

Abstract
Cellular (also termed ‘natural’) prion protein has been extensively studied for many years for its pathogenic role in prionopathies after misfolding. However, neuroprotective properties of the protein have been demonstrated under various scenarios. In this line, the involvement of the cellular prion protein in neurodegenerative diseases other than prionopathies continues to be widely debated by the scientific community. In fact, studies on knock-out mice show a vast range of physiological functions for the protein that can be supported by its ability as a cell surface scaffold protein. In this review, we first summarize the most commonly described roles of cellular prion protein in neuroprotection, including antioxidant and antiapoptotic activities and modulation of glutamate receptors. Second, in light of recently described interaction between cellular prion protein and some amyloid misfolded proteins, we will also discuss the molecular mechanisms potentially involved in protection against neurodegeneration in pathologies such as Alzheimer’s, Parkinson’s, and Huntington’s diseases.
Keywords: prion, Tau, Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, neuroprotection
1. Introduction
Pathogenic conformational changes of cellular prion protein (PrPC) generates a β-sheet-enriched isoform called PrPSc or prion (word derived from proteinaceous infectious particle) [1,2,3], the causal agent of prionopathies [1,2]. PrPC, the natural noninfective protein, is a cell surface glycoprotein linked to the membrane by a glycosylphosphatidylinositol (GPI) anchor, and is mainly located in lipid rafts. The protein is encoded by the Prnp gene and expressed in a wide range of tissues in mammals [4,5,6]. However, central nervous system (CNS) and lymphoid tissues express higher levels of PrPC, making them the best candidates to generate infectious prions. Prionopathies are a group of fatal neurodegenerative diseases (NDDs) that may present as genetic, infectious, or sporadic disorders. Kuru, Creutzfeldt–Jackob disease (CJD), Gertsmann–Straussler–Scheinker syndrome (GSS), and fatal familial insomnia (FFI) are diseases that occur in humans, while bovine spongiform encephalopathy (BSE) is found in cows, scrapie in sheep, and chronic wasting disease (CWD) in some members of the family Cervidae [7,8,9]. After the structural transition of PrPC, PrPSc acquires self-aggregative, spreading (intercellular propagation), and infective (understood as a synonym of contagious) properties [10,11].
In recent years, the scientific community has focused attention on defining the term “prion-like” or “prionoid” to describe other proteins with behavior similar to prions in terms of self-aggregation and spreading properties [10,11,12,13,14]. Proteins implicated in different NDDs, including Huntingtin [15], α-synuclein [16,17,18,19,20,21,22,23], amyloid-β (Aβ) [24,25,26], and tau [27,28,29,30,31], are currently seen in numerous studies as prion-like proteins. In fact, most of them display amino acid domains in their sequence that determine their specific self-aggregation properties [32,33]. However, the transmission of some of these amyloid proteins between individuals, although unlikely, is currently a relevant topic for discussion [34,35,36].
More relevantly, two or more of these proteinaceous species might coexist in particular NDDs or experimental models (i.e., [37,38,39,40,41,42,43,44,45]). Thus, the molecular interaction between them and their putative synergistic effects in affected patients are still under debate (i.e, tau and α-synuclein) [46]. However, the question arises when we try to ascertain natural PrPC functions and their specific roles in NDDs other than prionopathies because not only PrPSc (see above) but also PrPC can coexist with most of these amyloids in experimental NDD models and in brain affected tissue (i.e., [22,47,48,49,50]). In this sense, Figure 1A illustrates an example of the relevant colocalization between PrPC (green) and Aβ (red) in the postmortem frontal cortex of an Alzheimer’s disease (AD) patient in contrast to other prion-like proteins such as α-synuclein with little colocalization with Aβ-positive plaques in the same context; normal endogenous expression and function of PrPC may be largely compromised in NDDs [51,52]. In this respect, conflicting studies report neurotoxic roles of PrPC in particular NDDs while others point to a neuroprotective function of the protein in the same disease (i.e., AD), discussed below.
Figure 1.
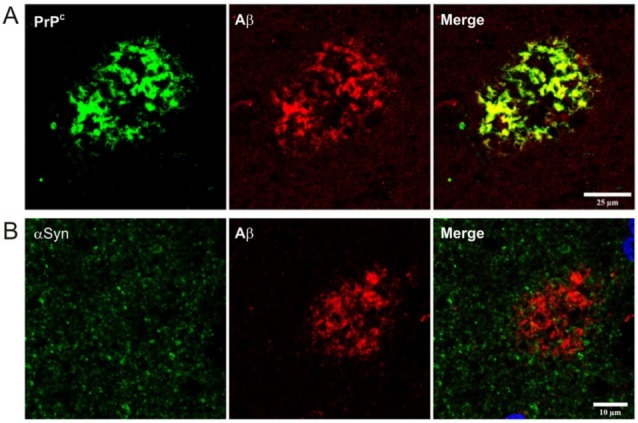
Confocal images of self-aggregative proteins in AD patients. (A) Double-labeling immunofluorescence of PrPC (clone 3F4 directed against aa 109–112 of prion protein, Merck Millipore) and Aβ (rabbit polyclonal antibody directed against the N-terminus 11-pyro E start point of human beta-amyloid, Novus Biologicals) showing colocalization of PrPC in Aβ deposits. (B) Double-labeling immunofluorescence of α-synuclein (clone 5C2 raised against recombinant alpha-synuclein aa 61–95 purified from E. coli, Labome) and Aβ (Novus). Note the absence of clear colocalization between these two proteins. Scale bar values are displayed in the Merge panels.
In the present review, far from arguing the pathogenicity of PrPSc or other prion-like proteins, which has already been done in several reviews (see, for instance, [53,54,55,56]), we will focus our attention on the neuroprotective role of PrPC and its putative implication in amyloid-associated NDDs.
2. PrPC and Neuroprotection
Since the generation of the first knock-out (Prnp0/0) mice of PrPC in 1992, referred to as Zürich 1 [57], researchers have used different Prnp0/0 mice in their studies (Zürich 1 [58], Edinburgh [59], and Zürich 3 [60]) to validate/reveal processes sustained by the functions of the protein (see also [61,62]). Moreover, the number of functions is paralleled by the numerous descriptions of PrPC interactions [63,64,65,66]. However, some attributed functions are controversial to interpret in biological terms [23,63,67,68]. We refer the reader to a compelling description of PrPC interactions [60,69,70] in order to evaluate the pitfalls associated with the genetic background of the Zürich 1 mice. However, one of the most frequently described functions in different experimental models is the participation of PrPC in neuroprotection. In these processes, the “positive” actions of the protein are linked to: i) particular PrPC activities and ii) PrPC interaction with counterpart actors modulating cell signaling cascades and mechanisms involved in neurotoxicity.
2.1. Antioxidant Activity
Membrane-anchored PrPC presents the ability to bind extracellular Cu2+ ions at the highly conserved octapeptide repeats (OR) region of the protein near the N-terminus [71,72,73,74]. This PrPC–Cu2+ interaction provides antioxidant properties to the protein as demonstrated by reducing copper-mediated oxidative stress [75]. In fact, Prnp0/0 mice display higher levels of oxidative stress markers in vivo compared to wild-type animals [76,77,78]. Moreover, in vitro studies have shown that PrPC overexpression in cultured non-neuronal cells results in decreased susceptibility to oxidative damage and toxicity induced by agents such as copper and hydrogen peroxide (H2O2) [79,80,81,82,83,84]. In fact, PrPC levels are increased in neuroblastoma (N2a) and HeLa cells after overload of extracellular copper [85]. Stress-mediated overexpression of PrPC might modulate superoxide dismutase (SOD) [86] and glutathione reductase (GR) activity [83], increasing antioxidant properties in treated cells.
The antioxidant role of PrPC has also been reported in vivo. In this regard, following ischemic insults, Prnp0/0 mice display larger affected regions with increased cell death than do wild-type mice [87]. In contrast, endogenous PrPC expression can protect against brain damage after traumatic brain injury in mice [88] and during stroke in rats [89,90,91]. In parallel, as a consequence of the lesion, Prnp expression is increased under oxidative stress conditions [92], and both increased mRNA and protein have been described in neurons located in the penumbra region in ischemic mice [87]. Lastly, the above-mentioned regulation has also been described in humans under oxidative damage or ischemia [87,93]. Overexpression of PrPC by affected brains should be considered an intrinsic response in an attempt to provide long-term neuroprotection, neurogenesis, or angiogenesis [94].
2.2. Antiapoptotic Activity
PrPC not only acts as an antioxidant protein but also exercises direct control on mitochondrial-associated apoptotic signaling. In this sense, overexpression of PrPC protects primary cultured neurons against Bax-mediated cell death [95]. Function-mapping studies have reported that the OR domain of PrPC is mandatory for antiapoptotic function, since deletion of this domain abolishes the protective function of the protein. Other antiapoptotic proteins such as Bcl-2 require the BH2 domain to interact with Bax protein and regulated permeabilization of the mitochondrial membrane [96]. Although PrPC does not contain BH2 domains and does not directly interact with Bax, it is able to bind with the C-terminal domain of Bcl-2 [97,98]. Moreover, ectopic expression of PrPC and Bcl-2 in Prnp0/0 cells suppresses apoptosis in serum-free conditions [80,99], suggesting the contribution of PrPC to antiapoptotic activity through caspase-dependent apoptotic pathways in mitochondria.
2.3. Regulation of Calcium Homeostasis and Ionotropic Glutamate Receptors by PrPC
Alterations in calcium metabolism have been extensively studied in acute injuries such as ischemia and in neurodegeneration. Relevantly, PrPC may regulate intracellular Ca2+ homeostasis [100,101,102]. For example, Krebs et al. showed that endoplasmic reticulum (ER) calcium stores respond to H2O2 in a PrPC-dependent way in neuronal cell cultures [103]. The data presented support the relevance of the OR domain of the protein in this function and the activation of a protective signaling cascade involving Src-like tyrosine kinase Fyn. Activated Fyn may further activate cellular phospholipases to generate inositol triphosphate (IP3), leading to the opening of ER-associated calcium channels [104].
In fact, a neuroprotective function of PrPC is also supported by several studies describing increased neuronal susceptibility to glutamate agonists in Prnp0/0 mice (see, for instance, [105,106,107]). In these studies, AMPA/KA receptor dysfunction, largely responsible for excitotoxicity, is highlighted by the absence of PrPC. Indeed, recent studies have also reported that PrPC binds to and modulates N-methyl-D-aspartate receptor (NMDAR) in cooperation with Cu2+ [108,109,110]. In addition, there is a susceptibility to stroke in rats with downregulation of Prnp and an increased expression of NR2B, a subunit of NMDAR implicated in excitotoxicity-induced neuronal apoptosis [111]. This suggests a functional regulation of ionotropic glutamate receptors by PrPC. The reader can find additional information about the mechanisms implicated in neuroprotection of excitotoxicity by PrPC in recent reviews [112,113].
2.4. Molecular Partners of PrPC for Interaction and Cell Signaling
The ability of PrPC to bind a variety of other molecules suggests the existence of different physiological roles, which may be context-specific. In this respect, PrPC could be considered a cell surface scaffold protein [70] as a means to explain the role of the protein as key in different signaling systems. However, it will be necessary to determine the biological significance of each interaction and the possibilities of response depending on these contacts. In terms of neuroprotection, PrPC transduces signals across the plasma membrane by binding to other plasma membrane molecules such as the laminin receptor and neural cell adhesion molecule. These interactions promote cell survival and neurite outgrowth [114,115]. In this sense, cell survival promoted by PrPC can involve the activation of both cyclic adenosine monophosphate/protein kinase A (cAMP/PKA) and the ERK1/2 signaling pathways [116], as well as activation of the phosphatidyl-inositol-3-kinase/Akt pathway [115]. In addition, Moulliet-Richard and colleagues proposed Fyn kinase as an initiator of the cascade leading to ERK1/2 activation by cross-linking of PrPC at the cell membrane with antibodies [117]. These and other results support the idea that dimer formation of PrPC is essential to its neuroprotective activity since antibody-mediated PrPC dimerization elicits rapid phosphorylation of ERK1/2 in cultured cells [118]. Moreover, in the same in vitro model, PrPC dimerization also promotes the recruitment of the cAMP responsive element-binding protein (CREB) transcription factor and the transcription of several genes with key functions in cellular protection and neuronal plasticity [119]. In addition, GSK3β, whose inhibition is neuroprotective, is a downstream target of PrPC dimerization signaling in serotonergic neuronal cells [120]. However, dimer formation of PrPC seems to be necessary but not sufficient for its stress protective activity [121].
Another neuroprotective pathway through PrPC binding with stress-inducible protein 1 (STI-1) has been reported by [122]. Moreover, Lopes and colleagues showed the effects on both neuritogenesis and survival in hippocampal neurons triggered by STI-1-PrPC interaction. In this regard, the neuritogenesis was found to be dependent only on mitogen-activated protein kinase (MAPK) activity, whereas cAMP-dependent PKA actions mediate neuroprotection [123]. In addition, PrPC cooperates with STI-1 to regulate SOD activity, and consequently cell survival [124]. In fact, a recent study demonstrates the neuritogenic potential of recombinant PrPC [125] which might trigger intracellular signaling cascades after its homophilic interaction with membrane-anchored PrPC. Indeed, intracellular endocytosed PrPC may interact with proteins involved in classical signaling pathways including the growth factor receptor-bound protein 2 (Grb2), an adaptor protein involved in neuronal survival [63,126].
2.5. Physiological Processing of PrPC and Neuroprotective Metabolites
PrPC can be proteolytically cleaved in the two structurally different regions of the protein at the plasmatic membrane. PrPC has a long, flexible N-terminal tail (residues 23–128) and a C-terminal globular domain that contains three α-helices and two parallel stranded β-sheets [127,128,129] (Figure 2) (the residue numbering refers to mouse PrP (moPrP)). These two regions follow physiological proteolytic processing by α-cleavage (approximately at aa 110), releasing the PrPN1 fragment (aa 23–110) and yielding the PrPC1 fragment tethered to the plasma membrane [130,131]. In addition, minor cleavage, termed β-cleavage, occurs at residues 90–91, releasing aa 23–89 or PrPN2 and PrPC2 [131] (Figure 3A). A third type of cleavage within the OR is induced by reactive oxygen species in the presence of Cu2+ [132]. One study [133] showed neuroprotective activity and anti-β-sheet-mediated corruption activity of PrPN1. In this sense, different studies reinforce this hypothesis [134,135,136], and neurotoxic consequences of the absence of α- or β-cleavage-derived forms of PrPC have been reported [137,138]. In fact, PrPC homodimerization stimulates its trafficking to the plasma membrane and α-cleavage with the consequent production of PrPN1 and PrPC1 [136].
Figure 2.

Schema of the PrPC sequence with its different domains showing the interaction of molecules described in this article. SP: signal peptide 1–22 aa; CC1: charged cluster 1,23–30 aa; OR: octarepeat region 60–91 aa; CC2: charged cluster 2, 95–110 aa; HR: hydrophobic region 112–133 aa; H1-3: α-Helix regions 143–452, 171–191, 199–221; GPIp: GPI anchor-signaling peptide. (Numbering based on the moPrP sequence.)
Figure 3.

Schematic representation of PrPC isoforms: (A) Cell surface PrPC and the four metabolites resulting from α- and β-cleavage (PrPN1, PrPC1, PrPN2, PrPC2, respectively). Homodimerization of PrPC enhanced α-cleavage and consequently the production of anti-β activity of PrPN1. (B) Cytosolic PrP (CyPrP) and the two transmembrane isoforms termed NtmPrP and CtmPrP with opposite sequence orientations with respect to the lumen of the endoplasmic reticulum. As the CC1 domain is not involved in the generation of different PrPC isoforms, it has not been included in this figure.
In addition, a great range of ligands binding to the N-terminal domain of PrPC are able to trigger rapid endocytosis of the protein. Ligand-induced internalization of PrPC may protect cells in different ways: through the transport and homeostasis of several ligands, including Cu2+ and hemin [75,139], by degradation of misfolded or inactive PrPC molecules [140], and by activating PrPC-mediated intracellular cell signaling after the stimuli [141,142,143]. In this regard, and under physiological conditions, endocytosis is essential to N-terminal PrPC function, although PrPN1 is able to elicit neuroprotective signals independently of internalization [134].
Alternatively, intracellular processing of PrPC could generate additional cytoplasmic forms. A cytosolic form (CyPrP) can be generated, probably as a result of retrotranslocation from the ER or from poor translocation into the ER [144]. CyPrP is proposed as being responsible for protection against Bax-mediated cell death [145,146]. In addition, PrPC presents two transmembrane isoforms, termed NtmPrP and CtmPrP, with opposite sequence orientations with respect to the lumen of the ER [147]. The single-pass transmembrane isoforms represent 2% of total PrPC inserts [148] (Figure 3B), and to date, no physiological function has been detailed for them. However, several studies have associated overexpression of CtmPrP with neurotoxicity [149,150].
3. Functions of PrPC during Aging and Neurodegeneration
In the healthy brain, there is a relationship between PrPC levels and aging. In fact, protein levels are reduced in older human brains [151]. In this sense, aging is associated with an increase in reactive oxygen species (ROS) which inversely correlates with PrPC levels. Moreover, oxidative stress is an important contributing factor in the pathogenesis of many human NDDs, such as prionopathies, Parkinson’s, Huntington’s, Alzheimer’s, and amyotrophic lateral sclerosis [152,153,154], which leads us to pose the question of what the role of PrPC is in each of these scenarios. In addition, as mentioned above, there is a specific interaction between the principal proteins implicated in these diseases and PrPC, for instance, tau [46,48], Aβ [155,156], and α-synuclein [22,23,157], reinforcing the notion of an active role for PrPC in these pathologies. Table 1 summarizes several studies describing examples of the neuroprotective role of PrPC in a number of neurodegenerative diseases and some nonspecific disorders. Please see text for opposing data.
Table 1.
Studies on contribution of PrPC to potential neuroprotection in neurodegenerative diseases.
| Disease | Finding | Model | Role of PrPC | Key Reference(s) |
|---|---|---|---|---|
| Alzheimer’s disease | Inhibition of BACE1 | In vitro | Decreases Aβ production | [158,159] |
| Binding of PrPN1 to Aβ | In vitro | Blocks transformation into ADDLs | [160,161] | |
| Binding to STI1 | In vitro | Decreases ADDLs toxicity | [162] | |
| Binding to Zn2+ | In vitro | Decreases Aβ aggregation | [163] | |
| Binding of PrPN1 to ADDLs | In vivo | Decreases ADDLs toxicity | [164] | |
| Prevention of cell death by Aβ | In vivo | Decreases caspase-3 and Bax/Bcl2 levels | [165] | |
| Increase in PrPN1 production in brain patients | Human samples | Blocks transformation into ADDLs | [160] | |
| Increase in brain regions prone to oxidative stress | Human samples | SOD and GR activity regulation | [166] | |
| Increase in initial stages of the disease | Human samples | Downregulates tau levels | [167] | |
| Huntington’s disease | Increase in proteasome activity | In vitro | Decreases HTT aggregation and toxicity | [139] |
| Amyotrophic lateral sclerosis | Induction of neuronal and glial survival signaling | In vivo | Antioxidant | [168] |
| Nonspecific disorder | Binding to Cu2+ | In vitro | Antioxidant | [75] |
| Modulation of SOD | In vitro | Antioxidant | [86] | |
| Modulation of GR | In vitro | Antioxidant | [83] | |
| Modulation of Bax function | In vitro | Antiapoptotic | [95] | |
| Regulation of Ca2+ homeostasis | In vitro | Reduces excitotoxicity | [101] | |
| Inhibition of NMDAR | In vitro | Reduces excitotoxicity | [108,109,169] | |
| PrP113-128 peptide | In vitro | Activates cAMP/PKA and MEK/Erk pathways | [116] | |
| PrP-Fc signaling | In vitro | Activates PI3K/Akt pathway | [75] | |
| Binding to STI1 | In vivo | Inhibits GSK3β activity and activates 7nAChR. All together induces neuroprotective signals. | [120,122,123,170] |
3.1. PrPC in Alzheimer’s Disease (AD) and Other Tauopathies
AD patients are characterized by a progressive cognitive decline and behavioral changes due to the dysregulation of two prion-like proteins: (i) Aβ, resulting from the abnormally processed amyloid precursor protein (APP) though greater activity of β-secretase-1 (BACE1), and (ii) tau, a microtubule-associated protein that promotes the polymerization and stabilization of microtubules (MT) under the regulatory control of several kinases and phosphatases [171,172]. As a result, the major histopathological hallmarks of the disease are the presence of senile plaques, enriched in Aβ, and neurofibrillary tangles (NFTs) containing hyperphosphorylated tau (e.g., [173,174]). The widely accepted amyloid cascade hypothesis posits that the generation of Aβ and its extracellular deposition in brain parenchyma triggers a sequence of events leading to tau dysfunction following the “staging” theory of disease progression [175,176]. Although higher toxic potential is actually attributed to Aβ-derived diffusible ligands (ADDLs) and not to insoluble forms of Aβ [177], tau is considered decisive for the progression of neurodegeneration [178], and the spreading of the tau pathology in affected individuals correlates well with memory impairment and dementia symptoms [179]. Other tauopathies, with different target cells (from neurons to astroglia or oligodendroglia) include Pick’s disease (PiD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and argyrophilic grain disease (AGD), among others [180,181]. See also [182,183] for recent classifications of tauopathies.
The tau gene (MAPT) is expressed in six isoforms as a result of mRNA alternative splicing in various combinations, distinguishable by the exclusion or inclusion of a repeat region of exon 10 that generates four microtubule-binding repeats (4R) or three (3R) tau, and both with either no (0N), one (1N), or two (2N) amino-terminal inserts [184]. In adult neurons, 3R and 4R tau isoforms are present to a similar degree, localized mainly in axons, but they are also present in the somatodendritic compartment of neurons underlying NFT formation and in AD. NFTs lead to increased MT instability, impaired axonal transport, and profound deficits in synaptic function. Among others, GSK3β and Cdk5 are the main kinases implicated in the phosphorylation of some tau epitopes described in AD [185,186,187,188]. In fact, several of these phosphorylated tau epitopes have been associated with in vitro cellular response to ADDLs [167,189,190].
In AD, there is a colocalization of PrPC and Aβ-containing senile plaques ([49] and Figure 1A). Moreover, PrPC and ADDLs interact specifically in AD patients’ brains [50,191,192], suggesting an active role for PrPC in the disease. In fact, Aβ oligomers influence PrPC trafficking and inhibit PrPC endocytosis [193], blocking BACE1 regulation by PrPC [194,195]. In addition, a study by Strittmatter’s laboratory pointed to ADDL–PrPC interaction as being responsible for neurodegeneration through regulation of glutamate receptors [50]. Later, other studies reinforced this hypothesis [196,197,198,199]. In the effort to explain the consequences of PrPC–ADDLs in glutamate receptor interaction, some groups have shown a physical connection between PrPC and ionotropic glutamate receptor NMDA [197,200] and metabotropic glutamate receptor 5 (mGluR5) [201] (reviewed in [202]). NMDA receptor activity is modulated by PrPC in a copper-dependent manner. Moreover, Um and colleagues linked the dysregulation of NMDA by ADDLs–PrPC with Fyn activation. In fact, Fyn has also been associated with PrPC [203] which presents direct binding to mGluR5, mGluR1, and NMDA receptors as well as tau [197,204,205,206,207], supporting a role for it in dysregulating NMDA- or mGluR5-mediated synaptic function as well as tau hyperphosphorylation induced by Aβ [208]. This was demonstrated by Lesne’s laboratory who showed that soluble Aβ binds to PrPC at neuronal dendritic spines where it forms a complex with Fyn and causes tau hyperphosphorylation [209], which may lead to cell death. Regarding synaptic changes and neuronal physiology, some studies reported different data from those reported by Strittmatter´s lab. Indeed, some authors have observed Aβ-induced depression of synaptic transmission in both wild-type and Prnp0/0 mouse slices [210], and others have found that Prnp+/+ and Prnp0/0 mice were equally susceptible to cognitive impairment after Aβ injection into the lateral ventricle [211]. Moreover, Aguzzi’s group has shown there to be no LTP impairment in APP/PS1 mice lacking Prnp [212]. In summary, although the binding of PrPC and ADDLs seems to be accepted, there are some differences in ascertaining whether this interaction affects only synaptic plasticity or cell death as well.
Below we describe several studies in which PrPC developed a neuroprotective role in AD through its natural function/s. For instance, it has been reported that overexpression of PrPC protects against Aβ-mediated cell death (i.e., caspase-3 activation) in mice via control of the Bax/Bcl-2 ratio and, over time, PrPC expression also prevents cognitive dysfunction [165]. In addition, Younan and colleagues have shown that PrPC inhibits fiber formation by trapping free ADDLs and causing disassembly of preformed Aβ fibrils [213]. The authors point to two charged clusters in the N-terminal domain of PrPC as being responsible for Aβ–PrPC binding: (aa 95–110) and (aa 23–27) [21,155,213]. At this point, it is important to keep in mind that the activity of PrPC is finely regulated by its dimerization and that PrPC homodimers stimulate the production of PrPN1, which in turn can bind to Aβ with high affinity, blocking transformation into soluble and toxic ADDLs [136,160,161,164]. In this sense, there is some evidence that α-cleavage (leading to PrPN1 and PrPC1) is increased in postmortem brains of AD patients, reinforcing this neuroprotective notion [160], and inhibition of PrPN1 production promotes AD progression [214]. The hydrophobic domain of the protein (amino acids 112–133), inside the N-terminal domain, is responsible for homodimer formation [121] and perhaps interferes with ADDL binding. In addition, although PrPC could mediate the toxicity of ADDLs [133,215], homodimerization and cleavage may be a common mechanism in preventing this.
In addition, some authors have recently shown that increased PrPC expression downregulates tau protein [167,216,217]. In this sense, we recently reported increased susceptibility of tau phosphorylation to ADDLs in primary cortical cultures lacking PrPC. Reported results indicate that increased PrPC between Braak I and IV stages renders lower tau and phospho-tau. In contrast, PrPC levels decreased at Braak V–VI stages which also correlates with increased amounts of tau and phospho-tau. Taken together, our observations suggest a protective role for PrPC in early stages of AD which may be extendable to other tauopathies [167]. In fact, tau pathology has been reported in a wide number of prionopathies such as sporadic CJD (sCJD) [35], GSS [218,219], and FFI [220], which showed lower PrPC levels due to the PrPC-to-pathogenic-prion conversion [221].
Regarding pathological phosphorylation of tau, some studies point to GSK3β kinase activity as a key element in neuroprotection, while GSK3β inhibition has been shown to play a pivotal role in synaptic plasticity and long-term potentiation (LTP) [222]. In this sense, PrPC–STI-1 interaction triggers reduction of GSK3β kinase activity which not only may affect tau phosphorylation but may also induce memory impairment [120]. Importantly, the interaction of STI-1 with PrPC was recently shown to hinder the binding of Aβ oligomers to PrPC, overcoming their toxicity [162].
As indicated by several studies, AD (in a broad sense) is characterized by neuroinflammation and oxidative stress [223]. In this sense, it is notable that levels of PrPC are increased between Braak I and IV stages [167], in contrast to decreased levels in advanced stages of the disease [221,224]. PrPC can reduce ROS by its intrinsic copper buffering roles and by modulating SOD1 and GR (see above). Elevated levels of PrPC have been reported to occur in brain regions prone to oxidative stress in AD, suggesting a possible antioxidant function in the disease [166]. In addition, increased expression of the PrPC in the first stages of AD [225,226] may promote competition between different ligands including Aβ (Figure 4). Along this line, zinc is another cation involved in the generation of ROS in neurons, and PrPC mediates uptake of extracellular zinc into neuronal cells [227]. Furthermore, zinc promotes Aβ aggregation and increases insoluble Aβ and its deposition in plaques in an AD mouse model [228,229]. In addition, synaptic zinc favors the attachment of Aβ to NMDAR, mediating its excitotoxicity [230]. This implies that the reduction in PrPC in the advanced AD brain migh result in decreased zinc uptake and, consequently, in an increase in the amount of zinc in the synaptic cleft, which would promote Aβ aggregation and synaptic targeting, thereby accelerating the neurodegenerative process.
Figure 4.

Proposal of a putative scenario for neuroprotective intervention of PrPC in AD: 1. Modulating ROS levels; 2. Inhibiting BACE1 activity; 3. Generating PrPN1; 4. Modulating glutamate receptors (both ionotropic (NMDAR) and metabotropic (mGluR5)); 5. Reducing phospho-tau levels through STI-1 interaction and GSK3β inhibition; 6. Reducing ROS levels through STI-1 interaction and consequent SOD modulation; 7. Executing anti-Bax activity; 8. Increasing Zn2+ uptake; and 9. Reducing tau levels. Number 10, in italics, represents the direct intervention of ADDLs in PrPC function, inhibiting its endocytosis and/or homodimerization, and competing with Cu2+ binding and homeostasis. TGN: Trans-Golgi Network.
PrPC has been shown to influence the processing of APP, lowering Aβ production through inhibition of BACE1, suggesting that PrPC functions are beneficial in AD [158,159]. In fact, protein and mRNA levels of PrPC correlate inversely with BACE-1 activity and Aβ levels [151,158,221,226,231,232]. Therefore, a decrease in PrPC levels at medium-late stages of AD may be a primary contributor to neurodegeneration and cognitive impairment.
3.2. Neuroprotective Role of PrPC in Huntington’s and Parkinson’s Diseases
Despite the paucity of data supporting this hypothesis, assorted evidence suggests that PrPC is a possible neuroprotective key in other diseases, since ROS and free radicals are important mediators of neurotoxicity in several other NDDs—for instance, HD and PD (see above), to which we will now turn.
Huntington’s disease (HD) is an inherited disorder which causes progressive neurodegeneration and which includes motor, cognitive, and psychiatric manifestations until inevitable death occurs [233]. The disease is caused by a polyglutamine (polyQ) expansion (encoded by a CAG repeat) of Huntingtin (HTT) protein. Mutated HTT gene is responsible for the aggregated polyQ, the main component of the proteinaceous deposits found in patient brains [234]. In fact, the age of onset of clinical manifestations is inversely correlated to the length of the polyQ expansion. HTT is expressed in a broad spectrum of neuronal and non-neuronal tissues [235]. Nevertheless, mutated HTT protein promotes progressive neurodegeneration of specific neuronal types, affecting particularly the caudate-putamen and neocortical regions of HD patient brains [236,237]. However, the mechanism of progressive neural loss has not been fully elucidated [238].
Parkinson’s disease (PD) is the second most common neurodegenerative disorder in the world, and it is characterized by the appearance of postural instability, bradykinesia, and tremor. These symptoms are associated with dopaminergic neurodegeneration of the substantia nigra pars compacta (SNc), which innervates basal ganglia and leads to loss of dopamine levels in the striatum [239,240]. Although cell death mechanisms are still unknown, great attention has been focused on α-synuclein because it is the major constituent of Lewy bodies, a principal hallmark of PD. In fact, the spread of fibrillar α-synuclein pathology from the brainstem to limbic and neocortical structures seems to be the strongest neuropathological correlate of emerging dementia and cognitive impairment in the disease [241].
In this scenario, the increased level of oxidatively modified proteins in PD leads to the impairment of several cellular functions [242,243]. We have already reported the importance of PrPC expression in modulating redox homeostasis [75]. In addition, some other common aspects suggest that PrPC may exert neuroprotective functions in these diseases. We already know the importance of biochemical interactions between PrPC and NMDAR or mGluR5 and their possible contribution in AD (see above). Along this line, dysregulation of glutamate receptors plays a role in both HD and PD (reviewed in [244,245]). The excess of glutamate is associated with NDDs, and it becomes excitotoxic by chronically activating both ionotropic and metabotropic glutamate receptors. As a result, an increase in intracellular Ca2+ promotes neuronal injury and cell death [246,247]. So, although changes in PRNP expression in early stages of PD and HD are unknown, we may speculate upon a putatively positive role of PrPC in inhibiting glutamate receptors in both diseases.
So, the mechanism that promotes excitotoxicity in HD is thought to be the increased redistribution of NMDAR to the extra-synaptic compartment. Indeed, NMDARs play a key role in neuronal cell death related to HD. Moreover, it has been demonstrated that mutated HTT protein leads to sensitization of the NMDAR, resulting in an increase in extracellular Ca2+ invading neurons and promoting excitotoxicity [245]. Furthermore, degeneration of dopaminergic neurons in SNc induces an increase in the activity of glutamatergic neurons in the subthalamic nucleus (STN) which is believed to contribute to the motor symptoms of PD. Group I mGluRs (mGluR1 and mGluR5) are widely expressed in the basal ganglia, especially at postsynaptic sites [248]. However, mGluR5 expression is higher than mGluR1. So, its role in PD motor deficits has been shown in a variety of preclinical studies [249,250]. In fact, antagonism of this receptor ameliorates motor deficits in animal models of PD [251,252]. In this line of research, a new topic of debate is emerging: the possible intervention of PrPC in inducing cognitive impairment through mGluR5 and NMDAR in synucleinopathies. As reported regarding ADDLs-PrPC interaction, Outeiro’s group has indicated that PrPC acts as a receptor for neurotoxic effects of oligomeric α-synuclein [23], although recent results contradict this [67].
As indicated above, neuroprotective PrPC cleaved-fragment PrPN1 binds to and antagonizes the toxicity of β-sheet rich oligomers. In this line, Wetzel’s group has reported a complex aggregation pathway for a polyQ containing the N-terminal 17 amino acids of HTT exon 1. In addition, they show the intermediate structures formed during aggregation of peptides [253]. A previous study showed a protective effect of PrPC in HTT pathology by reducing aggregation and associated toxicity in neuronal cells [139]. The authors suggest that PrPC protects cells from a reduction in proteasome activity by maintaining levels of ROS, thereby helping to prevent protein aggregation. It would be of interest to learn about the roles of full-length PrPC protein and cleaved fragments of the protein in the same model. In contrast, HD is considered a four-repeat tauopathy with tau nuclear rods [254]. In addition, GSK3β inhibitors prevent cellular toxicity caused by HD mutation [255]. Taking all the evidence together, it is tempting to posit that PrPC may have alternative roles in the disease through regulation of tau levels and modulation of GSK3β activity.
Despite the lack of research about intervention of PrPC in the α-synuclein aggregation process, there has been reported to be an increased tendency toward aggregation after oxidation of γ-synuclein, another member of the family that seeds α-synuclein aggregation [256]. In this context, an indirect role of PrPC in this process is plausible. Also, we and others have shown that PrPC is involved in the propagation and spreading of protofibrils of α-synuclein, with binding between the two proteins in Prnp-transfected HEK293 cells though residues located in the CC2 domain of PrPC [47,157,257]. Surprisingly, these are the same amino acids (95–110) involved in binding with Aβ [213]. Since Prnp expression is not mandatory for α-synuclein transport in the mouse brain [258], it is tempting to consider that role in the update and transport to be a collateral effect on the principal neuroprotective role of PrPC in the disease. In this respect, we also must recall that both our group and Aulic et al. [157] indicated that PrPC is a receptor for the fibrillar forms of α-synuclein [21,257,259]. However, as it has been reported that PrPC does not bind to oligomeric species of α-synuclein [67] in contrast to [23], additional studies are needed to ascertain whether oligomeric α-synuclein also mediates similar effects to ADDLs though PrPC interaction.
Finally, unpublished studies by our group indicate a tendency toward decreased levels of PrPC protein levels in the frontal cortex (area 8) in advanced PD patients (Braak stages 5 and 6) (Figure 5). These results may signal the importance of the protein in the progression of the pathology, as occurs in AD (see above).
Figure 5.

Graph representing densitometric study of PrPC immunoblot analysis in postmortem frontal cortex (Brodmann area 8) from PD patients at different stages compared to non-neurodegenerative cases (nND). Postmortem brain tissue was obtained from Hospital Clinic Brain Bank, following the Code of Ethics of the World Medical Association and the protocols of the local ethical committee. Each plot represents a quantitative level of PrPC standardized with actin level for each case. Data shows a progressive, albeit nonsignificant decline in PrPC levels in accordance with advance of the disease. Statistical analysis of the resulting data was performed using Anova (Kruskal–Wallis with Dunn multiparametric test) and Prism 8.0 (GraphPad Software, San Diego, CA, USA).
4. Concluding Remarks
The relationship between PrPC and other amyloids (oligomeric (ADDLs) and fibrillar forms (i.e., α-synuclein)) has been well established, and different roles of PrPC in AD have been described (see above). We argue for the role of PrPC in preventing the detrimental effects of the oligomeric species, especially at early stages of the neurodegenerative processes. In this review, we have focused our attention on analyzing a number of mechanisms through which PrPC may act as a neuroprotective molecule. In fact, we must not lose sight of the progression of the protein and its derivate fragments (PrPN1, for instance) in the evolution of diseases. A correlation of the symptoms with levels of PrPC expression may be an important element in increasing our understanding of the natural functions of the protein. In this review, we have also speculated about other diseases of which we do not have so much data, but regarding which it is nonetheless reasonable to posit that PrPC is expressed in order to slow down disease progression. Despite this, we cannot rule out the possibility that enhanced interaction between PrPC and the other proteins implicated in NDDs, such as Aβ and α-synuclein, results in fatal effects. Thus, a plausible intervention to avoid the progression of these diseases may involve blocking these specific interactions, thereby allowing the protein to maintain its natural function.
Acknowledgments
The authors thank Tom Yohannan for editorial advice.
Funding
This research was supported by grants from the Spanish Ministry of Science, Innovation and Universities MCIU/AEI/FEDER (RTI2018-099773-B-I00 and AGL2017-90665-REDT), the CERCA Programme, and the Commission for Universities and Research of the Department of Innovation, Universities, and Enterprise of the Generalitat de Catalunya (SGR2017-648) to JADR; and CIBERNED (CMED2018-2) to IF and JADR. The project leading to these results also received funding from “la Caixa” Foundation (ID 100010434) under the agreement LCF/PR/HR19/52160007 to JADR. L.L. is supported by a fellowship from the FPU Programme of MCIU.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Prusiner S.B. Prions. Proc. Natl. Acad. Sci. USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prusiner S.B. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 3.Bolton D.C., McKinley M.P., Prusiner S.B. Identification of a protein that purifies with the scrapie prion. Science. 1982;218:1309–1311. doi: 10.1126/science.6815801. [DOI] [PubMed] [Google Scholar]
- 4.Ford M.J., Burton L.J., Morris R.J., Hall S.M. Selective expression of prion protein in peripheral tissues of the adult mouse. Neuroscience. 2002;113:177–192. doi: 10.1016/S0306-4522(02)00155-0. [DOI] [PubMed] [Google Scholar]
- 5.Miele G., Alejo Blanco A.R., Baybutt H., Horvat S., Manson J., Clinton M. Embryonic activation and developmental expression of the murine prion protein gene. Gene Expr. 2003;11:1–12. doi: 10.3727/000000003783992324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nicolas O., Gavin R., del Rio J.A. New insights into cellular prion protein (PrPc) functions: The “ying and yang” of a relevant protein. Brain Res. Rev. 2009;61:170–184. doi: 10.1016/j.brainresrev.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 7.Aguzzi A., Sigurdson C., Heikenwaelder M. Molecular mechanisms of prion pathogenesis. Annu. Rev. Pathol. 2008;3:11–40. doi: 10.1146/annurev.pathmechdis.3.121806.154326. [DOI] [PubMed] [Google Scholar]
- 8.Collinge J. Prion diseases of humans and animals: Their causes and molecular basis. Annu. Rev. Neurosci. 2001;24:519–550. doi: 10.1146/annurev.neuro.24.1.519. [DOI] [PubMed] [Google Scholar]
- 9.Aguzzi A., Baumann F., Bremer J. The prion’s elusive reason for being. Annu. Rev. Neurosci. 2008;31:439–477. doi: 10.1146/annurev.neuro.31.060407.125620. [DOI] [PubMed] [Google Scholar]
- 10.Aguzzi A., Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron. 2009;64:783–790. doi: 10.1016/j.neuron.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez-Borges N., Erana H., Elezgarai S.R., Harrathi C., Gayosso M., Castilla J. Infectivity versus Seeding in Neurodegenerative Diseases Sharing a Prion-Like Mechanism. Int J. Cell Biol. 2013;2013:583498. doi: 10.1155/2013/583498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cushman M., Johnson B.S., King O.D., Gitler A.D., Shorter J. Prion-like disorders: Blurring the divide between transmissibility and infectivity. J. Cell Sci. 2010;123:1191–1201. doi: 10.1242/jcs.051672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ayers J.I., Cashman N.R. Prion-like mechanisms in amyotrophic lateral sclerosis. Handb. Clin. Neurol. 2018;153:337–354. doi: 10.1016/B978-0-444-63945-5.00018-0. [DOI] [PubMed] [Google Scholar]
- 14.Cascarina S.M., Ross E.D. Natural and pathogenic protein sequence variation affecting prion-like domains within and across human proteomes. BMC Genom. 2020;21:23. doi: 10.1186/s12864-019-6425-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren P.H., Lauckner J.E., Kachirskaia I., Heuser J.E., Melki R., Kopito R.R. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell Biol. 2009;11:219–225. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luk K.C., Kehm V.M., Zhang B., O’Brien P., Trojanowski J.Q., Lee V.M. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J. Exp. Med. 2012;209:975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masuda-Suzukake M., Nonaka T., Hosokawa M., Oikawa T., Arai T., Akiyama H., Mann D.M., Hasegawa M. Prion-like spreading of pathological alpha-synuclein in brain. Brain. 2013;136:1128–1138. doi: 10.1093/brain/awt037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aulic S., Le T.T., Moda F., Abounit S., Corvaglia S., Casalis L., Gustincich S., Zurzolo C., Tagliavini F., Legname G. Defined alpha-synuclein prion-like molecular assemblies spreading in cell culture. BMC Neurosci. 2014;15:69. doi: 10.1186/1471-2202-15-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee H.J., Patel S., Lee S.J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 2005;25:6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Desplats P., Lee H.J., Bae E.J., Patrick C., Rockenstein E., Crews L., Spencer B., Masliah E., Lee S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. USA. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Del Rio J.A., Ferrer I., Gavin R. Role of cellular prion protein in interneuronal amyloid transmission. Prog. Neurobiol. 2018;165–167:87–102. doi: 10.1016/j.pneurobio.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 22.Urrea L., Segura-Feliu M., Masuda-Suzukake M., Hervera A., Pedraz L., Garcia-Aznar J.M., Vila M., Samitier J., Torrents E., Ferrer I., et al. Involvement of Cellular Prion Protein in alpha-Synuclein Transport in Neurons. Mol. Neurobiol. 2018;55:1847–1860. doi: 10.1007/s12035-017-0451-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferreira D.G., Temido-Ferreira M., Vicente Miranda H., Batalha V.L., Coelho J.E., Szego E.M., Marques-Morgado I., Vaz S.H., Rhee J.S., Schmitz M., et al. alpha-synuclein interacts with PrP(C) to induce cognitive impairment through mGluR5 and NMDAR2B. Nat. Neurosci. 2017;20:1569–1579. doi: 10.1038/nn.4648. [DOI] [PubMed] [Google Scholar]
- 24.Nath S., Agholme L., Kurudenkandy F.R., Granseth B., Marcusson J., Hallbeck M. Spreading of neurodegenerative pathology via neuron-to-neuron transmission of beta-amyloid. J. Neurosci. 2012;32:8767–8777. doi: 10.1523/JNEUROSCI.0615-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Domert J., Rao S.B., Agholme L., Brorsson A.C., Marcusson J., Hallbeck M., Nath S. Spreading of amyloid-beta peptides via neuritic cell-to-cell transfer is dependent on insufficient cellular clearance. Neurobiol. Dis. 2014;65:82–92. doi: 10.1016/j.nbd.2013.12.019. [DOI] [PubMed] [Google Scholar]
- 26.Eisele Y.S., Obermuller U., Heilbronner G., Baumann F., Kaeser S.A., Wolburg H., Walker L.C., Staufenbiel M., Heikenwalder M., Jucker M. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science. 2010;330:980–982. doi: 10.1126/science.1194516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clavaguera F., Bolmont T., Crowther R.A., Abramowski D., Frank S., Probst A., Fraser G., Stalder A.K., Beibel M., Staufenbiel M., et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009;11:909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frost B., Ollesch J., Wille H., Diamond M.I. Conformational diversity of wild-type Tau fibrils specified by templated conformation change. J. Biol. Chem. 2009;284:3546–3551. doi: 10.1074/jbc.M805627200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sydow A., Mandelkow E.M. ‘Prion-like’ propagation of mouse and human tau aggregates in an inducible mouse model of tauopathy. Neurodegener. Dis. 2010;7:28–31. doi: 10.1159/000283479. [DOI] [PubMed] [Google Scholar]
- 30.Guo J.L., Lee V.M. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 2011;286:15317–15331. doi: 10.1074/jbc.M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clavaguera F., Akatsu H., Fraser G., Crowther R.A., Frank S., Hench J., Probst A., Winkler D.T., Reichwald J., Staufenbiel M., et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. USA. 2013;110:9535–9540. doi: 10.1073/pnas.1301175110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Michelitsch M.D., Weissman J.S. A census of glutamine/asparagine-rich regions: Implications for their conserved function and the prediction of novel prions. Proc. Natl. Acad. Sci. USA. 2000;97:11910–11915. doi: 10.1073/pnas.97.22.11910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sabate R., Rousseau F., Schymkowitz J., Ventura S. What makes a protein sequence a prion? PLoS Comput. Biol. 2015;11:e1004013. doi: 10.1371/journal.pcbi.1004013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jaunmuktane Z., Mead S., Ellis M., Wadsworth J.D., Nicoll A.J., Kenny J., Launchbury F., Linehan J., Richard-Loendt A., Walker A.S., et al. Evidence for human transmission of amyloid-beta pathology and cerebral amyloid angiopathy. Nature. 2015;525:247–250. doi: 10.1038/nature15369. [DOI] [PubMed] [Google Scholar]
- 35.Frontzek K., Lutz M.I., Aguzzi A., Kovacs G.G., Budka H. Amyloid-beta pathology and cerebral amyloid angiopathy are frequent in iatrogenic Creutzfeldt-Jakob disease after dural grafting. Swiss Med. Wkly. 2016;146:w14287. doi: 10.4414/smw.2016.14287. [DOI] [PubMed] [Google Scholar]
- 36.Kovacs G.G., Lutz M.I., Ricken G., Strobel T., Hoftberger R., Preusser M., Regelsberger G., Honigschnabl S., Reiner A., Fischer P., et al. Dura mater is a potential source of Abeta seeds. Acta Neuropathol. 2016;131:911–923. doi: 10.1007/s00401-016-1565-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hansen L.A., Masliah E., Terry R.D., Mirra S.S. A neuropathological subset of Alzheimer’s disease with concomitant Lewy body disease and spongiform change. Acta Neuropathol. 1989;78:194–201. doi: 10.1007/BF00688209. [DOI] [PubMed] [Google Scholar]
- 38.Race B., Phillips K., Kraus A., Chesebro B. Phosphorylated human tau associates with mouse prion protein amyloid in scrapie-infected mice but does not increase progression of clinical disease. Prion. 2016;10:319–330. doi: 10.1080/19336896.2016.1199313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Debatin L., Streffer J., Geissen M., Matschke J., Aguzzi A., Glatzel M. Association between deposition of beta-amyloid and pathological prion protein in sporadic Creutzfeldt-Jakob disease. Neurodegener. Dis. 2008;5:347–354. doi: 10.1159/000121389. [DOI] [PubMed] [Google Scholar]
- 40.Hainfellner J.A., Wanschitz J., Jellinger K., Liberski P.P., Gullotta F., Budka H. Coexistence of Alzheimer-type neuropathology in Creutzfeldt-Jakob disease. Acta Neuropathol. 1998;96:116–122. doi: 10.1007/s004010050870. [DOI] [PubMed] [Google Scholar]
- 41.Barcikowska M., Kwiecinski H., Liberski P.P., Kowalski J., Brown P., Gajdusek D.C. Creutzfeldt-Jakob disease with Alzheimer-type A beta-reactive amyloid plaques. Histopathology. 1995;26:445–450. doi: 10.1111/j.1365-2559.1995.tb00252.x. [DOI] [PubMed] [Google Scholar]
- 42.Leuba G., Saini K., Savioz A., Charnay Y. Early-onset familial Alzheimer disease with coexisting beta-amyloid and prion pathology. JAMA. 2000;283:1689–1691. doi: 10.1001/jama.283.13.1689-a. [DOI] [PubMed] [Google Scholar]
- 43.Alzualde A., Indakoetxea B., Ferrer I., Moreno F., Barandiaran M., Gorostidi A., Estanga A., Ruiz I., Calero M., van Leeuwen F.W., et al. A novel PRNP Y218N mutation in Gerstmann-Straussler-Scheinker disease with neurofibrillary degeneration. J. Neuropathol. Exp. Neurol. 2010;69:789–800. doi: 10.1097/NEN.0b013e3181e85737. [DOI] [PubMed] [Google Scholar]
- 44.Irwin D.J., Lee V.M., Trojanowski J.Q. Parkinson’s disease dementia: Convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat. Rev. Neurosci. 2013;14:626–636. doi: 10.1038/nrn3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cali I., Cohen M.L., Haik S., Parchi P., Giaccone G., Collins S.J., Kofskey D., Wang H., McLean C.A., Brandel J.P., et al. Iatrogenic Creutzfeldt-Jakob disease with Amyloid-beta pathology: An international study. Acta Neuropathol. Commun. 2018;6:5. doi: 10.1186/s40478-017-0503-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dasari A.K.R., Kayed R., Wi S., Lim K.H. Tau Interacts with the C-Terminal Region of alpha-Synuclein, Promoting Formation of Toxic Aggregates with Distinct Molecular Conformations. Biochemistry. 2019;58:2814–2821. doi: 10.1021/acs.biochem.9b00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Han J., Zhang J., Yao H., Wang X., Li F., Chen L., Gao C., Gao J., Nie K., Zhou W., et al. Study on interaction between microtubule associated protein tau and prion protein. Sci. China C Life Sci. 2006;49:473–479. doi: 10.1007/s11427-006-2019-9. [DOI] [PubMed] [Google Scholar]
- 48.Wang X.F., Dong C.F., Zhang J., Wan Y.Z., Li F., Huang Y.X., Han L., Shan B., Gao C., Han J., et al. Human tau protein forms complex with PrP and some GSS- and fCJD-related PrP mutants possess stronger binding activities with tau in vitro. Mol. Cell Biochem. 2008;310:49–55. doi: 10.1007/s11010-007-9664-6. [DOI] [PubMed] [Google Scholar]
- 49.Ferrer I., Blanco R., Carmona M., Puig B., Ribera R., Rey M.J., Ribalta T. Prion protein expression in senile plaques in Alzheimer’s disease. Acta Neuropathol. 2001;101:49–56. doi: 10.1007/s004010000271. [DOI] [PubMed] [Google Scholar]
- 50.Lauren J., Gimbel D.A., Nygaard H.B., Gilbert J.W., Strittmatter S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meyne F., Gloeckner S.F., Ciesielczyk B., Heinemann U., Krasnianski A., Meissner B., Zerr I. Total prion protein levels in the cerebrospinal fluid are reduced in patients with various neurological disorders. J. Alzheimers Dis. 2009;17:863–873. doi: 10.3233/JAD-2009-1110. [DOI] [PubMed] [Google Scholar]
- 52.Llorens F., Villar-Pique A., Schmitz M., Diaz-Lucena D., Wohlhage M., Hermann P., Goebel S., Schmidt I., Glatzel M., Hauw J.J., et al. Plasma total prion protein as a potential biomarker for neurodegenerative dementia: Diagnostic accuracy in the spectrum of prion diseases. Neuropathol. Appl. Neurobiol. 2019 doi: 10.1111/nan.12573. [DOI] [PubMed] [Google Scholar]
- 53.Aguzzi A., Lakkaraju A.K. Cell Biology of Prions and Prionoids: A Status Report. Trends Cell Biol. 2016;26:40–51. doi: 10.1016/j.tcb.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 54.Houston F., Andreoletti O. Animal prion diseases: The risks to human health. Brain Pathol. 2019;29:248–262. doi: 10.1111/bpa.12696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Asher D.M., Gregori L. Human transmissible spongiform encephalopathies: Historic view. Handb. Clin. Neurol. 2018;153:1–17. doi: 10.1016/B978-0-444-63945-5.00001-5. [DOI] [PubMed] [Google Scholar]
- 56.O’Carroll A., Coyle J., Gambin Y. Prions and Prion-like assemblies in neurodegeneration and immunity: The emergence of universal mechanisms across health and disease. Semin. Cell Dev. Biol. 2019 doi: 10.1016/j.semcdb.2019.11.012. [DOI] [PubMed] [Google Scholar]
- 57.Bueler H., Fischer M., Lang Y., Bluethmann H., Lipp H.P., DeArmond S.J., Prusiner S.B., Aguet M., Weissmann C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- 58.Rossi D., Cozzio A., Flechsig E., Klein M.A., Rulicke T., Aguzzi A., Weissmann C. Onset of ataxia and Purkinje cell loss in PrP null mice inversely correlated with Dpl level in brain. EMBO J. 2001;20:694–702. doi: 10.1093/emboj/20.4.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Manson J.C., Clarke A.R., Hooper M.L., Aitchison L., McConnell I., Hope J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol. Neurobiol. 1994;8:121–127. doi: 10.1007/BF02780662. [DOI] [PubMed] [Google Scholar]
- 60.Nuvolone M., Hermann M., Sorce S., Russo G., Tiberi C., Schwarz P., Minikel E., Sanoudou D., Pelczar P., Aguzzi A. Strictly co-isogenic C57BL/6J-Prnp-/- mice: A rigorous resource for prion science. J. Exp. Med. 2016;213:313–327. doi: 10.1084/jem.20151610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Steele A.D., Lindquist S., Aguzzi A. The prion protein knockout mouse: A phenotype under challenge. Prion. 2007;1:83–93. doi: 10.4161/pri.1.2.4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Del Rio J.A., Gavin R. Functions of the cellular prion protein, the end of Moore’s law, and Ockham’s razor theory. Prion. 2016;10:25–40. doi: 10.1080/19336896.2015.1126038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spielhaupter C., Schatzl H.M. PrPC directly interacts with proteins involved in signaling pathways. J. Biol. Chem. 2001;276:44604–44612. doi: 10.1074/jbc.M103289200. [DOI] [PubMed] [Google Scholar]
- 64.Schmitt-Ulms G., Legname G., Baldwin M.A., Ball H.L., Bradon N., Bosque P.J., Crossin K.L., Edelman G.M., DeArmond S.J., Cohen F.E., et al. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J. Mol. Biol. 2001;314:1209–1225. doi: 10.1006/jmbi.2000.5183. [DOI] [PubMed] [Google Scholar]
- 65.Zafar S., von Ahsen N., Oellerich M., Zerr I., Schulz-Schaeffer W.J., Armstrong V.W., Asif A.R. Proteomics approach to identify the interacting partners of cellular prion protein and characterization of Rab7a interaction in neuronal cells. J. Proteome Res. 2011;10:3123–3135. doi: 10.1021/pr2001989. [DOI] [PubMed] [Google Scholar]
- 66.Kuffer A., Lakkaraju A.K., Mogha A., Petersen S.C., Airich K., Doucerain C., Marpakwar R., Bakirci P., Senatore A., Monnard A., et al. The prion protein is an agonistic ligand of the G protein-coupled receptor Adgrg6. Nature. 2016;536:464–468. doi: 10.1038/nature19312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.La Vitola P., Beeg M., Balducci C., Santamaria G., Restelli E., Colombo L., Caldinelli L., Pollegioni L., Gobbi M., Chiesa R., et al. Cellular prion protein neither binds to alpha-synuclein oligomers nor mediates their detrimental effects. Brain. 2019;142:249–254. doi: 10.1093/brain/awy318. [DOI] [PubMed] [Google Scholar]
- 68.Azzalin A., Del Vecchio I., Chiarelli L.R., Valentini G., Comincini S., Ferretti L. Absence of interaction between doppel and GFAP, Grb2, PrPc proteins in human tumor astrocytic cells. Anticancer Res. 2005;25:4369–4374. [PubMed] [Google Scholar]
- 69.Nuvolone M., Kana V., Hutter G., Sakata D., Mortin-Toth S.M., Russo G., Danska J.S., Aguzzi A. SIRPalpha polymorphisms, but not the prion protein, control phagocytosis of apoptotic cells. J. Exp. Med. 2013;210:2539–2552. doi: 10.1084/jem.20131274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Linden R. The Biological Function of the Prion Protein: A Cell Surface Scaffold of Signaling Modules. Front. Mol. Neurosci. 2017;10:77. doi: 10.3389/fnmol.2017.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brown D.R., Qin K., Herms J.W., Madlung A., Manson J., Strome R., Fraser P.E., Kruck T., von Bohlen A., Schulz-Schaeffer W., et al. The cellular prion protein binds copper in vivo. Nature. 1997;390:684–687. doi: 10.1038/37783. [DOI] [PubMed] [Google Scholar]
- 72.Viles J.H., Cohen F.E., Prusiner S.B., Goodin D.B., Wright P.E., Dyson H.J. Copper binding to the prion protein: Structural implications of four identical cooperative binding sites. Proc. Natl. Acad. Sci. USA. 1999;96:2042–2047. doi: 10.1073/pnas.96.5.2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Evans E.G., Pushie M.J., Markham K.A., Lee H.W., Millhauser G.L. Interaction between Prion Protein’s Copper-Bound Octarepeat Domain and a Charged C-Terminal Pocket Suggests a Mechanism for N-Terminal Regulation. Structure. 2016;24:1057–1067. doi: 10.1016/j.str.2016.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nguyen X.T.A., Tran T.H., Cojoc D., Legname G. Copper Binding Regulates Cellular Prion Protein Function. Mol. Neurobiol. 2019;56:6121–6133. doi: 10.1007/s12035-019-1510-9. [DOI] [PubMed] [Google Scholar]
- 75.Vassallo N., Herms J. Cellular prion protein function in copper homeostasis and redox signalling at the synapse. J. Neurochem. 2003;86:538–544. doi: 10.1046/j.1471-4159.2003.01882.x. [DOI] [PubMed] [Google Scholar]
- 76.Brown D.R., Nicholas R.S., Canevari L. Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J. Neurosci. Res. 2002;67:211–224. doi: 10.1002/jnr.10118. [DOI] [PubMed] [Google Scholar]
- 77.Wong B.S., Liu T., Li R., Pan T., Petersen R.B., Smith M.A., Gambetti P., Perry G., Manson J.C., Brown D.R., et al. Increased levels of oxidative stress markers detected in the brains of mice devoid of prion protein. J. Neurochem. 2001;76:565–572. doi: 10.1046/j.1471-4159.2001.00028.x. [DOI] [PubMed] [Google Scholar]
- 78.Klamt F., Dal-Pizzol F., Conte da Frota M.L., Jr., Walz R., Andrades M.E., da Silva E.G., Brentani R.R., Izquierdo I., Fonseca Moreira J.C. Imbalance of antioxidant defense in mice lacking cellular prion protein. Free Radic Biol. Med. 2001;30:1137–1144. doi: 10.1016/S0891-5849(01)00512-3. [DOI] [PubMed] [Google Scholar]
- 79.Brown D.R., Schulz-Schaeffer W.J., Schmidt B., Kretzschmar H.A. Prion protein-deficient cells show altered response to oxidative stress due to decreased SOD-1 activity. Exp. Neurol. 1997;146:104–112. doi: 10.1006/exnr.1997.6505. [DOI] [PubMed] [Google Scholar]
- 80.Brown D.R., Besinger A. Prion protein expression and superoxide dismutase activity. Biochem. J. 1998;334 (Pt. 2):423–429. doi: 10.1042/bj3340423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kuwahara C., Takeuchi A.M., Nishimura T., Haraguchi K., Kubosaki A., Matsumoto Y., Saeki K., Matsumoto Y., Yokoyama T., Itohara S., et al. Prions prevent neuronal cell-line death. Nature. 1999;400:225–226. doi: 10.1038/22241. [DOI] [PubMed] [Google Scholar]
- 82.Withee J.L., Sen R., Cyert M.S. Ion tolerance of Saccharomyces cerevisiae lacking the Ca2+/CaM-dependent phosphatase (calcineurin) is improved by mutations in URE2 or PMA1. Genetics. 1998;149:865–878. doi: 10.1093/genetics/149.2.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.White A.R., Collins S.J., Maher F., Jobling M.F., Stewart L.R., Thyer J.M., Beyreuther K., Masters C.L., Cappai R. Prion protein-deficient neurons reveal lower glutathione reductase activity and increased susceptibility to hydrogen peroxide toxicity. Am. J. Pathol. 1999;155:1723–1730. doi: 10.1016/S0002-9440(10)65487-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nishimura T., Sakudo A., Nakamura I., Lee D.C., Taniuchi Y., Saeki K., Matsumoto Y., Ogawa M., Sakaguchi S., Itohara S., et al. Cellular prion protein regulates intracellular hydrogen peroxide level and prevents copper-induced apoptosis. Biochem. Biophys. Res. Commun. 2004;323:218–222. doi: 10.1016/j.bbrc.2004.08.087. [DOI] [PubMed] [Google Scholar]
- 85.Qin K., Zhao L., Ash R.D., McDonough W.F., Zhao R.Y. ATM-mediated transcriptional elevation of prion in response to copper-induced oxidative stress. J. Biol. Chem. 2009;284:4582–4593. doi: 10.1074/jbc.M808410200. [DOI] [PubMed] [Google Scholar]
- 86.Sakudo A., Lee D.C., Saeki K., Nakamura Y., Inoue K., Matsumoto Y., Itohara S., Onodera T. Impairment of superoxide dismutase activation by N-terminally truncated prion protein (PrP) in PrP-deficient neuronal cell line. Biochem. Biophys. Res. Commun. 2003;308:660–667. doi: 10.1016/S0006-291X(03)01459-1. [DOI] [PubMed] [Google Scholar]
- 87.McLennan N.F., Brennan P.M., McNeill A., Davies I., Fotheringham A., Rennison K.A., Ritchie D., Brannan F., Head M.W., Ironside J.W., et al. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am. J. Pathol. 2004;165:227–235. doi: 10.1016/S0002-9440(10)63291-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hoshino S., Inoue K., Yokoyama T., Kobayashi S., Asakura T., Teramoto A., Itohara S. Prions prevent brain damage after experimental brain injury: A preliminary report. Acta Neurochir. Suppl. 2003;86:297–299. doi: 10.1007/978-3-7091-0651-8_64. [DOI] [PubMed] [Google Scholar]
- 89.Shyu W.C., Lin S.Z., Chiang M.F., Ding D.C., Li K.W., Chen S.F., Yang H.I., Li H. Overexpression of PrPC by adenovirus-mediated gene targeting reduces ischemic injury in a stroke rat model. J. Neurosci. 2005;25:8967–8977. doi: 10.1523/JNEUROSCI.1115-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Spudich A., Frigg R., Kilic E., Kilic U., Oesch B., Raeber A., Bassetti C.L., Hermann D.M. Aggravation of ischemic brain injury by prion protein deficiency: Role of ERK-1/-2 and STAT-1. Neurobiol. Dis. 2005;20:442–449. doi: 10.1016/j.nbd.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 91.Weise J., Crome O., Sandau R., Schulz-Schaeffer W., Bahr M., Zerr I. Upregulation of cellular prion protein (PrPc) after focal cerebral ischemia and influence of lesion severity. Neurosci. Lett. 2004;372:146–150. doi: 10.1016/j.neulet.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 92.Williams W.M., Stadtman E.R., Moskovitz J. Ageing and exposure to oxidative stress in vivo differentially affect cellular levels of PrP in mouse cerebral microvessels and brain parenchyma. Neuropathol. Appl. Neurobiol. 2004;30:161–168. doi: 10.1111/j.1365-2990.2003.00523.x. [DOI] [PubMed] [Google Scholar]
- 93.Esiri M.M., Carter J., Ironside J.W. Prion protein immunoreactivity in brain samples from an unselected autopsy population: Findings in 200 consecutive cases. Neuropathol. Appl. Neurobiol. 2000;26:273–284. doi: 10.1046/j.1365-2990.2000.00239.x. [DOI] [PubMed] [Google Scholar]
- 94.Doeppner T.R., Kaltwasser B., Schlechter J., Jaschke J., Kilic E., Bahr M., Hermann D.M., Weise J. Cellular prion protein promotes post-ischemic neuronal survival, angioneurogenesis and enhances neural progenitor cell homing via proteasome inhibition. Cell Death Dis. 2015;6:e2024. doi: 10.1038/cddis.2015.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bounhar Y., Zhang Y., Goodyer C.G., LeBlanc A. Prion protein protects human neurons against Bax-mediated apoptosis. J. Biol. Chem. 2001;276:39145–39149. doi: 10.1074/jbc.C100443200. [DOI] [PubMed] [Google Scholar]
- 96.Yin X.M., Oltvai Z.N., Korsmeyer S.J. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature. 1994;369:321–323. doi: 10.1038/369321a0. [DOI] [PubMed] [Google Scholar]
- 97.Kurschner C., Morgan J.I. Analysis of interaction sites in homo- and heteromeric complexes containing Bcl-2 family members and the cellular prion protein. Brain Res. 1996;37:249–258. doi: 10.1016/0169-328X(95)00323-K. [DOI] [PubMed] [Google Scholar]
- 98.Kurschner C., Morgan J.I. The cellular prion protein (PrP) selectively binds to Bcl-2 in the yeast two-hybrid system. Brain Res. 1995;30:165–168. doi: 10.1016/0169-328X(95)00013-I. [DOI] [PubMed] [Google Scholar]
- 99.Kim B.H., Lee H.G., Choi J.K., Kim J.I., Choi E.K., Carp R.I., Kim Y.S. The cellular prion protein (PrPC) prevents apoptotic neuronal cell death and mitochondrial dysfunction induced by serum deprivation. Brain Res. 2004;124:40–50. doi: 10.1016/j.molbrainres.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 100.Herms J.W., Korte S., Gall S., Schneider I., Dunker S., Kretzschmar H.A. Altered intracellular calcium homeostasis in cerebellar granule cells of prion protein-deficient mice. J. Neurochem. 2000;75:1487–1492. doi: 10.1046/j.1471-4159.2000.0751487.x. [DOI] [PubMed] [Google Scholar]
- 101.Korte S., Vassallo N., Kramer M.L., Kretzschmar H.A., Herms J. Modulation of L-type voltage-gated calcium channels by recombinant prion protein. J. Neurochem. 2003;87:1037–1042. doi: 10.1046/j.1471-4159.2003.02080.x. [DOI] [PubMed] [Google Scholar]
- 102.Fuhrmann M., Bittner T., Mitteregger G., Haider N., Moosmang S., Kretzschmar H., Herms J. Loss of the cellular prion protein affects the Ca2+ homeostasis in hippocampal CA1 neurons. J. Neurochem. 2006;98:1876–1885. doi: 10.1111/j.1471-4159.2006.04011.x. [DOI] [PubMed] [Google Scholar]
- 103.Krebs B., Wiebelitz A., Balitzki-Korte B., Vassallo N., Paluch S., Mitteregger G., Onodera T., Kretzschmar H.A., Herms J. Cellular prion protein modulates the intracellular calcium response to hydrogen peroxide. J. Neurochem. 2007;100:358–367. doi: 10.1111/j.1471-4159.2006.04256.x. [DOI] [PubMed] [Google Scholar]
- 104.Jayaraman T., Ondrias K., Ondriasova E., Marks A.R. Regulation of the inositol 1,4,5-trisphosphate receptor by tyrosine phosphorylation. Science. 1996;272:1492–1494. doi: 10.1126/science.272.5267.1492. [DOI] [PubMed] [Google Scholar]
- 105.Rangel A., Burgaya F., Gavin R., Soriano E., Aguzzi A., Del Rio J.A. Enhanced susceptibility of Prnp-deficient mice to kainate-induced seizures, neuronal apoptosis, and death: Role of AMPA/kainate receptors. J. Neurosci. Res. 2007;85:2741–2755. doi: 10.1002/jnr.21215. [DOI] [PubMed] [Google Scholar]
- 106.Carulla P., Bribian A., Rangel A., Gavin R., Ferrer I., Caelles C., Del Rio J.A., Llorens F. Neuroprotective role of PrPC against kainate-induced epileptic seizures and cell death depends on the modulation of JNK3 activation by GluR6/7-PSD-95 binding. Mol. Biol. Cell. 2011;22:3041–3054. doi: 10.1091/mbc.e11-04-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Carulla P., Llorens F., Matamoros-Angles A., Aguilar-Calvo P., Espinosa J.C., Gavin R., Ferrer I., Legname G., Torres J.M., del Rio J.A. Involvement of PrP(C) in kainate-induced excitotoxicity in several mouse strains. Sci. Rep. 2015;5:11971. doi: 10.1038/srep11971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gasperini L., Meneghetti E., Pastore B., Benetti F., Legname G. Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid Redox Signal. 2015;22:772–784. doi: 10.1089/ars.2014.6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Khosravani H., Zhang Y., Tsutsui S., Hameed S., Altier C., Hamid J., Chen L., Villemaire M., Ali Z., Jirik F.R., et al. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J. Cell Biol. 2008;181:551–565. doi: 10.1083/jcb.200711002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Stys P.K., You H., Zamponi G.W. Copper-dependent regulation of NMDA receptors by cellular prion protein: Implications for neurodegenerative disorders. J. Physiol. 2012;590:1357–1368. doi: 10.1113/jphysiol.2011.225276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pham N., Dhar A., Khalaj S., Desai K., Taghibiglou C. Down regulation of brain cellular prion protein in an animal model of insulin resistance: Possible implication in increased prevalence of stroke in pre-diabetics/diabetics. Biochem. Biophys. Res. Commun. 2014;448:151–156. doi: 10.1016/j.bbrc.2014.04.071. [DOI] [PubMed] [Google Scholar]
- 112.Llorens F., Del Rio J.A. Unraveling the neuroprotective mechanisms of PrP (C) in excitotoxicity. Prion. 2012;6:245–251. doi: 10.4161/pri.19639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Black S.A., Stys P.K., Zamponi G.W., Tsutsui S. Cellular prion protein and NMDA receptor modulation: Protecting against excitotoxicity. Front. Cell Dev. Biol. 2014;2:45. doi: 10.3389/fcell.2014.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Graner E., Mercadante A.F., Zanata S.M., Forlenza O.V., Cabral A.L., Veiga S.S., Juliano M.A., Roesler R., Walz R., Minetti A., et al. Cellular prion protein binds laminin and mediates neuritogenesis. Brain Res. 2000;76:85–92. doi: 10.1016/S0169-328X(99)00334-4. [DOI] [PubMed] [Google Scholar]
- 115.Chen S., Mange A., Dong L., Lehmann S., Schachner M. Prion protein as trans-interacting partner for neurons is involved in neurite outgrowth and neuronal survival. Mol. Cell. Neurosci. 2003;22:227–233. doi: 10.1016/S1044-7431(02)00014-3. [DOI] [PubMed] [Google Scholar]
- 116.Chiarini L.B., Freitas A.R., Zanata S.M., Brentani R.R., Martins V.R., Linden R. Cellular prion protein transduces neuroprotective signals. EMBO J. 2002;21:3317–3326. doi: 10.1093/emboj/cdf324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mouillet-Richard S., Ermonval M., Chebassier C., Laplanche J.L., Lehmann S., Launay J.M., Kellermann O. Signal transduction through prion protein. Science. 2000;289:1925–1928. doi: 10.1126/science.289.5486.1925. [DOI] [PubMed] [Google Scholar]
- 118.Schneider B., Mutel V., Pietri M., Ermonval M., Mouillet-Richard S., Kellermann O. NADPH oxidase and extracellular regulated kinases 1/2 are targets of prion protein signaling in neuronal and nonneuronal cells. Proc. Natl. Acad. Sci. USA. 2003;100:13326–13331. doi: 10.1073/pnas.2235648100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pradines E., Loubet D., Schneider B., Launay J.M., Kellermann O., Mouillet-Richard S. CREB-dependent gene regulation by prion protein: Impact on MMP-9 and beta-dystroglycan. Cell Signal. 2008;20:2050–2058. doi: 10.1016/j.cellsig.2008.07.016. [DOI] [PubMed] [Google Scholar]
- 120.Hernandez-Rapp J., Martin-Lanneree S., Hirsch T.Z., Pradines E., Alleaume-Butaux A., Schneider B., Baudry A., Launay J.M., Mouillet-Richard S. A PrP(C)-caveolin-Lyn complex negatively controls neuronal GSK3beta and serotonin 1B receptor. Sci. Rep. 2014;4:4881. doi: 10.1038/srep04881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rambold A.S., Muller V., Ron U., Ben-Tal N., Winklhofer K.F., Tatzelt J. Stress-protective signalling of prion protein is corrupted by scrapie prions. EMBO J. 2008;27:1974–1984. doi: 10.1038/emboj.2008.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zanata S.M., Lopes M.H., Mercadante A.F., Hajj G.N., Chiarini L.B., Nomizo R., Freitas A.R., Cabral A.L., Lee K.S., Juliano M.A., et al. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 2002;21:3307–3316. doi: 10.1093/emboj/cdf325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lopes M.H., Hajj G.N., Muras A.G., Mancini G.L., Castro R.M., Ribeiro K.C., Brentani R.R., Linden R., Martins V.R. Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J. Neurosci. 2005;25:11330–11339. doi: 10.1523/JNEUROSCI.2313-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sakudo A., Lee D.C., Li S., Nakamura T., Matsumoto Y., Saeki K., Itohara S., Ikuta K., Onodera T. PrP cooperates with STI1 to regulate SOD activity in PrP-deficient neuronal cell line. Biochem. Biophys. Res. Commun. 2005;328:14–19. doi: 10.1016/j.bbrc.2004.12.132. [DOI] [PubMed] [Google Scholar]
- 125.Amin L., Nguyen X.T., Rolle I.G., D’Este E., Giachin G., Tran T.H., Serbec V.C., Cojoc D., Legname G. Characterization of prion protein function by focal neurite stimulation. J. Cell Sci. 2016;129:3878–3891. doi: 10.1242/jcs.183137. [DOI] [PubMed] [Google Scholar]
- 126.Lysek D.A., Wuthrich K. Prion protein interaction with the C-terminal SH3 domain of Grb2 studied using NMR and optical spectroscopy. Biochemistry. 2004;43:10393–10399. doi: 10.1021/bi0494828. [DOI] [PubMed] [Google Scholar]
- 127.Donne D.G., Viles J.H., Groth D., Mehlhorn I., James T.L., Cohen F.E., Prusiner S.B., Wright P.E., Dyson H.J. Structure of the recombinant full-length hamster prion protein PrP(29-231): The N terminus is highly flexible. Proc. Natl. Acad. Sci. USA. 1997;94:13452–13457. doi: 10.1073/pnas.94.25.13452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Riek R., Hornemann S., Wider G., Glockshuber R., Wuthrich K. NMR characterization of the full-length recombinant murine prion protein, mPrP(23-231) Febs Lett. 1997;413:282–288. doi: 10.1016/S0014-5793(97)00920-4. [DOI] [PubMed] [Google Scholar]
- 129.Riek R., Hornemann S., Wider G., Billeter M., Glockshuber R., Wuthrich K. NMR structure of the mouse prion protein domain PrP(121-321) Nature. 1996;382:180–182. doi: 10.1038/382180a0. [DOI] [PubMed] [Google Scholar]
- 130.Chen S.G., Teplow D.B., Parchi P., Teller J.K., Gambetti P., Autilio-Gambetti L. Truncated forms of the human prion protein in normal brain and in prion diseases. J. Biol. Chem. 1995;270:19173–19180. doi: 10.1074/jbc.270.32.19173. [DOI] [PubMed] [Google Scholar]
- 131.Mange A., Beranger F., Peoc’h K., Onodera T., Frobert Y., Lehmann S. Alpha- and beta- cleavages of the amino-terminus of the cellular prion protein. Biol. Cell. 2004;96:125–132. doi: 10.1016/j.biolcel.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 132.McMahon H.E., Mange A., Nishida N., Creminon C., Casanova D., Lehmann S. Cleavage of the amino terminus of the prion protein by reactive oxygen species. J. Biol. Chem. 2001;276:2286–2291. doi: 10.1074/jbc.M007243200. [DOI] [PubMed] [Google Scholar]
- 133.Resenberger U.K., Harmeier A., Woerner A.C., Goodman J.L., Muller V., Krishnan R., Vabulas R.M., Kretzschmar H.A., Lindquist S., Hartl F.U., et al. The cellular prion protein mediates neurotoxic signalling of beta-sheet-rich conformers independent of prion replication. EMBO J. 2011;30:2057–2070. doi: 10.1038/emboj.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Guillot-Sestier M.V., Sunyach C., Druon C., Scarzello S., Checler F. The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J. Biol. Chem. 2009;284:35973–35986. doi: 10.1074/jbc.M109.051086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Resenberger U.K., Winklhofer K.F., Tatzelt J. Neuroprotective and neurotoxic signaling by the prion protein. Top. Curr. Chem. 2011;305:101–119. doi: 10.1007/128_2011_160. [DOI] [PubMed] [Google Scholar]
- 136.Beland M., Motard J., Barbarin A., Roucou X. PrP(C) homodimerization stimulates the production of PrPC cleaved fragments PrPN1 and PrPC1. J. Neurosci. 2012;32:13255–13263. doi: 10.1523/JNEUROSCI.2236-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.McDonald A.J., Millhauser G.L. PrP overdrive: Does inhibition of alpha-cleavage contribute to PrP(C) toxicity and prion disease? Prion. 2014:8. doi: 10.4161/pri.28796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mitteregger G., Vosko M., Krebs B., Xiang W., Kohlmannsperger V., Nolting S., Hamann G.F., Kretzschmar H.A. The role of the octarepeat region in neuroprotective function of the cellular prion protein. Brain Pathol. 2007;17:174–183. doi: 10.1111/j.1750-3639.2007.00061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lee K.J., Panzera A., Rogawski D., Greene L.E., Eisenberg E. Celular prion protein protects (PrPC) protects neuronal cells from the effect of huntingtin aggregation. J. Cell Sci. 2007;120:2663–2671. doi: 10.1242/jcs.004598. [DOI] [PubMed] [Google Scholar]
- 140.Kiachopoulos S., Heske J., Tatzelt J., Winklhofer K.F. Misfolding of the prion protein at the plasma membrane induces endocytosis, intracellular retention and degradation. Traffic. 2004;5:426–436. doi: 10.1111/j.1398-9219.2004.00185.x. [DOI] [PubMed] [Google Scholar]
- 141.Prado M.A., Alves-Silva J., Magalhaes A.C., Prado V.F., Linden R., Martins V.R., Brentani R.R. PrPc on the road: Trafficking of the cellular prion protein. J. Neurochem. 2004;88:769–781. doi: 10.1046/j.1471-4159.2003.02199.x. [DOI] [PubMed] [Google Scholar]
- 142.Americo T.A., Chiarini L.B., Linden R. Signaling induced by hop/STI-1 depends on endocytosis. Biochem. Biophys. Res. Commun. 2007;358:620–625. doi: 10.1016/j.bbrc.2007.04.202. [DOI] [PubMed] [Google Scholar]
- 143.Caetano F.A., Lopes M.H., Hajj G.N., Machado C.F., Pinto Arantes C., Magalhaes A.C., Vieira Mde P., Americo T.A., Massensini A.R., Priola S.A., et al. Endocytosis of prion protein is required for ERK1/2 signaling induced by stress-inducible protein 1. J. Neurosci. 2008;28:6691–6702. doi: 10.1523/JNEUROSCI.1701-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mironov A., Jr., Latawiec D., Wille H., Bouzamondo-Bernstein E., Legname G., Williamson R.A., Burton D., DeArmond S.J., Prusiner S.B., Peters P.J. Cytosolic prion protein in neurons. J. Neurosci. 2003;23:7183–7193. doi: 10.1523/JNEUROSCI.23-18-07183.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Roucou X., Guo Q., Zhang Y., Goodyer C.G., LeBlanc A.C. Cytosolic prion protein is not toxic and protects against Bax-mediated cell death in human primary neurons. J. Biol. Chem. 2003;278:40877–40881. doi: 10.1074/jbc.M306177200. [DOI] [PubMed] [Google Scholar]
- 146.Lin D.T., Jodoin J., Baril M., Goodyer C.G., Leblanc A.C. Cytosolic prion protein is the predominant anti-Bax prion protein form: Exclusion of transmembrane and secreted prion protein forms in the anti-Bax function. Biochim. Et Biophys. Acta. 2008;1783:2001–2012. doi: 10.1016/j.bbamcr.2008.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hegde R.S., Mastrianni J.A., Scott M.R., DeFea K.A., Tremblay P., Torchia M., DeArmond S.J., Prusiner S.B., Lingappa V.R. A transmembrane form of the prion protein in neurodegenerative disease. Science. 1998;279:827–834. doi: 10.1126/science.279.5352.827. [DOI] [PubMed] [Google Scholar]
- 148.Rane N.S., Chakrabarti O., Feigenbaum L., Hegde R.S. Signal sequence insufficiency contributes to neurodegeneration caused by transmembrane prion protein. J. Cell Biol. 2010;188:515–526. doi: 10.1083/jcb.200911115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Stewart R.S., Piccardo P., Ghetti B., Harris D.A. Neurodegenerative illness in transgenic mice expressing a transmembrane form of the prion protein. J. Neurosci. 2005;25:3469–3477. doi: 10.1523/JNEUROSCI.0105-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chakrabarti O., Ashok A., Hegde R.S. Prion protein biosynthesis and its emerging role in neurodegeneration. Trends Biochem. Sci. 2009;34:287–295. doi: 10.1016/j.tibs.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Whitehouse I.J., Jackson C., Turner A.J., Hooper N.M. Prion protein is reduced in aging and in sporadic but not in familial Alzheimer’s disease. J. Alzheimers Dis. 2010;22:1023–1031. doi: 10.3233/JAD-2010-101071. [DOI] [PubMed] [Google Scholar]
- 152.Beal M.F. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann. Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- 153.Smith M.A., Rottkamp C.A., Nunomura A., Raina A.K., Perry G. Oxidative stress in Alzheimer’s disease. Biochim. Et Biophys. Acta. 2000;1502:139–144. doi: 10.1016/S0925-4439(00)00040-5. [DOI] [PubMed] [Google Scholar]
- 154.Wong B.S., Pan T., Liu T., Li R., Petersen R.B., Jones I.M., Gambetti P., Brown D.R., Sy M.S. Prion disease: A loss of antioxidant function? Biochem. Biophys. Res. Commun. 2000;275:249–252. doi: 10.1006/bbrc.2000.3158. [DOI] [PubMed] [Google Scholar]
- 155.Chen S., Yadav S.P., Surewicz W.K. Interaction between human prion protein and amyloid-beta (Abeta) oligomers: Role OF N-terminal residues. J. Biol. Chem. 2010;285:26377–26383. doi: 10.1074/jbc.M110.145516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nieznanski K., Surewicz K., Chen S., Nieznanska H., Surewicz W.K. Interaction between prion protein and Abeta amyloid fibrils revisited. Acs Chem. Neurosci. 2014;5:340–345. doi: 10.1021/cn500019c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Aulic S., Masperone L., Narkiewicz J., Isopi E., Bistaffa E., Ambrosetti E., Pastore B., De Cecco E., Scaini D., Zago P., et al. alpha-Synuclein Amyloids Hijack Prion Protein to Gain Cell Entry, Facilitate Cell-to-Cell Spreading and Block Prion Replication. Sci. Rep. 2017;7:10050. doi: 10.1038/s41598-017-10236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Parkin E.T., Watt N.T., Hussain I., Eckman E.A., Eckman C.B., Manson J.C., Baybutt H.N., Turner A.J., Hooper N.M. Cellular prion protein regulates beta-secretase cleavage of the Alzheimer’s amyloid precursor protein. Proc. Natl. Acad. Sci. USA. 2007;104:11062–11067. doi: 10.1073/pnas.0609621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Griffiths H.H., Whitehouse I.J., Baybutt H., Brown D., Kellett K.A., Jackson C.D., Turner A.J., Piccardo P., Manson J.C., Hooper N.M. Prion protein interacts with BACE1 protein and differentially regulates its activity toward wild type and Swedish mutant amyloid precursor protein. J. Biol. Chem. 2011;286:33489–33500. doi: 10.1074/jbc.M111.278556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Beland M., Bedard M., Tremblay G., Lavigne P., Roucou X. Abeta induces its own prion protein N-terminal fragment (PrPN1)-mediated neutralization in amorphous aggregates. Neurobiol. Aging. 2014;35:1537–1548. doi: 10.1016/j.neurobiolaging.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 161.Nieznanski K., Choi J.K., Chen S., Surewicz K., Surewicz W.K. Soluble prion protein inhibits amyloid-beta (Abeta) fibrillization and toxicity. J. Biol. Chem. 2012;287:33104–33108. doi: 10.1074/jbc.C112.400614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ostapchenko V.G., Beraldo F.H., Mohammad A.H., Xie Y.F., Hirata P.H., Magalhaes A.C., Lamour G., Li H., Maciejewski A., Belrose J.C., et al. The prion protein ligand, stress-inducible phosphoprotein 1, regulates amyloid-beta oligomer toxicity. J. Neurosci. 2013;33:16552–16564. doi: 10.1523/JNEUROSCI.3214-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Watt N.T., Griffiths H.H., Hooper N.M. Lipid rafts: Linking prion protein to zinc transport and amyloid-beta toxicity in Alzheimer’s disease. Front. Cell Dev. Biol. 2014;2:41. doi: 10.3389/fcell.2014.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Fluharty B.R., Biasini E., Stravalaci M., Sclip A., Diomede L., Balducci C., La Vitola P., Messa M., Colombo L., Forloni G., et al. An N-terminal fragment of the prion protein binds to amyloid-beta oligomers and inhibits their neurotoxicity in vivo. J. Biol. Chem. 2013;288:7857–7866. doi: 10.1074/jbc.M112.423954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Rial D., Piermartiri T.C., Duarte F.S., Tasca C.I., Walz R., Prediger R.D. Overexpression of cellular prion protein (PrP(C)) prevents cognitive dysfunction and apoptotic neuronal cell death induced by amyloid-beta (Abeta(1)(-)(4)(0)) administration in mice. Neuroscience. 2012;215:79–89. doi: 10.1016/j.neuroscience.2012.04.034. [DOI] [PubMed] [Google Scholar]
- 166.Voigtlander T., Kloppel S., Birner P., Jarius C., Flicker H., Verghese-Nikolakaki S., Sklaviadis T., Guentchev M., Budka H. Marked increase of neuronal prion protein immunoreactivity in Alzheimer’s disease and human prion diseases. Acta Neuropathol. 2001;101:417–423. doi: 10.1007/s004010100405. [DOI] [PubMed] [Google Scholar]
- 167.Vergara C., Ordonez-Gutierrez L., Wandosell F., Ferrer I., del Rio J.A., Gavin R. Role of PrP(C) Expression in Tau Protein Levels and Phosphorylation in Alzheimer’s Disease Evolution. Mol. Neurobiol. 2015;51:1206–1220. doi: 10.1007/s12035-014-8793-7. [DOI] [PubMed] [Google Scholar]
- 168.Steinacker P., Hawlik A., Lehnert S., Jahn O., Meier S., Gorz E., Braunstein K.E., Krzovska M., Schwalenstocker B., Jesse S., et al. Neuroprotective function of cellular prion protein in a mouse model of amyotrophic lateral sclerosis. Am. J. Pathol. 2010;176:1409–1420. doi: 10.2353/ajpath.2010.090355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Huang S., Chen L., Bladen C., Stys P.K., Zamponi G.W. Differential modulation of NMDA and AMPA receptors by cellular prion protein and copper ions. Mol. Brain. 2018;11:62. doi: 10.1186/s13041-018-0406-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Beraldo F.H., Arantes C.P., Santos T.G., Queiroz N.G., Young K., Rylett R.J., Markus R.P., Prado M.A., Martins V.R. Role of alpha7 nicotinic acetylcholine receptor in calcium signaling induced by prion protein interaction with stress-inducible protein 1. J. Biol. Chem. 2010;285:36542–36550. doi: 10.1074/jbc.M110.157263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Plattner F., Angelo M., Giese K.P. The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J. Biol. Chem. 2006;281:25457–25465. doi: 10.1074/jbc.M603469200. [DOI] [PubMed] [Google Scholar]
- 172.Vassar R., Bennett B.D., Babu-Khan S., Kahn S., Mendiaz E.A., Denis P., Teplow D.B., Ross S., Amarante P., Loeloff R., et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 173.Avila J. Tau aggregation into fibrillar polymers: Taupathies. Febs Lett. 2000;476:89–92. doi: 10.1016/S0014-5793(00)01676-8. [DOI] [PubMed] [Google Scholar]
- 174.Braak H., Braak E. Evolution of the neuropathology of Alzheimer’s disease. Acta Neurol. Scand. Suppl. 1996;165:3–12. doi: 10.1111/j.1600-0404.1996.tb05866.x. [DOI] [PubMed] [Google Scholar]
- 175.Hardy J.A., Higgins G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 176.Hardy J. The relationship between amyloid and tau. J. Mol. Neurosci. 2003;20:203–206. doi: 10.1385/JMN:20:2:203. [DOI] [PubMed] [Google Scholar]
- 177.Lambert M.P., Barlow A.K., Chromy B.A., Edwards C., Freed R., Liosatos M., Morgan T.E., Rozovsky I., Trommer B., Viola K.L., et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Serrano-Pozo A., Frosch M.P., Masliah E., Hyman B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect Med. 2011;1:a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Braak H., Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 180.Buee L., Delacourte A. Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP-17 and Pick’s disease. Brain Pathol. 1999;9:681–693. doi: 10.1111/j.1750-3639.1999.tb00550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Umeda Y., Taniguchi S., Arima K., Piao Y.S., Takahashi H., Iwatsubo T., Mann D., Hasegawa M. Alterations in human tau transcripts correlate with those of neurofilament in sporadic tauopathies. Neurosci. Lett. 2004;359:151–154. doi: 10.1016/j.neulet.2004.01.060. [DOI] [PubMed] [Google Scholar]
- 182.Hoglinger G.U., Respondek G., Kovacs G.G. New classification of tauopathies. Rev. Neurol. (Paris) 2018;174:664–668. doi: 10.1016/j.neurol.2018.07.001. [DOI] [PubMed] [Google Scholar]
- 183.Kovacs G.G. Tauopathies. Handb Clin. Neurol. 2017;145:355–368. doi: 10.1016/B978-0-12-802395-2.00025-0. [DOI] [PubMed] [Google Scholar]
- 184.Goedert M., Spillantini M.G., Jakes R., Rutherford D., Crowther R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 185.Burack M.A., Halpain S. Site-specific regulation of Alzheimer-like tau phosphorylation in living neurons. Neuroscience. 1996;72:167–184. doi: 10.1016/0306-4522(95)00546-3. [DOI] [PubMed] [Google Scholar]
- 186.Cruz J.C., Tsai L.H. Cdk5 deregulation in the pathogenesis of Alzheimer’s disease. Trends Mol. Med. 2004;10:452–458. doi: 10.1016/j.molmed.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 187.Hooper C., Killick R., Lovestone S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008;104:1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ferrer I., Gomez-Isla T., Puig B., Freixes M., Ribe E., Dalfo E., Avila J. Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer’s disease and tauopathies. Curr. Alzheimer Res. 2005;2:3–18. doi: 10.2174/1567205052772713. [DOI] [PubMed] [Google Scholar]
- 189.Bulbarelli A., Lonati E., Cazzaniga E., Gregori M., Masserini M. Pin1 affects Tau phosphorylation in response to Abeta oligomers. Mol. Cell. Neurosci. 2009;42:75–80. doi: 10.1016/j.mcn.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 190.De Felice F.G., Wu D., Lambert M.P., Fernandez S.J., Velasco P.T., Lacor P.N., Bigio E.H., Jerecic J., Acton P.J., Shughrue P.J., et al. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol. Aging. 2008;29:1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Dohler F., Sepulveda-Falla D., Krasemann S., Altmeppen H., Schluter H., Hildebrand D., Zerr I., Matschke J., Glatzel M. High molecular mass assemblies of amyloid-beta oligomers bind prion protein in patients with Alzheimer’s disease. Brain. 2014;137:873–886. doi: 10.1093/brain/awt375. [DOI] [PubMed] [Google Scholar]
- 192.Gunther E.C., Strittmatter S.M. Beta-amyloid oligomers and cellular prion protein in Alzheimer’s disease. J. Mol. Med. (Berl) 2010;88:331–338. doi: 10.1007/s00109-009-0568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Caetano F.A., Beraldo F.H., Hajj G.N., Guimaraes A.L., Jurgensen S., Wasilewska-Sampaio A.P., Hirata P.H., Souza I., Machado C.F., Wong D.Y., et al. Amyloid-beta oligomers increase the localization of prion protein at the cell surface. J. Neurochem. 2011;117:538–553. doi: 10.1111/j.1471-4159.2011.07225.x. [DOI] [PubMed] [Google Scholar]
- 194.Kellett K.A., Hooper N.M. Prion protein and Alzheimer disease. Prion. 2009;3:190–194. doi: 10.4161/pri.3.4.9980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Rushworth J.V., Griffiths H.H., Watt N.T., Hooper N.M. Prion protein-mediated toxicity of amyloid-beta oligomers requires lipid rafts and the transmembrane LRP1. J. Biol. Chem. 2013;288:8935–8951. doi: 10.1074/jbc.M112.400358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Gimbel D.A., Nygaard H.B., Coffey E.E., Gunther E.C., Lauren J., Gimbel Z.A., Strittmatter S.M. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J. Neurosci. 2010;30:6367–6374. doi: 10.1523/JNEUROSCI.0395-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.You H., Tsutsui S., Hameed S., Kannanaya kal T.J., Chen L., Xia P., Engbers J.D., Lipton S.A., Stys P.K., Zamponi G.W. Abeta neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc. Natl. Acad. Sci. USA. 2012;109:1737–1742. doi: 10.1073/pnas.1110789109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kudo W., Lee H.P., Zou W.Q., Wang X., Perry G., Zhu X., Smith M.A., Petersen R.B., Lee H.G. Cellular prion protein is essential for oligomeric amyloid-beta-induced neuronal cell death. Hum. Mol. Genet. 2012;21:1138–1144. doi: 10.1093/hmg/ddr542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Barry A.E., Klyubin I., Mc Donald J.M., Mably A.J., Farrell M.A., Scott M., Walsh D.M., Rowan M.J. Alzheimer’s disease brain-derived amyloid-beta-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J. Neurosci. 2011;31:7259–7263. doi: 10.1523/JNEUROSCI.6500-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Um J.W., Nygaard H.B., Heiss J.K., Kostylev M.A., Stagi M., Vortmeyer A., Wisniewski T., Gunther E.C., Strittmatter S.M. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012;15:1227–1235. doi: 10.1038/nn.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Um J.W., Strittmatter S.M. Amyloid-beta induced signaling by cellular prion protein and Fyn kinase in Alzheimer disease. Prion. 2013;7:37–41. doi: 10.4161/pri.22212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Hamilton A., Zamponi G.W., Ferguson S.S. Glutamate receptors function as scaffolds for the regulation of beta-amyloid and cellular prion protein signaling complexes. Mol. Brain. 2015;8:18. doi: 10.1186/s13041-015-0107-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Mattei V., Garofalo T., Misasi R., Circella A., Manganelli V., Lucania G., Pavan A., Sorice M. Prion protein is a component of the multimolecular signaling complex involved in T cell activation. Febs Lett. 2004;560:14–18. doi: 10.1016/S0014-5793(04)00029-8. [DOI] [PubMed] [Google Scholar]
- 204.Canu N., Filesi I., Pristera A., Ciotti M.T., Biocca S. Altered intracellular distribution of PrPC and impairment of proteasome activity in tau overexpressing cortical neurons. J. Alzheimers Dis. 2011;27:603–613. doi: 10.3233/JAD-2011-110446. [DOI] [PubMed] [Google Scholar]
- 205.Haas L.T., Kostylev M.A., Strittmatter S.M. Therapeutic molecules and endogenous ligands regulate the interaction between brain cellular prion protein (PrPC) and metabotropic glutamate receptor 5 (mGluR5) J. Biol. Chem. 2014;289:28460–28477. doi: 10.1074/jbc.M114.584342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Vilches S., Vergara C., Nicolas O., Sanclimens G., Merino S., Varon S., Acosta G.A., Albericio F., Royo M., Del Rio J.A., et al. Neurotoxicity of prion peptides mimicking the central domain of the cellular prion protein. PLoS ONE. 2013;8:e70881. doi: 10.1371/journal.pone.0070881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Goniotaki D., Lakkaraju A.K.K., Shrivastava A.N., Bakirci P., Sorce S., Senatore A., Marpakwar R., Hornemann S., Gasparini F., Triller A., et al. Inhibition of group-I metabotropic glutamate receptors protects against prion toxicity. PLoS Pathog. 2017;13:e1006733. doi: 10.1371/journal.ppat.1006733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hernandez-Rapp J., Martin-Lanneree S., Hirsch T.Z., Launay J.M., Mouillet-Richard S. Hijacking PrP(c)-dependent signal transduction: When prions impair Abeta clearance. Front. Aging Neurosci. 2014;6:25. doi: 10.3389/fnagi.2014.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Larson M., Sherman M.A., Amar F., Nuvolone M., Schneider J.A., Bennett D.A., Aguzzi A., Lesne S.E. The complex PrP(c)-Fyn couples human oligomeric Abeta with pathological tau changes in Alzheimer’s disease. J. Neurosci. 2012;32:16857–16871. doi: 10.1523/JNEUROSCI.1858-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 210.Kessels H.W., Nguyen L.N., Nabavi S., Malinow R. The prion protein as a receptor for amyloid-beta. Nature. 2010;466:E3–E4. doi: 10.1038/nature09217. discussion E4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Balducci C., Beeg M., Stravalaci M., Bastone A., Sclip A., Biasini E., Tapella L., Colombo L., Manzoni C., Borsello T., et al. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. USA. 2010;107:2295–2300. doi: 10.1073/pnas.0911829107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Calella A.M., Farinelli M., Nuvolone M., Mirante O., Moos R., Falsig J., Mansuy I.M., Aguzzi A. Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol. Med. 2010;2:306–314. doi: 10.1002/emmm.201000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Younan N.D., Sarell C.J., Davies P., Brown D.R., Viles J.H. The cellular prion protein traps Alzheimer’s Abeta in an oligomeric form and disassembles amyloid fibers. FASEB J. 2013;27:1847–1858. doi: 10.1096/fj.12-222588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Pietri M., Dakowski C., Hannaoui S., Alleaume-Butaux A., Hernandez-Rapp J., Ragagnin A., Mouillet-Richard S., Haik S., Bailly Y., Peyrin J.M., et al. PDK1 decreases TACE-mediated alpha-secretase activity and promotes disease progression in prion and Alzheimer’s diseases. Nat. Med. 2013;19:1124–1131. doi: 10.1038/nm.3302. [DOI] [PubMed] [Google Scholar]
- 215.Resenberger U.K., Winklhofer K.F., Tatzelt J. Cellular prion protein mediates toxic signaling of amyloid beta. Neurodegener. Dis. 2012;10:298–300. doi: 10.1159/000332596. [DOI] [PubMed] [Google Scholar]
- 216.Chen R.J., Chang W.W., Lin Y.C., Cheng P.L., Chen Y.R. Alzheimer’s Amyloid-beta Oligomers Rescue Cellular Prion Protein Induced Tau Reduction via Fyn Pathways. ACS Chem. Neurosci. 2013 doi: 10.1021/cn400085q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Schmitz M., Wulf K., Signore S.C., Schulz-Schaeffer W.J., Kermer P., Bahr M., Wouters F.S., Zafar S., Zerr I. Impact of the cellular prion protein on amyloid-beta and 3PO-tau processing. J. Alzheimers Dis. 2014;38:551–565. doi: 10.3233/JAD-130566. [DOI] [PubMed] [Google Scholar]
- 218.Ishizawa K., Mitsufuji T., Shioda K., Kobayashi A., Komori T., Nakazato Y., Kitamoto T., Araki N., Yamamoto T., Sasaki A. An autopsy report of three kindred in a Gerstmann-Straussler-Scheinker disease P105L family with a special reference to prion protein, tau, and beta-amyloid. Brain Behav. 2018;8:e01117. doi: 10.1002/brb3.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Matamoros-Angles A., Gayosso L.M., Richaud-Patin Y., di Domenico A., Vergara C., Hervera A., Sousa A., Fernandez-Borges N., Consiglio A., Gavin R., et al. iPS Cell Cultures from a Gerstmann-Straussler-Scheinker Patient with the Y218N PRNP Mutation Recapitulate tau Pathology. Mol. Neurobiol. 2017 doi: 10.1007/s12035-017-0506-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Jansen C., Parchi P., Capellari S., Strammiello R., Dopper E.G., van Swieten J.C., Kamphorst W., Rozemuller A.J. A second case of Gerstmann-Straussler-Scheinker disease linked to the G131V mutation in the prion protein gene in a Dutch patient. J. Neuropathol. Exp. Neurol. 2011;70:698–702. doi: 10.1097/NEN.0b013e3182270c54. [DOI] [PubMed] [Google Scholar]
- 221.Llorens F., Ansoleaga B., Garcia-Esparcia P., Zafar S., Grau-Rivera O., Lopez-Gonzalez I., Blanco R., Carmona M., Yague J., Nos C., et al. PrP mRNA and protein expression in brain and PrP in CSF in Creutzfeldt-Jakob disease MM1 and VV2. Prion. 2013:7. doi: 10.4161/pri.26416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Bradley C.A., Peineau S., Taghibiglou C., Nicolas C.S., Whitcomb D.J., Bortolotto Z.A., Kaang B.K., Cho K., Wang Y.T., Collingridge G.L. A pivotal role of GSK-3 in synaptic plasticity. Front. Mol. Neurosci. 2012;5:13. doi: 10.3389/fnmol.2012.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Guglielmotto M., Giliberto L., Tamagno E., Tabaton M. Oxidative stress mediates the pathogenic effect of different Alzheimer’s disease risk factors. Front. Aging Neurosci. 2010;2:3. doi: 10.3389/neuro.24.003.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Wadsworth J.D., Hill A.F., Beck J.A., Collinge J. Molecular and clinical classification of human prion disease. Br. Med. Bull. 2003;66:241–254. doi: 10.1093/bmb/66.1.241. [DOI] [PubMed] [Google Scholar]
- 225.McNeill A. A molecular analysis of prion protein expression in Alzheimer’s disease. Mcgill J. Med. 2004;8:7–14. [Google Scholar]
- 226.Rezaie P., Pontikis C.C., Hudson L., Cairns N.J., Lantos P.L. Expression of cellular prion protein in the frontal and occipital lobe in Alzheimer’s disease, diffuse Lewy body disease, and in normal brain: An immunohistochemical study. J. Histochem. Cytochem. 2005;53:929–940. doi: 10.1369/jhc.4A6551.2005. [DOI] [PubMed] [Google Scholar]
- 227.Watt N.T., Taylor D.R., Kerrigan T.L., Griffiths H.H., Rushworth J.V., Whitehouse I.J., Hooper N.M. Prion protein facilitates uptake of zinc into neuronal cells. Nat. Commun. 2012;3:1134. doi: 10.1038/ncomms2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Lee J.Y., Cole T.B., Palmiter R.D., Suh S.W., Koh J.Y. Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc. Natl. Acad. Sci. USA. 2002;99:7705–7710. doi: 10.1073/pnas.092034699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Bush A.I., Pettingell W.H., Multhaup G., d Paradis M., Vonsattel J.P., Gusella J.F., Beyreuther K., Masters C.L., Tanzi R.E. Rapid induction of Alzheimer A beta amyloid formation by zinc. Science. 1994;265:1464–1467. doi: 10.1126/science.8073293. [DOI] [PubMed] [Google Scholar]
- 230.Deshpande A., Kawai H., Metherate R., Glabe C.G., Busciglio J. A role for synaptic zinc in activity-dependent Abeta oligomer formation and accumulation at excitatory synapses. J. Neurosci. 2009;29:4004–4015. doi: 10.1523/JNEUROSCI.5980-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Whitehouse I.J., Miners J.S., Glennon E.B., Kehoe P.G., Love S., Kellett K.A., Hooper N.M. Prion protein is decreased in Alzheimer’s brain and inversely correlates with BACE1 activity, amyloid-beta levels and Braak stage. PLoS ONE. 2013;8:e59554. doi: 10.1371/journal.pone.0059554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Beyer N., Coulson D.T., Heggarty S., Ravid R., Hellemans J., Irvine G.B., Johnston J.A. Zinc transporter mRNA levels in Alzheimer’s disease postmortem brain. J. Alzheimers Dis. 2012;29:863–873. doi: 10.3233/JAD-2012-112105. [DOI] [PubMed] [Google Scholar]
- 233.Li S.H., Li X.J. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004;20:146–154. doi: 10.1016/j.tig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 234.Landles C., Bates G.P. Huntingtin and the molecular pathogenesis of Huntington’s disease. Fourth in molecular medicine review series. EMBO Rep. 2004;5:958–963. doi: 10.1038/sj.embor.7400250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Saudou F., Humbert S. The Biology of Huntingtin. Neuron. 2016;89:910–926. doi: 10.1016/j.neuron.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 236.De la Monte S.M., Vonsattel J.P., Richardson E.P., Jr. Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington’s disease. J. Neuropathol. Exp. Neurol. 1988;47:516–525. doi: 10.1097/00005072-198809000-00003. [DOI] [PubMed] [Google Scholar]
- 237.Vonsattel J.P., DiFiglia M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- 238.Strong T.V., Tagle D.A., Valdes J.M., Elmer L.W., Boehm K., Swaroop M., Kaatz K.W., Collins F.S., Albin R.L. Widespread expression of the human and rat Huntington’s disease gene in brain and nonneural tissues. Nat. Genet. 1993;5:259–265. doi: 10.1038/ng1193-259. [DOI] [PubMed] [Google Scholar]
- 239.Lee F.J., Liu F. Genetic factors involved in the pathogenesis of Parkinson’s disease. Brain Res. Rev. 2008;58:354–364. doi: 10.1016/j.brainresrev.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 240.Bartels A.L., Leenders K.L. Parkinson’s disease: The syndrome, the pathogenesis and pathophysiology. Cortex. 2009;45:915–921. doi: 10.1016/j.cortex.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 241.Goedert M., Spillantini M.G., Del Tredici K., Braak H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013;9:13–24. doi: 10.1038/nrneurol.2012.242. [DOI] [PubMed] [Google Scholar]
- 242.Butterfield D.A., Perluigi M., Sultana R. Oxidative stress in Alzheimer’s disease brain: New insights from redox proteomics. Eur. J. Pharm. 2006;545:39–50. doi: 10.1016/j.ejphar.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 243.Toulorge D., Schapira A.H., Hajj R. Molecular changes in the postmortem parkinsonian brain. J. Neurochem. 2016 doi: 10.1111/jnc.13696. [DOI] [PubMed] [Google Scholar]
- 244.Ribeiro F.M., Vieira L.B., Pires R.G., Olmo R.P., Ferguson S.S. Metabotropic glutamate receptors and neurodegenerative diseases. Pharm. Res. 2017;115:179–191. doi: 10.1016/j.phrs.2016.11.013. [DOI] [PubMed] [Google Scholar]
- 245.Lewerenz J., Maher P. Chronic Glutamate Toxicity in Neurodegenerative Diseases-What is the Evidence? Front. Neurosci. 2015;9:469. doi: 10.3389/fnins.2015.00469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Choi D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 247.Balazs R., Miller S., Romano C., de Vries A., Chun Y., Cotman C.W. Metabotropic glutamate receptor mGluR5 in astrocytes: Pharmacological properties and agonist regulation. J. Neurochem. 1997;69:151–163. doi: 10.1046/j.1471-4159.1997.69010151.x. [DOI] [PubMed] [Google Scholar]
- 248.Amalric M. Targeting metabotropic glutamate receptors (mGluRs) in Parkinson’s disease. Curr. Opin. Pharm. 2015;20:29–34. doi: 10.1016/j.coph.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 249.Breysse N., Baunez C., Spooren W., Gasparini F., Amalric M. Chronic but not acute treatment with a metabotropic glutamate 5 receptor antagonist reverses the akinetic deficits in a rat model of parkinsonism. J. Neurosci. 2002;22:5669–5678. doi: 10.1523/JNEUROSCI.22-13-05669.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Coccurello R., Breysse N., Amalric M. Simultaneous blockade of adenosine A2A and metabotropic glutamate mGlu5 receptors increase their efficacy in reversing Parkinsonian deficits in rats. Neuropsychopharmacology. 2004;29:1451–1461. doi: 10.1038/sj.npp.1300444. [DOI] [PubMed] [Google Scholar]
- 251.Phillips J.M., Lam H.A., Ackerson L.C., Maidment N.T. Blockade of mGluR glutamate receptors in the subthalamic nucleus ameliorates motor asymmetry in an animal model of Parkinson’s disease. Eur. J. Neurosci. 2006;23:151–160. doi: 10.1111/j.1460-9568.2005.04550.x. [DOI] [PubMed] [Google Scholar]
- 252.Ossowska K., Konieczny J., Wardas J., Pietraszek M., Kuter K., Wolfarth S., Pilc A. An influence of ligands of metabotropic glutamate receptor subtypes on parkinsonian-like symptoms and the striatopallidal pathway in rats. Amino Acids. 2007;32:179–188. doi: 10.1007/s00726-006-0317-y. [DOI] [PubMed] [Google Scholar]
- 253.Thakur A.K., Jayaraman M., Mishra R., Thakur M., Chellgren V.M., Byeon I.J., Anjum D.H., Kodali R., Creamer T.P., Conway J.F., et al. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat. Struct Mol. Biol. 2009;16:380–389. doi: 10.1038/nsmb.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Fernandez-Nogales M., Cabrera J.R., Santos-Galindo M., Hoozemans J.J., Ferrer I., Rozemuller A.J., Hernandez F., Avila J., Lucas J.J. Huntington’s disease is a four-repeat tauopathy with tau nuclear rods. Nat. Med. 2014;20:881–885. doi: 10.1038/nm.3617. [DOI] [PubMed] [Google Scholar]
- 255.Carmichael J., Sugars K.L., Bao Y.P., Rubinsztein D.C. Glycogen synthase kinase-3beta inhibitors prevent cellular polyglutamine toxicity caused by the Huntington’s disease mutation. J. Biol. Chem. 2002;277:33791–33798. doi: 10.1074/jbc.M204861200. [DOI] [PubMed] [Google Scholar]
- 256.Surgucheva I., Sharov V.S., Surguchov A. gamma-Synuclein: Seeding of alpha-synuclein aggregation and transmission between cells. Biochemistry. 2012;51:4743–4754. doi: 10.1021/bi300478w. [DOI] [PubMed] [Google Scholar]
- 257.Urrea L., Ferrer I., Gavin R., Del Rio J.A. The cellular prion protein (PrPC) as neuronal receptor for alpha-synuclein. Prion. 2017;11:226–233. doi: 10.1080/19336896.2017.1334748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Mao X., Ou M.T., Karuppagounder S.S., Kam T.I., Yin X., Xiong Y., Ge P., Umanah G.E., Brahmachari S., Shin J.H., et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science. 2016:353. doi: 10.1126/science.aah3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.De Cecco E., Legname G. The role of the prion protein in the internalization of alpha-synuclein amyloids. Prion. 2018;12:23–27. doi: 10.1080/19336896.2017.1423186. [DOI] [PMC free article] [PubMed] [Google Scholar]