

Abstract
Background
Hypertriglyceridemia (HTG) is a leading cause of acute pancreatitis. HTG can be caused by either primary (genetic) or secondary etiological factors, and there is increasing appreciation of the interplay between the two kinds of factors in causing severe HTG.
Objectives
The main aim of this study was to identify the genetic basis of hypertriglyceridemia-induced acute pancreatitis (HTG-AP) in a Chinese family with three affected members (the proband, his mother and older sister).
Methods
The entire coding and flanking sequences of LPL, APOC2, APOA5, GPIHBP1 and LMF1 genes were analyzed by Sanger sequencing. The newly identified LPL nonsense variant was subjected to functional analysis by means of transfection into HEK-293 T cells followed by Western blot and activity assays. Previously reported pathogenic LPL nonsense variants were collated and compared with respect to genotype and phenotype relationship.
Results
We identified a novel nonsense variant, p.Gln118* (c.351C > T), in the LPL gene, which co-segregated with HTG-AP in the Chinese family. We provided in vitro evidence that this variant resulted in a complete functional loss of the affected LPL allele. We highlighted a role of alcohol abuse in modifying the clinical expression of the disease in the proband. Additionally, our survey of 12 previously reported pathogenic LPL nonsense variants (in 20 carriers) revealed that neither serum triglyceride levels nor occurrence of HTG-AP was distinguishable among the three carrier groups, namely, simple homozygotes, compound heterozygotes and simple heterozygotes.
Conclusions
Our findings, taken together, generated new insights into the complex etiology and expression of HTG-AP.
Keywords: Gene-environment interaction, Genotype and phenotype relationship, Hypertriglyceridemia-induced acute pancreatitis, Lipoprotein lipase, LPL gene, Nonsense variant, Triglyceride
Introduction
Acute pancreatitis (AP) is an acute inflammatory disease that is characterized by local pancreatic inflammation and consequently systemic inflammatory response [1, 2]. Gallstones, alcohol abuse and massive hypertriglyceridemia (HTG) are generally thought to be three leading etiologies of AP worldwide [3]. However, unlike in Western countries, HTG, rather than alcohol abuse, is the second leading cause of AP in China [4]. Hypertriglyceridemia-induced acute pancreatitis (HTG-AP) is defined by serum triglyceride (TG) level exceeding 11.3 mmol/L (1000 mg/dL) or between 5.6 to 11.3 mmol/L (500~1000 mg/dL) together with lipemic serum [5, 6]. As compared to other etiologies, HTG-AP is usually more severe and has higher recurrence rate [7, 8].
According to the etiology, HTG can be divided into primary and secondary HTG. Secondary HTG is usually caused by metabolic syndrome, diabetes, alcohol consumption, obesity, chronic renal failure, etc. [9] Primary HTG is caused by genes defects related with TG metabolism, including lipoprotein lipase (LPL), apolipoprotein C-II (APOC2), apolipoprotein A-V (APOA5), glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1 (GPIHBP1) and lipase maturation factor 1 (LMF1). LPL is the key enzyme that catabolizes TG in non-hepatic tissues [10]. APOC2 and APOA5 act as essential LPL activators [11, 12]. LMF1 is involved in the folding and expression of LPL [13]. GPIHBP1 mediates the transmembrane transport and binding of LPL [14].
However, in most cases, the cause of HTG is complex [15]. Severe HTG was recently shown to be primarily polygenic [16], and there is increasing appreciation of the interplay between primary and secondary etiological factors in causing severe HTG [17, 18]. In this study, we reported a novel LPL nonsense variant in one typical Chinese family with HTG-AP history and discussed insights into the complex etiology of HTG-AP gleaned from the so far reported pathogenic LPL nonsense variants.
Methods
Ethical statement
This study was approved by the Ethics Committee of Jinling Hospital. Informed consent was obtained from all participants.
Family description
The male proband had been suffered from recurrent severe HTG-AP since 26 years old, respectively in 2003, 2007, 2014 and 2017. He has had hypertension for 7 years and abused alcohol for more than 5 years (250–350 g/d). His body mass index (BMI) was normal (22.7 kg/m2). His mother and older sister also respectively had one- and two-times onset of HTG-AP.
Sequencing of the LPL, APOC2, APOA5, GPIHBP1 and LMF1 genes
Genomic DNA was extracted from blood by the Gentra Puregene Blood kit (Qiagen, Dusseldorf, Germany) according to the manufacturer’s instructions. All exons and exon/intron boundaries of the LPL, APOA5, APOC2, LMF1 and GPIHBP1 genes were analyzed by sanger sequencing [18].
Population allele frequency reference and variant nomenclature
Population allele frequencies of variants found in this study were evaluated using the Genome Aggregation Database (gnomAD) genome dataset [19] via VarSome [20]. Variant nomenclature was in accordance with Human Genome Variation Society (HGVS) recommendations [21]. NM_000237.3 was used as the LPL mRNA reference sequence.
Plasma lipid profile analysis
Blood samples were taken from the proband after fasting for 12 h. Serum TG, TC, HDL, LDL levels were measured enzymatically on an automatic analyzer (Hitachi High-Tech, 7600–120, Japan).
Post-heparin LPL mass analysis
Post-heparin blood samples were collected into Na-EDTA tubes 10 min after intravenous heparin injection (60 IU/kg body weight) and fasting for 12 h. Post-heparin plasma LPL mass was detected by immunoassay using the Human LPL Elisa kit (TSZ Biological Trade, USA).
LPL activity analysis
LPL activity was in principle measured through detecting free fatty acid (FFA) concentration [22]. The reaction substrate, termed buffer A, was composed of 1 ml TG-rich serum (TG concentration, > 3000 mg/dL) from Gpihbp1-deficient mice (Gpihbp1−/−) [23], 0.18 g 10% fatty acid-poor bovine serum albumin (BSA) (Miles, West Haven, CT), 0.031 mg heparin, 0.012 g NaCl and 0.3 mmol Tris-HCl Buffer (pH 8.5), in a final volume of 5 mL. 5 μL buffer A were mixed with 5 μL serum from wild-type rats and 5 μL test sample, and incubated at 37 °C for 60 min. [Note that serum from either Gpihbp1-deficient mice or wild-type rats was pre-incubated for 10 min at 62.5 °C in order to inactivate any residual endogenous lipase activity.] FFA concentration was determined in triplicate on a spectrophotometer (Thermo Multiskan GO) using the Wako kit, NEFA-HR(2).
In the case of human serum test sample, the FFA concentration represented the total post-heparin lipase activities that comprised LPL and hepatic lipase (HL) activities. To correct for the contribution from HL, 1 M NaCl was added and incubated for 60 min, so that the LPL activity can be completely inhibited [24]. LPL activity was then calculated by the difference between total post-heparin lipase activity and HL activity. All assays were performed in triplicate.
Plasmid construction and transfection
Human wild-type and c.352C > T mutant LPL coding sequences were synthesized and cloned into pcDNA3.1 (Vigene Biosciences), respectively. Sequence accuracy of the inserts was confirmed by Sanger sequencing.
HEK-293 T cells (ATCC, CRL-3216) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, high glucose from Lonza, C11995500BT) containing 10% Fetal Bovine Serum (FBS) and 1% penicillin-streptomycin. Plasmids (1.5 μg/mL) were transiently transfected into HEK-293 T cells using Lipofectamine 3000 (Thermo, L3000015) in 6-well plates (Costar, 3516) according to the manufacturer’s instructions. After 6 h, the cells were changed into DMEM medium with 2% FBS. After 48 h, cells and medium were harvested separately. Proteins were extracted from cells solubilized in 70 μL RIPA (Beyotime, P0013E) with 7 μL protease inhibitor (PI, Roche, 4,693,116,001), and stored at − 20 °C. Protein concentration was determined by the BCA method. Cell medium was collected after inhibition with 20 U/mL heparin-DMEM (0.5 ml DMEM and 8 μL heparin (20 units/mL) for each well) for 30 min, centrifuged at 1000 r/min for 5 min, and the supernatant was stored at − 20 °C.
Western blot analysis
Cell proteins were mixed with SDS-PAGE Protein Loading Buffer and incubated at 95 °C for 5 min. Proteins were size-separated by SDS-PAGE (10% acrylamide gel, 130 V, 90 min), transferred onto a nitrocellulose membrane (220 mA, 120 min), blocked for 1 h with 5% BSA, and washed 3 times for 15 min with 0.2% Tris-Buffered Saline with Tween 20 (TBS-Tween). Membranes were incubated overnight with primary antibodies, washed 3 times for 15 min with 0.2% TBS-Tween, incubated for 1 h with HRP-conjugated secondary antibodies, and washed 3 times for 15 min with 0.2% TBS-Tween. After 5-min incubation with chemiluminescent HRP substrate (Thermo Scientific), bands were visualized by Chemidoc XRS System (Clinx Science Instruments, Shanghai, China) and analyzed by Image Lab Software (Clinx Science Instruments, Shanghai, China). The antibodies used were mouse anti-LPL (Santa, sc-73,646) (1:200 dilution), rabbit anti-GAPDH (Santa, sc-69,778) (1:2000 dilution), goat anti-rabbit IgG H&L (HRP) (Abcam, ab6721) (1:10000 dilution), and rabbit anti-mouse IgG H&L (HRP) (Abcam, ab6728) (1:5000 dilution).
Collation of previously published pathogenic LPL nonsense variants
Key words including “lipoprotein lipase”, “mutation”, “nonsense” and “termination” were used for searching previously reported pathogenic LPL nonsense variants in PubMed.
Results
Clinical findings and treatment of the proband
At his latest bout of AP in 2017, the proband was transferred into our severe acute pancreatitis therapy center in Jinling Hospital. His TG level was 71.3 mmol/L (6313.3 mg/dL) (Fig. 1) and his plasma was milky (Fig. 2a), fulfilling the definition of extreme HTG [25]. Physical examination revealed epigastric tenderness without rebound tenderness or Murphy’s sign. Laboratory examination revealed elevations in amylase level (446 U/L), white blood cell count (19.86 × 109/L), inflammation biomarkers CRP (263.4 mg/L), IL-6 (121.6 ng/L) and PCT (2.93 μg/L), and renal function biomarkers CRE (289 μmol/L) and BUN (13.3 mmol/L). Abdominal computed tomography showed evidence of AP (Fig. 2b). Moderate-severe AP was diagnosed in accordance with the 2012 revision of Atlanta classification [5].
Fig. 1.

A summary of the patient’s disease history and his TG levels and treatments since the fourth attack of AP. TG, triglyceride; AP, acute pancreatitis
Fig. 2.

Two clinical observations of the patient upon admission to our service. a Blood samples showing chylemia. b Abdominal computed tomography showing enlarged pancreas with adjacent water density shadow and disappearance of the space between the pancreas and peripancreatic adipose tissues (arrow)
The proband was treated with enzyme inhibitors, anti-infection agents and fasting. His TG level decreased rapidly to 17.8 mmol/L (1576.19 mg/dL) 2 days later, and was determined to be 5.5 mmol/L (487.05 mg/dL) when discharged on May 2nd, 2017. During his hospitalization, the patient well tolerated enteral nutrition and adhered to a low-fat diet plus the lipid-lowering drug fenofibrate (0.3 g/d).
In most of the follow-up period, the patient has taken 0.3 g/d fenofibrate, low-fat diet, dry out and exercise (1 h /day), keeping the TG level within the mild to moderate range (defined as 2~9.9 mmol/L in accordance with Dron et al. [16]). However, once he took 0.1 g/d fenofibrate, an obvious increase in TG level was observed (Fig. 1).
Genetic findings
Sequencing of the LPL, APOA5, APOC2, LMF1 and GPIHBP1 genes in the proband detected four gene variants, as one LPL nonsense variant (Fig. 3) and three LMF1 synonymous variants (Supplemental. Fig. S1). All three LMF1 synonymous variants are common in the general populations (Table 1) and therefore were excluded from further consideration. The LPL nonsense variant, p.Gln118* (c.351C > T), is absent from the gnomAD database (Table 1). The LPL p.Gln118*was also detected in the proband’s mother and sister (Fig. 3a), but not in others 256 unrelated HTG-AP patients. Additionally, the LPL p.Gln118* nonsense variant has not previously been reported.
Fig. 3.

Identification of a novel heterozygous nonsense variant in the LPL gene. a Family pedigree. Arrow indicates the proband. Patients with hypertriglyceridemia-induced acute pancreatitis are indicated by black symbols whilst clinically unaffected family members are indicated by empty symbols. LPL genotypes are provided for all subjects. wt, wild-type. b Sanger sequencing electropherogram showing the heterozygous C > T single nucleotide substitution at position c.352 of the LPL gene (indicated by arrow) that would change the codon for glutamine at position p.118 (underlined) to a stop codon (i.e., p.Gln118*)
Table 1.
Variants found in the present study
| Gene | mRNA reference | Variant | Allele frequency in gnomAD genome dataset | rs number | ||
|---|---|---|---|---|---|---|
| Nucleotide change | Amino acid change | East Asian population | All populations | |||
| LPL | NM_000237.3 | c.352C > T | p.Gln118* | 0 | 0 | No |
| LMF1 | NM_022773.3 | c.306G > A | p.Thr102= | 0.4006 | 0.2796 | rs3751667 |
| c.540G > A | p.Thr180= | 0.2352 | 0.1899 | rs2277892 | ||
| c.543G > A | p.Gly181= | 0.3007 | 0.2776 | rs2277893 | ||
LPL mass and activity in post-heparin plasma of the proband
LPL mass and activity in the proband’s post-heparin plasma were measured in September 2017, when his TG level was 2.1 mmol/L (185.84 mg/dL). The LPL mass and activity were respectively 47% (160.7 U/L vs. 302 U/L) and 37% (0.053 vs. 0.143 mEq/L) of the mean values of 15 normal controls.
In vitro analysis of the LPL p.Gln118* variant
LPL wild-type and p.Gln118* mutant expression plasmids were transiently transfected into HEK-293 T cells, respectively. LPL protein expression was analyzed by Western blot using transfected cell proteins and LPL activity was analyzed using transfected cell media. The LPL p.Gln118* mutant resulted in no detectable LPL activity and as shown in Fig. 4, the LPL p.Q118X resulted in undetectable LPL mass and activity, as compared to the positive results of LPL wild-type.
Fig. 4.
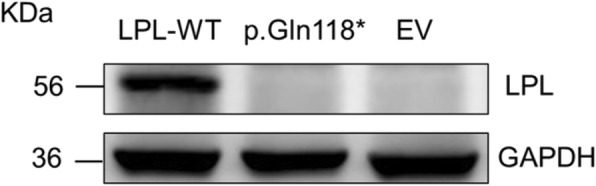
Functional characterization of the LPL p.Gln118* variant. Proteins prepared from the HEK293T cells that were transiently transfected with the LPL wild-type (WT) expression vector, the LPL p.Gln118* mutant expression vector and empty vector (EV), respectively, were used for Western blot analysis. GAPDH, loading control
Brief review of reported pathogenic LPL nonsense variants
Our literature search identified 18 papers that reported 12 distinct pathogenic LPL nonsense variants [26–43]. Variant zygosity, LPL mass and activity levels, TG level and disease outcomes in terms of HTG-AP were collected from each carrier whenever applicable (Table 2).
Table 2.
Summary of the pathogenic LPL nonsense variants so far reported in the literature
| Variant | Reference | Patient’s age | Country | Zygosity | The other variant in case of compound heterozygosity | LPL activity (% of normal) | LPL mass (% of normal) |
TG level (mmol/L) | AP (times) |
|---|---|---|---|---|---|---|---|---|---|
| p.Trp14* (c.42G > A) | Nakamura et al. (1996) [33] | 74 | Japan | Homozygote | NI | 0 | 18.5 | No | |
| Li et al. (2018) [41] | 61 | China | Compound heterozygote | p.Leu279Val (c.835C > G) | 39.9 | 48.60 | 38.6 | Yes (3) | |
| p.Cys54* (c.162C > A) | Chan et al. (2006) [37] | 7 d | China | Compound heterozygote | p.Leu279Val (c.835C > G) homozygote | NI | NI | 17.4 | NI |
| p.Tyr88* (c.264 T > A) | Ebara et al. (2001) [36] | 66 | Japan | Homozygote | 0 | 0 | 26.6 | No | |
| Gotoda et al. (1991) [27] | 3 m | Japan | Homozygote | 14 | 0 | 216 | No | ||
| Gotoda et al. (1992) [28] | 10 m | Japan | Compound heterozygote | p.Ala248LeufsTer4) (c.742del) | <5 | 0 | 46.2 | NI | |
| p.Trp91* (c.272G > A) | Sprecher et al. (1992) [30] | 3 | USA | Compound heterozygote | p.Ile221Thr (c.662 T > C) | 0.0 | 34.2 | > 226 | Yes (recurrent) |
| 23 | USA | Heterozygote | 60.2 | 37.6 | 1.4 | No | |||
| p.Tyr100* (c.300C > A) | Wilson et al. (1993) [31] | 43 | German and English-Irish ancestry | Compound heterozygote | p.Arg102Ser (c.306A > C) | 6 | 28.5 | 16.7 | Yes |
| p.Gln133* (c.397C > T) | Ishimura-Oka et al. (1992) [29] | 1 | English | Compound heterozygote | p.Trp113Arg (c.337 T > C) | 22.4 | NI | 7.9 | NI |
| Emi et al. (1990) [26] | 5 m | German and Polish ancestry | Homozygote | NI | NI | 56.5 | NI | ||
| p.Cys266* (c. 798C > A) | Takagi et al. (1994) [32] | 54 | Japan | Compound Heterozygote | p.Ser474*(p.1421C > G) | 55.1 | 61.6 | 4.5 | NI |
| p.Tyr289* (c.867C > A) | Evans et al. (2011) [39] | 44 | German | Compound heterozygote | p.Asp36Asn (c.106G > A) | NI | NI | 11.6 | NI |
| p.Cys291* (c.873C > A) | Jap et al. (2003) [43] | 46 | China | Compound heterozygote | p.Leu279Val(c.835C > G) | 9 | 13 | 51.6 | Yes (>10) |
| p.Tyr329* (c.987C > A) | Bertolini et al. (2000) [35] | 7 | Italy | Homozygote | 0 | 0 | 29.8 | Yes | |
| Hegele et al. (2018) [40] | NI | UK | Homozygote | 0.1 | NI | NI | NI | ||
| p.Trp409* (c.1227G > A) | Gotoda et al. (1991) [27] | 6 m | Japan | Homozygote | 15.6 | 5.4 | 67.9 | NI | |
| Takagi et al. (1999) [34] | 5 m | Japan | Compound heterozygote | p.Gly215Glu (c.644G > A) | <10 | <10 | 48.1 | No | |
| Suga et al. (1998) [42] | 40 | Japan | Homozygote | <1 | <1 | 37.6 | Yes | ||
| p.Trp421* (c.1262G > A) | Hooper et al. (2008) [38] | 43 | Australia | Compound heterozygote | p.Gly215Glu (c.644 G > A) | Very low | NI | 32.2 | Yes (recurrent) |
AP Acute pancreatitis, d days, m Months, LPL Lipoprotein lipase, NI Not informative, TG Triglyceride
Discussion
In this study, we reported a novel heterozygous nonsense variant in the LPL gene, p.Gln118* (c.351C > T), in one typical Chinese family with HTG-AP history. Presumably, this variant should cause a complete functional loss of the affected LPL allele due to its significant truncation of the 475 amino acid protein. Indeed, the LPL mass and activity in the proband’s plasma revealed a roughly 50% reduction as compared to normal controls. And in vitro, the results confirmed that the p.Gln118* mutant resulted in undetectable LPL protein and activity. Taken together, LPL p.Gln118* could be a novel and pathogenic LPL gene variant.
In this typical HTG-AP family, the proband, his mother and sister all had the LPL p.Gln118* nonsense variant, and separately had four, one and two times of HTG-AP onset. Moreover, HTG-AP was milder in the mother and older sister than the proband, as mild compared to moderate-severe. This variable clinical expression may be, at least partly, explained by the existence of an established secondary etiological factor, alcohol abuse, in the proband but not in the diseased mother and older sister. Although interplay between primary and secondary etiological factors in causing HTG-AP has been described in the literature [17, 18], to our best knowledge, the present study is the first to demonstrate the possible effect of alcohol abuse in modifying the clinical expression of a pathogenic genetic variant in the context of a HTG-AP family that exhibited a mode of monogenic inheritance. In this regard, it is pertinent to mention that, given the apparent effect of alcohol abuse in inducing or worsening HTG-AP, the proband has been required to quit alcohol since May 2017.
In this study, both in vivo and in vitro results showed that the LPL p.Gln118* nonsense variant could be pathogenic as resulting in complete functional loss of LPL mass and activity. Moreover, we briefly reviewed all the reported LPL nonsense variants together with the clinical features, as to evaluate the complex etiology of HTG or HTG-AP from a perspective of genotype and phenotype relationship. As shown in Table 2, the reported 12 LPL nonsense variants were detected in 20 subjects worldwide. As in detail, 8 were homozygotes, 2 were heterozygotes and 10 were compound heterozygotes. As expected, simple homozygotes showed no or barely detectable LPL mass and activity in the proband. The compound heterozygotes showed variable LPL mass and activity levels from zero to 50% of normal (depending upon the functional effect of the variant in trans). And, the heterozygotes showed LPL mass and activity levels that were around 50% of normal. We found correlation between mutation status and TG levels or occurrence of HTG-AP, as most informative adult patients had suffered recurrent AP with extreme high TG level, however, some patient is quite special. Taken p.Trp14* (c.42G > A) for example, the patient was had homozygote LPL variant, a 74-years-old Japanese with a complete LPL deficiency, never developed AP whilst the compound heterozygote, a 61-years-old Chinese whose LPL mass was ~ 50% of normal, had three times of AP (Table 1). Above all, the zygosity and AP occurrence of all the nonsense patients reported before emphasized again the complex etiology and expression of HTG-AP. Findings from this comparative analysis provide new evidence suggesting that new layers of complexity, beyond known genetic risk factors, predispose to, or prevent, the development of HTG-AP. Nowadays, there are some new genetic drugs for these LPL deficient patients like alipogene tiparvovec, LCQ908 etc., beyond the dietary management and usual pharmacologic therapies, these new genetic treatments can be certainly promising and effective therapy basing on the patients’ genetic background [44].
Conclusion
To sum up, in this report, a novel LPL p.Gln118* (c.351C > T) variant was detected in one typical Chinese family with HTG-AP history, and had been verified to be pathogenic as resulting complete loss of LPL function in vitro. In particular, we highlighted a role of alcohol abuse in modifying the clinical expression of the disease in the proband. Moreover, we briefed reviewed all reported LPL nonsense variants, together with the phenotype, which may give new insights into the complex etiology of HTG-AP.
Supplementary information
Additional file 1: Figure S1. Sanger sequencing electropherograms showing the three heterozygous LMF1 synonymous variants detected in the proband.
Acknowledgements
The authors are indebted to all doctors and researchers for the follow-up assessment and data collection during the study from the severe acute pancreatitis care center of Jinling Hospital, Medical School of Nanjing University.
Abbreviations
- LPL
Lipoprotein lipase
- TG
Triglyceride
- HTG
Hypertriglyceridemia
- HTG-AP
Hypertriglyceridemia-induced acute pancreatitis
- FFA
Free fatty acid
- HEK-293 T
Human embryonic kidney 293 T
- AP
Acute pancreatitis
- WB
Western blotting
- APOC2
Apolipoprotein C-II
- APOA5
Apolipoprotein A-V
- LMF1
Lipase maturation factor 1
- GPIHBP1
Glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1
- BMI
Body mass index
- MSAP
Moderate severe acute pancreatitis
- AKI
Acute kidney injury
- ANC
Acute necrotic collection
- TC
Total cholesterol
- HDL
High density lipoprotein
- LDL
Low density lipoprotein
- PCR
Polymerase chain reaction
- PHLA
Post-heparin lipase activity
- BSA
Bovine serum albumin
- HL
Hepatic lipase
- EV
Empty vector
- ATCC
American Tissue Culture Collection
- DMEM
Dulbecco’s Modified Eagle’s Medium
- FBS
Fetal Bovine Serum
- PBS
Phosphate buffer saline
- PI
Protease inhibitor
Authors’ contributions
All authors contributed extensively to the work presented in this paper. Conceptualization, XYL, NP and QY; Investigation, LK, BY and TZH; Formal Analysis, WWC, XLS and ZGF; Writing – Original Draft Preparation, LXY and NP; Writing – Review & Editing, YQ, YHW, GL and JMC; Supervision, WQL and JSL. All authors have read and approved the final version of this manuscript.
Funding
This study was supported by the Key Research and Development Program Foundation of Jiangsu Province of China (BE2015685 and BE2016749) and the National Natural Science Foundation of China (Nos. 81570584, 81670588 and 81870441).
Availability of data and materials
Data for the analyses are available from the corresponding author on request.
Ethics approval and consent to participate
The study was approved by the ethics committee of the Jinling Hospital, Medical School of Nanjing University. Written informed consent was obtained from each participant.
Consent for publication
Consent for publication was obtained from the patient.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Xiao-Yao Li and Na Pu contributed equally to this work.
Contributor Information
Xiao-Yao Li, Email: lixiaoyaonju@163.com.
Na Pu, Email: punayeah@163.com.
Wei-Wei Chen, Email: cww1984@126.com.
Xiao-Lei Shi, Email: 15950561608@163.com.
Guo-fu Zhang, Email: njuzgf@163.com.
Lu Ke, Email: kkb9832@163.com.
Bo Ye, Email: bobmaster819@126.com.
Zhi-Hui Tong, Email: njzyantol@hotmail.com.
Yu-Hui Wang, Email: wangyuhui2009@bjmu.edu.cn.
George Liu, Email: vangeorgeliu@gmail.com.
Jian-Min Chen, Email: jian-min.chen@univ-brest.fr.
Qi Yang, Email: yangqi_nj@163.com.
Wei-Qin Li, Email: njzy_pancrea@163.com.
Jie-Shou Li, Email: lijieshou2013@sohu.com.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12944-020-01249-z.
References
- 1.Tenner S, Baillie J, DeWitt J, Vege SS. American College of Gastroenterology guideline: management of acute pancreatitis. Am J Gastroenterol. 2013;108:1400–1415. doi: 10.1038/ajg.2013.218. [DOI] [PubMed] [Google Scholar]
- 2.Peery AF, Crockett SD, Murphy CC, et al. Burden and Cost of Gastrointestinal, Liver, and Pancreatic Diseases in the United States: Update 2018. Gastroenterology. 2019;156:254. doi: 10.1053/j.gastro.2018.08.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forsmark Ch E, Vege SS, Wilcox CM. Acute Pancreatitis. N Engl J Med. 2017;376:598–599. doi: 10.1056/NEJMc1616177. [DOI] [PubMed] [Google Scholar]
- 4.Li X, Ke L, Dong J, et al. Significantly different clinical features between hypertriglyceridemia and biliary acute pancreatitis: a retrospective study of 730 patients from a tertiary center. BMC Gastroenterol. 2018;18:89. doi: 10.1186/s12876-018-0821-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banks PA, Bollen TL, Dervenis C, et al. Classification of acute pancreatitis--2012: revision of the Atlanta classification and definitions by international consensus. Gut. 2013;62:102–111. doi: 10.1136/gutjnl-2012-302779. [DOI] [PubMed] [Google Scholar]
- 6.Scherer J, Singh VP, Pitchumoni CS, Yadav D. Issues in hypertriglyceridemic pancreatitis: an update. J Clin Gastroenterol. 2014;48:195–203. doi: 10.1097/01.mcg.0000436438.60145.5a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yin G, Cang X, Yu G, et al. Different clinical presentations of Hyperlipidemic acute pancreatitis: a retrospective study. Pancreas. 2015;44:1105–1110. doi: 10.1097/MPA.0000000000000403. [DOI] [PubMed] [Google Scholar]
- 8.Adiamah A, Psaltis E, Crook M, Lobo DN. A systematic review of the epidemiology, pathophysiology and current management of hyperlipidaemic pancreatitis. Clin Nutr. 2018;37:1810–1822. doi: 10.1016/j.clnu.2017.09.028. [DOI] [PubMed] [Google Scholar]
- 9.Lewis GF, Xiao C, Hegele RA. Hypertriglyceridemia in the genomic era: a new paradigm. Endocr Rev. 2015;36:131–147. doi: 10.1210/er.2014-1062. [DOI] [PubMed] [Google Scholar]
- 10.Olivecrona G. Role of lipoprotein lipase in lipid metabolism. Curr Opin Lipidol. 2016;27:233–241. doi: 10.1097/MOL.0000000000000297. [DOI] [PubMed] [Google Scholar]
- 11.Kei AA, Filippatos TD, Tsimihodimos V, Elisaf MS. A review of the role of apolipoprotein C-II in lipoprotein metabolism and cardiovascular disease. Metab Clin Exp. 2012;61:906–921. doi: 10.1016/j.metabol.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 12.Sharma V, Forte TM, Ryan RO. Influence of apolipoprotein A-V on the metabolic fate of triacylglycerol. Curr Opin Lipidol. 2013;24:153–159. doi: 10.1097/MOL.0b013e32835c8c1a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doolittle MH, Ehrhardt N, Peterfy M. Lipase maturation factor 1: structure and role in lipase folding and assembly. Curr Opin Lipidol. 2010;21:198–203. doi: 10.1097/MOL.0b013e32833854c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davies BS, Beigneux AP, Barnes RH, 2nd, et al. GPIHBP1 is responsible for the entry of lipoprotein lipase into capillaries. Cell Metab. 2010;12:42–52. doi: 10.1016/j.cmet.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hegele RA, Ginsberg HN, Chapman MJ, et al. The polygenic nature of hypertriglyceridaemia: implications for definition, diagnosis, and management. Lancet Diabetes Endocrinol. 2014;2:655–666. doi: 10.1016/S2213-8587(13)70191-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dron JS, Wang J, Cao H, et al. Severe hypertriglyceridemia is primarily polygenic. J Clin Lipidol. 2019;13:80–88. doi: 10.1016/j.jacl.2018.10.006. [DOI] [PubMed] [Google Scholar]
- 17.Serveaux Dancer M, Di Filippo M, Marmontel O, et al. New rare genetic variants of LMF1 gene identified in severe hypertriglyceridemia. J Clin Lipidol. 2018;12:1244–1252. doi: 10.1016/j.jacl.2018.06.018. [DOI] [PubMed] [Google Scholar]
- 18.Chen WW, Yang Q, Li XY, et al. Identification of a novel and heterozygous LMF1 nonsense mutation in an acute pancreatitis patient with severe hypertriglyceridemia, severe obesity and heavy smoking. Lipids Health Dis. 2019;18:68. doi: 10.1186/s12944-019-1012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kopanos C, Tsiolkas V, Kouris A, et al. VarSome: the human genomic variant search engine. Bioinformatics (Oxford, England) 2019;35:1978–1980. doi: 10.1093/bioinformatics/bty897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.den Dunnen JT, Dalgleish R, Maglott DR, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37:564–569. doi: 10.1002/humu.22981. [DOI] [PubMed] [Google Scholar]
- 22.Di Filippo M, Marcais C, Charriere S, et al. Post-heparin LPL activity measurement using VLDL as a substrate: a new robust method for routine assessment of plasma triglyceride lipolysis defects. PLoS One. 2014;9:e99721. doi: 10.1371/journal.pone.0096482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan Y, Li Y, Gao L, et al. Development of a novel model of hypertriglyceridemic acute pancreatitis in mice. Sci Rep. 2017;7:40799. doi: 10.1038/srep40799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zemankova K, Makoveichuk E, Vlasakova Z, Olivecrona G, Kovar J. Acute alcohol consumption downregulates lipoprotein lipase activity in vivo. Metab Clin Exp. 2015;64:1592–1596. doi: 10.1016/j.metabol.2015.08.016. [DOI] [PubMed] [Google Scholar]
- 25.Berglund L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97:2969–2989. doi: 10.1210/jc.2011-3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emi M, Hata A, Robertson M, Iverius PH, Hegele R, Lalouel JM. Lipoprotein lipase deficiency resulting from a nonsense mutation in exon 3 of the lipoprotein lipase gene. Am J Hum Genet. 1990;47:107–111. [PMC free article] [PubMed] [Google Scholar]
- 27.Gotoda T, Yamada N, Kawamura M, et al. Heterogeneous mutations in the human lipoprotein lipase gene in patients with familial lipoprotein lipase deficiency. J Clin Invest. 1991;88:1856–1864. doi: 10.1172/JCI115507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gotoda T, Yamada N, Murase T, et al. A newly identified null allelic mutation in the human lipoprotein lipase (LPL) gene of a compound heterozygote with familial LPL deficiency. Biochim Biophys Acta. 1992;1138:353–356. doi: 10.1016/0925-4439(92)90015-F. [DOI] [PubMed] [Google Scholar]
- 29.Ishimura-Oka K, Faustinella F, Kihara S, Smith LC, Oka K, Chan L. A missense mutation (Trp86----Arg) in exon 3 of the lipoprotein lipase gene: a cause of familial chylomicronemia. Am J Hum Genet. 1992;50:1275–1280. [PMC free article] [PubMed] [Google Scholar]
- 30.Sprecher DL, Kobayashi J, Rymaszewski M, et al. Trp64----nonsense mutation in the lipoprotein lipase gene. J Lipid Res. 1992;33:859–866. [PubMed] [Google Scholar]
- 31.Wilson DE, Hata A, Kwong LK, et al. Mutations in exon 3 of the lipoprotein lipase gene segregating in a family with hypertriglyceridemia, pancreatitis, and non-insulin-dependent diabetes. J Clin Invest. 1993;92:203–211. doi: 10.1172/JCI116551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takagi A, Ikeda Y, Mori A, et al. A newly identified heterozygous lipoprotein lipase gene mutation (Cys239-->stop/TGC972-->TGA; LPLobama) in a patient with primary type IV hyperlipoproteinemia. J Lipid Res. 1994;35:2008–2018. [PubMed] [Google Scholar]
- 33.Nakamura T, Suehiro T, Yasuoka N, et al. A novel nonsense mutation in exon 1 and a transition in intron 3 of the lipoprotein lipase gene. J Atheroscler Thromb. 1996;3:17–24. doi: 10.5551/jat1994.3.17. [DOI] [PubMed] [Google Scholar]
- 34.Takagi A, Ikeda Y, Tachi K, Shinozuka T, Yamamoto A. Identification of compound heterozygous mutations (G188E/W382X) of lipoprotein lipase gene in a Japanese infant with hyperchylomicronemia: the G188E mutation was newly identified in Japanese. Clin Chim Acta. 1999;285:143–154. doi: 10.1016/S0009-8981(99)00116-3. [DOI] [PubMed] [Google Scholar]
- 35.Bertolini S, Simone ML, Pes GM, et al. Pseudodominance of lipoprotein lipase (LPL) deficiency due to a nonsense mutation (Tyr302>term) in exon 6 of LPL gene in an Italian family from Sardinia (LPL (Olbia)) Clin Genet. 2000;57:140–147. doi: 10.1034/j.1399-0004.2000.570209.x. [DOI] [PubMed] [Google Scholar]
- 36.Ebara T, Okubo M, Horinishi A, Adachi M, Murase T, Hirano T. No evidence of accelerated atherosclerosis in a 66-yr-old chylomicronemia patient homozygous for the nonsense mutation (Tyr61-->stop) in the lipoprotein lipase gene. Atherosclerosis. 2001;159:375–379. doi: 10.1016/S0021-9150(01)00510-X. [DOI] [PubMed] [Google Scholar]
- 37.Chan AO, But WM, Lau GT, Tse WY, Shek CC. A novel nonsense mutation in the LPL gene in a Chinese neonate with hypertriglyceridemia. Clin Chim Acta. 2006;368:120–124. doi: 10.1016/j.cca.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 38.Hooper AJ, Crawford GM, Brisbane JM, et al. Familial lipoprotein lipase deficiency caused by known (G188E) and novel (W394X) LPL gene mutations. Ann Clin Biochem. 2008;45:102–105. doi: 10.1258/acb.2007.007080. [DOI] [PubMed] [Google Scholar]
- 39.Evans D, Arzer J, Aberle J, Beil FU. Rare variants in the lipoprotein lipase (LPL) gene are common in hypertriglyceridemia but rare in type III hyperlipidemia. Atherosclerosis. 2011;214:386–390. doi: 10.1016/j.atherosclerosis.2010.11.026. [DOI] [PubMed] [Google Scholar]
- 40.Hegele RA, Berberich AJ, Ban MR, et al. Clinical and biochemical features of different molecular etiologies of familial chylomicronemia. J Clin Lipidol. 2018;12:920–927.e924. doi: 10.1016/j.jacl.2018.03.093. [DOI] [PubMed] [Google Scholar]
- 41.Li X, Yang Q, Shi X, et al. Compound but non-linked heterozygous p.W14X and p.L279 V LPL gene mutations in a Chinese patient with long-term severe hypertriglyceridemia and recurrent acute pancreatitis. Lipids Health Dis. 2018;17:144. doi: 10.1186/s12944-018-0789-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suga S, Tamasawa N, Kinpara I, et al. Identification of homozygous lipoprotein lipase gene mutation in a woman with recurrent aggravation of hypertriglyceridaemia induced by pregnancy. J Intern Med. 1998;243:317–321. doi: 10.1046/j.1365-2796.1998.00306.x. [DOI] [PubMed] [Google Scholar]
- 43.Jap TS, Jenq SF, Wu YC, Chiu CY, Cheng HM. Mutations in the lipoprotein lipase gene as a cause of hypertriglyceridemia and pancreatitis in Taiwan. Pancreas. 2003;27:122–126. doi: 10.1097/00006676-200308000-00003. [DOI] [PubMed] [Google Scholar]
- 44.Fogacci F, Cicero AFG. Gene targeting for chylomicronemia syndrome: the brave new world. Atherosclerosis. 2018;269:254–255. doi: 10.1016/j.atherosclerosis.2017.12.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Sanger sequencing electropherograms showing the three heterozygous LMF1 synonymous variants detected in the proband.
Data Availability Statement
Data for the analyses are available from the corresponding author on request.