

Abstract
Single-cell multi-omics technologies are rapidly evolving, prompting both methodological advances and biological discoveries at an unprecedented speed. Gene regulatory network modeling has been used as a powerful approach to elucidate the complex molecular interactions underlying biological processes and systems, yet its application in single-cell omics data modeling has been met with unique challenges and opportunities. In this review, we discuss these challenges and opportunities, and offer an overview of the recent development of network modeling approaches designed to capture dynamic networks, within-cell networks, and cell–cell interaction or communication networks. Finally, we outline the remaining gaps in single-cell gene network modeling and the outlooks of the field moving forward.
Keywords: cell–cell communication networks, dynamic networks, network modeling, single-cell omics, single-cell RNA sequencing, within-cell networks
Introduction
Network modeling has long been employed as a powerful tool to understand and interpret complex biological systems, with networks themselves serving both as a computational framework and a major data type. Networks depict biological systems as nodes and edges, where nodes represent biological entities such as genes, proteins, metabolites, phenotypic traits, cells, environmental exposures, or even gut bacteria, and edges represent the relationships between nodes such as regulator–effector connections, statistical correlations, physical binding, and enzymatic or metabolic reactions (Figure 1A). As the amount and types of biological data continue to grow at an exponential rate, so too do the number and types of biological networks including protein–protein interaction networks [1], metabolic networks [2], genetic interaction networks [3], gene/transcriptional regulatory networks (GRNs) [4], and cell signaling networks [5]. While different network models possess inherent strengths and limitations depending on their underlying assumptions, they share the common feature of being graphical models which describe information flow in biological systems to help understand and interpret the underlying biological processes.
Figure 1. Overview of molecular networks and single-cell network modeling approaches.
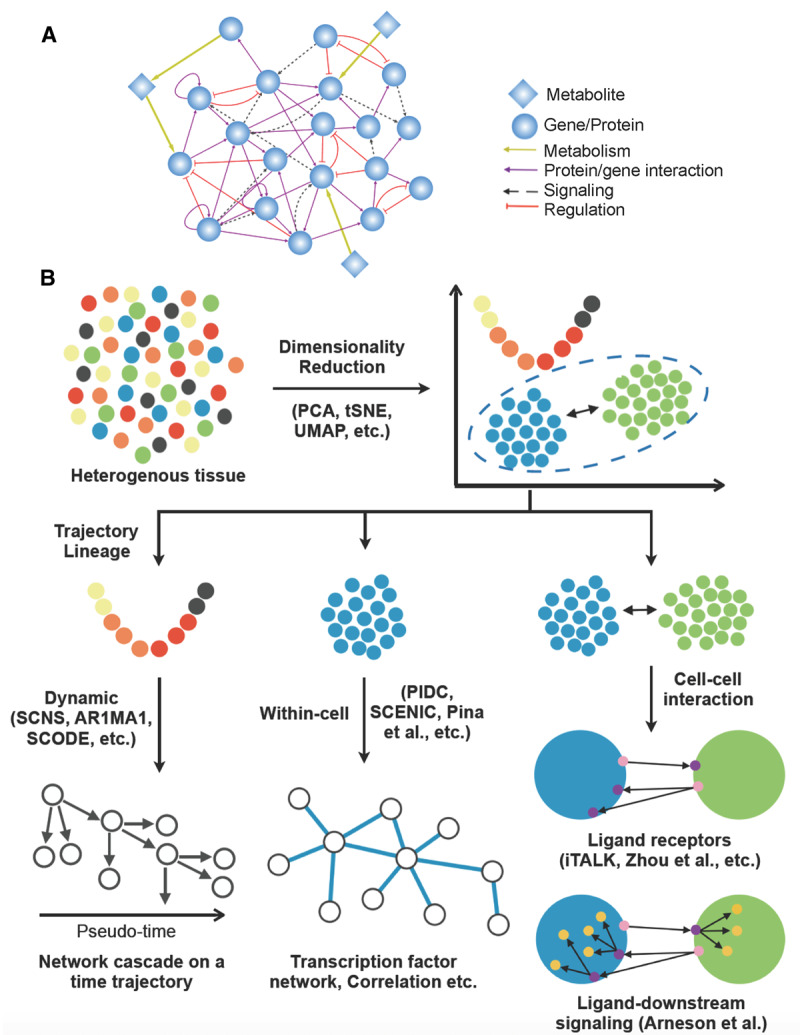
(A) Various types of network nodes and edges (connections) in a molecular network. (B) Concepts and example methods of current single-cell network modeling approaches. Transcriptomes of single cells from tissue samples are first processed and clustered using dimension reduction techniques, followed by cell identity determination using known cell markers. Cell populations in various dynamic states can be ordered using pseudo-time and trajectory analyses, and the pseudo-time information can be used in dynamic network modeling. Gene networks within a given cell population and cell–cell communication networks can also be reconstructed based on various assumptions and algorithms.
Network modeling has seen extensive applications over the past decades to help understand key biological processes and regulators of health and disease. In particular, the enormous complexity of human physiology and pathophysiology demands a systems level understanding of how biological molecules interact within individual cells and tissues and between cells and tissues to maintain homeostasis, and how perturbations of these interactions lead to diseases. The omnigenic disease model, which states that all genes interacting in networks can contribute to complex diseases, is increasingly recognized and accepted [6]. These conceptual frameworks match perfectly with the capacity of network biology, and, therefore, it is not surprising to witness the increasing use of network modeling approaches in essentially all fields of biology. For example, many genetic variants can influence disease, each through very small effects which make biological interpretation difficult. These convoluted genetic effects can be better understood through their relationships in transcriptional and signaling networks and biological pathways [6,7]. Our group and others have leveraged network models for the interpretation of genetic causes of complex diseases [8–20]. Similarly, networks can be used to understand the molecular cascades involved in various environmental causes of diseases [21–23]. For example, by integrating genetic association with tissue-specific GRNs, Chella Krishnan et al. [20] found that numerous genetic variants associated with nonalcoholic fatty liver disease affect diverse biological pathways ranging from lipid metabolism, immune system, cell cycle, transcriptional regulation to insulin signaling, Notch signaling, and oxidative phosphorylation that interact in GRNs in the liver and adipose tissues. Based on network topology, they identified key regulators involved in mitochondrial function at the center of disease pathways and subnetworks. In another study, network modeling of genetic risks of cardiovascular disease and type 2 diabetes using tissue-specific GRNs revealed shared and disease-specific networks and regulators [10]. In a systematic effort, Greene et al. [16] constructed 144 tissue/cell-specific networks and used these networks to predict and understand lineage-specific responses to IL1B stimulation, tissue-specific activities and functions of LEF1, connection of Parkinson's disease with other diseases, and the tissue context and genes involved in hypertension.
While network-based approaches have furthered our understanding of complex diseases, it is important to note that the majority of the network methodologies and applications have primarily relied upon omics data derived from bulk tissues. At the bulk tissue level, many methodologies and algorithms have been developed for network modeling, primarily focusing on predicting GRNs within [24–28] and between tissues [29–31] with reasonable accuracy. However, bulk tissues like non-parenchymal cells in the liver are comprised of heterogeneous cell populations including Kupffer cells, sinusoidal endothelial cells, and hepatocyte satellite cells, all with distinct functions associated with unique gene regulatory profiles [32]. Given the heterogeneous nature of tissues, bulk tissue networks primarily represent the average activities of all cell populations which can be dominated by the most abundant cell types. Therefore, tissue networks cannot capture the unique behaviors of individual cell populations and how cells interact to perform higher-level tissue functions.
The recent explosion of high-throughput single-cell omics technologies brings exciting possibilities to model dynamic, within-, and between-cell gene networks to elucidate the processes underlying cell development, functional state, and cell–cell communications that are missing from bulk tissue networks. These single-cell omics technologies give us the unprecedented ability to examine the transcriptional, protein, and epigenomic profiles at single-cell resolution, which are necessary to tease apart the regulatory and functional relationships of biomolecules within individual cells or cell types, and between cell populations. In theory, similar framework and methodologies that have been used for bulk tissue network modeling could be extended to single-cell data to uncover the regulatory mechanisms governing functions within and between cells. However, as suggested by Chen and Mar in their recent study [33], the models for bulk tissue may not be well suited to overcome the unique challenges introduced by single-cell data.
Here, we discuss the existing network modeling approaches developed for bulk tissue omics data, the unique challenges imposed by single-cell omics data for use in network modeling, the recent development of approaches for network models which make use of single-cell data, and their key underlying algorithms, advantages, and disadvantages. Lastly, we discuss the remaining gaps to be overcome and where we see the field headed to achieve a more efficient and accurate modeling of gene regulatory networks based on single-cell omics data.
Commonly used GRN modeling approaches for bulk tissue data
Common GRN methods that have been developed and optimized for bulk tissue data are generally based on correlation, regression, ordinary differential equations (ODEs), mutual information, Gaussian graphical models, and Bayesian approaches [24–28]. For example, a correlation-based method named weighted gene coexpression network analysis (WGCNA) is the most commonly used methodology [34,35]. WGCNA is used to find clusters (or modules) of genes which are highly correlated and usually represent tightly regulated genes involved in similar biological pathways or functions. Although coexpression-based methods are computationally efficient and less dependent on assumptions, these methods mainly group genes involved in similar functions or under similar regulation, but cannot infer directionality or direct regulatory relations and require integration with other information to facilitate interpretability [28]. Regression-based methods such as GENIE3 resolve networks by determining the most predictive subset of genes for each network gene based on regression models [36]. These methods perform well for linear cascades but not for feed-forward loops. For methods based on mutual information, such as ARACNE [37] and CLR [38], network structure is determined by the degree of dependencies between pairs of genes. These mutual information-based network methods can infer directionality and potential causality, and can predict feed-forward loops more accurately but have limited performance with linear cascades. Bayesian network (BN) modeling approaches offer flexible frameworks to incorporate and integrate multi-omics data as prior information to infer causal and directional gene–gene interactions [39,40]. A BN encodes conditional dependencies between genes, where each gene is determined by the values of its parent nodes. To improve accuracy, BNs search through the multivariate space of possible graphs which comes at the expense of higher computation cost [25,41] and the lack of guarantee that optimal topology can be detected. The different commonly used GRN inference algorithms come with different pros and cons, and the integration of multiple methods can compensate for the disadvantages inherent to each method and provide a better interpretation of the data [27]. It is important to note that these methods were optimized for bulk tissue-level data that generally conform to standard data distributions and have few missing values.
Single-cell technologies and data structures
Leveraging the recently developed single-cell technologies, we are now able to examine the transcriptional (DropSeq [42], inDrop [43], 10× Genomics, SmartSeq v4, Mars-Seq [44], Seq-Well [45], SPLiT-seq [46], sci-RNA-seq [47]), protein (CITE-seq [48]), and epigenomic profiles such as open chromatin (scATAC-seq [49]) and methylation landscapes [50]. These single-cell technologies bring about exciting possibilities to explore biology at an unprecedented resolution and scale. The most popular and widely available technologies for interrogating single cells in a high-throughput manner are single-cell RNA sequencing (scRNAseq). Typically, these high-throughput single-cell transcriptome technologies are based on counting transcript fragments from the 3′-end which are then aligned to reference genomes. The resulting data structure which aggregates gene counts for each single cell is called a digital gene expression (DGE) matrix. For other data types, similar cell by marker (e.g. protein, chromatin location, and methylation sites) matrices form the main data structures. Although the projection of single-cell epigenome onto single-cell transcriptome [51] has been performed, the integration of multi-omics data for GRN modeling has not been attempted to our knowledge and is a future direction for methodological development. Multi-omics data can be incorporated in many ways, including constructing a single network with edge confidence extrapolated across omics layers and multiple networks from individual omics layers with interactions between the layers drawn from correlative relations or known functional relevance. For example, open chromatin located in the promoter or enhancer regions of particular genes would allow a directed edge to be drawn between the scATAC-seq and scRNAseq layers; proteome data may help infer interactions between proteins and provide information on regulatory proteins such as transcription factors (TFs) and epigenomic regulators that regulate the transcriptome and epigenome. In this review, we will focus on scRNAseq data as these are the most abundant single-cell data type investigated for GRN modeling.
Performance of existing GRN approaches designed for bulk tissue data in single-cell network modeling
Recently, Chen and Mar [33] evaluated the ability of five generalized network reconstruction methods that were commonly used for bulk tissue data in network construction using both empirical and simulated single-cell data. The methods used in their analyses included partial correlation, BN, GENIE3, ARACNE, and CLR [36–38,52]. Using precision-recall and receiver operating characteristic curves to evaluate whether each method can accurately recapitulate a reference network, it was found that all methods failed to perform significantly better than random generation in both simulated and experimental single-cell datasets. Additionally, there was limited overlap in network predictions between methods. These findings suggest a lack of generalizability and applicability of the existing methods for network construction based on single-cell data. However, caution is needed to interpret such comparison results as the validity of the gold standard reference network used and the quality evaluation metric can significantly influence the comparison results.
Unique challenges, opportunities, and considerations in network modeling of scRNAseq data
The potential lack of performance of existing methods can result from the unique challenges related to data sparsity, distribution, and increased data dimension and capacity [53–55]. First, for scRNAseq using recent high-throughput platforms, due to the miniscule amount of mRNA present in a single-cell and current technological limitations, most entries in DGE matrices are zeros which result in a very sparse matrix, making the direct extension of methods designed for bulk tissue data difficult. Importantly, although these zeros can be a result of stochastic gene expression in individual cells (biological zeros), they do not necessarily mean the absence of mRNA molecules but rather the result of low technical sensitivity for moderate to lowly expressed genes, termed dropouts. Of note, read count-based scRNAseq is zero-inflated, whereas scRNAseq incorporating unique molecular identifier (UMI) counts has been found to possess ‘non-zero-inflated’ features resulting in different distributions compared with the read count-based technologies [56]. The discrepancies in the underlying data distributions in read count and UMI-based scRNAseq demand the implementation of novel methods that consider these different technologies in the future [57,58].
In an attempt to assign values to dropouts, many single-cell imputation methods such as MAGIC [59], scImpute [60], DrImpute [61], SAVER [62], BISCUIT [63], ScUnif [64], PBLR [65], and deepImpute [66] have been developed in recent years; however, the performance of these methods varies significantly [67]. In benchmarking, scImpute and DrImpute succeeded in simulated data but failed when faced with non-collinear empirical data, while both SAVER and BISCUIT could only consistently impute dropouts with near-zero values [67]. Additionally, the primary metrics used to measure performance (e.g. rand index or mutual information) benchmark the ability of these methods to define cell clusters; it is unclear how these imputed values may affect network structures. Without a consensus and experimental validation of the results from these imputation methods, caution is needed when using imputed data for network construction. A straightforward and intuitive approach taken by Han et al. [68] used subsets of cells from the same cell type and averaged the non-zero values of each gene across cells from each subset to derive a ‘supercell’ gene expression matrix, which is less inflated with zero values and may prove more biologically relevant. It is important to note that this practice will reduce the cell numbers and sacrifice statistical power.
A second challenge, related to the dropout issue in single-cell data, is the nonstandard data distribution patterns. The high number of dropout values significantly skews the data distribution from unimodal such as a Gaussian distribution to multimodal distributions, which violate the statistical assumptions underlying most of the classic GRN modeling approaches. Careful assessment of the data distribution patterns and tailoring of appropriate statistical methods are needed for single-cell network construction. Several statistical methods such as zero-inflated factor analysis (ZIFA) [69] and ZINB-WaVE (Zero-Inflated Negative Binomial-based Wanted Variation Extraction) [70] have been developed to specifically model the zero-inflated single-cell data distribution. ZIFA is a dimensionality reduction method based on the assumption that lowly expressed genes are more likely to result in dropouts than those that are highly expressed. ZIFA extends factor analysis by incorporating a model of the dropout rate as an exponential decay based on the mean non-zero expression. However, there are limitations to ZIFA in that it models strictly zero measurements and cannot account for near-zero values. Additionally, ZIFA has an underlying linear transformation framework; however, nonlinear dimensionality reduction techniques like t-SNE and UMAP have been demonstrated to be useful in the interpretation of single-cell data, so the extension of zero-inflation modeling to these nonlinear approaches could be useful. ZINB-WaVE is another dimensionality reduction technique which uniquely models the count nature of scRNAseq data and offers normalization using a sample-level intercept and flexible gene-level and sample-level covariate incorporation to address batch effects and sequence composition effects (e.g. gene length or GC content). To address the zero inflation and overdispersion of the data, ZINB-WaVE modifies a standard negative binomial distribution which does not fit the data well, with a term that gives the probability of observing a 0 instead of an actual count. Although ZINB-WaVE is primarily demonstrated as a dimensionality reduction technique for single-cell data, the authors suggest that the low-dimensional representation can be used in downstream analyses like clustering or pseudo-time. Recently, Townes et al. [58] found that multinomial methods outperform other current practices in feature selection and dimension reduction. Consideration of these alternative statistical methods in GRN inference may prove useful. It should be noted that these statistical methods have been developed for read count data and may not be suitable for UMI-based single-cell datasets as they have different underlying data distributions which are not zero-inflated.
Thirdly, it is essential that the field masters the ability to correct for confounding factors and extrapolate data acquired from multiple experiments into one common atlas. Challenges arise as the composition of cellular data remains variable across batches and studies, and even when batches contain the same cell types, the cell number and transcriptional states of individual cell types can vary significantly due to procedural noise (tissue dissociation, sorting, and reagent batches), scRNAseq platforms (e.g. 10× Genomics vs. Dropseq), and chemistry versions (version 2 vs. 3 of 10× Genomics). Much like using batch correction within the bulk tissue setting to adjust confounding factors, the integration of data sets produced by different experiments or even labs are invaluable, as it bolsters the statistical strength and reproducibility. Methods originally developed for bulk tissue batch correction such as limma [71] and ComBat [72] have been applied in the batch correction of single-cell data [73–76]; however, there have been studies which demonstrate the limitations of applying these methods developed for bulk data to both simulated and real single-cell data [77]. Recently, significant progress has been made in this area, yielding methods developed specifically for single-cell batch correction such as canonical correlation analysis (CCA) [78] and mnnCorrect [77] and methods for cell-type identification based on a labeled reference dataset such as scmap [79] and singleR [80]. However, it is important to proceed to downstream analyses like GRN construction with caution after applying a batch correction method to single-cell data, and it is necessary to understand the underlying algorithms and assumptions. Methods like CCA and mnnCorrect only leverage the highly variable genes which are shared across datasets for integration and return a corrected gene expression matrix which only contains the variable genes used for integration. These genes primarily define cell-type-specific markers, and the process of CCA inherently introduces dependencies between genes and violates the assumptions of statistical tests used for downstream analysis like differential expression, so the authors of CCA caution against using CCA for more than conserved cell-type identification across datasets. Broadly speaking, batch correction methods developed for bulk data perform more poorly in batch correction, whereas methods developed for single-cell data are more accurate at clustering cell types from different batches but may not be extended to downstream analyses. Therefore, there is a need for the development of methods which can do both.
Lastly, compared with bulk tissue data which usually consists of experimental group IDs, sample IDs, and feature measurements, single-cell data also present increased dimensionality and data volume by adding tens of cell types and thousands of cells from each sample. Such increases in dimensionality and data volume not only make network modeling more complex and computationally expensive, but also bring new possibilities from the biological perspective that are beyond the capacity of existing methods. In addition to the typical question of how genes are organized and interacting in a network, one can address many new provocative questions. For example, what defines a cell type? How are genes organized in each cell type? How different are the network architectures between cell types? What are the relationships between cells — do they come from the same or different lineages and how do the lineages evolve? Are there different states of the same cell type? What gene regulatory circuitry determines a cell state? How do cells transition from one state to another? Which cells communicate with one another to determine higher-level functions, and through which genes and pathways do they communicate? Many of these new questions are not considered or readily addressable by the existing methods designed for bulk tissues. In addition to offering the opportunities to answer these important questions, the cell–cell variability or heterogeneity in the large numbers of cells measured in each sample also provides sufficient information to construct within-sample or profile-specific networks [55]. Such networks describe the GRN of a single biological sample, which is not possible for bulk tissue profiling data. In other words, the ability to exploit the large cell number dimension allows GRNs to be constructed for each sample based on its constituent cell profiles, which can be used to derive consensus networks across samples to enhance accuracy.
Recent methods for scRNAseq GRN modeling
Recognizing the need for new GRN modeling methods for single-cell data, many approaches have been recently developed primarily based on scRNAseq data. We categorize these methods based on fundamental biological questions (dynamic modeling, within-cell networks, and cell–cell interaction networks; Figure 1B), followed by the specific biological assumptions (e.g. TF to target interactions, ligand–receptor interactions) and algorithms (e.g. coexpression, regression, ODEs, Bayesian, and Boolean), as summarized in Table 1.
Table 1. Summary of single-cell network modeling approaches.
| Category | Example methods | Underlying biological assumption | Algorithmic basis | Advantages | Limitations |
|---|---|---|---|---|---|
| Dynamic network (extensively reviewed in refs [53–55]) | SCNS [81] | Single-gene changes between cell transition states can inform on gene regulatory relations | Boolean | Does not rely on prior knowledge. Has a web UI. Resulting models are executable and can be used to make predictions | Need data discretization; limit to small numbers of genes; regulatory relations need to follow Boolean rules |
| SCODE [82] | TF expression dynamics (pseudo-time) and TF regulatory relations (GENEI3) | ODE; Bayesian model selection | Estimate relational expression efficiently using linear regression; reduction of time complexity; fast algorithm | Need dimension reduction first for computing speed and memory feasibility; assumes that all cells are on the same trajectory; optimization is computationally intractable | |
| GRISLI [83] | Variability in scRNAseq data caused by cell cycle, states, etc. allows the inference of pseudo-time associated with each individual cell | ODE | Makes no restrictive assumption on the gene network structure; can consider multiple trajectories; fast algorithm | Has to estimate the velocity of each individual cell using information from neighbors | |
| SINCERITIES [84] | Changes in the expression of a TF will alter the expression of target genes | Ridge regression and partial correlation analysis | Low computational complexity and able to handle large-scale data | Requires scRNAseq data at multiple time points. Restricted to TFs and their targets to infer edges | |
| Scribe [85] | Cell ordering can be improved with time-series or cell velocity estimations | RDI | Outperforms other pseudo-time methods given time-series data. Can be applied to any data type if the data structure is appropriate | Requires time-ordered gene expression profiles or velocity estimation from introns and exons | |
| AR1MA1-VBEM [40] | The cell differentiation process or response to external stimulus reveals the hierarchical structure of the transcriptome | First-order autoregressive moving-average and variational Bayesian expectation-maximization | Weighted interactions between genes along psuedotime. Model used accounts for noisy data | Data are expressed as fold changes between timepoints/conditions or scaled by housekeeping genes | |
| SCINGE [86] | Learned target regulator genes can be used to assign each cell to their progress along a trajectory | Granger causality | Smooths irregular pseudo-times and missing expression values | Near random performance for predicting targets of individual regulators | |
| SoptSC [87] | Similarities between whole transcriptomes of single cells can be used to order them | Cells ordered by minimum paths on weighted cluster-to-cluster graph derived from cell similarity matrix | Includes comprehensive single-cell workflow; leverages information from other parts of the workflow to improve performance | Cannot be run with other tools, have run the full workflow to get pseudo-time inference | |
| Within-cell or cell population network | SCENIC [88] | TF target-based regulation | Combining TF regulatory relations (GENIE3) with TF-binding motif analysis | Robust against dropouts, get a TF score for individual cells (no averaging of cells). | Limited to TF-based relations |
| Pina et al. [89] | TFs drive lineage commitment | Odds ratio for on/off gene associations and spearmen correlation for expression levels associations | Robust to dropouts | Based on single-cell multiplex qRT-PCR, may be difficult to extend the method to sparse single-cell data (selected 44 genes to test) | |
| Iacono et al. [90] | Coexpression is regulated by TFs, cofactors, and signaling molecules which can be captured with gene–gene correlations | Pearson correlation using z-score-transformed counts | Can compute correlations at the single-cell level and it is robust to dropouts and noise inherent to single-cell data | Networks are very dense (some have millions of significant edges) | |
| PIDC [39,91] | Gene regulatory information reflected in dependencies in the expression patterns of genes | Partial information decomposition using gene trios | Compared with correlation, captures more complicated gene dependencies | Networks are influenced by data discretization, choice of mutual information estimator, method developed for sc-qPCR data, may not be extendable to higher throughput and sparser scRNAseq data | |
| Jackson et al. [92] | Deletion of TFs combined with experimental conditions allows for the inference of gene relationships | MTL to leverage cross-dataset commonalities and incorporate prior knowledge | Does not require sophisticated normalization of single-cell data or imputation. Able to combine multiple conditions/datasets for more accurate inference. TF deletions give strong causal link to affected genes | Requires single-cell data with TF deletions and/or environmental perturbations | |
| Wang et al. [93] | Gene perturbations allow for inference of causal relationships | Scoring of conditional independence test to identify optimal DAG | Gives causal relationships between genes | Requires interventional data. No loops allowed in DAG | |
| ACTION [94] | Functional identity of cells is determined by a weak, but specifically expressed set of genes which are mediated by TFs | Kernel-based cell similarity and geometric approach to identify primary functions | Robust to dropout and does not require averaging. Identifies functions unique to cell types | Requires TFs and their targets. Only provides TF-driven networks | |
| SINCERA [95] | TF target-based regulation | First-order conditional dependence on gene expression to construct a DAG | Key TFs identified using multiple importance metrics | Only considers TFs and their targets. Requires genes/TFs to be DEGs or expressed in >80% of cells | |
| Cell–cell communication network | iTALK [96] | Ligand–receptor interactions | Threshold ranked list of genes from two cell types for ligand–receptor pairs | Allows for the inference of directionality of interaction | Requires curation of ligand–receptor interactions (not all interactions are known). Average expression at the cell-type level (no longer single cell). Cannot reveal novel interactions beyond known ligand–receptor knowledge |
| Zhou et al. [97] | Ligand–receptor interactions | Expression of ligand and corresponding receptor more than three standard deviations greater than the mean | Allows for the inference of directionality of interaction | Requires curation of ligand–receptor interactions (not all interactions are known). Average expression at the cell-type level (no longer single cell) | |
| Kumar et al. [98] | Ligand–receptor interactions | Product of the average expression of ligand and corresponding receptor | Allows for the inference of directionality of interaction. Interaction score gives the strength of interaction (rather than just significance) | Requires curation of ligand–receptor interactions (not all interactions are known). Average expression at the cell-type level (no longer single cell) | |
| Arneson et al. [99] | Ligand to downstream signaling | Coexpression of ligand genes in source cells with other genes in target cells | Use secreted ligands as a guidance for directional inference between cell populations | Gene expression is summarized to the cell population level and coexpression is at the sample level, requiring large sample sizes | |
| SoptSC [87] | Ligand–receptor interactions | Likelihood estimate of the interaction between two cells based on expression of the ligand, receptor, and downstream pathway target genes (including expression direction). Consensus signaling network derived from all cells in each cluster | Incorporates target genes of pathways and their directionality. Computes interaction likelihood at the single-cell level and summarizes across all cells in the cluster for higher confidence | Requires curation of ligand–receptor interactions and their downstream pathways | |
| scTensor [100] | Ligand–receptor interactions | Tensor decomposition with cell–cell interactions as hypergraphs | Allows L–R pairs to function across multiple cell-type pairs (not restricted to a single-cell-type pair), which is more reflective of underlying biology | Requires curation of ligand–receptor interactions. Averages single cells to the cell-type level |
The most straightforward algorithm is coexpression in which the likelihood of a gene interacting with another depends on the strength of their pairwise correlation coefficients. Though computationally tractable, most of these methods do not provide directionality and likely infer functional relatedness rather than direct regulation. More complex methods include ODEs, Boolean networks, and BNs, each with advantages and limitations, as discussed earlier. Boolean networks require discretization of gene expression values and apply Boolean functions to describe regulatory interactions, which likely result in oversimplification. ODE-based methods involve using linear, nonlinear, or piecewise differential equations to model the dynamic nature of mRNA content in a continuous, rather than discrete, manner. A BN is a directed acyclic graph (DAG) that integrates prior information to guide its gene–gene interaction predictions and is probabilistic in nature. Lastly, information theory measures describe statistical dependencies between biological entities and include entropy, the notion that information is quantified based on the uncertainty of a random variable, and mutual information, in which the observation of one random variable can inform on or reduce the uncertainty of another random variable. This measure produces more general correlations that allow capturing of nonlinear dependencies and is commonly employed in network inference.
Of note, with new methodologies being developed at rapid speed, it is not possible to exhaustively document all available methodologies. Here, we highlight the broad categories for single-cell GRN modeling and discuss example methods to illustrate the concepts and make note of their advantages and potential limitations. We also exclude methods that were developed based on data from older low-throughput single-cell platforms such as single-cell qPCR, which do not share the same challenges as sparse high-throughput scRNAseq.
Dynamic networks
To date, the majority of the scRNAseq-based GRN modeling approaches were designed to address dynamic cell-state transition (Figure 1B), as scRNAseq data contain information from asynchronous cell populations which show temporal dynamics, allowing for the mapping of cellular transitions on a pseudo-time scale [101,102]. Common models for expression dynamics or pseudo-time estimates assume that cellular changes (i.e. development, activation, and deactivation) progress along a continuous curve or an idealized tree and that each intermediate stage is short and captured through the sequencing of large numbers of cells. Under these assumptions, computational modeling can infer the trajectories of cellular dynamics, which can be derived based on known regulatory relations such as TF target information, similarities in gene expression, and RNA velocity represented by immature and mature mRNA content [60]. However, it is important to note that the simultaneous presence of various cellular states at a given snapshot does not represent real time course for the inference of sequential or lineage information. Therefore, incorporating pseudo-time may not necessarily improve GRN construction.
To date, more than 50 methods have been developed for trajectory inference to derive pseudo-time information, and these have been reviewed and compared previously [101,102]. Pseudo-time ordering lends directionality and interaction-type information for dynamic GRN modeling [40,82,83,85,86,103,104]. Such pseudo-time information is integrated with commonly used network construction algorithms outlined above such as correlation [88,94,95], ODE [82,83,103,104], Boolean [105,106], BN [39,40], information theory [91], and other methods [107].
Many of the dynamic GRN methods have been extensively reviewed by others [53–55], and we only discuss a few examples of the different categories here. A Boolean network method, SCNS, is based on single-gene changes between ordered cells where cells have been discretized into an on/off state [81]. Another method SCODE uses a linear ODE, a pseudo-time estimation that assumes all cells are on the same trajectory, and a TF-based framework to model TF dynamics that captures regulatory relationship between genes [82]. Building on this, GRISLI was recently developed, which uses a similar approach to SCODE, but considers multiple cell trajectories, does not assume a network structure, and has faster computation times [83]. GRISLI first estimates the velocity of each cell, followed by solving a sparse regression problem to relate the gene expression of a cell to its velocity profile to estimate the GRN. An information theory-based method, SINCERITIES, utilizes Granger Causality for directionality information and quantifies temporal changes in the expression of each gene between two subsequent (pseudo)timepoints [82]. Changes in TF expression are used to predict changes in corresponding genes in the next time window using ridge regression, with edge direction and sign inferred using partial correlation analysis on the expression of every gene pair. SCINGE also uses kernel-based Granger Causality regression on ordered single-cell data to predict regulator-target gene interactions and then ranks the predicted interactions by aggregating the regression results [86]. An additional method is PIPER [107], which uses local Poisson graphical modeling to more effectively capture network changes during cellular differentiation and highlight the key TFs that drive these changes. A BN inference approach, AR1MA1-VBEM (Variational Bayesian Expectation-Maximization), applies a first-order autoregressive moving-average (AR1MA1) model to fit the time-series with a linear model that represents observations as combinations of the data at the previous timepoint and a noise term, and uses a VBEM framework that utilizes variational calculus to optimize the marginal likelihood and posterior distributions of network models [40]. Scribe [85] is another recently developed method, which uses restricted directed information (RDI) [108] to infer causal GRNs by borrowing linked time-series data or inferred cell velocity from intronic (indicative of immature RNA) and exonic reads. The authors demonstrate that Scribe outperforms other pseudo-time methods when true time-series data are available; however, the performance of all methods suffers dramatically when temporal information for the measurements is lost. Interestingly, Deshpande et al. [86] recently compared various methods and found that incorporating pseudo-time does not always lead to better performance but can hurt network reconstruction in certain cases. As discussed earlier, this is likely due to issues in the assumptions of pseudo-time methods.
Within-cell population networks
The second category of methods focuses on modeling GRNs within-cell populations without considering cell trajectories or dynamics. These methods include coexpression and TF-based [88,94,95], coexpression and TF-independent [89,90,109], and information theory [91] (Table 1 and Figure 1B). This is in line with the basic concepts underlying GRN modeling of gene–gene interactions for a tissue, except here single-cell data are modeled for specific cell populations.
Similar to dynamic network modeling, the simplest approach for modeling GRNs within-cell populations is based on coexpression. Here, the coexpression methods are divided into two groups: those which utilize prior information in the form of TFs and those that do not. For the methods which are TF-independent, the likelihood of a gene interacting with another depends on the strength of their pairwise correlation coefficients and all possible gene pairs are considered. In the TF-based methods, genes are grouped into modules based on those with the strongest pairwise correlation coefficients with the different TFs or are segregated into potential interactions based on prior literature or motif evidence. A more sophisticated approach for defining GRNs within-cell populations, which can capture nonlinear gene dependencies, is partial information decomposition which is derived from information theory. Here, the information provided by pairs of genes is used to quantify unique, shared, and synergistic information about a third gene across all sets of three genes to infer a network structure.
Several correlation-based methods have been developed that compare the gene expression patterns between known or predicted TFs and target genes, or between all genes. For instance, SCENIC couples gene coexpression with TF-binding motif analyses of modules of coexpressed genes to identify GRN modules, predict TF regulators, and identify single-cell level activity of putative TF targets (called regulons) [88]. The activity of the regulons can be used to cluster cell types, compare network conservation, and identify important cell states and GRNs involved in disease. Another method SINCERA is a full analytical pipeline for processing scRNAseq data. It first identifies candidate TFs and their targets for each cell type [95]. The interactions between two TFs or a TF and a target gene are then determined using first-order conditional dependence on gene expression [110], and the key TFs in each GRN are identified by integrating six different node importance metrics. An additional coexpression-based GRN method, ACTION, uses a novel archetype orthogonalization approach to construct cell-type-specific GRNs based on the key assumption that the functional identity of a cell is determined by a set of weak, but specifically expressed genes which are mediated by a set of TFs [94]. ACTION describes each cell as a set of ‘cellular functions’ in high dimensional space and the number of these functions is determined using a non-parametric approach. The genes which are unique to each cellular function are determined using orthogonalization and the role of TFs in controlling the genes in these cellular functions is assessed. The TF and associated target genes within a cellular function serve to constitute the network.
Pina et al. [89] and, more recently, Iacono et al. [90] also utilize coexpression but build global GRNs that are not limited to TF target relations [89,90]. The former calculates pairwise Spearman rank correlations between all sets of genes across cells within a cell type to infer cell-type GRNs in hematopoiesis, and significant pairwise associations were identified using the odds ratio of linearly transformed expression data. Iacono et al. [109] used a Pearson correlation-based method which first transforms the expression values using bigSCale to derive a z-score for each gene using a probabilistic model to account for noise and variability inherent to single-cell data. Pairwise correlations of z-scores are used to construct GRNs. The use of z-scores boosts the number of significant gene-to-gene correlations.
To reveal complex gene dependencies not afforded by simpler correlation strategies, GRN inference methods have employed techniques from information theory. Specifically, PIDC uses partial information decomposition to find the unique information provided by any pair of two genes across all other possible genes [80]. The confidence of an edge between two genes is the sum of the scores of those genes across all other genes in the set. This multivariate information approach makes use of the large sample size present in single-cell analyses to identify nonlinear dependencies between pairs of genes by leveraging a third gene.
Cell–cell communication networks
The basic functions of a given heterogeneous tissue are determined not only by the activities of individual cell types within the tissue, but also by the intimate communications and coordination among cell populations. For instance, neurons and astrocytes interact to ensure essential brain functions [111], and immune cells interact with adipocytes in the adipose tissue to regulate energy metabolism and thermogenesis [112]. As such, cell–cell communication is a critical biological question yet has not been comprehensively addressed due to the previous lack of high-throughput, high-resolution singe-cell data. The unique ability of single-cell approaches to simultaneously capture numerous cells of diverse cell types makes it feasible to model cell–cell communication networks (Table 1 and Figure 1B). The underlying assumption for modeling such networks is that communications between cells can be captured by molecular patterns measured in individual cell populations. For example, a pair of communicating cells may express genes and proteins involved in a particular function (e.g. one expressing a ligand and another expressing the corresponding receptor to trigger signaling pathways) in a coordinated fashion.
Early attempts to model cell–cell communication networks have been primarily based on the concept of gene coexpression with or without the consideration of ligand–receptor interaction information. The underlying assumption is that gene correlation patterns between cells reflect true biological interactions. The validity of this assumption has been supported by evidence at the level of tissue–tissue interactions. For instance, gene coexpression between brain regions can recapitulate the functionally derived interactions of the mouse brain connectome [113], and gene coexpression between five different mouse tissues revealed novel endocrine factors mediating the communications which are subsequently validated with experiments [30].
Coexpression methods were quickly adapted to single-cell data when Han et al. built cell–cell connections based on the similarities in gene expression profiles across cell types [68]. However, such networks more likely reflect the similarities between cell types rather than interactions or communications. To modify the classical coexpression framework, ligand–receptor-based methods have been proposed which rely on the assumption that a significant portion of cell–cell communication occurs via the release of chemical molecules from one cell that bind to receptors of another cell. Utilizing this assumption allows ligand–receptor-based methods to construct reliable biology-based directed networks. However, it comes at the expense of heavily limiting the set of potential genes in an inherently sparse data modality. It is important to note that coexpression-based analyses typically utilize Pearson's correlation coefficient, which may not be suitable for read-based single-cell datasets due to the zero-inflated nature and unique distribution patterns. When using coexpression-based analysis on single-cell data, it is important that data transformation and appropriate statistics are taken into consideration.
There are several methods illustrating cell–cell communication via ligand–receptor interactions. Zhou et al. [97] curated a list of >25000 known ligand–receptor pairs to examine their changes in the transcriptomes of ∼4000 melanoma cells. To determine if a pair of cells were communicating, the ligand and corresponding receptor had to be expressed above a certain tunable threshold in the two cell types. Similarly, Kumar et al. [98] focused on ∼1800 literature-based ligand–receptor pairs, but implemented a different scoring scheme that considers the product of average receptor expression and average ligand expression in the respective cell types under examination. Ported as an R package with a data visualization tool, iTALK is another new ligand–receptor interaction-based network construction method [96]. iTALK identifies ligand–receptor pairs (from a database of >2600 pairs) between two cell types by interrogating the list of ranked genes derived from average expression (single timepoint/condition) or differentially expressed genes (multiple timepoints/conditions) for each cell type and the list of ligand–receptor pairs in the iTALK database. Additionally, iTALK is able to use metadata (e.g. timepoints, groups, and cohorts) to find cell–cell interaction changes by identifying differentially expressed ligand–receptor pairs. Similarly, Smillie et al. [114] have used thousands of literature-supported receptor–ligand interactions from the FANTOM5 database to identify cell–cell interactions by requiring that genes are cell marker genes or differentially expressed genes to call significant interactions between cells. In most ligand–receptor approaches, ligand–receptor pairs are restricted to communicating cell types; however, in scTensor, Tsuyuzaki et al. [100] took a more flexible approach where no such restrictions are made. In scTensor, cell–cell interactions are represented as hypergraphs which describe directed edges of ligand–receptor pairs determined using tensor decomposition. A recent method proposed by Vento-Tormo et al. [115] also considers secreted molecules as well as cell-surface molecules and uses a permutation-based approach to find enriched ligand–receptor pairs between cell types. To achieve this, the authors developed CellPhoneDB, a public repository of ligand–receptor interactions curated from public resources of protein–protein interactions, which includes the subunit composition of ligands and receptors to fully represent their interactions. For proteins which are comprised of multiple subunits, expression of all subunits is required to infer accurate interactions.
The above methods all focus exclusively on ligand–receptor pairs which heavily limit the putative genes to sets of literature-curated gene pairs which can inform on cell–cell communication. Previously, a less restrictive modeling approach that dissects tissue–tissue communication networks [30] based on the coexpression of genes encoding secreted peptides from a source tissue and all genes in a target tissue has been developed. Arneson et al. [99] adopted this concept to build cell–cell communication network maps in the hippocampus of sham mice versus mice with traumatic brain injury, revealing extensive rewiring of networks in brain injury. This method infers connections between cells based on the assumption that one cell communicates with another cell by secreting signaling molecules that bind to their receptors on the target cell to trigger downstream molecular events in the target cell. As such, it is likely that coexpression exists between the genes that encode secreted signaling molecules (i.e. the ligands) in the source cell type and the receptors as well as the downstream pathway genes in the target cell type. Additional methods can broaden the scope of cell–cell interactions beyond ligand–receptor-based relationships by considering the patterns among all expressed genes between cell types, although the biological interpretation of this approach is less straightforward.
Hybrid methods
Although most GRN methods tackle either dynamic or within-cell or between-cell networks, Wang et al. [87] have proposed SoptSC, a unifying framework to conduct single-cell analysis starting from gene expression matrices to basic analytical workflows (e.g. normalization, clustering, dimensionality reduction, and identifying cell marker genes) and subsequently to infer both cell–cell communication networks and pseudotemporal ordering/lineage. The key premise underlying SoptSC is that the structured cell-to-cell similarity matrix can help improve the network inference steps. The similarity matrix is also used for pseudotemporal ordering by finding the shortest path between cells on a weighted cluster-to-cluster graph. To infer cell–cell signaling networks, the likelihood estimate of the interaction between two cells is calculated based on the expression of ligand–receptor pairs and the directionality of downstream pathway target genes. A consensus network between clusters/cell types is generated by summarizing the probability of signaling between all cells of any two cell types.
Gene perturbation networks
All of the above methods utilize assumptions regarding information flow such as TF cascades and ligand–receptor relationships, without direct causal information. Single-cell data containing gene perturbation information are extremely useful for providing causal information for GRN construction, as targeted perturbation of a gene is the source or trigger of downstream responses of other genes. A method proposed by Jackson et al. [92] leverages gene deletion mutants. Specifically, they pooled 72 different yeast strains across 12 different genotypes (TF deletions) and 11 different conditions to generate scRNAseq data for 38 000 cells. In addition to the expression data, this method uses prior information from TF targets and biophysical parameters like TF activity and mRNA decay rates to construct a GRN using a multitask learning (MTL) framework [116]. This allows for the integration of information across different conditions and experiments that explains the relationships between the TF perturbations and the observed gene expression changes. By directly deleting TFs, the authors have created a valuable dataset which could serve as a useful benchmark for other single-cell network inference methods. Leveraging single-cell data from Perturb-seq [117], which combines CRISPR/Cas9-mediated gene perturbation with single-cell sequencing to generate high-throughput interventional gene expression data, Wang et al. [93] proposed an algorithm for inferring causal DAGs. The algorithm is based on Greedy SP which restricts the permutation-based DAG search space, and potential network scores are evaluated using the Greedy Interventional Equivalence Search [118]. To further extend this research on causal network inference, Wang et al. [119] introduced a method which could identify differences between DAGs inferred from different datasets. The same group has also demonstrated that soft interventions used in Perturb-seq, such as those that cause partial disruption of gene dependencies (e.g. RNAi or CRISPR-mediated gene activation), provide the same amount of causal information as hard interventions (e.g. CRISPR/Cas9-mediated gene deletions), which result in complete disruptions, despite being less invasive [120].
Performance assessment of single-cell GRN modeling methods
Chen and Mar recently applied a few single-cell network modeling methods including SCENIC [88], SCODE [82], and PIDC [39,91] to both simulated and empirical single-cell datasets to assess their ability to capture known network interactions. They found that there was low agreement between methods. However, as each method has unique assumptions and may not be designed to capture similar interactions, agreement between methods is not necessarily appropriate to assess performance. Another comparison study that examined the performance of multiple network inference methods that incorporate pseudo-time information, such as SCINGE, SCODE, and SINCERITIES, also indicates that many regulator-target predictions can be near random for each of the methods tested [86].
These findings call for the refinement of single-cell network modeling approaches and comprehensive revaluation of the performance of existing single-cell GRN methods. On the other hand, the ligand–receptor framework that is driven by a biological assumption along with data-driven gene coexpression appears to be promising for cell–cell communication network modeling. For example, modeling with this approach to scRNAseq data recapitulated known cell–cell interactions within the hippocampus [99].
Remaining gaps and future directions
Single-cell multi-omics profiling technologies are rapidly evolving, bringing revolutionary forces to improve our understanding of the basic unit of life, the cell, as well as the cross-talks between cells in physiological and pathological conditions. Major progresses have been made to more accurately classify cell types, correct for confounding factors, and delineate cell lineages and cell-state transitions. However, these advances are not sufficient to bring a complete understanding of the regulatory machinery underlying the functions of individual cell populations and cell–cell interactions that determine higher-level tissue functions. Existing methodologies to model gene networks that were optimized for bulk tissue data either perform poorly for single-cell data or cannot accommodate the new biological questions brought about by single-cell data, and methods that efficiently and accurately model the outpour of single-cell data into comprehensive GRN maps are still in infancy. In particular, novel network methods that are designed to address the unique challenges of single-cell data, such as data sparsity, multimodal distribution, and higher dimensionality, are still in great need. The data sparsity issue can be addressed through the improvement of single-cell technologies to enhance signal capture, or by more accurate imputation methods that are supported by strong experimental validation data. These efforts will help mitigate the issues associated with nonstandard data distribution that limits the utilization of existing network methodologies. Alternatively, methods built on more appropriate statistics and algorithms that can better accommodate dropout values and the unique data distribution are warranted.
Another critical and less highlighted gap in network modeling of single-cell data is the missing spatial information to restrain the modeling space. Many of the current high-throughput single-cell sequencing methods lack the ability to maintain the spatial identity of individual cells, which reduces one's ability to resolve cell networks accurately, particularly during development where development layers are in close proximity. Various high-throughput fluorescence in situ hybridization (FISH) methods have been developed as tools to resolve spatial information [121–128]. The spatial distances between pairs of single cells can be used as a prior to construct more sophisticated and accurate network models under the assumption that cells which are located closer together are more likely to communicate. This assumption is supported by the recent discovery of localization of ligand-producing cells directly adjacent to target cells expressing the corresponding receptor [129]. Another key advantage of single-molecule FISH-based methods is that they are extremely quantitative and do not suffer from dropouts which plague high-throughput single-cell sequencing-based methods. The absence of dropouts allows for accurate single-cell level interrogation of network predictions. With the spatial single-cell methods, it is also possible to combine phenotypes (i.e. behavior) with cellular activation (i.e. cFos) to integrate into the model under the assumption that cells which are active during a particular phenotype or stimulus are more likely to be communicating. This approach has been previously used by Moffitt et al. [130] to identify sets of neurons activated during parenting. Therefore, coupling single-cell sequencing approaches with high-throughput single-molecule imaging has enormous potential to improve network modeling at single-cell resolution. Despite the potential, there are limitations and complications involved in using spatial data to construct GRNs. First, cell segmentation of single-molecule FISH-based methods is non-trivial and GRN construction is impossible without it. Additionally, a single image carries limited representation of the dynamic cellular landscape. In fact, many of these technologies can only achieve the imaging depth of a single cell, so it is essentially a two-dimensional snapshot at a given time which may not capture cellular dynamics outside of the imaged plane and time frame.
At present, the majority of the methods are designed for scRNAseq, and methods incorporating other single-cell omics scales (genetic, epigenetic, and protein) are needed [55]. This faces the same challenge that has been encountered by bulk tissue GRN inference, and recent progresses in multi-omics integration and modeling may offer guidance for single-cell multi-omics modeling [131–134].
Lastly, the accuracy of predicted networks from empirical data is difficult to assess, as high-throughput validation through perturbing predicted regulators in single cells in vivo is more challenging than that for whole-body knockout or knockdown. On a positive note, new high-throughput gene perturbation technologies such as Perturb-seq in combination with scRNAseq have the potential to generate insight into true casual relationships between genes and cells. Data from such platforms can serve as more appropriate benchmarking datasets to assess the predictions of existing network methods by testing how well each method can retrieve the true regulatory or interactive relations known from the perturbation-response experiments. Along the same line, use of known, experimentally validated gene–gene, cell–cell circuits from the literature can be used to benchmark the methods. Even in the absence of validated network connections, a community-based approach can be employed to improve the network performance in silico by combining multiple inferred networks from various methods to obtain consensus networks. This approach has been shown to be invaluable for improving the quality of the predicted networks [27,91,135,136].
In summary, we are entering a golden era where biological discoveries can be made at an unprecedented resolution and throughput. Network modeling of single-cell multi-omics data represents one of the key tools to unlock the convoluted molecular mechanisms underlying pathophysiology and guide precision medicine. Despite numerous challenges, the field is rapidly evolving and ample opportunities for methodological innovations await to more accurately depict the molecular maps of cells in health and disease.
Summary
Single-cell omics data offer unique challenges and opportunities for molecular network modeling.
Significant progress has been made to dissect the dynamic, within-cell, and between-cell gene regulatory networks.
Performance of current methods await further evaluation.
Significant gaps remain in the development of network modeling approaches that can accommodate unique statistical challenges, diverse omics domains, and spatial information.
Abbreviations
- AR1MA1
autoregressive moving-average
- BN
Bayesian network
- CCA
canonical correlation analysis
- DAG
directed acyclic graph
- DGE
digital gene expression
- FISH
fluorescence in situ hybridization
- GIES
Greedy Interventional Equivalence Search
- GRNs
gene/transcriptional regulatory networks
- MTL
multitask learning
- ODE
ordinary differential equation
- RDI
restricted directed information
- TFs
transcription factors
- UMI
unique molecular identifier
- VBEM
Variational Bayesian Expectation-Maximization
- WGCNA
weighted gene coexpression network analysis
- ZIFA
zero-inflated factor analysis
Author Contribution
M.B., D.A., J.D., Y.-W.C., Z.S., and X.Y. drafted and edited the manuscript.
Funding
D.A. is funded by the UCLA Dissertation Year Fellowship. X.Y. is funded by the National Institutes of Health Grants DK104363 and NS103088. Y.-W.C. is supported by the UCLA Hyde Fellowship.
Competing Interests
The Authors declare that there are no competing interests associated with the manuscript.
References
- 1.Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J. et al. (2014) STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 10.1093/nar/gku1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Terzer M., Maynard N.D., Covert M.W. and Stelling J. (2009) Genome-scale metabolic networks. Wiley Interdisciplinary Reviews: Systems Biology and Medicine 1, 285–297 10.1002/wsbm.37 [DOI] [PubMed] [Google Scholar]
- 3.Costanzo M., VanderSluis B., Koch E.N., Baryshnikova A., Pons C., Tan G. et al. (2016) A global genetic interaction network maps a wiring diagram of cellular function. Science 353, aaf1420 10.1126/science.aaf1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vlaic S., Conrad T., Tokarski-Schnelle C., Gustafsson M., Dahmen U., Guthke R. et al. (2018) Modulediscoverer: identification of regulatory modules in protein–protein interaction networks. Sci. Rep. 8, 433 10.1038/s41598-017-18370-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thurley K., Wu L.F. and Altschuler S.J. (2018) Modeling cell-to-cell communication networks using response-time distributions. Cell Syst. 6, 355–367.e5 10.1016/j.cels.2018.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boyle E.A., Li Y.I. and Pritchard J.K. (2017) An expanded view of complex traits: from polygenic to omnigenic. Cell 169, 1177–1186 10.1016/j.cell.2017.05.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Costanzo M., Kuzmin E., van Leeuwen J., Mair B., Moffat J., Boone C. et al. (2019) Global genetic networks and the genotype-to-phenotype relationship. Cell 177, 85–100 10.1016/j.cell.2019.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mäkinen V.-P., Civelek M., Meng Q., Zhang B., Zhu J., Levian C. et al. (2014) Integrative genomics reveals novel molecular pathways and gene networks for coronary artery disease. PLoS Genet. 10, e1004502 10.1371/journal.pgen.1004502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shu L., Zhao Y., Kurt Z., Byars S.G., Tukiainen T., Kettunen J. et al. (2016) Mergeomics: multidimensional data integration to identify pathogenic perturbations to biological systems. BMC Genomics 17, 874 10.1186/s12864-016-3198-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shu L., Chan K.H.K., Zhang G., Huan T., Kurt Z., Zhao Y. et al. (2017) Shared genetic regulatory networks for cardiovascular disease and type 2 diabetes in multiple populations of diverse ethnicities in the United States. PLoS Genet. 13, e1007040 10.1371/journal.pgen.1007040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kurt Z., Barrere-Cain R., LaGuardia J., Mehrabian M., Pan C., Hui S.T. et al. (2018) Tissue-specific pathways and networks underlying sexual dimorphism in non-alcoholic fatty liver disease. Biol. Sex Differ. 9, 46 10.1186/s13293-018-0205-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao Y., Blencowe M., Shi X., Shu L., Levian C., Ahn I.S. et al. (2019) Integrative genomics analysis unravels tissue-specific pathways, networks, and key regulators of blood pressure regulation. Front. Cardiovasc. Med. 6, 21 10.3389/fcvm.2019.00021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao Y., Jhamb D., Shu L., Arneson D., Rajpal D.K. and Yang X. (2019) Multi-omics integration reveals molecular networks and regulators of psoriasis. BMC Syst. Biol. 13, 8 10.1186/s12918-018-0671-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calabrese G.M., Mesner L.D., Stains J.P., Tommasini S.M., Horowitz M.C., Rosen C.J. et al. (2017) Integrating GWAS and co-expression network data identifies bone mineral density genes SPTBN1 and MARK3 and an osteoblast functional module. Cell Syst. 4, 46–59.e4 10.1016/j.cels.2016.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carlin D.E., Demchak B., Pratt D., Sage E. and Ideker T. (2017) Network propagation in the cytoscape cyberinfrastructure. PLoS Comput. Biol. 13, e1005598 10.1371/journal.pcbi.1005598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greene C.S., Krishnan A., Wong A.K., Ricciotti E., Zelaya R.A., Himmelstein D.S. et al. (2015) Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 47, 569–576 10.1038/ng.3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krishnan A., Zhang R., Yao V., Theesfeld C.L., Wong A.K., Tadych A. et al. (2016) Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder. Nat. Neurosci. 19, 1454–1462 10.1038/nn.4353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoon S., Nguyen H.C.T., Yoo Y.J., Kim J., Baik B., Kim S. et al. (2018) Efficient pathway enrichment and network analysis of GWAS summary data using GSA-SNP2. Nucleic Acids Res. 46, e60 10.1093/nar/gky175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hui S.T., Kurt Z., Tuominen I., Norheim F., C Davies R., Pan C. et al. (2018) The genetic architecture of diet-induced hepatic fibrosis in mice. Hepatology 68, 2182–2196 10.1002/hep.30113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chella Krishnan K., Kurt Z., Barrere-Cain R., Sabir S., Das A., Floyd R. et al. (2018) Integration of multi-omics data from mouse diversity panel highlights mitochondrial dysfunction in non-alcoholic fatty liver disease. Cell Syst. 6, 103–115.e7 10.1016/j.cels.2017.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meng Q., Ying Z., Noble E., Zhao Y., Agrawal R., Mikhail A. et al. (2016) Systems nutrigenomics reveals brain gene networks linking metabolic and brain disorders. EBioMedicine 7, 157–166 10.1016/j.ebiom.2016.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meng Q., Zhuang Y., Ying Z., Agrawal R., Yang X. and Gomez-Pinilla F. (2017) Traumatic brain injury induces genome-wide transcriptomic, methylomic, and network perturbations in brain and blood predicting neurological disorders. EBioMedicine 16, 184–194 10.1016/j.ebiom.2017.01.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shu L., Meng Q., Diamante G., Tsai B., Chen Y.-W., Mikhail A. et al. (2018) Prenatal bisphenol A exposure in mice induces multitissue multiomics disruptions linking to cardiometabolic disorders. Endocrinology 160, 409–429 10.1210/en.2018-00817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bansal M., Belcastro V., Ambesi-Impiombato A. and di Bernardo D. (2007) How to infer gene networks from expression profiles. Mol. Syst. Biol. 3, 78 10.1038/msb4100158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chai L.E., Loh S.K., Low S.T., Mohamad M.S., Deris S. and Zakaria Z. (2014) A review on the computational approaches for gene regulatory network construction. Comput. Biol. Med. 48, 55–65 10.1016/j.compbiomed.2014.02.011 [DOI] [PubMed] [Google Scholar]
- 26.Karlebach G. and Shamir R. (2008) Modelling and analysis of gene regulatory networks. Nat. Rev. Mol. Cell Biol. 9, 770–780 10.1038/nrm2503 [DOI] [PubMed] [Google Scholar]
- 27.Marbach D., Costello J.C., Kuffner R., Vega N.M., Prill R.J., Camacho D.M. et al. (2012) Wisdom of crowds for robust gene network inference. Nat. Methods 9, 796–804 10.1038/nmeth.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Smet R. and Marchal K. (2010) Advantages and limitations of current network inference methods. Nat. Rev. Microbiol. 8, 717 10.1038/nrmicro2419 [DOI] [PubMed] [Google Scholar]
- 29.Dobrin R., Zhu J., Molony C., Argman C., Parrish M.L., Carlson S. et al. (2009) Multi-tissue coexpression networks reveal unexpected subnetworks associated with disease. Genome Biol. 10, R55 10.1186/gb-2009-10-5-r55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seldin M.M., Koplev S., Rajbhandari P., Vergnes L., Rosenberg G.M., Meng Y. et al. (2018) A strategy for discovery of endocrine interactions with application to whole-body metabolism. Cell Metab. 27, 1138–1155.e6 10.1016/j.cmet.2018.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Talukdar H.A., Foroughi Asl H., Jain R.K., Ermel R., Ruusalepp A., Franzen O. et al. (2016) Cross-tissue regulatory gene networks in coronary artery disease. Cell Syst. 2, 196–208 10.1016/j.cels.2016.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malik R., Selden C. and Hodgson H. (2002) The role of non-parenchymal cells in liver growth. Semin. Cell Dev. Biol. 13, 425–431 10.1016/S1084952102001301 [DOI] [PubMed] [Google Scholar]
- 33.Chen S. and Mar J.C. (2018) Evaluating methods of inferring gene regulatory networks highlights their lack of performance for single cell gene expression data. BMC Bioinformatics 19, 232 10.1186/s12859-018-2217-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langfelder P. and Horvath S. (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 10.1186/1471-2105-9-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang B. and Horvath S. (2005) A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 4, Article17 10.2202/1544-6115.1128 [DOI] [PubMed] [Google Scholar]
- 36.Huynh-Thu V.A., Irrthum A., Wehenkel L. and Geurts P. (2010) Inferring regulatory networks from expression data using tree-based methods. PLoS ONE 5, e12776 10.1371/journal.pone.0012776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Margolin A.A., Nemenman I., Basso K., Wiggins C., Stolovitzky G., Favera R.D. et al. (2006) ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics 7, S7 10.1186/1471-2105-7-S1-S7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Greenfield A., Madar A., Ostrer H. and Bonneau R. (2010) DREAM4: combining genetic and dynamic information to identify biological networks and dynamical models. PLoS ONE 5, e13397 10.1371/journal.pone.0013397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Filippi S. and Holmes C.C. (2017) A Bayesian nonparametric approach to testing for dependence between random variables. Bayesian Anal. 12, 919–938 10.1214/16-BA1027 [DOI] [Google Scholar]
- 40.Sanchez-Castillo M., Blanco D., Tienda-Luna I.M., Carrion M. and Huang Y. (2017) A Bayesian framework for the inference of gene regulatory networks from time and pseudo-time series data. Bioinformatics 34, 964–970 10.1093/bioinformatics/btx605 [DOI] [PubMed] [Google Scholar]
- 41.Faith J.J., Hayete B., Thaden J.T., Mogno I., Wierzbowski J., Cottarel G. et al. (2007) Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 5, e8 10.1371/journal.pbio.0050008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Macosko E.Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M. et al. (2015) Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 10.1016/j.cell.2015.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klein A.M., Mazutis L., Akartuna I., Tallapragada N., Veres A., Li V. et al. (2015) Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 10.1016/j.cell.2015.04.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jaitin D.A., Kenigsberg E., Keren-Shaul H., Elefant N., Paul F., Zaretsky I. et al. (2014) Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343, 776–779 10.1126/science.1247651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gierahn T.M., Wadsworth M.H. II, Hughes T.K., Bryson B.D., Butler A., Satija R. et al. (2017) Seq-Well: portable, low-cost RNA sequencing of single cells at high throughput. Nat. Methods 14, 395–398 10.1038/nmeth.4179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosenberg A.B., Roco C.M., Muscat R.A., Kuchina A., Sample P., Yao Z. et al. (2018) Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182 10.1126/science.aam8999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cao J., Packer J.S., Ramani V., Cusanovich D.A., Huynh C., Daza R. et al. (2017) Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661–667 10.1126/science.aam8940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stoeckius M., Hafemeister C., Stephenson W., Houck-Loomis B., Chattopadhyay P.K., Swerdlow H. et al. (2017) Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 10.1038/nmeth.4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buenrostro J.D., Wu B., Litzenburger U.M., Ruff D., Gonzales M.L., Snyder M.P. et al. (2015) Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 10.1038/nature14590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karemaker I.D. and Vermeulen M. (2018) Single-cell DNA methylation profiling: technologies and biological applications. Trends Biotechnol. 36, 952–965 10.1016/j.tibtech.2018.04.002 [DOI] [PubMed] [Google Scholar]
- 51.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M. et al. (2018) Comprehensive integration of single cell data. bioRxiv 10.1016/j.cell.2019.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scutari M. (2010) Learning Bayesian networks with the bnlearn R package. J. Stat. Softw. 35, 22 10.18637/jss.v035.i03 [DOI] [Google Scholar]
- 53.Babtie A.C. and Stumpf M.P.H. (2017) How to deal with parameters for whole-cell modelling. J. R. Soc. Interface 14, 20170237 10.1098/rsif.2017.0237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fiers M., Minnoye L., Aibar S., Bravo Gonzalez-Blas C., Kalender Atak Z. and Aerts S. (2018) Mapping gene regulatory networks from single-cell omics data. Brief. Funct. Genomics 17, 246–254 10.1093/bfgp/elx046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Todorov H., Cannoodt R., Saelens W. and Saeys Y. (2019) Network inference from single-cell transcriptomic data. Methods Mol. Biol. 1883, 235–249 10.1007/978-1-4939-8882-2_10 [DOI] [PubMed] [Google Scholar]
- 56.Svensson V. (2019) Droplet scRNA-seq is not zero-inflated. bioRxiv, 582064 10.1101/582064 [DOI] [PubMed] [Google Scholar]
- 57.Hafemeister C. and Satija R. (2019) Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. bioRxiv, 576827 10.1101/576827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Townes F.W., Hicks S.C., Aryee M.J. and Irizarry R.A. (2019) Feature selection and dimension reduction for single cell RNA-seq based on a multinomial model. bioRxiv, 574574 10.1101/574574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.van Dijk D., Sharma R., Nainys J., Yim K., Kathail P., Carr A.J. et al. (2018) Recovering gene interactions from single-cell data using data diffusion. Cell 174, 716–729.e27 10.1016/j.cell.2018.05.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li W.V. and Li J.J. (2018) An accurate and robust imputation method scImpute for single-cell RNA-seq data. Nat. Commun. 9, 997 10.1038/s41467-018-03405-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gong W., Kwak I.-Y., Pota P., Koyano-Nakagawa N. and Garry D.J. (2018) Drimpute: imputing dropout events in single cell RNA sequencing data. BMC Bioinformatics 19, 220 10.1186/s12859-018-2226-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang M., Wang J., Torre E., Dueck H., Shaffer S., Bonasio R. et al. (2018) SAVER: gene expression recovery for single-cell RNA sequencing. Nat. Methods 15, 539–542 10.1038/s41592-018-0033-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Prabhakaran S., Azizi E., Carr A. and Pe'er D. (2016) Dirichlet process mixture model for correcting technical variation in single-cell gene expression data. JMLR Workshop Conf. Proc. 48, 1070–1079 PMID: [PMC free article] [PubMed] [Google Scholar]
- 64.Zhu L., Lei J., Devlin B. and Roeder K. (2017) A unified statistical framework for single cell and bulk sequencing data. bioRxiv, 206532 10.1101/206532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang L. and Zhang S. (2018) PBLR: an accurate single cell RNA-seq data imputation tool considering cell heterogeneity and prior expression level of dropouts. bioRxiv, 379883 10.1101/379883 [DOI] [Google Scholar]
- 66.Arisdakessian C., Poirion O., Yunits B., Zhu X. and Garmire L.X. (2018) Deepimpute: an accurate, fast and scalable deep neural network method to impute single-cell RNA-seq data. bioRxiv, 353607 10.1101/353607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang L. and Zhang S. (2018) Comparison of computational methods for imputing single-cell RNA-sequencing data. IEEE/ACM Trans. Comput. Biol. Bioinform., 1 10.1109/TCBB.2018.2848633 [DOI] [PubMed] [Google Scholar]
- 68.Han X., Wang R., Zhou Y., Fei L., Sun H., Lai S. et al. (2018) Mapping the mouse cell atlas by Microwell-Seq. Cell 172, 1091–1107.e17 10.1016/j.cell.2018.02.001 [DOI] [PubMed] [Google Scholar]
- 69.Pierson E. and Yau C. (2015) ZIFA: dimensionality reduction for zero-inflated single-cell gene expression analysis. Genome Biol. 16, 241 10.1186/s13059-015-0805-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Risso D., Perraudeau F., Gribkova S., Dudoit S. and Vert J.-P. (2018) A general and flexible method for signal extraction from single-cell RNA-seq data. Nat. Commun. 9, 284 10.1038/s41467-017-02554-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W. et al. (2015) Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johnson W.E., Li C. and Rabinovic A. (2007) Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 10.1093/biostatistics/kxj037 [DOI] [PubMed] [Google Scholar]
- 73.Messmer T., von Meyenn F., Savino A., Santos F., Mohammed H., Lun A.T.L. et al. (2019) Transcriptional heterogeneity in naive and primed human pluripotent stem cells at single-cell resolution. Cell Rep. 26, 815–824.e4 10.1016/j.celrep.2018.12.099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Grive K.J., Hu Y., Shu E., Grimson A., Elemento O., Grenier J.K. et al. (2019) Dynamic transcriptome profiles within spermatogonial and spermatocyte populations during postnatal testis maturation revealed by single-cell sequencing. PLoS Genet. 15, e1007810 10.1371/journal.pgen.1007810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Loo L., Simon J.M., Xing L., McCoy E.S., Niehaus J.K., Guo J. et al. (2019) Single-cell transcriptomic analysis of mouse neocortical development. Nat. Commun. 10, 134 10.1038/s41467-018-08079-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guo M., Du Y., Gokey J.J., Ray S., Bell S.M., Adam M. et al. (2019) Single cell RNA analysis identifies cellular heterogeneity and adaptive responses of the lung at birth. Nat. Commun. 10, 37 10.1038/s41467-018-07770-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Haghverdi L., Lun A.T., Morgan M.D. and Marioni J.C. (2018) Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421 10.1038/nbt.4091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Butler A., Hoffman P., Smibert P., Papalexi E. and Satija R. (2018) Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 10.1038/nbt.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kiselev V.Y., Yiu A. and Hemberg M. (2018) Scmap: projection of single-cell RNA-seq data across data sets. Nat. Methods 15, 359–362 10.1038/nmeth.4644 [DOI] [PubMed] [Google Scholar]
- 80.Aran D., Looney A.P., Liu L., Wu E., Fong V., Hsu A. et al. (2019) Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20, 163 10.1038/s41590-018-0276-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Woodhouse S., Piterman N., Wintersteiger C.M., Göttgens B. and Fisher J. (2018) SCNS: a graphical tool for reconstructing executable regulatory networks from single-cell genomic data. BMC Syst. Biol. 12, 59 10.1186/s12918-018-0581-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Matsumoto H., Kiryu H., Furusawa C., Ko M.S., Ko S.B., Gouda N. et al. (2017) SCODE: an efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation. Bioinformatics 33, 2314–2321 10.1093/bioinformatics/btx194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aubin-Frankowski P.-C. and Vert J.-P. (2018) Gene regulation inference from single-cell RNA-seq data with linear differential equations and velocity inference. bioRxiv, 464479 10.1101/464479 [DOI] [PubMed] [Google Scholar]
- 84.Papili Gao N., Ud-Dean S.M., Gandrillon O. and Gunawan R. (2017) SINCERITIES: inferring gene regulatory networks from time-stamped single cell transcriptional expression profiles. Bioinformatics 34, 258–266 10.1093/bioinformatics/btx575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Qiu X., Rahimzamani A., Wang L., Mao Q., Durham T., McFaline-Figueroa J.L. et al. (2018) Towards inferring causal gene regulatory networks from single cell expression measurements. bioRxiv, 426981 10.1101/426981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Deshpande A., Chu L.-F., Stewart R. and Gitter A. (2019) Network inference with granger causality ensembles on single-cell transcriptomic data. bioRxiv, 534834 10.1101/534834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang S., MacLean A.L. and Nie Q. (2018) SoptSC: similarity matrix optimization for clustering, lineage, and signaling inference. bioRxiv, 168922 [Google Scholar]
- 88.Aibar S., González-Blas C.B., Moerman T., Imrichova H., Hulselmans G., Rambow F. et al. (2017) SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083 10.1038/nmeth.4463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pina C., Teles J., Fugazza C., May G., Wang D., Guo Y. et al. (2015) Single-cell network analysis identifies DDIT3 as a nodal lineage regulator in hematopoiesis. Cell Rep. 11, 1503–1510 10.1016/j.celrep.2015.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Iacono G., Massoni-Badosa R. and Heyn H. (2019) Single-cell transcriptomics unveils gene regulatory network plasticity. Genome Biol. 20, 110 10.1186/s13059-019-1713-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chan T.E., Stumpf M.P.H. and Babtie A.C. (2017) Gene regulatory network inference from single-cell data using multivariate information measures. Cell Syst. 5, 251–267.e3 10.1016/j.cels.2017.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jackson C.A., Castro D.M., Saldi G.-A., Bonneau R. and Gresham D. (2019) Gene regulatory network reconstruction using single-cell RNA sequencing of barcoded genotypes in diverse environments. bioRxiv, 581678 10.1101/581678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang Y., Solus L., Yang K. and Uhler C. (2017) Permutation-based causal inference algorithms with interventions. In Advances in Neural Information Processing Systems, pp. 5822–5831
- 94.Mohammadi S., Ravindra V., Gleich D.F. and Grama A. (2018) A geometric approach to characterize the functional identity of single cells. Nat. Commun. 9, 1516 10.1038/s41467-018-03933-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Guo M., Wang H., Potter S.S., Whitsett J.A. and Xu Y. (2015) SINCERA: a pipeline for single-cell RNA-seq profiling analysis. PLoS Comput. Biol. 11, e1004575 10.1371/journal.pcbi.1004575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang Y., Wang R., Zhang S., Song S., Jiang C., Han G. et al. (2019) iTALK: an R Package to characterize and illustrate intercellular communication. bioRxiv, 507871 10.1101/507871 [DOI] [Google Scholar]
- 97.Zhou J.X., Taramelli R., Pedrini E., Knijnenburg T. and Huang S. (2017) Extracting intercellular signaling network of cancer tissues using ligand-receptor expression patterns from whole-tumor and single-cell transcriptomes. Sci. Rep. 7, 8815 10.1038/s41598-017-09307-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kumar M.P., Du J., Lagoudas G., Jiao Y., Sawyer A., Drummond D.C. et al. (2018) Analysis of single-cell RNA-seq identifies cell-cell communication associated with tumor characteristics. Cell Rep. 25, 1458–1468.e4 10.1016/j.celrep.2018.10.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Arneson D., Zhang G., Ying Z., Zhuang Y., Byun H.R., Ahn I.S. et al. (2018) Single cell molecular alterations reveal target cells and pathways of concussive brain injury. Nat. Commun. 9, 3894 10.1038/s41467-018-06222-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tsuyuzaki K., Ishii M. and Nikaido I. (2019) Uncovering hypergraphs of cell-cell interaction from single cell RNA-sequencing data. bioRxiv, 566182 10.1101/566182 [DOI] [Google Scholar]
- 101.Saelens W., Cannoodt R., Todorov H. and Saeys Y. (2019) A comparison of single-cell trajectory inference methods. Nat. Biotechnol. 37, 547–554 10.1038/s41587-019-0071-9 [DOI] [PubMed] [Google Scholar]
- 102.Babtie A.C., Chan T.E. and Stumpf M.P. (2017) Learning regulatory models for cell development from single cell transcriptomic data. Curr. Opin. Syst. Biol. 5, 72–81 10.1016/j.coisb.2017.07.013 [DOI] [Google Scholar]
- 103.Ocone A., Haghverdi L., Mueller N.S. and Theis F.J. (2015) Reconstructing gene regulatory dynamics from high-dimensional single-cell snapshot data. Bioinformatics 31, i89–i96 10.1093/bioinformatics/btv257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wei J., Hu X., Zou X. and Tian T. (2016) Inference of genetic regulatory network for stem cell using single cells expression data 2016 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), IEEE [Google Scholar]
- 105.Lim C.Y., Wang H., Woodhouse S., Piterman N., Wernisch L., Fisher J. et al. (2016) BTR: training asynchronous Boolean models using single-cell expression data. BMC Bioinformatics 17, 355 10.1186/s12859-016-1235-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Moignard V., Woodhouse S., Haghverdi L., Lilly A.J., Tanaka Y., Wilkinson A.C. et al. (2015) Decoding the regulatory network of early blood development from single-cell gene expression measurements. Nat. Biotechnol. 33, 269–276 10.1038/nbt.3154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mukherjee S., Carignano A., Seelig G. and Lee S.I. (2018) Identifying progressive gene network perturbation from single-cell RNA-seq data. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2018, 5034–5040 [DOI] [PubMed] [Google Scholar]
- 108.Rahimzamani A. and Kannan S. (2016) Network inference using directed information: the deterministic limit 2016 54th Annual Allerton Conference on Communication, Control, and Computing (Allerton), IEEE [Google Scholar]
- 109.Iacono G., Mereu E., Guillaumet-Adkins A., Corominas R., Cusco I., Rodriguez-Esteban G. et al. (2018) bigSCale: an analytical framework for big-scale single-cell data. Genome Res. 28, 878–890 10.1101/gr.230771.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lèbre S. (2009) Inferring dynamic genetic networks with low order independencies. Stat. Appl. Genet. Mol. Biol. 8, 1–38 10.2202/1544-6115.1294 [DOI] [PubMed] [Google Scholar]
- 111.Bélanger M., Allaman I. and Magistretti P.J. (2011) Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 14, 724–738 10.1016/j.cmet.2011.08.016 [DOI] [PubMed] [Google Scholar]
- 112.Rajbhandari P., Thomas B.J., Feng A.-C., Hong C., Wang J., Vergnes L. et al. (2018) IL-10 signaling remodels adipose chromatin architecture to limit thermogenesis and energy expenditure. Cell 172, 218–233.e17 10.1016/j.cell.2017.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fulcher B.D. and Fornito A. (2016) A transcriptional signature of hub connectivity in the mouse connectome. Proc. Natl Acad. Sci. U.S.A. 113, 1435–1440 10.1073/pnas.1513302113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Smillie C.S., Biton M., Ordovas-Montanes J., Sullivan K.M., Burgin G., Graham D.B. et al. (2018) Rewiring of the cellular and inter-cellular landscape of the human colon during ulcerative colitis. bioRxiv, 455451 10.1101/455451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vento-Tormo R., Efremova M., Botting R.A., Turco M.Y., Vento-Tormo M., Meyer K.B. et al. (2018) Single-cell reconstruction of the early maternal–fetal interface in humans. Nature 563, 347–353 10.1038/s41586-018-0698-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Castro D.M., De Veaux N.R., Miraldi E.R. and Bonneau R. (2019) Multi-study inference of regulatory networks for more accurate models of gene regulation. PLoS Comput. Biol. 15, e1006591 10.1371/journal.pcbi.1006591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dixit A., Parnas O., Li B., Chen J., Fulco C.P., Jerby-Arnon L. et al. (2016) Perturb-Seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 167, 1853–1866.e17 10.1016/j.cell.2016.11.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hauser A. and Bühlmann P. (2012) Characterization and greedy learning of interventional Markov equivalence classes of directed acyclic graphs. J. Mach. Learn. Res. 13, 2409–2464 [Google Scholar]
- 119.Wang Y., Squires C., Belyaeva A. and Uhler C. (2018) Direct estimation of differences in causal graphs. In Advances in Neural Information Processing Systems, pp. 3770–3781
- 120.Yang K.D., Katcoff A. and Uhler C. (2018) Characterizing and learning equivalence classes of causal dags under interventions. preprint arXiv, 180206310 [Google Scholar]
- 121.Femino A.M., Fogarty K., Lifshitz L.M., Carrington W. and Singer R.H. (2003) Visualization of single molecules of mRNA in situ. Methods Enzymol. 361, 245–304 10.1016/S0076-6879(03)61015-3 [DOI] [PubMed] [Google Scholar]
- 122.Shah S., Lubeck E., Zhou W. and Cai L. (2017) seqFISH accurately detects transcripts in single cells and reveals robust spatial organization in the hippocampus. Neuron 94, 752–758.e1 10.1016/j.neuron.2017.05.008 [DOI] [PubMed] [Google Scholar]
- 123.Chen K.H., Boettiger A.N., Moffitt J.R., Wang S. and Zhuang X. (2015) RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 10.1126/science.aaa6090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lovatt D., Ruble B.K., Lee J., Dueck H., Kim T.K., Fisher S. et al. (2014) Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nat. Methods 11, 190–196 10.1038/nmeth.2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Simone N.L., Bonner R.F., Gillespie J.W., Emmert-Buck M.R. and Liotta L.A. (1998) Laser-capture microdissection: opening the microscopic frontier to molecular analysis. Trends Genet. 14, 272–276 10.1016/S0168-9525(98)01489-9 [DOI] [PubMed] [Google Scholar]
- 126.Ke R., Mignardi M., Pacureanu A., Svedlund J., Botling J., Wahlby C. et al. (2013) In situ sequencing for RNA analysis in preserved tissue and cells. Nat. Methods 10, 857–860 10.1038/nmeth.2563 [DOI] [PubMed] [Google Scholar]
- 127.Lee J.H., Daugharthy E.R., Scheiman J., Kalhor R., Ferrante T.C., Terry R. et al. (2015) Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat. Protoc. 10, 442–458 10.1038/nprot.2014.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rodriques S.G., Stickels R.R., Goeva A., Martin C.A., Murray E., Vanderburg C.R. et al. (2019) Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 10.1126/science.aaw1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Eng C.-H.L., Lawson M., Zhu Q., Dries R., Koulena N., Takei Y. et al. (2019) Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235–239 10.1038/s41586-019-1049-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Moffitt J.R., Bambah-Mukku D., Eichhorn S.W., Vaughn E., Shekhar K., Perez J.D. et al. (2018) Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362, eaau5324 10.1126/science.aau5324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kim M. and Tagkopoulos I. (2018) Data integration and predictive modeling methods for multi-omics datasets. Mol. Omics 14, 8–25 10.1039/C7MO00051K [DOI] [PubMed] [Google Scholar]
- 132.Zarayeneh N., Ko E., Oh J.H., Suh S., Liu C., Gao J. et al. (2017) Integration of multi-omics data for integrative gene regulatory network inference. Int. J. Data Min. Bioinform. 18, 223–239 10.1504/IJDMB.2017.087178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Huang S., Chaudhary K. and Garmire L.X. (2017) More is better: recent progress in multi-omics data integration methods. Front. Genet. 8, 84 10.3389/fgene.2017.00084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Arneson D., Shu L., Tsai B., Barrere-Cain R., Sun C. and Yang X. (2017) Multidimensional integrative genomics approaches to dissecting cardiovascular disease. Front. Cardiovasc. Med. 4, 8 10.3389/fcvm.2017.00008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Marbach D., Prill R.J., Schaffter T., Mattiussi C., Floreano D. and Stolovitzky G. (2010) Revealing strengths and weaknesses of methods for gene network inference. Proc. Natl Acad. Sci. U.S.A. 107, 6286–6291 10.1073/pnas.0913357107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hill S.M., Heiser L.M., Cokelaer T., Unger M., Nesser N.K., Carlin D.E. et al. (2016) Inferring causal molecular networks: empirical assessment through a community-based effort. Nat. Methods 13, 310 10.1038/nmeth.3773 [DOI] [PMC free article] [PubMed] [Google Scholar]