

Résumé
La recherche antivirale est une discipline encore récente et le nombre de molécules disponibles pour lutter contre les infections virales demeure insuffisant. Pourtant, tant les pathologies causées par des virus endémiques ou émergents que l’existence de résistance de certains virus aux antiviraux rendent indispensables une recherche constante de nouvelles molécules antivirales. Parallèlement à la mise au point des molécules traditionnelles tels que les analogues nucléosidiques, dont l’efficacité n’est plus à démontrer, l’industrie pharmaceutique se tourne aujourd’hui vers de nouvelles solutions antivirales telles que les peptides, qui constituent un nouveau champ d’exploration pour la thérapie. Les progrès importants et récents de disciplines comme la génomique, la protéomique ou la biologie structurale ont permis d’améliorer notre connaissance fondamentale du monde viral. Ces progrès peuvent être mis à profit pour créer de façon rationnelle de nouveaux médicaments plus sélectifs et plus efficaces. L’identification et la mise au point de ces molécules nécessitent l’utilisation de techniques récentes comme le criblage à haut-débit de banques de composés combinatoires où l’utilisation de nouveaux outils bioinformatiques. Cette revue a pour but de présenter certaines méthodes récentes de mise au point de molécules antivirales.
Keywords: Antiviraux, double hybride, phage display, drug design, ARN interférence
Summary
Antiviral research is a recent discipline and the number of molecules available to fight against viral infections remains still insufficient. However, both diseases caused by emerging endemic viruses and the existence of resistance from some viruses against antiviral make necessary a constant search for new antiviral drugs. Parallel to the development of traditional molecules such as nucleoside analogues, whose effectiveness is well demonstrated, pharmaceutical industry is now turning to new solutions such as antiviral peptides, which constitute a new exploration field in therapy. The recent progress in disciplines such as genomics, proteomics and structural biology have improved our fundamental understanding of the viral world. These advances can be used to efficiently create new drugs more selective and more effective. Identification and development of these molecules require the use of newer techniques such as high-throughput screening of combinatorial compound libraries and the use of new bioinformatics tools. This review aims to present some recent methods for the development of antiviral molecules.
Keywords: Antiviral, two hybrid, phage display, drug design, RNA interference
1. Introduction
1.1. 50 ans de recherche antivirale
Les maladies infectieuses causées par des agents viraux représentent un important problème de Santé publique dans le monde entier. L’une des premières molécules antivirales mise à disposition, la 5’ – iodo-2’ – desoxyuridine (IduviranR), a été découverte en 1959 [1], il s’agissait d’un analogue nucléosidique de la thymidine utilisé contre les infections herpétiques [2]. D’autres molécules ont suivi et lors des premières années, la recherche antivirale s’est surtout concentrée sur les analogues de nucléosides [2, 3]. Dans les débuts de la lutte antivirale, la recherche de molécule se faisait souvent sans connaissances précises de la cible et sans recherche d’une action antivirale spécifique. Pour certaines d’entre elles, le mécanisme d’action demeure toujours incompris ou controversé à l’heure actuelle (ribavirine) [3].
De nos jours, la recherche de composés antiviraux a pris un nouvel essor et les scientifiques tentent de mettre au point des molécules de façon plus rationnelle. Ces molécules doivent cibler des étapes clés du cycle viral, qu’il faut avoir décryptées au préalable grâce à une recherche amont sur les relations structure-fonction des protéines virales ainsi que sur l’étude de leurs éventuels partenaires cellulaires. Les virus ayant une réplication intracellulaire, ces molécules doivent pouvoir pénétrer dans la cellule avant d’y inhiber spécifiquement le cycle viral [4]. Elles doivent d’autre part répondre aux exigences de n’importe quelle substance thérapeutique (propriétés ADME : absorption, distribution, métabolisme, excrétion) : un faible poids moléculaire, une bonne solubilité, une élimination rapide de l’organisme, peu d’effets secondaires, une production facile et peu coûteuse et une administration aisée [5]. La découverte de l’acicloviracyclovir en 1977 (anti-herpétique) et de sa très grande spécificité en est un bon exemple [6]. Les pathologies d’étiologie virale touchant très durement les pays en voie de développement, il est essentiel que ces molécules puissent être produites et commercialisées aux coûts les plus faibles possibles [7, 8]. La recherche d’un large spectre antiviral est donc également une caractéristique très prisée par les concepteurs d’agents antiviraux [9]. Enfin, considérant d’une part la faible part d’essais cliniques aboutissant in fine à la mise sur le marché d’une molécule thérapeutique, et d’autre part la flexibilité génétique des virus qui leur permet d’ « échapper » à la pression antivirale, il est nécessaire de disposer de technologies permettant de sélectionner constamment de nouveaux candidats et de pouvoir évaluer rapidement leur éventuel potentiel antiviral [10]. Ces critères sont autant de conditions que les nouvelles techniques de mises au point d’antiviraux doivent prendre en compte.
En théorie, toutes les étapes de l’infection sont potentiellement attractives et peuvent être ciblées lors de la mise au point d’agents antiviraux : la pénétration, la libération des génomes viraux dans la cellule, la transcription/réplication, la traduction, l’assemblage ou la libération des virus néoformés [11]. Les molécules ciblant les étapes internes du cycle viral sont confrontées à une difficulté supplémentaire : pouvoir pénétrer dans la cellule pour y exercer leur pouvoir inhibiteur. Certaines protéines cellulaires essentielles au cycle viral peuvent aussi constituer des cibles potentielles. Pour obtenir le spectre d’action le plus large possible, les traitements doivent cibler en priorité les étapes génériques du cycle [12].
1.2. Les peptides antiviraux, une nouvelle voie prometteuse
L’arsenal antiviral disponible concerne surtout des virus à fort potentiel économique tels que les virus des hépatites B (VHB) et C (VHC), de l’immunodéficience humaine (VIH), de l’herpès (VHS) ou de la grippe. Jusqu’à aujourd’hui, les sociétés pharmaceutiques se sont plutôt concentrées sur les inhibiteurs d’activités enzymatiques virales ou sur des molécules interférant avec les récepteurs, donc avec l’entrée virale. Déstabiliser les interactions protéine-protéine entre protéines virales ou protéines virales et cellulaires constitue un concept plus récent qui offre de nombreuses cibles prometteuses [13, 14]. Pour interférer avec ces liaisons, les peptides sont d’excellents outils qui, comparés aux molécules utilisées traditionellement, présentent des caractéristiques thérapeutiques trés intéressantes [5]. Ils sont ainsi plus spécifiques et plus efficaces car mimant la plus petite partie fonctionnelle possible d’une protéine, moins toxiques puisque leurs produits de dégradation sont des acides aminés, et enfin, ils s’accumulent peu dans les tissus en raison de leur grande biodégradabilité par les peptidases de l’organisme [5]. Les inconvénients potentiels sont leur faible capacité à traverser les barrières membranaires, leur courte demi-vie, des modes d’actions souvent complexes, un coût de production élevé, et enfin une mauvaise assimilation par voie orale qui contraint souvent à administrer les peptides thérapeutiques par voie intraveineuse [5, 15]. Des modifications sont cependant possibles pour pallier ces inconvénients [5] : ainsi l’utilisation de D-peptides résistants aux protéases (contrairement aux L-peptides) augmente substantiellement leur demi-vie dans le sérum et les petits D-peptides peuvent être absorbés efficacement par voie orale [14].
Certains peptides peuvent être issus d’organismes vivants (d’origine végétale ou animale), la biodiversité offrant une grande quantité de substances naturelles utilisable comme médicaments ou précurseurs [3, 8]. La littérature regorge d’études sur les capacités thérapeutiques de telles molécules. Parmi eux, les dermaseptines (sécrétées par la peau de certaines grenouilles) sont des peptides amphiphiles de 28 à 34 acides aminés (aa) de longueur, présentant une activité antimicrobienne à large spectre contre les levures, les bactéries, les champignons, des parasites intracellulaires et les virus enveloppés comme le VIH ou le virus de l’herpès simplex de type 1 (VHS-1) [16], [17], [18].
Les peptides thérapeutiques peuvent également être d’origine synthétique comme le T20 (enfurvirtide), un peptide dérivé de la séquence de la protéine gp41 du VIH-1 et utilisé pour combattre l’infection par ce virus en empêchant son entrée dans la cellule [19].
L’objet de cette revue est de présenter certaines méthodes récentes utilisées pour isoler des peptides à activité antivirale ou les sélectionner par des méthodes de criblages à haut débit.
2. Les nouvelles approches antivirales
Le choix de cibles d’intérêt et communes à différents virus dans la perspective d’obtenir des antiviraux potentiellement à large spectre est la première étape d’une stratégie antivirale. Par exemple, le complexe ribonucléoprotéique de transcription/réplication (RNP), dont la structure est similaire chez les virus à ARN négatif (ARN -) non segmenté (familles des Rhabdoviridae, des Paramyxoviridae et des Filoviridae) ou segmenté (familles des Orthomyxoviridae et des Bunyaviridae) et qui fonctionne de manière typique et autonome par rapport aux polymérases cellulaires, constitue une cible de choix. Ceci d’autant plus que ce type de polymérase virale n’a pas d’homologue chez l’homme, ce qui laisse espérer de faibles effets secondaires pour ces traitements potentiels.
Une stratégie attractive pour inhiber les complexes RNP consiste à dissocier ou à inhiber leur formation par des peptides interférant avec les interactions protéines-protéines qui cimentent structurellement et fonctionnellement le complexe [20, 21]. Dans cette optique, on peut procéder selon deux grands types d’approches :
-
1.
une approche combinatoire basée sur le criblage à haut débit de banques de molécules plus ou moins aléatoires ;
-
2.
une approche plus cognitive basée sur la connaissance de la structure des composants viraux et des interactions qu’ils établissent au sein du complexe.
2.1. Les approches combinatoires
Les approches de « drug discovery » font appel à des techniques permettant de cribler à haut débit (HTS : « high throughput screening ») des banques de composés biologiques naturels ou générés par chimie combinatoire (acides nucléiques, peptides, protéines) de très grande diversité de façon à isoler rapidement des candidats antiviraux [22]. Ces approches sont particulièrement utiles lorsqu’un manque de connaissances concernant la protéine cible rend inapplicables des techniques plus rationnelles [13].
2.1.1. Le criblage par double-hybride
Découverte en 1989, par Fields et ses collaborateurs [23], la technique de double hybride permet de détecter une interaction physique entre deux protéines dans le noyau de la levure Saccharomyces cerevisiae. C’est aussi un bon outil pour l’identification de « ligands » susceptibles d’interférer avec les interactions protéine-protéine au sein des complexes réplicatifs viraux.
Cette technique repose sur le fait que les facteurs de transcription eucaryotes sont des protéines possédant deux domaines distincts : le domaine de liaison à l’ADN (BD pour « binding domain ») et le domaine d’activation de la transcription (AD pour « activating domain »). BD et AD restent actifs pour initier la transcription même si ils sont exprimés séparément mais rapprochés l’un de l’autre autour de la région promotrice ( figure 1 ). Le système double hybride tire avantage de cette propriété en séparant physiquement AD et BD et en fusionnant chacun d’entre eux à une des deux protéines dont on veut rechercher l’interaction. La force de cette interaction est évaluée en co-transfectant les vecteurs exprimant AD-protéine X (appât) et BD-protéine Y (proie) dans une levure contenant un gène rapporteur (comme lacZ) sous le contrôle d’un promoteur (comme Gal4) régulé par le facteur de transcription AD-BD. AD-protéine X va alors se lier au promoteur. Si les protéines X et Y ont de l’affinité l’une pour l’autre BD-protéine Y va alors entrer en contact avec AD-protéine X et se trouver en position d’activer la transcription du gène rapporteur.
Figure 1.
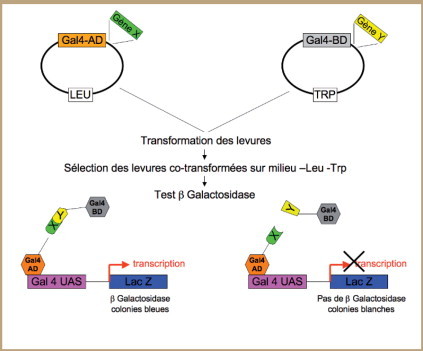
Principe du double hybride.
Il existe plusieurs gènes rapporteurs possibles, comme des marqueurs d’auxotrophie qui permettent la croissance des levures sur un milieu sélectif (par exemple le gène His3 qui permet aux levures de se développer dans un milieu sélectif dépourvu d’histidine) ou bien des marqueurs colorimétriques (par exemple le gène de la ß-galactosidase qui colore les levures en bleu en présence de X-gal). Ainsi l’intensité de l’expression du marqueur utilisé dépend de la force de l’interaction éventuelle entre l’appât et la proie.
Le double hybride est utilisable pour cribler les protéines codées par une banque d’ADNc pour leur affinité vis-à-vis d’une cible protéique donnée, et permet ainsi de détecter à haut débit des partenaires (intéracteurs) potentiels. Ces banques d’ADNc peuvent coder des protéines partenaires cellulaires ou de plus petites molécules (peptides). Dans le second cas, ceci permet l’isolement de ligands qui pourront servir à déstabiliser les complexes protéiques viraux et donc notamment, à inhiber la réplication virale [21, 24]. Le double hybride offre des avantages importants pour le criblage de telles banques : (1) il s’agit d’une méthode puissante, relativement simple à mettre en place et peu onéreuse ; (2) les levures assurent des modifications post traductionnelles proches de celles des autres cellules eucaryotes ; (3) enfin, le plasmide codant le peptide d’intérêt est facilement récupérable à partir de la colonie de levure et il est alors facile d’en déterminer la séquence. Cette technique présente cependant aussi des inconvénients : (1) les nombreux faux positifs générés par le système, ce qui oblige à confirmer chaque interaction trouvée ; (2) le fait que les interactions AD-BD doivent se dérouler dans le noyau de S. cerevisae ; (3) ce qui induit une autre limite, la grande difficulté à exprimer et donc à tester les protéines/peptides interagissant avec les protéines virales membranaires.
Les banques de peptides testées ont à la fois une meilleure affinité et une meilleure chance de préserver leur structure (donc leur effet) une fois déconnectés des domaines AD ou BD, lorsqu’ils présentent un certain état de contrainte intrinsèque que lorsqu’ils sont linéaires. De courts peptides autocontraints naturels, actifs pharmacologiquement, ont servi de modèles pour concevoir certaines banques. En particulier le squelette des conotoxines, architecturé autour de résidus « cystéine » (cys) régulièrement disposés, ou le squelette « proline » (pro) des lebocines, ont été utilisées pour construire des banques de peptides aléatoires autocontraints. Celles-ci ont notamment été criblées contre la phosphoprotéine P du virus de la rage, un cofacteur indispensable du complexe de transcription/réplication [25]. Cette approche a permis d’isoler des peptides montrant une forte affinité pour la P, dont 4 (3 « cys » et 1 « pro ») montrent une forte activité antivirale in vitro spécifique au virus rabique. Une alternative consiste à utiliser des banques d’aptamères peptidiques [26]. Il s’agit de molécules s’inspirant des immunoglobulines, constituées par une boucle peptidique de séquence aléatoire dont la structure est contrainte par une plateforme protéique (thioredoxine A). Le criblage de ces banques combinatoires par la technique du double hybride a permis d’isoler des peptides inhibant des interactions fonctionnelles [27] et possédant notamment une activité antivirale contre le VHB, le virus de la mosaïque dorée de la tomate (TGMV), le virus de la vaccine ou bien encore induisant l’apoptose de cellules infectées par le papillomavirus humain de type 16 (HPV16) ( tableau I ).
Tableau I.
Exemple de mise au point de molécules inhibant l’infection par différents virus (la nomenclature de l’ICTV figure entre parenthèses) grâce aux méthodes combinatoires de double hybride et de phage display ou grâce aux méthodes « cognitives » de drug design et d’ARN interférence (liste non exhaustive).
| Technique | Virus | Référence |
|---|---|---|
| Double hybride |
Rage (RABV) |
Réal et al. J Virol 2004; 78:7410-7 |
| Hépatite B (HBV) |
Butz et al. Oncogene 2001;20:6579-86 |
|
| Mosaïque dorée de la tomate (TGMV) |
Lopez-Ochoa et al. J Virol 2006;80:5841-53 |
|
| Vaccine (VACV) |
Saccucci et al. Antiviral Res 2009;82:134-40 |
|
| Papillomavirus humain de type 16 (HPV16) |
Butz et al. Proc Natl Acad Sci USA 2000;97:6693-7 | |
| Nauenburg et al. FASEB J 2001;15:592-4 | ||
| Immunodéficience humaine (HIV) |
Tavassoli et al. ACS Chem Biol 2008;3:757-64 |
|
| Phage display |
Bronchite infectieuse aviaire (IBV) |
Peng et al. Sci China C Life Sci 2006, 49:158-163. |
| Rotavirus humain (RV) |
Yao et al. Sheng Wu Gong Cheng Xue Bao 200;23:403-8 |
|
| Syndrome des points blancs de la crevette (WSSV) |
Yi et al. J Gen Virol 2003;84:2545-53 |
|
| Maladie de Newcastle (NDV) |
Chia et al. Peptides 2006; 27:1217-25 | |
| Ramanujam et al. Arch Virol 2002;,147:981-93 | ||
| Ozawa et al. J Vet Med Sci 2005;67:1237-41 | ||
| Maladie hémorragique de la carpe (GCHV) |
Wang et al. Virus Res 2000;67:119-25 |
|
| West Nile (WNV) |
Bai et al. J Virol 2007;81:2047-55 |
|
| Hépatite B (HBV) |
Deng et al. J Virol 2007;81:4244-54. | |
| Dyson et al. Proc Natl Acad Sci USA 1995;92:2194-8 | ||
| Papillomavirus humain de type 16 (HPV16) |
Fujii et al. Clin Cancer Res 2003;9:5423-8 |
|
| Hépatite C (HCV) |
Kim et al. J Microbiol Biotechnol 2008;18:328-33 |
|
| Immunodéficience humaine 1 (HIV-1) |
Desjobert et al. Biochemistry 2004;43:13097-105 | |
| Sticht et al. Nat Struct Mol Biol 2005;12:671-7 | ||
| Stangler et al. Biol Chem 2007;388:611-5 | ||
| Sin nombre (SNV), Hantaan (HTNV), Prospect hill (PHV) |
Hall et al. Antimicrob Agents Chemother 2008;52:2079-88 | |
| Larson et al. J Virol 2005;79:7319-26 | ||
| Stomatite vésiculeuse (VSV) |
Zhang et al. Acta Pharmacol Sin 2008;29:634640 |
|
| Influenza A H9N2 |
Rajik et al. Virol J 2009;6:74 |
|
| Drug design |
Immunodéficience humaine 1 (HIV-1) |
Schramm et al. Biochem Biophys Res Commun 1993;194:595-600 |
| Rao et al. J Biomol Struct Dyn 2002;20:31-8 | ||
| Coronavirus du syndrome respiratoire aigu sévère (SARS CoV) |
Du et al. Med Chem 2005;1:209-13 |
|
| Hépatite C (HCV) |
Venkatraman et al. J Med Chem 2005;48:5088-91 |
|
| Interférence Arn | Immunodéficience humaine 1 (HIV-1) |
Rossi et al. Biotechniques 2006;Suppl:25-9 |
| Virus respiratoire syncitial (RSV) |
Bitko et al. Nat Med 2005;11:50-5 | |
| Barik et al. Virus Res 2004;102:27-35 | ||
| West Nile (WNV) |
Ong et al. Antiviral Res 2006;72:216-23 |
|
| Hépatite B (HBV) |
Yang et al. Antiviral Res 2007;73:24-30 |
|
| Hépatite C (HCV) |
Trejo-Avila et al. Ann Hepatol 2007;6:14-80 |
|
| Entérovirus 71 (EV) |
Wu et al. J Virol Methods 2009;159:233-8 |
|
| Influenza A |
Ge et al. Proc Natl Acad Sci USA 2004;101:8676-81 |
|
| Coronavirus du syndrome respiratoire aigu sévère (SARS cov) |
Zhang et al. Intervirology 2007;50:63-70 |
|
| Dengue (DENV) |
Adelman et al. J Virol 2002;76:12925-33 | |
| Caplen et al. Mol Ther 2002;6:243-51 | ||
| Poliovirus (PV) |
Gitlin et al. Nature 2002;418:430-4 |
|
| Ebola (EBOV) |
Groseth et al. J Infect Dis 2007;196 Suppl 2:S382-9 |
|
| Rage (RABV) | Brandao et al. Braz J Infect Dis 2007;11:224-5 | |
| Fu et al. Antisense Nucleic Acid Drug Dev 1996;6:87-93 |
Une variante de la technique de double hybride, le reverse double hybride, permet de cribler finement une interaction fonctionnelle entre 2 molécules dans le but de les déstabiliser. Dans ce cas, l’interaction entre deux protéines n’induit pas un gène rapporteur mais un gène toxique qui conduit à la mort de la levure. Ainsi, seules les molécules inhibant l’interaction permettent la survie des levures [22]. Ce système permet de cribler facilement des banques de petites molécules ou de peptides capables d’inhiber l’interaction.
Le principe du double hybride et du reverse double hybride peut également être transposé chez la bactérie. Ceci a notamment permis la sélection d’un peptide cyclique inhibant le bourgeonnement du VIH ( tableau 1 ). Les avantages du double hybride bactérien sont sa rapidité, sa simplicité de manipulation et le fait qu’il n’exige pas que les protéines soient adressées dans le noyau. En revanche, les bactéries ne réalisent pas les modifications post-traductionnelles eucaryotes des peptides/protéines.
2.1.2. Le criblage par phage display
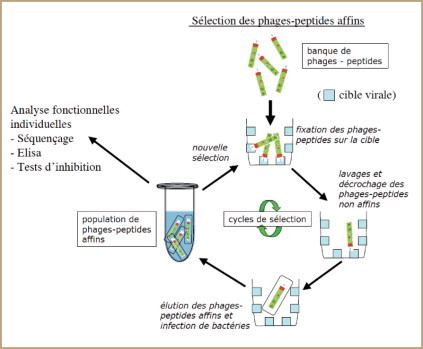
Découverte par Georges Smith (1985) [28], la technique de phage display est un procédé approprié à la découverte de nouvelles molécules thérapeutiques et notamment d’antiviraux. Elle consiste à exprimer un répertoire protéique aléatoire ou semi-aléatoire à la surface de bactériophages, en fusion avec le domaine NH2-terminal d’une des protéines de surface du phage [28, 29]. Les phages recombinants ainsi obtenus sont soumis à sélection afin d’isoler ceux présentant les meilleures affinités pour une cible choisie. Celle-ci peut être de nature protéique (anticorps, enzyme, récepteur) ou non protéique (acide nucléique par exemple) [29] ( figure 2 ). Le processus de sélection se compose de plusieurs cycles (généralement 3 à 5), comportant chacun des étapes de capture, lavage, élution et amplification. La répétition de ces étapes permet d’enrichir fortement la proportion de phages exposant une protéine ou un peptide interagissant fortement et spécifiquement avec la cible. Les phages sélectionnés sont ensuite testés individuellement dans des systèmes fonctionnels.
Figure 2.
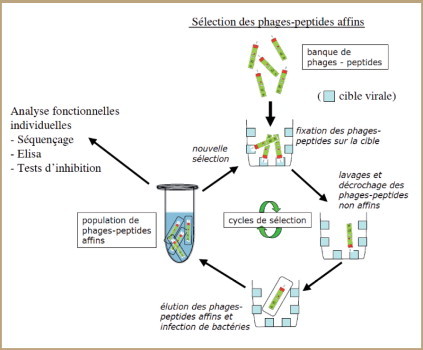
Principe du phage display.
Les bactériophages couramment utilisés sont des phages filamenteux infectant E. coli tels que les phages M13, fd, f1, T4 et T7 [30, 31], ayant pour avantage de se multiplier à des titres extrêmement élevés (1012-1013 pfu/ml) et de posséder un génome ADN simple, facilement manipulable, ce qui rend possible la création de banques de très grande diversité [32, 33]. En général, les banques sont composées de 107-109 clones, une diversité maximale étant toujours recherchée [34] car la probabilité de sélectionner des molécules d’intérêt, c’est-à-dire montrant une forte affinité pour la cible, est proportionnelle à la taille de la banque, donc à la diversité du répertoire moléculaire.
Les banques générées pour la recherche d’antiviraux sont généralement constituées de peptides de 6 à 15 aa, conçues de façon à en maximiser la diversité [32]. Les premières banques réalisées présentaient des peptides courts et linéaires, pouvant adopter un grand nombre de conformations et ainsi s’adapter à des structures variées. Toutefois cette flexibilité peut pénaliser l’affinité pour la cible une fois le peptide déconnecté de la protéine du phage qui le porte. C’est pourquoi comme pour le double hybride, des banques dans lesquelles les peptides ont une structure contrainte au départ grâce à des séquences semi-aléatoires (contenant un squelette pré-organisé limitant le degré de liberté et privilégiant certaines conformations) ont été construites. Les « boucles » peptidiques formées présentent alors les séquences variables (peptides cycliques). De tels peptides sont généralement plus affins et plus spécifiques [29, 35], ont une entropie conformationnelle réduite et sont donc moins dépendants du mode de présentation.
Différentes méthodes peuvent être utilisées pour cloner les gènes codant les peptides de la banque en fusion avec la protéine du bactériophage [34].
La première consiste à cloner le gène du (poly)peptide d’intérêt en fusion avec la protéine de capside de surface du phage, directement dans le génome phagique. Ainsi toutes les copies de la protéine de surface portent le peptide d’intérêt (mode d’exposition multivalent) [34].
La seconde méthode est plus sophistiquée puisque le gène de fusion (poly)peptide-protéine de surface est cloné dans un vecteur d’expression (phagemide), possédant une origine de réplication et un signal d’encapsidation propres [36]. En infectant des bactéries par un phage helper, on apporte en trans les protéines nécessaires à l’encapsidation, la réplication et à l’assemblage du génome phagique. En conséquence, l’expression du peptide est dans la majorité des cas réalisée à raison d’un peptide par phage (mode d’exposition monovalent, ce qui limite les effets d’avidité et augmente la stringence des sélections) [37]. Le système monovalent est plus adapté à la caractérisation précise d’un peptide inhibiteur de grande affinité [37, 38].
Les molécules cibles peuvent être criblées en phase solide dans des conditions semblables aux tests ELISA (revêtement dans des puits de plastique) ou en fusion avec de la biotine capturée sur des billes magnétiques recouvertes de streptavidine [30, 32].
La méthode de phage display présente deux intérêts majeurs. D’une part, elle permet de cribler un très grand nombre de molécules différentes (jusqu’à 1 010) dans un volume réduit (quelques μL). D’autre part, elle permet une correspondance phénotype/génotype car le gène codant la protéine (ou le peptide) exposée à la surface est directement isolé et séquencé à partir de l’ADN du phage [29]. En outre, l’utilisation d’un virus pour développer une stratégie de présentation présente en elle-même d’autres intérêts majeurs (étapes de clonage simplifiées, stockage facile à 4 °C ; machinerie d’infection virale très efficace) et contrairement aux banques de composés issus de la chimie de synthèse, qui du fait de leur consommation et de la dégradation ont tendance à s’épuiser au cours du temps, les banques de phage display peuvent être régénérées facilement et régulièrement [29, 39].
Ses principaux inconvénients sont l’existence de biais possibles dans le processus de sélection [29]. Ainsi des clones toxiques ou possédant un caractère qui diminuerait l’infectiosité des phages, leur sécrétion, ou encore la croissance des bactéries sont contre-sélectionnés et disparaissent rapidement de la banque, indépendamment du processus de sélection. De plus, le taux d’expression de certaines séquences peptidiques dans E. coli peut être variable (en fonction de la fréquence d’utilisation des codons), ce qui peut aussi constituer un biais dans la sélection [40].
D’autre part, certains motifs peptidiques riches en résidus hydrophobes sont susceptibles d’interagir non spécifiquement avec la phase solide (plastique par exemple) [41]. Enfin, il peut arriver, comme mentionné antérieurement, que les peptides candidats, une fois synthétisés, présentent une activité différente de celle qu’ils manifestent lorsqu’ils se trouvent à la surface du phage.
Malgré toutes ces limites, le phage display a déjà été utilisé plusieurs fois avec succès en matière de recherche antivirale et a permis la découverte de peptides inhibiteurs isolés par criblage de diverses cibles d’origines virales [42] ( tableau I ).
-
•
Inhibiteur de protéine virales impliquées dans l’entrée du virus : la découverte de peptides inhibant l’entrée des virus a été réalisée : (1) soit en utilisant le virus entier, purifié et immobilisé ce qui a permis d’isoler des peptides inhibant l’entrée du virus de la bronchite infectieuse aviaire (IBV), du rotavirus humain, du virus du syndrome des points blancs de la crevette (WSSV), du virus de la maladie de Newcastle (NDV), ou encore du virus hémorragique de la carpe herbivore (GCHV) ; (2) soit en utilisant un élément sous-unitaire du virus et notamment la protéine d’enveloppe purifiée. Ainsi, des peptides inhibant l’infection par le virus West-Nile (WNV), par le virus influenza A et par le virus de la dengue ont été caractérisés ; des peptides inhibiteurs liant le domaine Pré-S de la protéine de d’enveloppe LHBs du VHB ont également été caractérisés par cette approche.
-
•
Inhibiteurs de protéines impliquées dans des complexes viraux intracellulaires, notamment ceux responsables de la réplication virale : ce type d’approche demeure un défit en raison notamment de la moindre accessibilité des cibles pour la délivrance d’un éventuel peptide inhibiteur. C’est peut-être pour cette raison, que l’essentiel des travaux réalisés par cette approche a concerné des pathologies virales à fort potentiel commercial ; VHB, HPV16, VHC, et évidemment VIH.
-
•
Inhibiteurs de partenaires cellulaires des protéines virales : cette approche consiste à cibler une protéine cellulaire jouant un rôle dans l’infection virale ou la réponse immunitaire antivirale. Elle est particulièrement adaptée contre des virus présentant une fréquence de mutation élevée et donc un potentiel à développer des résistances aux molécules dirigées contre les protéines virales. Elle a été utilisée avec succès pour isoler des peptides inhibant l’infection par le VIH, le virus Sin Nombre (VSN), le virus Hantaan (VHTN), le virus de Prospect Hill (VPH) ou encore le virus de la stomatite vésiculeuse (VSV).
En conclusion, le phage display permet donc de sélectionner des motifs peptidiques qui peuvent servir de base pour le développement d’un peptide antiviral plus élaboré ou encore de modèle pour générer des sous-banques dérivées des premières, reprenant les caractéristiques des meilleurs peptides isolés pour ainsi améliorer progressivement l’affinité par criblages et sélections successives.
2.2. Les approches cognitives : une focalisation sur la cible
Ces approches ont été rendues possibles grâce au développement des outils puissants que sont la génomique, la protéomique, la transcriptomique, la cristallographie, et la bioinformatique en particulier. L’application de tout ou partie de cet arsenal technique a grandement amélioré la connaissance des séquences des génomes viraux, l’identification des éléments responsables de l’étiologie et de la pathologie ainsi que l’établissement de la structure et de l’interactome (l’ensemble des interactions entre une protéine donnée et ses partenaires protéiques) des protéines responsables, isolées ou en complexe. L’ensemble de ces connaissances a permis l’élaboration de méthodes de ciblage rationnelles visant à perturber des interactions protéine-protéine qui sont a priori essentielles au processus infectieux [21, 22, 24]. Le potentiel antiviral des molécules ainsi sélectionnées est ensuite testé dans un système fonctionnel adapté, soit in vitro (cellulaire ou tissulaire), soit in vivo chez l’animal.
2.2.1. Le « drug design » : l’apport de l’informatique à la conception de nouveaux médicaments
Il s’agit d’utiliser les connaissances disponibles sur les structures et les interactions entre les différentes protéines des complexes viraux ainsi qu’entre les protéines virales et leurs partenaires cellulaires, afin de les déstabiliser dans une perspective antivirale. Ainsi, en se basant sur la connaissance des caractéristiques de la cible biologique, on peut mettre au point de petits peptides mimant des zones essentielles des protéines virales. Ils peuvent agir comme compétiteurs et empêcher la formation des complexes ou les dissocier. Ce concept appliqué à différents virus a déjà donné de bons résultats d’inhibition de la réplication virale [43, 44].
L’affinité d’un peptide pour sa cible dépend de certains paramètres comme la complémentarité de leur structure, de leurs fonctions chimiques ou de leurs charges. Souvent, les peptides interacteurs isolés par cette approche ne constituent pas le médicament final, mais leurs caractéristiques peuvent alors servir de base pour entreprendre des analyses de peptidomimétisme, c’est-à-dire pour développer des molécules plus simples et/ou plus petites, mimant les propriétés d’interaction du peptide et donc ses propriétés bioactives.
Le drug design est donc un moyen rationnel prisé pour proposer de nouveaux médicaments ou améliorer ceux existant, et cela de façon plus rapide et moins coûteuse que par les méthodes classiques de caractérisations de nouvelles molécules. Le grand nombre de protéines virales dont la structure macromoléculaire a été identifiée durant ces dernières années a permis de potentialiser de nombreuses nouvelles cibles thérapeutiques. La modélisation moléculaire utilise des outils bioinformatiques (in silico) et les applique aux données issues de la biologie moléculaire et structurale, permettant ainsi de prédire et de visualiser les propriétés physico-chimiques des protéines virales et les modes d’interaction/fixation avec leurs partenaires viraux ou cellulaires. On peut ainsi reproduire en trois dimensions la forme et les propriétés des molécules candidates et de leur cible (flexibilité, encombrement, forces d’attraction et de répulsion s’exerçant entre elles) puis simuler l’action de molécules virtuelles sur ces cibles ( figure 3 ). Cette structure virtuelle va permettre la réalisation d’un pharmacophore [5], c’est-à-dire une empreinte chimico-spatiale d’un ligand caractérisé, possédant ses capacités de liaison à la cible et utilisable comme base (optimisée par la suite) pour la mise au point d’une molécule thérapeutique [45].
Figure 3.
Dégradation de l’ARN ciblé.
De nombreux logiciels de modélisation sont aujourd’hui disponibles et permettent l’automatisation (et l’utilisation de plus en plus systématique par les industriels) de cette démarche. Plusieurs façons de procéder existent en fonction de l’état des connaissances cible/ligand disponibles [46].
Si les structures 3D de la cible et d’un ligand sont connues, on cherche à potentialiser ce ligand, à rechercher un nouveau ligand ou à « concevoir virtuellement » un inhibiteur dans la topographie de l’interface cible-ligand. La situation idéale est de réaliser une co-cristallisation de la cible avec son ligand naturel. Si le cristal de la protéine cible est disponible, il est même possible de le tremper dans une solution d’inhibiteur et de visualiser ensuite la position de la molécule dans le cristal [3]. Cela permet de cribler des banques de molécules à la recherche de celles s’adaptant au mieux chimiquement et structurellement au site de liaison. Ainsi, l’accès à la structure tridimensionnelle du complexe protéine/ligand permet la conception virtuelle et rationnelle de nouvelles petites molécules capables de se fixer avec une grande affinité et donc de bloquer ces sites actifs. Cette approche est appelée « structure based drug design » (SBDD). Les logiciels réalisent alors les étapes de docking (positionnement) puis de scoring (classification en fonction de l’affinité). Plusieurs programmes de docking sont utilisés pour ce travail (Dock, AutoDock, Gold, Slide, Glide, Affinity, FlexX, etc. [5, 47]. Si la structure de la cible et de son site actif sont connus mais qu’aucun ligand n’est disponible, on utilise alors une approche dite « de novo » qui va générer automatiquement de nouvelles molécules dans le site de liaison de la protéine ciblée [21]. Les molécules sont ici construites pour s’adapter au mieux aux propriétés des résidus présents à la surface de la poche du site actif. En revanche, lorsque ni ligand ni site actif ne sont connus pour une cible, la situation est plus complexe et il faut alors procéder à une analyse individuelle des sites potentiellement actifs.
Si la structure de la cible est inconnue (beaucoup de cibles sont difficiles à purifier et à cristalliser), il faut alors procéder préalablement à une reconstruction théorique de celle-ci par homologie de structure avec une protéine connue pour être proche de la cible et dont la structure est disponible [10]. En revanche, si un ligand est connu, on peut l’utiliser pour créer directement un pharmacophore et utiliser ses caractéristiques (charges, hydrophobicité, encombrement stérique, etc.) pour dessiner des molécules similaires. On parle alors de « ligand based drug design » (LBDD) [48].
Enfin, dans un second temps, le drug design permet d’optimiser virtuellement l’affinité de molécules ainsi conçues ou isolées de banques combinatoires, et même de potentialiser des molécules plus anciennes et déjà disponibles sur le marché. Dans le cadre de la recherche de peptides thérapeutiques, la modélisation moléculaire apporte aussi des solutions aux limites pharmacologiques de leur utilisation in vivo. Une fois le « squelette » du peptide choisi, le drug design, en prédisant in silico leur structure, leur affinité et leur stabilité, va permettre d’améliorer des paramètres tels que leur capacité à traverser les membranes ou leur demi-vie ou bien encore aider à comprendre leur mécanisme d’action [5].
Le drug design a permis la mise sur le marché de médicaments bien connus comme le zanamivir (Relenza®) et l’oseltamivir (Tamiflu®) qui ont été conçus d’après la structure 3D de la neuraminidase du virus de la grippe [3] ainsi que les anti-protéases qui sont utilisées en trithérapie contre le VIH ou des inhibiteurs du VHS. L’utilisation du drug design pour la conception de peptides antiviraux reste pour le moment encore sous exploitée mais a déjà permis des succès notables contre des virus comme le VIH, l’hépatite C ou bien encore contre le virus du syndrome respiratoire aigu sévère (SRAS) ( tableau I ). Actuellement, cette technologie est mise en pratique à grande échelle pour essayer de trouver des molécules antivirales contre les virus à ARN (projet européen VIZIER : Viral Enzymes Involved In Replication) [49].
La modélisation moléculaire peut aussi servir à réaliser un criblage virtuel de banques de molécules virtuelles (in silico) d’une diversité bien plus importante que celle permise par les banques de composés biologiques ou chimiques. Les logiciels disponibles effectuent toutes les étapes en commençant par le positionnement des molécules dans le site actif de la cible, puis en procédant au calcul des énergies d’interaction et des affinités, et finalement au calcul des « scores » permettant de déterminer les meilleurs candidats (leads) qui constituent un modèle optimisable du futur médicament [10]. Avant l’apparition de cette méthode, il fallait tester des milliers de molécules pour espérer en isoler une possédant des propriétés thérapeutiques adaptées.
Le drug design est donc un procédé très économique qui peut être avantageusement utilisé lors d’études préliminaires de façon à ne passer aux études fonctionnelles (plus longues et coûteuses bien qu’indispensables) que lors des tests de confirmation des propriétés antivirales des molécules.
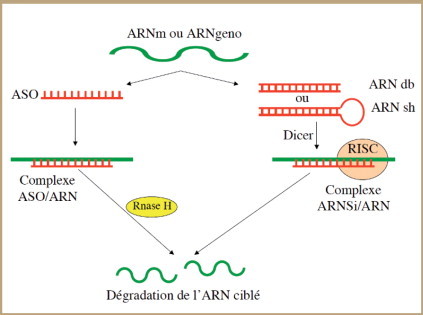
2.2.2. L’interférence ARN
Découvert en 1998 chez les plantes [50], l’ARN interférence fait appel non pas à des peptides interférant mais à de petits acides nucléiques. Ici la cible n’est donc pas la protéine elle-même mais son ARNm. Ces acides nucléiques sont utilisées pour interférer avec l’ARN, selon un mécanisme de « silencing » spécifique de séquences et sont susceptibles de détruire spécifiquement un ARNm cible. Elles permettent donc une inhibition très efficace et surtout très spécifique de l’expression d’un gène, ce qui en fait un outil idéal pour l’inhibition de la réplication virale [51] et cela à de faibles doses [52]. On distingue deux principales sortes d’interférences ARN.
-
1.
Les ARNdb et les ARNsh (short hairpin) sont de courts fragments d’ARN double brin (en général 21-23 nucléotides) qui, une fois dans la cellule, vont être clivés par une enzyme appelée Dicer (RNAses de type III), en petites molécules, les siRNA [53, 54]. Le brin sens est dégradé pendant que le brin antisens est utilisé pour cibler l’ARNm pour le « silencing ». En s’appariant par complémentarité avec leur ARNm cible, ils forment un ARN double brin qui va être pris en charge par le complexe multiprotéique RISC (RNA-Induced Silencing Complex), ce qui conduit à la dégradation de l’ARNm cible qui n’est donc plus traduit en protéine ( figure 4 ).
-
2.
Les oligonucléotides antisens (ASO) qui sont des brins d’ADN simple brin d’environ 20 nucléotides complémentaires d’une cible ARN avec laquelle ils vont former un dimère ARN/ADN détruit ensuite par les RNAses H cellulaires [55].
Figure 4.
Principe du drug design.
Ces molécules doivent souvent être présentes en large excès pour être efficaces et d’autres contraintes limitent leur utilisation comme molécules thérapeutiques in vivo, comme leur extrême sensibilité aux nucléases, leur toxicité, leur instabilité dans le sérum, les voies de délivrance et les résistances développées par certains virus [52].
L’une des premières utilisations fructueuses de ces molécules comme agent antiviral date 2001, et avait pour cible le virus respiratoire syncytial (VRS) [56] en ciblant l’ARNm de la phosphoprotéine P. Depuis, ce procédé a été utilisé avec succès pour inhiber la réplication virale de différents virus responsables de graves pathologies [51, 55] ( tableau I ). Le nombre et la diversité des virus ciblés illustre bien le potentiel de cette technique pour la recherche antivirale. Parmi eux on peut citer l’exemple du VIH, du virus West-Nile, des virus des hépatites B et C, du VRS, d’entérovirus, du virus de la grippe, du virus SARS-CoV, du virus de la dengue, du virus de la polio, du virus Ebola ou bien encore du virus de la rage.
L’utilisation de l’ARN interférence in vivo est limitée par un certain nombre de contraintes [52] comme la grande sensibilité de ce type de molécules aux nucléases ou leur instabilité dans le sérum, mais la principale limite de l’interférence ARN en tant que thérapie antivirale demeure la délivrance des molécules d’ARN à l’intérieur des cellules cibles [58]. Les efforts futurs vont ainsi devoir se concentrer sur la mise au point de vecteurs efficaces et non toxiques, capables de délivrer ces molécules en concentration suffisante, dans des organes parfois difficilement accessibles. Plusieurs solutions sont explorées comme l’utilisation de vecteurs lipidiques « stable nucleic acid-lipid particle » (SNALP) [59], de vecteurs lentiviraux [51] ou bien la fusion avec des peptides porteurs comme par exemple un motif issu de la glycoprotéine rabique, qui permet de délivrer par la voie intraveineuse des siRNA dans le système nerveux central en passant la barrière hémato-encéphalique [60]. Enfin, comme c’est le cas avec les autres stratégies antivirales, certains virus peuvent développer des mécanismes d’échappement à l’interférence ARN [57, 61, 62].
3. Conclusion
Alors que très peu d’antiviraux étaient disponibles il y a seulement 30 ans de ça, on trouve aujourd’hui sur le marché plus d’une cinquantaine de molécules antivirales. Les progrès majeurs réalisés dans les années 1980, tant dans la compréhension des mécanismes moléculaires de la réplication virale que dans les techniques de criblage à haut débit, associés aux avancées rapides en informatique ont permis d’accélérer grandement la découverte de nouvelles molécules. Le choix de la cible est une étape importante et si les enzymes virales impliquées dans la réplication demeurent les principales cibles des antiviraux, les chercheurs s’intéressent aujourd’hui à toutes les étapes du cycle viral et aux protéines associées (fusion, libération) y compris les partenaires cellulaires des protéines virales, élargissant ainsi le champs d’action des thérapies antivirales. Deux grands types d’approches sont utilisés : d’une part les connaissances accumulées grâce aux analyses structurales des protéines permettent la conception puis la synthèse de molécules efficaces plus rapidement et plus économiquement que les anciennes méthodes ; d’autre part, l’automatisation et les progrès réalisés dans les techniques de criblages de molécules candidates parmi des banques combinatoires de très grandes diversités ont ouvert de nouvelles perspectives dans la lutte antivirale. Ces deux approches doivent être utilisées en synergie, chacune d’entre elles pouvant servir de point de départ et proposer des candidats qui seront alors optimisés par l’autre approche. De plus, si les techniques pour isoler à partir de banques ou pour concevoir virtuellement des molécules antivirales permettent aujourd’hui d’obtenir rapidement des candidats inhibiteurs, il y a un besoin essentiel à développer et à disposer de tests fonctionnels fiables et simples comme les réplicons sub-génomiques, qui permettent l’étude des complexes de réplication viraux sans utiliser le virus lui-même [63], [64], [65], [66], pour tester et valider en aval les propriétés antivirales et la toxicité des molécules isolées par ces techniques.
Les thérapies antivirales ainsi que les méthodes permettant de les mettre au point progressent donc de façon notable, mais malheureusement, il existe encore de nombreuses failles dans le bouclier antiviral.
Tout d’abord les molécules disponibles aujourd’hui ne permettent pas d’éliminer des virus en phase de latence, responsables d’infections chroniques. En effet, les antiviraux sont surtout actifs lors des phases actives de la réplication, ce qui contraint à traiter en continu en cas de d’infection latente. L’apparition chez les virus de résistance aux traitements antiviraux ainsi que l’émergence de nouveaux virus à fort potentiel pathogène nécessitent un effort constant pour renouveler l’arsenal des molécules antivirales disponibles, y compris contre des pathologies négligées et à moindre potentiel économique que le VIH ou les hépatites. En effet, beaucoup de maladies d’origine virale sont encore de nos jours oubliées dans la recherche antivirale car jugées peu rentables par l’industrie pharmaceutique.
Conflit d’intérêt : aucun.
Références
- 1.Prusoff WH. Synthesis and biological activities of iododeoxyuridine, an analog of thymidine. Biochim Biophys Acta. 1959;32:295–296. doi: 10.1016/0006-3002(59)90597-9. [DOI] [PubMed] [Google Scholar]
- 2.De Clercq E, Field HJ. Antiviral prodrugs - the development of successful prodrug strategies for antiviral chemotherapy. Br J Pharmacol. 2006;147:1–11. doi: 10.1038/sj.bjp.0706446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Canard B. Les perspectives offertes par la biologie structurale à haut débit dans le domaine des médicaments antiviraux. Virologie. 2005;9:257–260. doi: 10.1684/vir.2011.2116. [DOI] [PubMed] [Google Scholar]
- 4.Galasso GJ. Les agents antiviraux dans la lutte contre les maladies à virus. Bulletin de l’Organisation mondiale de la santé. 1982;60:313–324. [Google Scholar]
- 5.Decaffmeyer M, Thomas A, Brasseur R. Les médicaments peptidiques: mythe ou réalité ? Biotechnol Agron Soc. 2008;12:2057. [Google Scholar]
- 6.Elion GB, Furman PA, Fyfe JA, de Miranda P, Beauchamp L, Schaeffer HJ. Selectivity of action of an antiherpetic agent, 9-(2-hydroxyethoxymethyl) guanine. Proc Natl Acad Sci USA. 1977;74:5716–5720. doi: 10.1073/pnas.74.12.5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knobel DL, Cleaveland S, Coleman PG, Fevre EM, Meltzer MI, Miranda ME. Re-evaluating the burden of rabies in Africa and Asia. Bull World Health Organ. 2005;83:360–368. [PMC free article] [PubMed] [Google Scholar]
- 8.Massé N, Selisko B, Malet H, Peyrane F, Debarnot C, Decroly E. Le virus de la dengue : cibles virales et antiviraux. Virologie. 2007;11:121–133. doi: 10.1684/vir.2011.8912. [DOI] [PubMed] [Google Scholar]
- 9.Aman MJ, Kinch MS, Warfield K, Warren T, Yunus A, Enterlein S. Development of a broad-spectrum antiviral with activity against Ebola virus. Antiviral Res. 2009;83:245–251. doi: 10.1016/j.antiviral.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 10.Lambert-van der Brempt C. La modélisation moléculaire, une aide à la conception de nouveaux médicaments. Méd Thérap. 2004;10:244–251. [Google Scholar]
- 11.Leyssen P, De Clercq E, Neyts J. Molecular strategies to inhibit the replication of RNA viruses. Antiviral Res. 2008;78:9–25. doi: 10.1016/j.antiviral.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bray M. Highly pathogenic RNA viral infections: challenges for antiviral research. Antiviral Res. 2008;78:1–8. doi: 10.1016/j.antiviral.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 13.Vicent MJ, Perez-Paya E, Orzaez M. Discovery of inhibitors of protein-protein interactions from combinatorial libraries. Curr Top Med Chem. 2007;7:83–95. doi: 10.2174/156802607779318307. [DOI] [PubMed] [Google Scholar]
- 14.Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. Potent D-peptide inhibitors of HIV-1 entry. Proc Nal Acad Sci USA. 2007;104:16828–16833. doi: 10.1073/pnas.0708109104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Edwards CM, Cohen MA, Bloom SR. Peptides as drugs. QJM. 1999;92:1–4. doi: 10.1093/qjmed/92.1.1. [DOI] [PubMed] [Google Scholar]
- 16.Belaid A, Aouni M, Khelifa R, Trabelsi A, Jemmali M, Hani K. In vitro antiviral activity of dermaseptins against herpes simplex virus type 1. J Med Virol. 2002;66:229–234. doi: 10.1002/jmv.2134. [DOI] [PubMed] [Google Scholar]
- 17.Lorin C, Saidi H, Belaid A, Zairi A, Baleux F, Hocini H. The antimicrobial peptide dermaseptin S4 inhibits HIV-1 infectivity in vitro. Virology. 2005;334:264–275. doi: 10.1016/j.virol.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 18.Zairi A, Tangy F, Saadi S, Hani K. In vitro activity of dermaseptin S4 derivatives against genital infections pathogens. Regul Toxicol Pharmacol. 2008;50:353–358. doi: 10.1016/j.yrtph.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 19.Raffi F. Enfuvirtide, premier inhibiteur de fusion dans le traitement de l’infection par le virus de l’immunodéficience humaine: mécanisme d’action et pharmacocinétique. Méd Malad Infect. 2004;34:3–7. [PubMed] [Google Scholar]
- 20.Loregian A, Palu G. Disruption of the interactions between the subunits of herpesvirus DNA polymerases as a novel antiviral strategy. Clin Microbiol Infect. 2005;11:437–446. doi: 10.1111/j.1469-0691.2005.01149.x. [DOI] [PubMed] [Google Scholar]
- 21.Loregian A, Marsden HS, Palu G. Protein-protein interactions as targets for antiviral chemotherapy. Rev Med Virol. 2002;12:239–262. doi: 10.1002/rmv.356. [DOI] [PubMed] [Google Scholar]
- 22.Vidal M, Endoh H. Prospects for drug screening using the reverse two-hybrid system. Trends Biotechnol. 1999;17:374–381. doi: 10.1016/s0167-7799(99)01338-4. [DOI] [PubMed] [Google Scholar]
- 23.Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 24.Loregian A, Palu G. Disruption of protein-protein interactions: towards new targets for chemotherapy. J Cell Physiol. 2005;204:750–762. doi: 10.1002/jcp.20356. [DOI] [PubMed] [Google Scholar]
- 25.Real E, Rain JC, Battaglia V, Jallet C, Perrin P, Tordo N. Antiviral drug discovery strategy using combinatorial libraries of structurally constrained peptides. J Virol. 2004;78:7410–7417. doi: 10.1128/JVI.78.14.7410-7417.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colas P, Cohen B, Jessen T, Grishina I, McCoy J, Brent R. Genetic selection of peptide aptamers that recognize and inhibit cyclin-dependent kinase 2. Nature. 1996;380:548–550. doi: 10.1038/380548a0. [DOI] [PubMed] [Google Scholar]
- 27.Geyer CR, Brent R. Selection of genetic agents from random peptide aptamer expression libraries. Methods Enzymol. 2000;328:171–208. doi: 10.1016/s0076-6879(00)28398-5. [DOI] [PubMed] [Google Scholar]
- 28.Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 29.Souriau C, Hua T, Lefranc M, Weill M. Présentation à la surface de phages filamenteux: les multiples applications du phage display. Médecine/Sciences. 1998;14:300–309. [Google Scholar]
- 30.Falciani C, Lozzi L, Pini A, Bracci L. Bioactive peptides from libraries. Chem Biol. 2005;12:417–426. doi: 10.1016/j.chembiol.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 31.Scott JK, Smith GP. Searching for peptide ligands with an epitope library. Science. 1990;249:386–390. doi: 10.1126/science.1696028. [DOI] [PubMed] [Google Scholar]
- 32.Devlin JJ, Panganiban LC, Devlin PE. Random peptide libraries: a source of specific protein binding molecules. Science. 1990;249:404–406. doi: 10.1126/science.2143033. [DOI] [PubMed] [Google Scholar]
- 33.Sergeeva A, Kolonin MG, Molldrem JJ, Pasqualini R, Arap W. Display technologies: application for the discovery of drug and gene delivery agents. Adv Drug Deliv Rev. 2006;58:1622–1654. doi: 10.1016/j.addr.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brissette R, Goldstein NI. The use of phage display peptide libraries for basic and translational research. Methods Mol Biol. 2007;383:203–213. doi: 10.1007/978-1-59745-335-6_13. [DOI] [PubMed] [Google Scholar]
- 35.Ladner RC. Constrained peptides as binding entities. Trends Biotechnol. 1995;13:426–430. doi: 10.1016/S0167-7799(00)88997-0. [DOI] [PubMed] [Google Scholar]
- 36.Sidhu SS. Engineering M13 for phage display. Biomol Eng. 2001;18:57–63. doi: 10.1016/s1389-0344(01)00087-9. [DOI] [PubMed] [Google Scholar]
- 37.O’Connell D, Becerril B, Roy-Burman A, Daws M, Marks JD. Phage versus phagemid libraries for generation of human monoclonal antibodies. J Mol Biol. 2002;321:49–56. doi: 10.1016/s0022-2836(02)00561-2. [DOI] [PubMed] [Google Scholar]
- 38.Lowman HB. Bacteriophage display and discovery of peptide leads for drug development. Annu Rev Biophys Biomol Struct. 1997;26:401–404. doi: 10.1146/annurev.biophys.26.1.401. [DOI] [PubMed] [Google Scholar]
- 39.Burritt JB, Bond CW, Doss KW, Jesaitis AJ. Filamentous phage display of oligopeptide libraries. Anal Biochem. 1996;238:1–13. doi: 10.1006/abio.1996.0241. [DOI] [PubMed] [Google Scholar]
- 40.Rodi DJ, Soares AS, Makowski L. Quantitative assessment of peptide sequence diversity in M13 combinatorial peptide phage display libraries. J Mol Biol. 2002;322:1039–1052. doi: 10.1016/s0022-2836(02)00844-6. [DOI] [PubMed] [Google Scholar]
- 41.Adey NB, Mataragnon AH, Rider JE, Carter JM, Kay BK. Characterization of phage that bind plastic from phage-displayed random peptide libraries. Gene. 1995;156:27–31. doi: 10.1016/0378-1119(95)00058-e. [DOI] [PubMed] [Google Scholar]
- 42.Castel G, Heyd B, Tordo N. Le phage display: une nouvelle arme pour la recherche antivirale. Virologie. 2009;13:93–102. doi: 10.1684/13-2.2021.14902. [DOI] [PubMed] [Google Scholar]
- 43.Ghanem A, Mayer D, Chase G, Tegge W, Frank R, Kochs G. Peptide-mediated interference with influenza A virus polymerase. J Virol. 2007;81:7801–7804. doi: 10.1128/JVI.00724-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hrobowski YM, Garry RF, Michael SF. Peptide inhibitors of dengue virus and West Nile virus infectivity. Virol J. 2005;2:49. doi: 10.1186/1743-422X-2-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grohmann D, Corradi V, Elbasyouny M, Baude A, Horenkamp F, Laufer SD. Small molecule inhibitors targeting HIV-1 reverse transcriptase dimerization. Chembiochem. 2008;9:916–922. doi: 10.1002/cbic.200700669. [DOI] [PubMed] [Google Scholar]
- 46.Laguerre M. Molecular modelling and drug design. Bull Cancer. 2007;94:F115–127. [PubMed] [Google Scholar]
- 47.Ghosh S, Nie A, An J, Huang Z. Structure-based virtual screening of chemical libraries for drug discovery. Curr Opin Chem Biol. 2006;10:194–202. doi: 10.1016/j.cbpa.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 48.Bacilieri M, Moro S. Ligand-based drug design methodologies in drug discovery process: an overview. Curr Drug Discov Technol. 2006;3:155–165. doi: 10.2174/157016306780136781. [DOI] [PubMed] [Google Scholar]
- 49.Coutard B, Gorbalenya AE, Snijder EJ, Leontovich AM, Poupon A, De Lamballerie X. The VIZIER project: preparedness against pathogenic RNA viruses. Antiviral Res. 2008;78:37–46. doi: 10.1016/j.antiviral.2007.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 51.Morris KV, Rossi JJ. Antiviral applications of RNAi. Curr Opin Mol Ther. 2006;8:115–121. [PubMed] [Google Scholar]
- 52.Haasnoot J, Westerhout EM, Berkhout B. RNA interference against viruses: strike and counterstrike. Nat Biotechnol. 2007;25:1435–1443. doi: 10.1038/nbt1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elbashir SM, Lendeckel W, Tuschl T. RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev. 2001;15:188–200. doi: 10.1101/gad.862301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 55.Spurgers KB, Sharkey CM, Warfield KL, Bavari S. Oligonucleotide antiviral therapeutics: antisense and RNA interference for highly pathogenic RNA viruses. Antiviral Res. 2008;78:26–36. doi: 10.1016/j.antiviral.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bitko V, Barik S. Phenotypic silencing of cytoplasmic genes using sequence-specific double-stranded short interfering RNA and its application in the reverse genetics of wild type negative-strand RNA viruses. BMC Microbiol. 2001;1:34. doi: 10.1186/1471-2180-1-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Radhakrishnan SK, Layden TJ, Gartel AL. RNA interference as a new strategy against viral hepatitis. Virology. 2004;323:173–181. doi: 10.1016/j.virol.2004.02.021. [DOI] [PubMed] [Google Scholar]
- 58.Ge Q, Eisen HN, Chen J. Use of siRNAs to prevent and treat influenza virus infection. Virus Res. 2004;102:37–42. doi: 10.1016/j.virusres.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 59.Zimmermann TS, Lee AC, Akinc A, Bramlage B, Bumcrot D, Fedoruk MN. RNAi-mediated gene silencing in non-human primates. Nature. 2006;441:111–114. doi: 10.1038/nature04688. [DOI] [PubMed] [Google Scholar]
- 60.Kumar P, Wu H, McBride JL, Jung KE, Kim MH, Davidson BL. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448:39–43. doi: 10.1038/nature05901. [DOI] [PubMed] [Google Scholar]
- 61.Mallory AC, Reinhart BJ, Bartel D, Vance VB, Bowman LH. A viral suppressor of RNA silencing differentially regulates the accumulation of short interfering RNAs and micro-RNAs in tobacco. Proc Natl Acad Sci USA. 2002;99:15228–15233. doi: 10.1073/pnas.232434999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Westerhout EM, Ooms M, Vink M, Das AT, Berkhout B. HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic Acids Res. 2005;33:796–804. doi: 10.1093/nar/gki220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gu B, Ouzunov S, Wang L, Mason P, Bourne N, Cuconati A. Discovery of small molecule inhibitors of West Nile virus using a high-throughput sub-genomic replicon screen. Antiviral Res. 2006;70:39–50. doi: 10.1016/j.antiviral.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 64.Lo MK, Tilgner M, Shi PY. Potential high-throughput assay for screening inhibitors of West Nile virus replication. J Virol. 2003;77:12901–12906. doi: 10.1128/JVI.77.23.12901-12906.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pelet T, Miazza V, Mottet G, Roux L. High throughput screening assay for negative single stranded RNA virus polymerase inhibitors. J Virol Methods. 2005;128:29–36. doi: 10.1016/j.jviromet.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 66.Randall G, Grakoui A, Rice CM. Clearance of replicating hepatitis C virus replicon RNAs in cell culture by small interfering RNAs. Proc Natl Acad Sci USA. 2003;100:235–240. doi: 10.1073/pnas.0235524100. [DOI] [PMC free article] [PubMed] [Google Scholar]