

Abstract
Despite the enthusiasm surrounding cancer immunotherapy, most small cell lung cancer (SCLC) patients show very modest response to immune checkpoint inhibitor monotherapy treatment. There is therefore growing interest in combining immune checkpoint blockade (ICB) with chemotherapy and other treatments to enhance ICB efficacy. Based on favorable clinical trial results, chemotherapy and immunotherapy combinations have been recently approved by the Food and Drug Administration (FDA) for the frontline treatment for SCLC. Here, we show that combined treatment of SRA737, an oral CHK1 inhibitor, and anti-PD-(L)1 leads to an anti-tumor response in multiple cancer models, including SCLC. We further show that combining low, non-cytotoxic doses of gemcitabine with SRA737+anti-PD-(L)1 significantly increased anti-tumorigenic CD8+ cytotoxic T-cells, dendritic cells and M1 macrophage populations in an SCLC model. This regimen also led to a significant decrease in immunosuppressive M2 macrophage and MDSC populations, as well as an increase in the expression of the Type I interferon gene, IFNβ, and chemokines, CCL5 and CXCL10. Given that anti-PD-(L)1 drugs have recently been approved as monotherapy and in combination with chemotherapy for the treatment of SCLC, and that SRA737+low dose gemcitabine (LDG) regimen is currently in clinical trials for SCLC and other malignancies, our preclinical data provide a strong rational for combining this regimen with inhibitors of PD-(L)1 pathway.
Keywords: SCLC, DNA damage response, immune checkpoint blockade, low dose gemcitabine
Introduction
For decades, chemotherapy remained the only systemic treatment option for small cell lung cancer (SCLC), the most aggressive form of lung cancer, until the recent introduction of immunotherapy 1. Immune checkpoint blockade (ICB), such as antibodies targeting programmed cell death protein 1 (PD-1) or programmed death ligand 1 (PD-L1) and cytotoxic T-lymphocyte associated protein 4 (CTLA4), have shown activity in only a minority of SCLC patients 2. Therefore, there is a growing interest in combining ICB with chemotherapy and other treatments to enhance their efficacy.
Recently, the results from the phase III ImPower133 trial 3, testing the combination of anti-PD-L1 (atezolizumab) plus chemotherapy (platinum and etoposide) versus chemotherapy alone, demonstrated a modest but significant increase in overall survival (12.3 versus 10.3 months, p=0.007) and progression-free survival (5.2 versus 4.3 months, p=0.02) with the addition of atezolizumab 3. Based on this study, the chemotherapy and immunotherapy combination has now been Food and Drug Administration (FDA) approved for the frontline treatment of extensive stage SCLC. This highlights the need to better understand the extent to which chemotherapy may enhance ICB response; and whether new approaches beyond cytotoxic chemotherapy may represent more optimal treatment regimen(s) for immunotherapy combinations.
We have previously shown that SCLC exhibits increased expression of CHK1 and other DNA damage response (DDR) proteins 4, 5. SCLC is characterized by an almost universal loss of TP53 and RB1 resulting in high levels of replication stress (RS) and an increased reliance on checkpoint kinase 1 (CHK1). Replication stress (RS) is caused by slowing or stalling of the replication fork in response to various disruptions such as depletion of dNTP pools, replication-transcription collision, or DNA damage. CHK1 is considered a master regulator of RS which temporarily arrests the cell cycle and manages replication origin firing, thereby preventing excessive DNA damage and increasing the overall survival fitness of the tumor cells6. SRA737 is an oral small molecule inhibitor of CHK1 that is currently being tested in clinical trials as monotherapy and in combination with low dose gemcitabine (LDG) in SCLC and other cancers (NCT0279964, NCT02797977). Gemcitabine, a ribonucleotide reductase (RNR) inhibitor leads to dNTP depletion and fork stalling, even at subtherapeutic concentrations. The combination of SRA737 with LDG represents a unique approach to combining chemotherapy with targeted agents. Instead of using standard dose chemotherapy to induce cancer cell death, the LDG is used for its RS-inducing properties to increase reliance of cancer cells on CHK1 and therefore potentiate SRA737’s intrinsic cytotoxicity and immuno-stimulatory activities.
Recently, DDR inhibitors were shown to activate cGAS, which induces an innate immune response through STING signaling 7, 8. Moreover, DDR inhibition was shown to induce PD-L1 expression and synergize with anti-PD-L1 in vivo 8–10. Previous data from our group further demonstrated that CHK1 inhibition enhances anti-tumor immunity when combined with PD-L1 blockade in SCLC 8.
In the current study, we explored the efficacy of SRA737 in combination with anti-PD-L1 or anti-PD-1 (anti-PD-(L)1) in SCLC and other cancer models in vivo. We further investigated the treatment regimen of SRA737+LDG in combination with anti-PD-L1 in SCLC. We identified a new role for SRA737+LDG combination in inducing T-cell infiltration in tumors and in modulating macrophage phenotype switching. Our results support a novel treatment strategy for SCLC patients combining a low dose of the RS-inducing chemotherapy agent, gemcitabine, and a DDR-inhibitor, SRA737, with immunotherapy.
Materials and methods
Cell lines and characterization
Cell lines were purchased from ATCC or Sigma Aldrich and, unless the studies were performed within 6 months from purchase, the cell lines were tested and authenticated by short tandem repeat profiling (DNA Fingerprinting) within 6 months of the study. Cells were routinely tested for Mycoplasma species before any experiments were performed.
Cell lines used for in vivo studies at Crown Biosciences were purchased from ATCC (HT-29), NIH (Pan-02), FDCC (MC38) and RIKEN (MBT-2).
SCLC murine cell line derived from Trp53, p130, Rb1 (RPP) conditional knockout mice was a gift from Dr. Julien Sage, Stanford University, CA, USA.
Cell culture
Cell lines were grown in RPMI, unless otherwise mentioned by the provider, with 10% fetal bovine serum and antibiotics, cultured at 37°C in a humidified chamber with 5% CO2. All cell lines included in the study were profiled at passage 4–8 to abrogate the heterogeneity introduced by long-term culture.
Chemical compounds
SRA737 was provided by Sierra Oncology. Gemcitabine was acquired from The MD Anderson Cancer Center pharmacy. Anti-PD-L1 antibodies (clone 10F.9G2) and anti-PD-1 antibodies (clone RMP1–14) were purchased from BioXcell.
Cell proliferation assay
SCLC cell lines were incubated with dimethyl sulfoxide (vehicle control), SRA737 or gemcitabine for 96 hours at nine distinct concentrations, with the maximum dose being 10μM. A CellTiter-Glo luminescent cell viability assay (Promega) was performed after 96 hours of treatment as per manufacturer’s specifications. IC50 values were estimated using Drexplorer, as previously described 11 .
RNA isolation
RNA was isolated using the Direct-zol RNA MiniPrep Kit (Zymo Research, cat# R2050) according to the manufacturer’s instructions. RNA concentrations were determined using a NanoDrop 2000 UV-Vis spectrophotometer (Thermo Scientific).
Quantitative PCR (qPCR)
Reverse transcription reactions were carried out using SuperScript III First-Strand Synthesis SuperMix (Invitrogen, cat# 18080–400) according to the manufacturer’s instructions.
Real-time PCR was done using SYBR Select Master Mix (Life Technologies, cat# 4472908) according to the manufacturer’s protocol. Primers were purchased from Sigma (USA); the details of the primers are given in Table S1. Triplicate PCR reactions were run on ABI (7500 Fast Real Time PCR System) according to the manufacturer’s instructions. The comparative Ct method using the average 2ΔΔCT value for each set of triplicates was used, and the average of the biological replicates was calculated. Negative controls were included for every primer set, and GAPDH was used as the positive control.
Preparation of protein lysates
Protein lysate was collected from sub confluent cultures after 24-hr in full-serum media (10% fetal bovine serum [FBS]). The lysates were collected as described previously 4.
Western blot analysis
Western blot analysis was performed using SDS-PAGE followed by transfer to nitrocellulose membrane using the BioRad Gel system. Membranes were incubated in the following primary antibodies (1:1000) overnight: PD-L1 (CST), total and phospho- (S366) STING (CST), total and phospho- (S396) IRF3 (CST), phospho-ϒH2AX (CST) and Actin (Sigma). Secondary anti-rabbit, HRP-linked antibodies were purchased from Cell Signaling Biosciences and detected using the Chemidoc imaging system, image captured with Image Studio Version 3.1 software
Reverse-phase protein array (RPPA)
RPPAs were printed from lysates as previously described. The quality of the antibodies and validated by western blots and correlation of protein levels in previous RPPA experiments were determined, as previously described. The RPPA samples were analyzed as described before4.
Tumor growth assessment
Female B6129F1 mice used for RPP xenograft studies were obtained from Taconic Biosciences. Female C57BL/6 mice used for MC38 and Pan02 xenograft studies, female C3H mice used for MBT-2 xenograft study, and female BALB/c nude mice used in HT-29 xenograft studies were obtained from Shanghai Lingchang Bio-Technology Co. Ltd. All animal studies in B6129F1 mice were conducted under a protocol approved by an Institutional Animal Care and Use Committee. Cells (1×106) in 0.2 mL of a 1:1 mixture of medium and matrigel were injected subcutaneously into the right flank of each mouse. In the case of MC38, Pan02, MBT-2, and HT-29, the studies were conducted at Crown Bioscience, Taicang, China where 1×106, 3×106, 4×105 and 3×106 tumor cells dissolved in PBS, respectively, were injected subcutaneously into the right flank. In all xenograft studies, tumors were evaluated twice weekly for the duration of the study. Tumor volumes were measured on all mice three times per week and calculated (width2 × length × 0.4) using manual calipers. Once the average tumor size was in the range of 120–150 mm3, mice were randomized into dosing groups. In the case of RPP xenografts, stratified sampling was used by assigning three animals per group for short-term reverse-phase protein array analysis and long-term treatment. Dosing schedules and duration varied depending on the study. Mice were weighed three times per week for the duration of the study, and a decrease in body weight >15% was considered indicative of a toxic dose. The Student t-test was used to determine statistical significance between compound- and vehicle-treated groups.
Flow cytometry
Single-cell suspensions were prepared and stained according to standard protocols for flow cytometry with antibodies listed in Table S2. For intracellular staining, cells were fixed and permeabilized with BD Cytofix/Cytoperm (BD Biosciences). The data were acquired on a Fortessa or Calibur platform (BD Biosciences) and analyzed with FlowJo software (version 7.6; Tree Star). For analyzing the abundance and the function of CD4+ or CD8+ TILs, single-cell suspensions were prepared from tumors and spleen and stained; the staining of spleen cells was used as the reference for lymphocyte gating, then CD3+ cells were gated, and then CD4+ or CD8+ population was analyzed.
Histologic analysis
For immunohistochemistry, cryosections (8 μm) of tumor tissues were fixed with acetone and stained with antibody against mouse CD4 (Sinobiological, Cat.no. 50134-R001) and CD8 (eBioscience, Cat. No. 14–0808-82) and horseradish peroxidase-conjugated secondary antibody. The image of the entire stained tumor section was scanned with an Olympus BX41 microscope and a semi-quantitative immunohistochemical score was determined by visual examination of the entire stained section of the tumor. The composite score represents a sum of the score for the highest intensity of the staining in any area of the tumor section (scored 0–3) and the score for the prevalence of staining across the entire section of the tumor (scored 0–3). Therefore, the composite score reflects both the prevalence and the intensity of the staining across the entire tumor section (scored 0–6).
Micronuclei Assay
Cells were cultured as discussed earlier and treated with or without SRA737 (1μM). Cytochalasin B was added at the 20th hour to each culture to give a final concentration of 3 μg/ml and the culture was incubated at 37 °C for up to 24hrs. After 24hrs incubation, the cells were centrifuged at 1000 rpm for 5 min. The supernatant was removed and the pellet was treated with weak hypotonic solution (0.075 M KCl/0.9 % Saline, 1:9) and incubated at 37 °C for 5 min. After this, the cells were centrifuged and the pellets were fixed in fresh fixative (methanol:acetic acid, 3:1). Cells were dropped onto glass slides and were prepared and stained with ProLong® Gold Antifade Mountant with DAPI for scoring. At least 1000 multinucleated cells, following the standard specifications, were scored for each slide. In the case of EMT-6 cells, micronuclei formation assays were performed at Phenovista Biosciences, San Diego. The cells were treated with either 1μM or 5μM SRA737 for 24h followed by fixing with 4%PFA for 20min at RT directly in the culture wells, washing with PBS and staining with Hoechst (1:1000). The plates were imaged at 40X using the Thermo Fisher Scientific CellInsight CX7 imaging system and the the micronuclei algorithm was adjusted to score all nuclei that are <55% of the size of the main nucleus. 25 fields per well were imaged and the data was averaged across 8 replicates.
Statistics
Flow cytometry statistical analyses were performed with GraphPad Prism 5.0 software. Significant differences (p<0.05) between two groups were identified by Student’s t-tests. The protein expression from cell lines was compared by t test between models sensitive to SRA737 (IC50<5μM; n=13) and those that were resistant (IC50>5μM; n=31).
Results
Human and murine cancer cells show sensitivity to CHK1 inhibition by SRA737
To investigate the in vitro activity of SRA737, we screened 51 SCLC cell lines (including one murine derived model) and a panel of additional cancer types that included NSCLC, pancreatic, colon and bladder cancer cell lines (representing cancer types in ongoing CHK1/DDR clinical trials) using proliferation assays. The SCLC cell lines demonstrated a range of sensitivity to SRA737 and were classified according to the half-maximal inhibitory concentration (IC50) into sensitive (IC50<5μM; n=15) and resistant (IC50>5μM; n=35) (Fig. 1A and S1A).
Figure 1.

(A) SRA737 IC50 values in a panel of 51 SCLC cell lines, seven NSCLC cell lines, three other cancer type cell lines (one pancreatic-SW1990, one colon-SNUC1 and one bladder-5637 cancer cell line) indicate a wide range of sensitivities in vitro.
(B) Representative image showing micronuclei (MN) formation following SRA737 treatment in SCLC cell line H69, after DAPI staining.
(C) Quantification of MN formation shows a significant increase of MN following 24h treatment with SRA737 in multiple cell lines (two human SCLC cell lines, H69 and H446 cells, and one SCLC murine cell line RPP, two NSCLC cell lines, Calu-1 and Calu-6, and one murine breast cancer cell lines, EMT6/P). Data presented as mean ± SD and p values by t-test ***p<0.001, **p<0.01, *p<0.05.
(D) Immunoblot analysis shows increased PD-L1 protein expression in a panel of SCLC cell lines treated with 1 μM SRA737 for 24 hours.
(E) SRA737 treatment for 72 hours augments PD-L1 cell surface expression, as measured by flow cytometry, in SCLC cell lines. Data presented as mean ± SD and p values by t-test ***p<0.001.
(F) Immunoblot analysis of protein markers of the STING pathway, including total and phospho STING (S366), total and phospho IRF3 (S396), in lysates collected from two SCLC cell lines treated with 1 μM SRA737 for 24 and 72 hours.
(G) Quantitative PCR (qPCR) measurement of CCL5, CXCL10 and IFNβ mRNA expression in three SCLC cell lines treated with 1 μM SRA737 or 24 and 72 hours. Data presented as mean ± SD and p values by t-test ***p<0.001.
In the additional cancer types, we observed a range of sensitivity with some lines demonstrating low to sub-micromolar sensitivity, such as pancreatic cancer line SW1990 (IC50=0.7μM), colon cancer line SNU-C1 (IC50=1.3μM), bladder cancer line 5637 (IC50=2.1μM) and NSCLC cancer lines (A549, Calu1, Calu6, H1299, H1573, H1944, H1993 IC50 range 0.8μM to >9.6μM) (Fig. 1A). Overall, these findings demonstrate a strong intrinsic anti-cancer activity of SRA737 in vitro.
Having identified a range of in vitro responses to SRA737 in SCLC, we next investigated potential protein biomarkers of response. Using available proteomic profiles from 44 of the cell lines, we found significantly higher levels of cMYC protein in cell lines sensitive to SRA737; whereas resistant cell lines had higher expression of Bcl-2, cKit and E-cadherin (Fig. S1B and S1C). This finding is consistent with our previous observation that high cMYC expression is an important predictor of sensitivity to the CHK1 inhibitor prexasertib 5, 12, and the well documented role of cMYC in driving intrinsic RS in tumors.
Based on prior data suggesting that CHK1 targeting can induce DNA damage and cytogenetic stress, we then evaluated SRA737-induced cytogenetic stress using a micronuclei (MN) assay. Treatment of SCLC human cell lines (H69 and H446) and murine cell line derived from Trp53, p130, Rb1 (RPP) conditional knockout mice; breast cancer cell EMT6/P; and NSCLC cells Calu1 and Calu6, with SRA737 (1μM) for 24 hours led to significant increase in MN frequency in all treated cells (Fig 1C). In addition, in all SCLC cell lines tested, treatment with SRA737 (1μM, 72 hours) significantly increased total PD-L1 levels (by western blot) (Fig. 1D) and cell surface PD-L1 expression (by fluorescence-activated cell sorting (FACS)) (Fig 1E).
As our group and others have demonstrated that DDR inhibitors induce micronuclei formation, which is associated with release of cytosolic DNA and STING pathway activation 8, we tested whether SRA737 also causes STING activation. Indeed, SRA737 treatment in H1694 and H847 cells caused a time-dependent activation of the STING pathway (pSTING_S366, STING, pIRF3_S396 and IRF3) (Fig. 1F). Transcriptional targets of IRF3, including Type I interferon, IFNβ, 13, 14 and inflammatory cytokines, CXCL10 and CCL5, were also induced by SRA737 treatment (Fig. 1G). These data indicate that, similar to other DDR inhibitors, SRA737 demonstrates intrinsic anti-proliferative activity as well as the ability to induce immunomodulatory factors in multiple cancer models.
SRA737 augments ICB-induced anti-tumor response in SCLC
Next, we treated B6129F1-immunocompetent flank RPP tumor-bearing mice15 with SRA737 (100mg/kg) at two frequency schedules of either 3 days on-treatment followed by 4 days off-treatment (3/7) or 5 days on-treatment followed by 2 days off-treatment (5/7). Anti-PD-L1 (300μg)8 was administered on the third day of every treatment week either alone or in combination with SRA737 (n = 10 per group; Fig 2A). Mice treated with anti-PD-L1 alone showed no anti-tumor response in this model (Fig 2A). Animals treated with single agent SRA737 showed significant reduction in tumor growth as compared to either vehicle or anti-PD-L1 treatment alone, with SRA737 schedule of 5/7 (Treatment/Vehicle (T/C)=0.27; p<0.001) being moderately more effective than SRA737 schedule of 3/7 (T/C=0.32; p<0.001). In contrast to the single agent activities, combination of SRA737 (5/7) and anti-PD-L1 resulted in a complete inhibition of tumor growth within one week of treatment (T/C=0.064; p<0.001) (Fig 2A).
Figure 2.

(A)B6129F1 immunocompetent mice bearing RPP tumors on their flank were treated with SRA737 (100mg/kg, 3/7 days or 5/7 days) and/or anti-PD-L1 (300μg, 1/7 days) (n = 10 per group). Tumor growth curves are shown as the mean tumor volume +/− standard error of the mean (SEM).
(B)Immunoblot analysis of tumors resected at Day 7 from a cohort of mice treated with vehicle, SRA737 (5/7), anti-PD-L1 or SRA737 (5/7) + anti-PD-L1. STING pathway activation was determined by immunoblotting for pSTING_S366, total STING, pTBK1_S172, total TBK1, cGAS, pIRF3_S396, and total IRF3.
(C) Quantitative PCR (qPCR) measurement of IFNβ, CXCL10 and CCL5 mRNA expression in SCLC tumors resected as described in B.
Data presented as mean ± SD and p values by t-test * p<0.05, **p<0.01, ***p<0.001.
Having observed activation of the STING pathway following SRA737 treatment in vitro (Fig 1F), we sought to confirm these findings with SRA737+/−anti-PD-L1 treatment in a cohort of SCLC tumors resected after one week of treatment. Western blot analysis showed that treatment with SRA737 (100mg/kg; 5/7) either alone or in combination with anti-PD-L1 led to significant activation of the STING pathway (pSTING_S366, pTBK1_S172, cGAS, and pIRF3_S396) (Fig 2B). Furthermore, SRA737 (100mg/kg; 5/7) treatment led to an enhanced expression of Type I interferon gene, IFNβ, and chemokines, CXCL10 and CCL5, in the tumors after one week (Fig 2C). In summary, we observed anti-tumor activity when SRA737 was combined with ICB in this model of SCLC. Furthermore, SRA737 both as a single agent and in the combination treatment led to an activation of the STING pathway.
SRA737 augments ICB-induced anti-tumor response in multiple cancers
Next, we tested several syngeneic models representing the other cancer types from Fig 1A, namely colon, bladder and pancreatic cancers. Tumor cells were implanted into the flank of immunocompetent C57BL/6 or C3H mice and once tumor volumes reached 150–200 mm3, mice were randomized to treatment with either monotherapy SRA737 (100mg/kg; 5/7), anti-PD-1 (10 mg/kg; 2/7 days) or the combination. Similarly to anti-PD-L1 treatment in the SCLC model, monotherapy treatment with anti-PD1 showed very limited activity in the colon (MC38 tumor growth inhibition (TGI) = 23%), bladder (MBT-2 TGI = 18%) and pancreatic (Pan02 TGI = 25%) models, while SRA737 monotherapy (100mg/kg, 5/7) induced moderate tumor growth inhibition in all models (MC38 TGI = 48%; MBT-2 TGI = 53%; Pan02 TGI = 59%). However, combination treatment of SRA737 and anti-PD1 resulted in a substantial additive improvement in efficacy in all three models (MC38 TGI = 76%; MBT-2 TGI = 69%; Pan02 TGI = 70%) (Fig. 3A–C).
Figure 3.
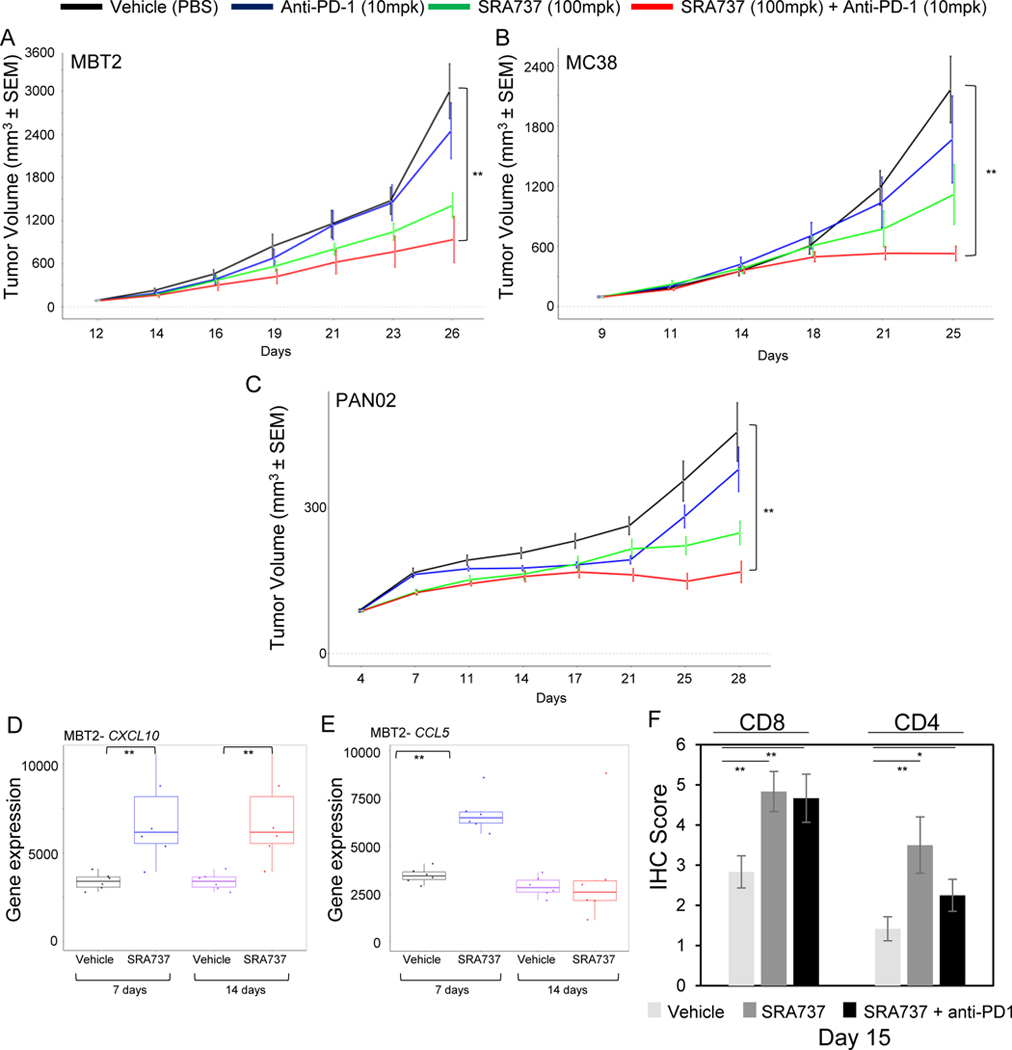
(A-C)Immunocompetent mice bearing bladder cancer tumors MBT-2 (A), colon cancer tumors MC38 (B), and pancreatic cancer tumors Pan02 (C) on their flanks were treated with vehicle, SRA737 (100mg/kg; 5/7 days), anti-PD1 (10mg/kg; BIW i.p.) or their combination. Tumor growth curves are shown as mean tumor volumes +/− SEM. **p<0.01.
(D-E)Nanostring analysis of CXCL10 and CCL5 mRNA expression in MBT-2 tumors resected after 7 or 14 days of treatment with SRA737 (100 mg/kg; 5/7 days). Data presented as mean ± SD and p values by t-test **p<0.01.
(F) Quantification of CD4+ and CD8+ IHC staining in tumors resected from MC38 colon model, following 14 days of treatment with a modified treatment schedule of SRA737 (100mg/kg; 7 days on/ 7 days off) +/− anti-PD-1 (10mg/kg; day 3,7 and 11).
As we observed STING activation and increased expression of IFN response genes following SRA737 treatment in SCLC model, we further explored the effect of SRA737 treatment on the expression of IFN-responsive inflammatory chemokines in MC38 and MBT-2 models. We treated mice with SRA737 (100mg/kg; 5/7) for either one or two weeks and analyzed the expression of CCL5 and CXCL10 in extracted tumors. In MBT-2 tumors, CXCL10 levels showed a significant induction after both 7 days and 14 days of SRA737 treatment (p<0.01) (Fig. 3D). CCL5 was significantly induced after 7 days of treatment (p<0.01), while no significant difference was observed at day 14 (Fig. 3E). In the MC38 model, CXCL10 levels were increased (although not significantly) only after 14 days of treatment (Fig. S2A), while expression of CCL5 was modestly, but not statistically-significantly reduced after 7 days (Fig. S2B). This data further supported the potential of SRA737 to induce the expression of immunomodulatory chemokines and warranted further exploration of the infiltration of immune cells into the tumors.
To assess recruitment of infiltrating immune cells, MC38 tumors were harvested 15 days after treatment initiation with SRA737 (100mg/kg) with a modified treatment schedule of 7on/7off either alone or in combination with anti-PD-1 (Day 3, 7 and 11). The infiltration of CD4+ and CD8+ cells was determined by immunohistochemistry staining of tumor sections harvested at Day 15. SRA737 treatment either alone or in combination with anti-PD1 led to a marked increase in CD4+ and CD8+ cells infiltrating into the tumors (Fig 3F). These results further demonstrated the ability of SRA737 to modulate the anti-tumor immune response and warranted further exploration of the strategies that maximize the intrinsic and immune-mediated anti-tumor activity of SRA737 in combination with immune checkpoint blockade.
Adding gemcitabine chemotherapy to SRA737 enhances the effect of PD-L1 blockade by modulating the immune microenvironment in SCLC
Gemcitabine-based chemotherapy has been used for relapsed SCLC patients, but its activity is limited to a minority of patients when used as a monotherapy at standard doses1, highlighting the limitations of this treatment paradigm. In contrast to standard cytotoxic doses, low doses of gemcitabine induce RS and increase reliance on CHK1 without overt DNA damage and cytotoxicity. This mechanism synergizes with CHK1 inhibition to induce potent tumor cell death while also avoiding the potential for overlapping toxicities.
To explore this combination further, we tested the SRA737+LDG in the HT-29 in vivo model of colon cancer. In order to mimic the treatment schedule used in the current SRA737+LDG clinical trial (NCT02797977), mice were treated with RS-inducing LDG (40mg/kg) on the first day, followed by inhibition of CHK1 by SRA737 (50mg/kg) for two consecutive days and a dose holiday for the last 4 days of the week. This combination led to a strong inhibition of tumor growth after 3 weeks of treatment (TGI = 83%) highlighting the intrinsic anti-tumor activity of this combination in HT-29 immunodeficient model (Fig. S3A).
Next, we expanded our study to an immunocompetent SCLC model to explore both the intrinsic and immune-mediated anti-tumor effects of SRA737+LDG regimen in combination with anti-PD-L1. We first determined that SCLC cell lines H209, H524 and RPP were resistant to monotherapy gemcitabine in vitro (Fig. S3B), with the RPP tumor model described above selected for further in vivo testing. RPP tumor bearing B6129F1 immunocompetent mice were treated with LDG (40mg/kg, 1/7, first day of week), SRA737 (100mg/kg, 2/7 first and second days of week) and anti-PD-L1 (300μg, 1/7, third day of week) as single agents or in combination. SRA737+LDG regimen mimicked the schedule currently being used in the clinical trial described above with or without anti-PD-L1 on the third day of a weekly cycle. We did not observe a significant anti-tumor effect with any single agent treatments (anti-PD-L1, SRA737 or LDG), and only a moderately delayed tumor growth with combined SRA737 and PD-L1 treatment (Fig 4A). However, we observed remarkable tumor regressions when we combined SRA737+LDG with anti-PD-L1. All mice achieved some level of tumor regressions and 80% of mice (8/10) had complete tumor regressions which were sustained up to 60 days post treatment (Fig 4A).
Figure 4.

(A) RPP tumor bearing B6129F1 immunocompetent mice were treated with anti-PD-L1 (300μg, 1/7), LDG (40mg/kg, 1/7), SRA737 (100mg/kg, 2/7) and the combinations. Tumor growth curves are shown as mean tumor volume +/− SEM, ***p<0.001.
(B-G)SCLC tumors harvested at Day 21 and profiled by FACS analysis for CD3+CD45+CD8+ cytotoxic T-cells (B), PD1+TIM3+exhausted T-cells (C), CD86+ CD68+iNOS+ M1 macrophages (D), Arg1+CD68+M2 macrophages (E), F4/80- MHCII+ GR1-dendritic cells (F) and MDSCs (G). P-values were determined by ANOVA, ns, no significance; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
(H) qPCR measurement of IFNβ, CXCL10 and CCL5 mRNA expression in RPP tumors resected from mice after 21 days of treatment as described in A. Data are presented as mean ± SD and p values were determined by t-test * p<0.05, **p<0.01, ***p<0.001.
A cohort of RPP tumors (n=8) were resected after 21 days of treatment and analyzed by multicolor flow cytometry for the changes in tumor infiltrating lymphocytes (T-cell panel) (Fig S3C). Although we have previously shown that CHK1 targeting activates T-cells in SCLC8 , little is known about the effects on tumor-associated macrophages (TAMs) in this cancer. Here, we evaluated the effects of the SRA737+LDG+anti-PD-L1 regimen on antigen presenting cells (APC panel) including macrophages, dendritic cells and granulocytes (Fig. S3D). Treatment with SRA737+LDG with and without addition of anti-PD-L1 significantly increased CD3+ and CD8+ total T-cell infiltration (Fig S3E; 4B) as compared to vehicle or single agent treatments. Additionally, the triple combination resulted in a significant decrease in the infiltration of CD4+ helper T-cells (Fig S2F), CD25+/FOXP3+ regulatory T-cells (Fig. S2G), and PD1+/TIM3+ exhausted CD8+ T cell population (p<0.001 for all) as compared to vehicle or single agent treatments (Fig 4C). Interestingly, in the APC panel, the triple combination led to significant increase in the M1 macrophage population with a decrease in the M2 macrophage population (Fig. 4D and 4E). Furthermore, we observed an increase of dendritic cells (Fig. 4F) and a decrease of MDSC immune subset (Fig. 4G) post-LDG+SRA737+anti-PD-L1 treatment. However, there was no significant change in the granulocyte population in any treatment groups (Fig. S2H).
The effect of chemotherapies, either as single agents or when combined with ICB or DDR inhibitors, on the immune microenvironment is not fully understood in SCLC. Hence, we next aimed to investigate the effect of LDG, SRA737 and anti-PD-L1 treatments and their combinations on the innate immune signaling through the IFN pathway. LDG and SRA737 administered as single agents had no significant effect on transcription of Type I interferon gene, IFNβ, or the chemokines (CCL5 or CXCL10) in RPP tumors resected at Day 21 (Fig. 4H). In contrast, the combined treatment of SRA737+LDG led to significantly increased expression of IFNβ, CCL5 and CXCL10 (Fig. 4H). Considering the limited anti-tumor benefit when SRA737 was combined with anti-PD-L1 in RPP model, we did not expect to see any remarkable activation of STING pathway with this combination. As anticipated, we observed a very modest enhancement of IFNβ, CCL5 or CXCL10 expression with the SRA737+anti-PD-L1 combination (Fig. 4H). However, SRA737+LDG in combination with anti-PD-L1 led to a significantly enhanced expression of IFNβ, CXCL10 and CCL5 in SCLC tumors (Fig. 4H). In summary, these data show that SRA737+LDG regimen in combination with anti-PD-L1 leads to a dramatic anti-tumor activity accompanied by the establishment of a strong anti-tumor immune microenvironment in SCLC.
Discussion:
The present study explores the combination of SRA737, an oral CHK1 inhibitor with or without anti-PD-(L)1 in multiple cancer models where DDR inhibitors are currently under clinical investigation. We show that the combined treatment of SRA737 and anti-PD-(L)1 leads to anti-tumor response in multiple cancer models, including SCLC. We further show that combining low dose non-cytotoxic gemcitabine, a known RS-inducing agent, with SRA737+anti-PD-L1 can significantly increase anti-tumorigenic CD8+ cytotoxic T-cell, dendritic cell and M1 macrophage populations. Also, the regimen led to significant decrease in immunosuppressive M2 macrophage and MDSC populations in an SCLC model. The triple combination also increased the expression of the Type 1 interferon gene, IFNβ and chemokines CCL5 and CXCL10.
Despite having a very high mutational burden, SCLC is relatively immunosuppressed 16. We and others have shown that targeting the DDR pathway is an effective therapeutic strategy for SCLC 4, 5, 12, 17–22. Furthermore, we have recently shown that DDR pathway targeting potentiates the anti-tumor immune response of PD-L1 blockade through activation of the STING/TBK1/IRF3 pathway-induced activation of cytotoxic T lymphocytes 8. In this study, we demonstrated immuno-modulatory effects of SRA737 treatment through activation of STING and interferon signaling and induction of expression of PD-L1 and inflammatory chemokines, CCL5 and CXCL10 in vitro and in vivo. We further demonstrated that SRA737 potentiates the anti-tumor activity of ICB in multiple cancer models which, we believe, suggests that this combination may have potential for translational impact in several cancer types. Comparison of different SRA737 treatment schedules in combination with anti-PD-(L)1 highlights the need for careful optimization of treatment dosing in order to maximize the intrinsic anti-tumor and immune-modulating activities of SRA737.
We further demonstrated that the combination of SRA737 with LDG shows striking activity in combination with ICB in SCLC and warrants further mechanistic investigation. In the present study, we examined for the first time the effects of LDG either as single agent or in combination with ICB and SRA737 on T-cell infiltration and APC subsets. We observed that the combination of LDG+SRA737+anti-PD-L1 leads to enhanced T-cell infiltration with coincident decrease of T-cell exhaustion. We also demonstrated a significant effect of this triple combination on APC subsets in SCLC. We found that LDG+SRA737+anti-PD-L1 modulated macrophage polarization leading to an increase of the M1 subtype and a decrease in the M2 subtype. Macrophages have previously been shown to play a crucial role in ICB resistance and the balance of the M1 and M2 macrophages is believed to effect the long term efficacy of anti-PD1 blockade in multiple cancer types. Here we show that the SRA737+LDG regimen in combination with anti-PD-L1 enhances dendritic cells in the tumor and leads to a decrease in pro-tumorigenic MDSC populations. Most notably, the establishment of the strong anti-tumor immune microenvironment observed when SRA737 is combined with low dose gemcitabine opens an opportunity for a highly efficacious and tolerable treatment regimen with a potentially distinct mechanism from that of standard chemotherapy or DDR inhibitors alone. Further studies are warranted to determine if this approach would be beneficial with other chemotherapies, such as low dose platinum-based doublets, in combination with ICB and DDR inhibitors. While the RPP SCLC model used in the immune-competent animal experiments represents only part of the heterogeneity of SCLC12, 23, our previous work with olaparib and prexasertib in combination with anti-PD-L18 suggests that our observations are more broadly applicable to SCLC. As with many other cancer types, pre-clinical studies are limited to only a few immune-competent models of SCLC, consequently further validation of the combinations described here, will likely need to take place in a clinical setting.
Given that anti-PD-(L)1 drugs have recently been approved as monotherapy and in combination with chemotherapy for the treatment of SCLC, and that the SRA737+LDG regimen is well tolerated in clinical trials in SCLC (NCT02797977), our preclinical data provide a strong rationale for combining this regimen with inhibitors of PD-(L)1 pathway in the clinic.
Supplementary Material
Acknowledgments
Financial Support:This work was supported by: Research funding from Sierra Oncology (LAB); NIH/NCI award U01-CA213273 (JVH, LAB); NIH/NCI award 1-R01-CA207295 (LAB); NIH/NCI CCSG P30-CA016672 (shRNA and ORFeome Core, Boinformatics Shared Resource); The University of Texas-Southwestern and MD Anderson Cancer Center Lung SPORE (5 P50 CA070907) (JVH, LAB); NIH/NCI T32 CA009666 (CMG); the Lung Cancer Research Foundation (TS); through generous philanthropic contributions to The University of Texas MD Anderson Lung Cancer Moon Shot Program (JVH, JW, LAB); MD Anderson Cancer Center Physician Scientist Award (LAB); and The Rexanna Foundation for Fighting Lung Cancer (JVH, LAB).
Conflict of Interest:
J.V.H serves on the advisory committees of AstraZeneca, Boehringer Ingelheim, Exelixis, Genentech, GSK, Guardant Health, Hengrui, Lilly, Novartis, Spectrum, EMD Serono, and Synta; received research support from AstraZeneca, Bayer, GlaxoSmithKline, Spectrum; receives royalties and licensing fees from Spectrum.
L.A.B. serves on advisory committees for AstraZeneca, AbbVie, GenMab, BergenBio, Pharma Mar, SA, Sierra Oncology, and has research support from AbbVie, AstraZeneca, GenMab, Sierra Oncology, Tolero Pharmaceuticals.
S.M., R.J.H., B.S., M.P.H., C.A.H. are full time employees of Sierra Oncology.
References
- 1.NCCN. http://www.nccn.org/professionals/physician_gls/pdf/sclc.pdf.
- 2.Antonia SJ, Lopez-Martin JA, Bendell J, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol 2016;17:883–895. [DOI] [PubMed] [Google Scholar]
- 3.Horn L, Mansfield AS, Szczesna A, et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med 2018;379:2220–2229. [DOI] [PubMed] [Google Scholar]
- 4.Byers LA, Wang J, Nilsson MB, et al. Proteomic Profiling Identifies Dysregulated Pathways in Small Cell Lung Cancer and Novel Therapeutic Targets Including PARP1. Cancer Discov 2012;2:798–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sen T, Tong P, Stewart CA, et al. CHK1 Inhibition in Small-Cell Lung Cancer Produces Single-Agent Activity in Biomarker-Defined Disease Subsets and Combination Activity with Cisplatin or Olaparib. Cancer Res 2017;77:3870–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kotsantis P, Petermann E, Boulton SJ. Mechanisms of Oncogene-Induced Replication Stress: Jigsaw Falling into Place. Cancer Discov 2018;8:537–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mackenzie KJ, Carroll P, Martin CA, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017;548:461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sen T, Rodriguez BL, Chen L, et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harding SM, Benci JL, Irianto J, et al. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017;548:466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiao S, Xia W, Yamaguchi H, et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin Cancer Res 2017;23:3711–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tong P, Coombes KR, Johnson FM, et al. drexplorer: A tool to explore dose-response relationships and drug-drug interactions. Bioinformatics 2015;31:1692–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cardnell RJ, Li L, Sen T, et al. Protein expression of TTF1 and cMYC define distinct molecular subgroups of small cell lung cancer with unique vulnerabilities to aurora kinase inhibition, DLL3 targeting, and other targeted therapies. Oncotarget 2017;8:73419–73432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009;461:788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun L, Wu J, Du F, et al. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013;339:786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jahchan NS, Dudley JT, Mazur PK, et al. A drug repositioning approach identifies tricyclic antidepressants as inhibitors of small cell lung cancer and other neuroendocrine tumors. Cancer Discov 2013;3:1364–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang W, Hodkinson P, McLaren F, et al. Small cell lung cancer tumour cells induce regulatory T lymphocytes, and patient survival correlates negatively with FOXP3+ cells in tumour infiltrate. Int J Cancer 2012;131:E928–937. [DOI] [PubMed] [Google Scholar]
- 17.Cardnell RJ, Feng Y, Diao L, et al. Proteomic markers of DNA repair and PI3K pathway activation predict response to the PARP inhibitor BMN 673 in small cell lung cancer. Clin Cancer Res 2013;19:6322–6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cardnell RJ, Feng Y, Mukherjee S, et al. Activation of the PI3K/mTOR Pathway following PARP Inhibition in Small Cell Lung Cancer. PloS one 2016;11:e0152584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Bono J, Ramanathan RK, Mina L, et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov 2017;7:620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pietanza MC, Waqar SN, Krug LM, et al. Randomized, Double-Blind, Phase II Study of Temozolomide in Combination With Either Veliparib or Placebo in Patients With Relapsed-Sensitive or Refractory Small-Cell Lung Cancer. J Clin Oncol 2018;36:2386–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sen T, Tong P, Diao L, et al. Targeting AXL and mTOR Pathway Overcomes Primary and Acquired Resistance to WEE1 Inhibition in Small-Cell Lung Cancer. Clin Cancer Res 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stewart CA, Tong P, Cardnell RJ, et al. Dynamic variations in epithelial-to-mesenchymal transition (EMT), ATM, and SLFN11 govern response to PARP inhibitors and cisplatin in small cell lung cancer. Oncotarget 2017;8:28575–28587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rudin CM, Poirier JT, Byers LA, et al. Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat Rev Cancer 2019;19:289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.