| GZ01 | AY278489 |
| BJ01 | AY278488 |
| BJ02 | AY278487 |
| BJ03 | AY278490 |
| BJ04 | AY279354 |
| Coronavirus isolates | |
| MHV strain 2 | AF201929.1 |
| MHV Penn 97-1 | AF208066 |
| MHV | NC_001846 |
| MHV strain ML-10 | AF208067 |
| Bovine CoV | NC_003045.1 |
| BCoV Quebac strain | AF220295.1 |
| AIBV | NC_001451.1 |
| TGV | NC_002306.2 |
| TGV | Z34093.1 |
| HCoV 229E | NC_002645.1 |
| PEDV strain C | AF35311.1 |
| PEDV | NC_003436.1 |
| Rat SDAV | AF207551.1 |
| Porcine HEV | AY078417.1 |
Associations between the members of the coronaviridae family to the SARS virus were assessed by comparing overlapping fragments of the SIN2500 genomic sequence against a database of coronavirus sequences. To calculate regional homologies with sister coronaviruses we used sequences within sliding 200 base pair windows sampled in a tiled fashion every 100 base pairs across the viral genomes using the blast BLASTN program from WU-BLAST (Washington University, St Louis, MO, USA).19 In the heat map that was generated, the SARS genomic fragments are plotted along the horizontal axis in the order they appear in the genome, and the corresponding fragments from other coronaviruses are placed vertically. The brightness of a pixel corresponds to the strength of the match between a SARS fragment and a coronavirus genome; the smaller the p value, the brighter the pixel. At p=1 it is black, and the brightness is proportional to log (1÷p) until p is less than 10−11, when it is white. The panel shows the accession numbers of the coronavirus sequences used.
Statistical analysis
In the analysis of the common sequence variations, the probability of co-occurrence of multiple polymorphisms or mutations in an isolate was used as a measure of significance. We used 13 of the samples (we excluded sample BJ04 because of substantial missing sequence information) and restricted our attention to 26 140 loci at which nucleotides were determined in all 13 samples. The null hypothesis was that the nucleotides at these loci were obtained by mutating a single consensus sequence independently at random at each position of each sequence. The mutation rate was estimated from the data by the fraction of positions in the various genomes that differed from the consensus sequence obtained by taking the most frequent nucleotide at each position. We also tested a weaker null hypothesis, in which the mutations taking place at any given locus in different samples are independent, but arbitrary dependence between the loci is possible subject to this constraint. Details of the analytical approach are described in webappendix 1 (http://image.thelancet.com/extras/03art4454webappendix1.pdf) and on our website (http://www.gis.astar.edu.sg/homepage/toolssup.jsp).
Phylogenetic analysis of the SARS viral genomes was done with paup * 20 with the maximum probability criterion. We used the default variable settings, with one exception: the substitution rates were estimated from the data. A separate phylogenetic analysis done with CLUSTALW18 gave the same structure.
Role of the funding source
The sponsor of the study had no role in study design, data collection, data analysis, data interpretation, or in the writing of the report.
Results
We have sequenced the complete genomes of SARS-CoV from five Singapore isolates derived from one index case (SIN2500), three primary cases (SIN2677, SIN2748, and SIN2774), and one secondary case (SIN2679). These sequences showed that the genomes of SARS-CoV isolated in Singapore are comprised of 29 711 bases, with the exception of a five-nucleotide deletion in strain SIN2748 and a six-nucleotide deletion in SIN2677. Initial BLAST analysis suggested that the Singapore SARS virus is similar to, but distinct from, the group 2 coronaviruses in the Coronaviridae family of enveloped and positive-stranded RNA viruses. As in the recently sequenced SARS-CoV (HKU39849, CUHK-W1, TOR2, and URBANI), the Singapore SARS virus contains 11 predicted open reading frames that encode 23 putative mature proteins with known and unknown functions. Most of the non-structural proteins seem to be encoded in the first half of the genome including nsp 1 and nsp2, with putative proteinase function and nsp9 RNA-dependent RNA polymerase, whereas most of the structural proteins such as spike, membrane, envelop, and nucleocapsid are located in the second half of the genome (figure 1 , webfigure: http://image.thelancet.com/extras/03art4454webfigure.pdf). The haemagglutinin esterase, which is common in the group 2 coronavirus, is missing in the SARS-CoV genome, suggesting that some of the non-structural genes are dispensable in coronaviruses.
Figure 1.

Genome structure of SARS-CoV
NSP=Non-structural proteins. S=spike protein. E=small envelop protein. N=nucleocapsid. M=Membrane protein. MHV=murine hepatitis virus. MD=metal binding. 3CL-PRO=3C-like proteinase. The top scale shows the approximate nucleotide position along SARS-CoV genome (golden bar) determined from the Singapore isolate SIN2500. The arrows on top of the genome bar map the locations where nucleotide sequence variations, present in two or more isolates, were detected. The two open triangles point to the locations where the two multi-nucleotide deletions occurred. The SARS genome is predicted to encode 23 putative mature proteins (blue bars). The arrows on top of the protein bars indicate the location of aminoacid changes.
Although the genome organisation of SARS-CoV is similar to that of other coronaviruses, SARS-CoV is only distantly related to any coronavirus member, irrespective of species specificity, at both nucleotide and aminoacid levels (figure 2 ). In assessing the homology with coronaviridae genomes from other species with the SARS-CoV, sequence conservation seems to be restricted to the middle part of the genome, between bases 14 000 and 21 000, where the RNA-dependent polymerase and several uncharacterised proteins (eg, orf1ab:nsp 10–13) are located. The remainder of the genome, especially at the 5’and 3’regions, diverges from other strains at the nucleotide and aminoacid level.
Figure 2.

Homology of SARS-CoV genome sequence (SIN2500) to other coronaviruses
A heat map created by comparing overlapping fragments of the SARS-CoV genome sequence against a database of coronavirus sequences. The SARS fragments are plotted along the horizontal axis in the order they appear in the genome, and the other coronaviruses are plotted vertically. The brightness of a pixel corresponds to the strength of the match between a SARS fragment and a coronavirus genome; the smaller the p value, the lighter the pixel.
We aligned the five complete genome sequences of Singapore SARS-CoV isolates and the nine SARS-CoV genomes from outside of Singapore, which were sequenced by others, and investigated the genetic variations between these 14 genomes (webappendix 1). In total, there were 127 single nucleotide sequence variations, one deletion of six nucleotides (nt 27782–27787) in strain SIN2677, and one deletion of five nucleotides (nt 27810–27814) in strain SIN2748 (webappendix 2, http://image.thelancet.com/extras/03art4454webappendix2.pdf). Both these deletion sites were in the noncoding sequences between an uncharacterised open reading frame and the nucleocapsid protein. Of the 127 base substitutions, 94 changed the aminoacid sequence (webappendix 2). The mutations were in the following open reading frames: orf1 a polyprotein, orf1a RNA-polymerase, orf1ab: nsp 10 to nsp 13, spike glycoprotein, membrane nucleocapsid, and several uncharacterised putative proteins.
To eliminate mutational noise induced in culture from real strain differences, we reanalysed the data using a probabilistic approach. Mutations that might have been artifacts of cell culture would occur only once in our survey, whereas sequence variants associated with common ancestry should be seen in multiple isolates. Of the 127 sequence variations in the 14 isolates, 16 variant loci were identified in two or more isolates, and eight were seen in three or more isolates (figure 3 ). With the more stringent criterion, four loci recurred five or more times in the 14 SARS-CoVs analysed: C/T polymorphisms at position 9404 resulting in a valine to alanine change in orf1 a (nsp 1); position 19 084 leading to an isoleucine to threonine change in orf1ab (nsp 11); position 22 222 changing an isoleucine residue to threonine in the S 1 portion of the spike protein, and position 27 827 which is in a non-coding region (figure 3). In addition, a T/G polymorphism is noted at position 17 564 changing orf1ab (nsp 10, helicase domain) from an aspartic acid to a glutamic acid. Sequence variants at these four loci segregate together as a specific genotype. For example, isolates CUHKW1, GZDI, BJ01, BJ02, BJ03, BJ04 all have the configuration C:G:C:C at those nucleotide positions, whereas isolates SIN2500, SIN2774, SIN2679, SIN2677, TOR2, URBANI, HKU39849 have the configuration T:T:T:T (figure 3). Assuming that all base substitutions were random events propagated in the Vero cells, the probability of four specific nucleotide changes occurring concurrently is very low. The significance is at p<10−60 when the null hypothesis is that each locus in each sample mutates independently, and p<1015 when dependence is allowed among the loci. The C:G:C:C and T:T:T:T genotypes are, therefore, very unlikely to have emerged by chance, and might be evidence for the first genetic signature of strain differences in the SARS virus. All isolates with the T:T:T:T genotype were linked to infection acquired at the Hotel M in Hong Kong,18 whereas none of the C:G:C:C genotype isolates had this association (figure 3). Phylogenetic analysis based on the common variant sequences (defined as present in two or more isolates) confirmed that the cases associated with exposure in the Hotel M formed a cluster that was distinct from the other isolates (figure 4 ). On the basis of the molecular and contact history, we have reconstructed a probable lineage map of the SARS-CoV infections investigated here (figure 5 ).
Figure 3.

Sequence comparisons of the 14 available SARS-CoV genomes
Only those variant sequences (red) that were present in at least two independent sequences are shown. See webappendix2 for complete list of all variant nucleotides. The frequency of appearance of each variant nucleotide is presented. Highlighted in yellow are four sequence positions that define two distinct genotypic variants of SARS-CoV. The position of each nucleotide is based upon the URBANI SARS-CoV sequence and the corresponding encoded protein or uncharacterised open reading frames (ORF) are indicated. The effect, if any, on the encoded aminoacid (AA) is described. Nucleotides that are missing (-) or ambiguous (N) in the genome sequences are indicated and the length of each genome sequence is given. The patients’countries of origin and association of patient with Hotel M are provided for all viral genome sequences, and the relative order of transmission is provided for the Singapore cases.
Figure 4.
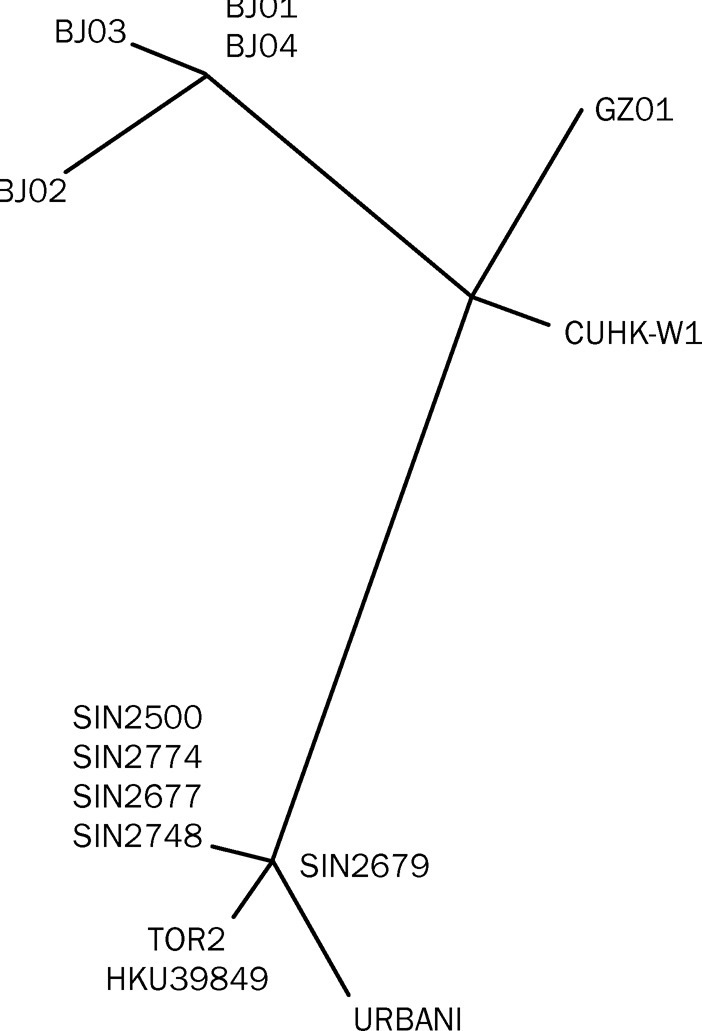
Molecular relations between the 14 SARS-CoV Isolates
Phylogenetic trees obtained by applying PAUP* to complete genome sequences of all 14 SARS-CoV samples. The tree was built with only sequence variants that occurred at least twice.
Figure 5.

Clinical relations between the 14 SARS-CoV isolates
The routes of transmissions are shown for the 14 viral isolates that have been sequenced, indicated with solid boxes. Solid arrows=routes of transmission from Hotel M are known be direct. Broken arrow=direct relation information is not available. Patient A is the Hong Kong index patient who travelled from Guangdong to Hong Kong and transmitted SARS to others at Hotel M, who then travelled to Singapore, Canada, and Vietnam, thus becoming index cases in those countries.15 The routes of transmission from Guangdong are unknown and shown as dotted arrows. *Dashed boxes are routes of transmission that are uncertain.
In addition to this four-locus genotype, four other common variant sequences (all occurring three to four times in the 14 isolates) seem to further define subgroups geographically (figure 3). The variant sequences at position 19 084 seem to distinguish the Singaporean isolates from all others. Although all nine isolates from outside Singapore showed a C at this position, four of the five isolates from Singapore had a T. The only reversion from T to a C in SIN2679 (a secondary contact case) might be the result of a backmutational event potentially occurring during the passage of the virus. Taking into account missing data, the polymorphisms at nucleotide positions 9854, 19 838, and 27 243 all segregate with isolates identified specifically in Beijing. Thus, these common sequence polymorphisms might be useful in identifying the differential source of a SARS viral infection.
Discussion
Although the genome organisation of SARS-CoV is similar to that of other coronaviruses, the SARS-CoV sequence is only distantly related to any coronavirus member. Results of earlier reports4 suggested that SARS CoV more closely resembled the cow coronavirus and the mouse hepatitis virus by comparing a conserved 215 aminoacid segment of the polymerase protein product. However, when taking into account the entire genomic sequence, the strength of the associations was reduced, confirming the reports of others that the SARS coronavirus is a completely new pathogenic strain that does not arise from a simple recombination of known existing strains.2, 21
Since the S1 subunit of the spike protein is the major antigenic moiety for coronaviruses and is not an essential structural protein, it is prone to high mutation rates as the virus evolves in host populations. That the S1 region did not seem to have excessive numbers of base substitutions suggests that the viral isolates have not been subject to immunological selection.22, 23 However, because all samples were from viral cultures propagated in Vero cells, some, if not most, of these 129 mutations might have occurred during in-vitro expansion and not because of host pressures.24 In addition, some of the available nucleotide substitutions might have been the result of sequencing errors since several of the sequences were submitted in draft form. To reduce the effects of these technical artifacts, we restricted our analysis to the 16 loci with recurrent mutations among the 14 isolates. These loci are the sequence variants most likely to have been resident in human populations.
Of special interest are the nucleotide changes in four of these loci (positions 9404, 19 084, 22 222, and 27 827) that recurred five or more times. The base substitutions at these locations are highly restricted and segregate together as specific genotypes (C:G:C:C vs T:T:T:T). Thus, it is highly unlikely that the C:G:C:C and T:T:T:T genotypes emerged by chance. Rather, we believe this to be evidence for the first genetic signature of strain differences in the SARS virus. All isolates with the T:T:T:T genotype were linked to infection acquired at the Hotel M in Hong Kong,15 whereas none of the C:G:C:C genotype isolates had this association.
The index case from Singapore (from which the Singaporean infections described herein were derived) acquired the SARS-CoV infection while staying at Hotel M. The TOR2 virus, cultured in Canada, and the HKU39849 isolate from Hong Kong, were from patients who became infected through contact at Hotel M, although perhaps not directly. The URBANI SARS CoV isolate was from a physician infected by a patient who contracted SARS while staying at Hotel M. Isolates CUHKW1, GZD1, BJ01, BJ02. BJ03, BJ04, however, came from patients with no known linkage with Hotel M and, on the whole, were derived later than the Hotel M linked set. Our results showed that the cases associated with exposure in the Hotel M formed a cluster that was distinct from the other isolates.
In addition to this four locus genotype, the variant sequences at position 19 084 distinguished the Singaporean isolates from all others. There also seems to be a signature for the North China isolates at positions 9854, 19838, and 27,243. Thus, the common sequence polymorphisms might be useful in identifying the differential source of a SARS viral infection.
Whether any of these common polymorphisms will result in biological and clinical differences remains to be determined. However, the common mutation in position 22 222 changing an isoleucine residue to threonine in the important antigenic region of the spike protein might be relevant. Mutations in this region of the SARS-CoV genome can arise because of selective pressure from host immune responses. That an isoleucine is present in all Hotel M linked isolates whereas a threonine at the same position in the major antigenic protein is found in all other geographically distinct isolates suggests that such non-conservative aminoacid changes have occurred to evade immunological pressures.
The SARS viral epidemic has placed a substantial strain on the health and economic status of nations. Understanding the nature of this virus and deriving methods to control the epidemic are very important. Our results show several molecular facets of the SARS coronavirus pertinent to public-health management of this epidemic. Its novelty as a human pathogen suggests that most populations might be immunologically naive to its infection. The discovery of genotypes linked to geographic and temporal clusters of infectious contacts suggests that molecular signatures can be used to refine contact histories.
Acknowledgments
Acknowledgments
We thank Prasanna Kolatkar, Marie Wong, Liu Jianjun, and Joshy George for their help in this project. The authors are members of the GIS and are therefore funded by the Agency for Science Technology and Research of Singapore.
Contributors
A E Ling organised and directed the in-vitro investigations of the biology of the SARS virus. K P Chan provided viral cultures, and L L. Oon and S Y SeThoe did the molecular diagnostic analysis and extraction of the viral nucleic acids. N M Lee did the electron microscopy of the initial viral samples. Y S Leo was the chief infectious disease clinician caring for the patients. Sequence determination and analysis was done by Y Ruan, C Wei, H Thoreau, P Ng, K Chiu, L Lim, T Zhang, E E C Ren, L Stanton, P Long, and E T Liu.
Conflict of interest statement
None declared.
GLOSSARY
- blast
The basic local alignment search tool is a system for searching similar sequences against all available sequence databases irrespective of whether the query is DNA or protein sequences. The BLAST programs consist of blastn, blastp, tblast, tblastn, tblastx, PSI-BLAST, MEGABLAST, and so on. The most improved point of this software is its high searching speed
- clustalw
CLUSTALW is a multiple sequence alignment tool that is commonly used in the bioinformatics community. It produces global multiple sequence alignments through three major phases: pairwise alignment, guide tree construction, and multiple alignment. The guide tree generated by CLUSTALW is an estimate, of relations between sequences much like a phylogenetic tree
- paup*
The Phylogenetic Analysis Using Parsimony (PAUP*) is a well established package for phylogenetic tree construction. It uses various methods, including parsimony, maximum likelihood, and distance methods to estimate phylogenetic relations.
- phred/phrap/consed
Bioinformatics tools to assemble shotgun sequences, which are available from University of Washington, Phred=base=calling program; Phrap=assembly program for shotgun sequences; Consed=UNIX-based graphic editor for Phrap sequence assemblies
- positive-stranded rna viruses
Some viruses, such as coronaviruses, carry their genetic material as RNA rather then the more typical DNA-based genomes. Positive-stranded RNA (also called plus-stranded) indicates that the single stranded RNA genome is of the same sense as coding messenger RNA
References
- 1.WHO Cumulative number of reported probable cases of severe acute respiratory syndrome (SARS) www.who.int/csr/sarscountry/2003_04_24/en/ (accessed April 19, 2003).
- 2.Drosten C, Gunther S, Preiser W. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003 doi: 10.1056/NEJMoa030747. (published online April 10, 10.1056/NEJMoa030781). [DOI] [PubMed] [Google Scholar]
- 3.Ksiazek TG, Erdman D, Goldsmith CS. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003 doi: 10.1056/NEJMoa030781. (published online April 10, DOI: 10.1056/NEJMoa030747). [DOI] [PubMed] [Google Scholar]
- 4.Peiris JSM, Lai ST, Poon LLM. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuo L, Godeke GJ, Raamsman MJ, Masters PS, Rottier PJ. Retargeting of coronavirus by substitution of the spike glycoprotein ectodomain: crossing the host cell species barrier. J Virol. 2000;74:1393–1406. doi: 10.1128/jvi.74.3.1393-1406.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanchez CM, Izeta A, Sanchez-Morgado JM. Targeted recombination demonstrates that the spike gene of transmissible gastroenteritis coronavirus is a determinant of its enteric tropism and virulence. J Virol. 1999;73:7607–7618. doi: 10.1128/jvi.73.9.7607-7618.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phillips JJ, Chua MM, Lavi E, Weiss SR. Pathogenesis of chimeric MHV4/MHV-A59 recombinant viruses: the murine coronavirus spike protein is a major determinant of neurovirulence. J Virol. 1999;73:7752–7760. doi: 10.1128/jvi.73.9.7752-7760.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergmann CC, Yao Q, Lin M, Stohlman SA. The JHM strain of mouse hepatitis virus induces a spike protein-specific Db-restricted cytotoxic T cell response. J Gen Virol. 1996;77:315–325. doi: 10.1099/0022-1317-77-2-315. [DOI] [PubMed] [Google Scholar]
- 9.Gomez N, Carrillo C, Salinas J, Parra F, Borca MV, Escribano JM. Expression of immunogenic glycoprotein S polypeptides from transmissible gastroenteritis coronavirus in transgenic plants. Virology. 1998;249:352–358. doi: 10.1006/viro.1998.9315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackwood MW, Hilt DA. Production and immunogenicity of multiple antigenic peptide (MAP) constructs derived from the S1 glycoprotein of infectious bronchitis virus (IBV) Adv Exp Med Biol. 1995;308:213–219. doi: 10.1007/978-1-4615-1899-0_35. [DOI] [PubMed] [Google Scholar]
- 11.Ndifuna A, Waters AK, Zhou M, Collison EW. Recombinant nucleocapsid protein is potentially an inexpensive, effective serodiagnostic reagent for IBV. J Virol Methods. 1998;70:37–44. doi: 10.1016/S0166-0934(97)00170-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Callenbaut P, Enjuanes L, Pensaert M. An adenovirus recombinant expression the spike glycoprotein of porcine respiratory coronavirus is immunogenic in swine. J Gem Virol. 1998;77:309–313. doi: 10.1099/0022-1317-77-2-309. [DOI] [PubMed] [Google Scholar]
- 13.Homberger FR. Nucleotide sequence comparison of the membrane protein genes of three enterotropic strains of mouse hepatitis virus. Virus Res. 1994;31:49–56. doi: 10.1016/0168-1702(94)90070-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.WHO Severe acute respiratory syndrome (SARS) Wkly Epidemiol Rec. 2003;78:81–88. http://www.who.int/wer/pdf/2003/wer7812.pdf (accessed April 27, 2003). [PubMed] [Google Scholar]
- 15.US CDC, WHO Update: Outbreak of severe acute respiratory syndrome, worldwide. MMWR Morb Mortal Wkly Rep. 2003;52:269–272. ( www.cdc.gov/mmwr/preview/mmwrhtml/mm5212a1.htm accessed March 28, 2003). [PubMed] [Google Scholar]
- 16.www.who.int/cdsdiagnostics (accessed April 5, 2003).
- 17.Michael Smith Genome Science Centre. www.bcgsc.bc.ca (accessed April 13, 2003).
- 18.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL-W—Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gish W. WU-BLAST (1996–2003) 2002. http://blast.wustl.edu (accessed April 25, 2003) [Google Scholar]
- 20.Swofford DL. PAUP. Phylogenetic analysis using parsimony (and other methods). Version 4. Sinauer Associates; Sunderland, Massachusetts: 2003. [Google Scholar]
- 21.Marra MA, Jones SJM, Astell CR, et al. The genome sequence of the SARS-associated coronavirus. Published online May 1, 2003: http://www.sciencemag.org/cgi/rapidpdf/1085953vl.pdf (accessed May 7, 2003).
- 22.Rowe CL, Fleming JO, Nathan MJ, Sgro JY, Palmenberg AC, Baker SC. Generation of coronavirus spike deletion variants by highfrequency recombination at regions of predicted RNA secondary structure. J Virol. 1997;71:6183–6190. doi: 10.1128/jvi.71.8.6183-6190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee CW, Jackwood MW. Origin and evolution of Georgia 98 (GA98), a new serotype of avian infectious bronchitis virus. Virus Res. 2001;80:33–39. doi: 10.1016/s0168-1702(01)00345-8. [DOI] [PubMed] [Google Scholar]
- 24.Bush RM, Smith CB, Cox NJ, Fitch WM. Effects of passage history and sampling bias on phylogenetic reconstruction of human influenza A evolution. Proc Natl Acad Sci USA. 2000;97:6974–6980. doi: 10.1073/pnas.97.13.6974. [DOI] [PMC free article] [PubMed] [Google Scholar]