

Abstract
Heterozygous mutations in PTEN, which encodes a negative regulator of the mTOR and β-catenin signaling pathways, cause macrocephaly/autism syndrome. However, the neurobiological substrates of the core symptoms of this syndrome are poorly understood. Here, we investigate the relationship between cerebral cortical overgrowth and social behavior deficits in conditional Pten heterozygous female mice (Pten cHet) using Emx1-Cre, which is expressed in cortical pyramidal neurons and a subset of glia. We found that conditional heterozygous mutation of Ctnnb1 (encoding β-catenin) suppresses Pten cHet cortical overgrowth, but not social behavioral deficits, whereas conditional heterozygous mutation of Mtor suppresses social behavioral deficits, but not cortical overgrowth. Neuronal activity in response to social cues and excitatory synapse markers are elevated in the medial prefrontal cortex (mPFC) of Pten cHet mice, and heterozygous mutation in Mtor, but not Ctnnb1, rescues these phenotypes. These findings indicate that macroscale cerebral cortical overgrowth and social behavioral phenotypes caused by Pten haploinsufficiency can be dissociated based on responsiveness to genetic suppression of Ctnnb1 or Mtor. Furthermore, neuronal connectivity appears to be one potential substrate for mTOR-mediated suppression of social behavioral deficits in Pten haploinsufficient mice.
Keywords: PTEN, macrocephaly, social behavior, synapses, network activity
Lay Summary:
A subgroup of individuals with autism display overgrowth of the head and the brain during development. Using a mouse model of an autism risk gene, Pten, that displays both brain overgrowth and social behavioral deficits, we show here that that these two symptoms can be dissociated. Reversal of social behavioral deficits in this model is associated with rescue of abnormal synaptic markers and neuronal activity.
Introduction
Autism spectrum disorder (ASD) is a group of neuro-developmental disorders characterized by deficits in social interaction and communication, as well as the presence of repetitive behaviors and restricted interests. It occurs in roughly 1% of general population [Lyall et al., 2017]. Although the cause of ASD is still unknown, the relative risks of individuals developing ASD are proportional to the percentage of genome shared with a person with ASD [Bourgeron, 2015]. Reports of relatively large head size in some children with autism date back to Leo Kanner’s original description of the disorder [Kanner, 1943], and increased head circumference is a replicated finding in a subset of individuals with ASD [Fidler, Bailey, & Smalley, 2000; Lainhart et al., 1997]. However, the relationship between brain overgrowth and the behavioral and cognitive symptoms of ASD is unclear.
Mutations in the tumor suppressor gene PTEN (phosphatase and tensin homolog) are a cause of autism and macrocephaly (head circumference > 2 SD above normal), with an incidence of ~7%–17% among individuals with autism and macrocephaly [Butler et al., 2005; Buxbaum et al., 2007; McBride et al., 2010; Varga, Pastore, Prior, Herman, & McBride, 2009]. The PTEN mutations identified to date are generally heterozygous mutations that reduce PTEN protein level [Frazier et al., 2015; Rodríguez-Escudero et al., 2011] and cause macrocephaly/autism syndrome (OMIM #605309) in an autosomal dominant manner. PTEN encodes a phosphatidylinositol-3,-4,5-triphosphate 3-phosphatase that acts as a negative regulator of the mTOR and β-catenin signaling pathways [Hay & Sonenberg, 2004; Persad et al., 2016; Zhang & Yu, 2010]. Both mTOR and β-catenin have independently been shown to regulate brain growth, as mice with conditional knockout of Mtor or Ctnnb1 exhibit brain undergrowth [Brault et al., 2001; Ka, Condorelli, Woodgett, & Kim, 2014]. We previously reported that both germline and conditional Pten haploinsufficient mice show brain overgrowth [Chen, Huang, Séjourné, Clipperton-Allen, & Page, 2015; Page, Kuti, Prestia, & Sur, 2009], and that this phenotype can be rescued by haploinsufficiency for Ctnnb1 [Chen et al., 2015]. In addition, we have also shown that Pten haploinsufficient mice have social behavioral deficits [Clipperton-Allen & Page, 2014; Huang, Chen, & Page, 2016; Page et al., 2009], and that reducing mTOR-S6K signaling rescues this phenotype [Huang et al., 2016]. One possible substrate for this effect is hyperconnectivity of projections from the medial prefrontal cortex (mPFC) to basolateral amygdala (BLA), and hyperactivity in response to social cues in the mPFC and BLA in Pten haploinsufficient mice [Huang et al., 2016].
The observation that haploinsufficiency for Ctnnb1 can suppress brain overgrowth in conditional Pten heterozygous mice (Pten cHet) prompted us to test whether this can also rescue social behavioral deficits. Here, we show that while cortical overgrowth in Pten cHet mice is rescued by haploinsufficiency for Ctnnb1, but not Mtor, the social behavioral deficit is rescued by haploinsufficiency for Mtor, but not Ctnnb1. This suggests that brain overgrowth and social behavioral phenotypes in Pten cHet mice are dissociable based on responsiveness to Ctnnb1 or Mtor mutations. Analysis of connectivity and activity in response to social cues in these mutant mice reveals that increased vGluT1 levels in the prefrontal cortex and the hyperactivity in response to social cues in the mPFC and BLA shown in Pten cHet mice are rescued by haploinsufficiency for Mtor, but not Ctnnb1. Taken together, our results indicate that increased mPFC activity in response to social cues and vGluT1 levels appear to be key substrates for social behavioral deficits in a mouse model of Pten haploinsufficiency.
Materials and Methods
Mice
All mouse lines were described previously and were obtained from the Jackson Laboratory. These include Emx1tm1(cre)Krj (Emx1-Cre+, stock number 005628), Ptentm1Hwu (Ptenloxp/loxp, stock number 006440), Mtortm1.2Koz (Mtorloxp/loxp, stock number 011009), and Ctnnb1tm2Kem (Ctnnb1loxp/loxp, stock number 004152). Male Ptenloxp/loxp; Mtorloxp/+ mice were crossed with female Emx1-Cre+/− mice to generate control, Pten cHet, and Pten; Ctnnb1 cHet mice. Mouse genotypes were confirmed by PCR using genomic DNA obtained from ear samples. Mice age 2–4 months old in a C57BL/6J background were used in this study. For all experiments, 2–3 cohorts of transgenic animals and littermate controls were used. All animal experiments were conducted in accordance with NIH and AAALAC guidelines and were approved by The Scripps Research Institute’s Institutional Animal Care and Use Committee. All mice used in this study were female.
Immunohistochemistry
Mice were perfused with 4%PFA, and brains were then postfixed in 4% PFA for overnight following by incubating in a 20% sucrose/PBS solution at 4 C for 3–5 days, and embedded in Tissue-Tek OCT compound (Sakura). Coronal sections were collected on Superfrost/Plus slides and immunostained with the following antibodies. Primary antibodies used in this study include vGluT1 (1:2,000, Millipore, AB5905) and vesicular GABA transporter (vGAT) (1:1,000, Synaptic System, 131013). Alexa Fluor 488, 594, and 647 conjugated secondary antibodies (Life Technologies) were used in this study. Immunofluorescent brain sections were counterstained with Prolong Gold with DAPI (Life Technologies). For c-fos staining, avidin-biotin complex (ABC) kit (Vector Lab, PK-6101) and 3,3′ Diaminobenzidine (DAB) (Life Technologies, #34001) were used. Images were obtained with an Olympus VS120 microscope and processed using the VS-DESKTOP software (Olympus).
c-fos immunoreactive neurons were quantified manually using the VS-DESKTOP software (Olympus) in the mPFC (prelimbic cortex, PrL) of coronal section (Bregma 1.98, 1.94, and 1.78), and in the BLA of coronal section (Bregma −1.46, −1.58, and −1.70). For each region, the number of c-fos+ cells was quantified in three coronal brain sections from both left and right hemispheres, and averaged within each animal.
vGluT1 and vGAT puncta were quantified using the ITCN plugin of ImageJ in layer 1 of mPFC and BLA. For each region, the number of vGluT1 and vGAT puncta was quantified in three coronal brain sections from both left and right hemispheres, and averaged within each animal.
Cell Size Analysis
Brain sections were stained with NeuroTrace green fluorescent Nissl stain (1:100 dilution). Regions of interest (ROIs) were cropped from the primary somatosensory cortex barrel field (coronal section) using Photoshop (Adobe). Each layer (layer II–IV, V, and VI) was further cropped according to anatomical boundaries as judged by fluorescent Nissl stain. Cell soma size of all neurons in layer V was measured using ImageJ (NIH). The freehand tool in ImageJ was used to contour the boundary of neuronal soma. Three ROIs were measured in each animal.
Mouse Behavior Tests
Female mice age 2–4 months old in C57BL6J background were used for behavioral tests. For all behavioral tests, 2–3 cohorts of transgenic animals and littermate controls were used. Each cohort was tested independently.
Three-chamber social approach test.
Female mice were housed under a reversed 12:12 hr light cycle for at least 2 weeks before testing. Mice were put into the apparatus for 5 min to habituate on each of the 2 days preceding the test, and then returned to the housing room. During the test, each mouse was put into the three-chamber apparatus for 5 min for habituation, followed by 10 min with a stimulus mouse inside a tube in one chamber and an empty tube in the chamber on the other side (the center chamber was left empty). The percent time spent in each of the three chambers, containing either a mouse in a tube, an empty tube, or nothing (in the center) was recorded during the 10-min social approach trial.
Social exposure.
Female mice were individually housed in a home cage containing a perforated acrylic tube for 7 days before the test. On the test day, a novel conspecific female mouse was placed in the tube within the home cage of the subject female mouse. The subject mouse was allowed to explore the stimulus mouse until it reached 30 sec of sniffing time. In this way, each mouse was engaged in a similar amount of social interaction time. After the stimulus mouse was removed, the subject mouse remained in the home cage for additional 2 hr before being perfused.
Statistical Analysis
Prior studies using similar experiments were used to determine sample size. One-way analyses of variance (ANOVAs) were used to assess genotype effects, and planned comparison independent-sample t-tests compared Emx1-Cre+; Ptenloxp/+ mice to each genotype, including control, Emx1-Cre+; Ptenloxp/+; Mtorloxp/+, and Emx1-Cre+; Ptenloxp/+; Ctnnb1loxp/+ mice. Tukey’s post hoc tests were used where appropriate. The significant or nonsignificant stats were illustrated in the graphs for the following comparisons. Control & Emx1-Cre+; Ptenloxp/+ mice. Emx1-Cre+; Ptenloxp/+ & Emx1-Cre+; Ptenloxp/+; Mtorloxp/+ mice. Emx1-Cre+; Ptenloxp/+ & Emx1-Cre+; Ptenloxp/+; Ctnnb1loxp/+ mice. For the three-chamber social approach test, paired-sample t-tests were used to compare time spent in the chambers containing a mouse in a tube and an empty tube separately for each genotype. All statistics were performed using Graphpad, with significance set at P < 0.05. Throughout the article, values represent means, error bars indicate SEMs, and N values refer to biological replicates. All measurements and testing were performed blind to genotype or experimental manipulations.
Results
Brain Overgrowth Is Rescued by Heterozygous Mutation in Ctnnb1
We used Emx1-Cre, which is broadly expressed in cerebral cortical pyramidal neurons, astrocytes, and oligodendrocytes [Gorski et al., 2002], to generate mice conditionally heterozygous (cHet) for Pten alone or with additional mutations in either Mtor or Ctnnb1. Our previous study showed that male Pten cHet mice (Emx1-Cre+; Ptenloxp/+) exhibit brain overgrowth, and that this phenotype is suppressed in male mice cHet for Pten and Ctnnb1 (Emx1-Cre+; Ptenloxp/+; Ctnnb1loxp/+), but not for Pten and Mtor1 (Emx1-Cre+; Ptenloxp/+; Mtorloxp/+), while cHet for Ctnnb1 or Mtor alone has no significant effect on overall brain mass [Chen et al., 2015]. Although the brain over-growth phenotype is not suppressed by heterozygous deletion of Mtor, it is important to note that mTOR signaling, as reflected by p-S6, is transiently elevated in Pten Het mice and corrected by deleting one copy of Raptor [Huang et al., 2016], an essential component of mTOR complex. Since social approach deficits are most consistent in female Pten haploinsufficient mice in our experience [Huang et al., 2016; Page et al., 2009], we used female mice in the current study. Consistent with the results in male mice, we found that haploinsufficiency for Ctnnb1, but not Mtor, rescues brain overgrowth shown in female Pten cHet mice (Fig. 1), suggesting that the brain overgrowth phenotype, and its responsiveness to heterozygous Ctnnb1 mutation, is not sex-specific.
Figure 1.

Brain overgrowth in Pten conditional heterozygous mice is rescued in Pten and Ctnnb1 double conditional heterozygous mice. Graph showing brain mass in Control, Emx1-Cre+; Ptenloxp/+, Emx1-Cre+; Ptenloxp/+; Mtorloxp/+, and Emx1-Cre+; Ptenloxp/+; Ctnnb1 loxp/+ mice. One-way ANOVA and Tukey’s posthoc tests were used. F(3,12) = 7.89, P = 0.004. *P < 0.05, and **P < 0.01. n.s. indicates no significant difference. N = 4 animals per genotype.
Social Behavioral Deficits Are Rescued by Heterozygous Mutation of Mtor
We have previously shown that Emx1-Cre+; Ptenloxp/+ mice exhibit both brain overgrowth and social behavioral deficits [Chen et al., 2015; Huang et al., 2016], and these were confirmed in the current study (Figs. 1 and 2). Moreover, the brain overgrowth shown in Emx1-Cre+; Ptenloxp/+ mice was suppressed in Emx1-Cre+; Ptenloxp/+; Ctnnb1loxp/+ mice, prompting us to test whether social behavioral deficits are also suppressed in these animals. We therefore used the three-chamber social approach assay to test social behavior in the following genotypes: Emx1-Cre+; Ptenloxp/+, Emx1-Cre+; Ptenloxp/+; Mtorloxp/+, and Emx1-Cre+; Ptenloxp/+; Ctnnb1loxp/+ mice. We found that the three-chamber social approach deficit shown in Emx1-Cre+; Ptenloxp/+ mice was rescued in Emx1-Cre+; Ptenloxp/+; Mtorloxp/+, but not Emx1-Cre+; Ptenloxp/+; Ctnnb1loxp/+ mice (Fig. 2A,B). This suggests that brain overgrowth and social behavioral deficits caused by Pten mutation are genetically dissociable phenotypes. To test for a possible confounding factor of abnormal locomotion, we measured the average velocity of the mice during the 10 min social approach test. Basic locomotion was not altered among genotypes, as reflected by velocity (Fig. 2C).
Figure 2.
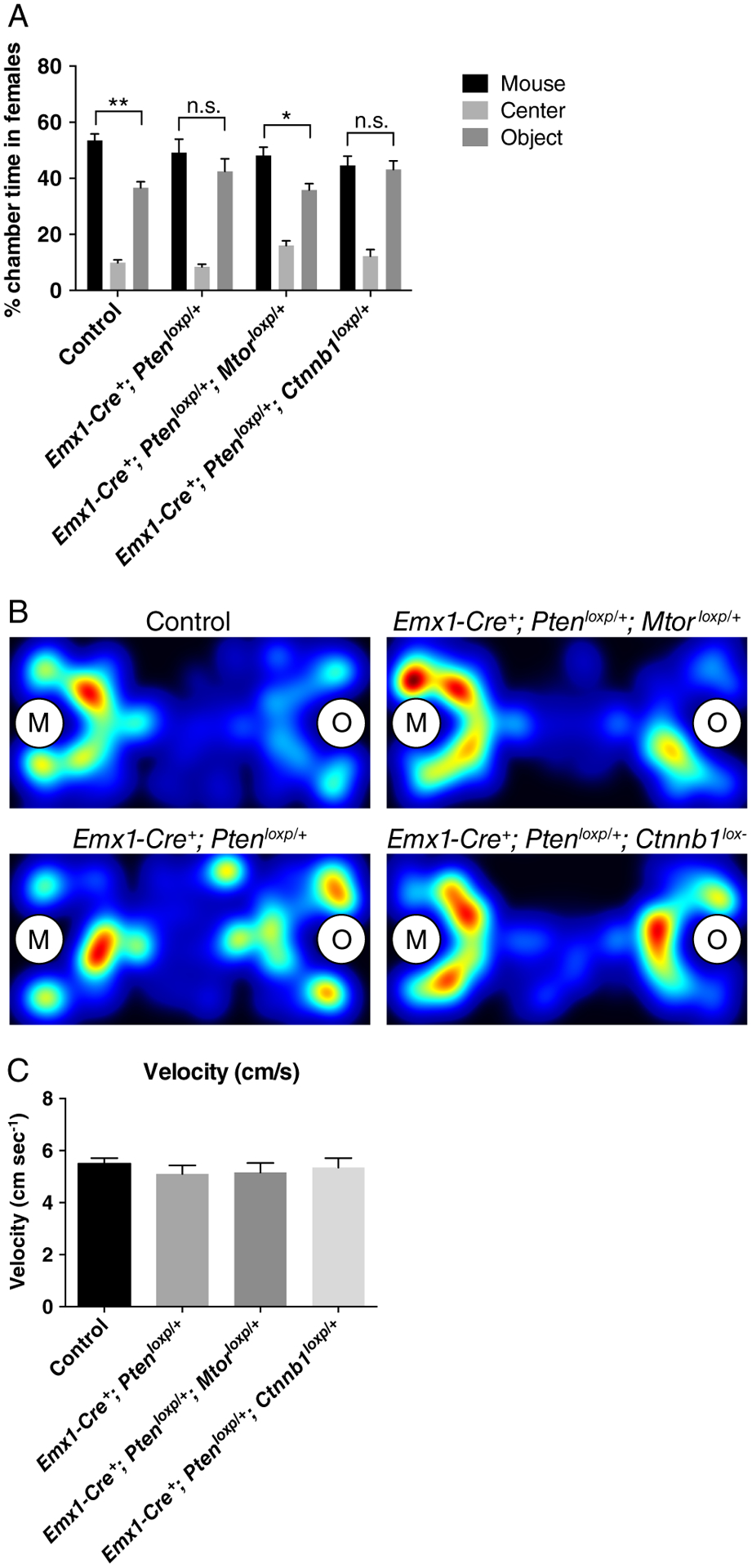
Three-chamber social approach deficits in Pten conditional heterozygous mice are corrected in Pten and Mtor double conditional heterozygous mice. (A) Graph showing percent time female mice spent in each chamber. Paired t-tests were used to compare time spent in the chambers containing a mouse in a tube (Mouse chamber, M) and an empty tube (Object chamber, O) separately for each genotype. *P < 0.05, and **P < 0.01. n.s. indicates no significant difference. (B) Example heat maps showing the time spent in each chamber. M indicates mouse chamber, and O indicates object chamber. (C) Graph showing the velocity of the mice during the 10 min sociability test. Velocity is not altered across genotypes. One-way ANOVA was used. F(3,41) = 0.32, P = 0.81. N = 12 Control, 12 Emx1-Cre+; Ptenloxp/+, 9 Emx1-Cre+; Ptenloxp/+; Mtorloxp/+, and 12 Emx1-Cre+; Ptenloxp/+; Ctnnb1 loxp/+ mice.
Elevated vGluT1 Puncta in Pten Mutant mPFC Is Rescued by Heterozygous Mutation of Mtor
We have previously reported evidence for structural hyperconnectivity between the prefrontal cortex (PFC) and amygdala, brain regions implicated in social information processing [Adolphs, 2010; Grossmann, 2013] and ASDs [Amaral, Schumann, & Nordahl, 2008; Donovan & Basson, 2017], and we found that suppressing the activity of neurons that project from the PFC to the amygdala can rescue social behavioral deficits in Pten haploinsufficient mice [Huang et al., 2016]. To determine whether markers of synaptic vesicle proteins are altered in Pten cHet mice, we immunostained for vesicular glutamate transporter 1 (vGluT1), a marker for excitatory synapses, and vGAT, a marker for inhibitory synapses, in the PrL and BLA. Layer 1 of the PrL, where the apical dendrites of layer 5 neurons receive presynaptic inputs, was selected to perform the analysis. Measuring vGluT1 and vGAT puncta in layer 1 of the PrL (Fig. 3A), we found that Emx1-Cre+; Ptenloxp/+ mice exhibited elevated density ofvGluT1, but not vGAT, puncta in outer layer 1 (Fig. 3B–F), and that this phenotype is rescued in Emx1-Cre+; Ptenloxp/+; Mtorloxp/+, but not Emx1-Cre+; Ptenloxp/+; Ctnnb1loxp/+ mice(Fig. 3B–F). This suggests that the excitatory synaptic vesicle proteins are likely elevated in response to Pten mutation, and that reducing mTOR, but not β-catenin, levels rescues the elevated excitatory synaptic vesicle proteins. We also measured vGluT1 and vGAT puncta in the BLA (Fig. 3G), and found that neither vGluT1 nor vGAT puncta are altered across genotypes (Fig. 3H,I). In summary, elevated vGluT1 level in the prefrontal cortex of Emx1-Cre+; Ptenloxp/+ mice is rescued by reducing mTOR, but not β-catenin, levels. This is consistent with the rescue of social behavioral deficits in Emx1-Cre+; Ptenloxp/+; Mtorloxp/+, but not Emx1-Cre+; Ptenloxp/+; Ctnnb1loxp/+ mice, suggesting that altered synaptic connectivity, including in the mPFC, may be a substrate for mTOR-mediated rescue of social behavioral deficits in Pten cHet mice.
Figure 3.

Increased vGluT1 puncta in Pten conditional heterozygous mice is corrected in Pten and Mtor double conditional heterozygous mice. (A) Cartoon and an image showing the staining of the vesicular glutamate transporter 1 (vGluT1) and the vesicular GABA transporter (vGAT) in the prelimbic cortex (PrL). vGluT1 and vGAT puncta were quantified separately in outer and inner layer 1. Scale bar: 50 μm. (B) Representative images of vGluT1 and vGAT staining in the PrL of each genotype. Scale bar: 10 μm. (C–F) Quantification of vGluT1 and vGAT puncta in outer (C and D) and inner (E and F) layer 1 of PrL. N = 4 animals per genotype. (G) Cartoon and an image showing the staining of the vGluT1 and vGAT in the basolateral amygdala (BLA). Scale bar: 50 μm. (H and I) Quantification of vGluT1 (H) and vGAT (I) puncta in the BLA. N = 4 animals each genotype. (J) Representative images showing fluorescent Nissl staining of layerV cells in the primary somatosensory cortex. Scale bar: 50 μm. (K) Quantification of layer V cell soma size. N = 5 animals per genotype. For all quantification graphs, one-way ANOVA was used, and Tukey’s post hoc tests were used where appropriate. *P < 0.05, and **P < 0.01. n.s. indicates no significant difference. F(3, 12) = 8.25, P = 0.003 (C); F(3, 12) = 1.01, P = 0.421 (D); F(3, 12) = 0.641, P = 0.603 (E); F(3, 12) = 0.337, P = 0.799 (F); F(3, 12) = 1.95, P = 0.176 (H); F(3, 12) = 1.94, P = 0.177 (I); F(3, 16) = 7.986, P = 0.002 (K).
Our previous study showed that reducing mTOR signaling rescued hypertrophy in layer 5 cells in germline Pten haploinsufficient mice [Huang et al., 2016]. To determine whether layer 5 cells are hypertrophic in the genotypes used in the current study, we performed fluorescent Nissl staining and measured soma size in layer 5 cells. We found that layer 5 cell soma size was enlarged in Emx1-Cre+; Ptenloxp/+ mice (Fig. 3J,K), and that this phenotype was rescued in both Emx1-Cre+; Ptenloxp/+; Mtorloxp/+ and Emx1-Cre+; Ptenloxp/+; Ctnnb1loxp/+ mice (Fig. 3J,K).Since social behavioral deficits are present in Emx1-Cre+; Ptenloxp/+; Ctnnb1loxp/+ mice, but not Emx1-Cre+; Ptenloxp/+; Mtorloxp/+ mice, this result indicates that hypertrophy of layer 5 neuron soma may be dissociable from social behavioral deficits caused by Pten haploinsufficiency.
Neuronal Hyperactivity in Response to Social Cues Is Rescued by Heterozygous Mutation in Mtor
Our previous study showed that germline Pten haploinsufficient mice display neuronal hyperactivity in the PFC and amygdala in response to social cues [Huang et al., 2016]. To investigate neuronal activity in response to social cues in the genotypes used in this study, we exposed subject mice to novel stimulus mice, and measured the c-fos+ neurons in the PrL and BLA (Fig. 4A). Since our previous study found that the number of c-fos+ neurons in the PFC and amygdala was correlated with social interaction time [Huang et al., 2016], as reflected by sniffing time, we controlled the sniffing time during the social exposure period, restricting it to 30 sec (Fig. 4A). We found that the number of c-fos+ cells is elevated in Emx1-Cre+; Ptenloxp/+ mice compared to control mice in the PrL and BLA (Fig. 4B–D), and that this phenotype is rescued in Emx1-Cre+; Ptenloxp/+; Mtorloxp/+, but not Emx1-Cre+; Ptenloxp/+; Ctnnb1loxp/+ mice (Fig. 4B–D). This is consistent with the rescue of social behavioral deficits in Emx1-Cre+; Ptenloxp/+; Mtorloxp/+, but not Emx1-Cre+; Ptenloxp/+; Ctnnb1loxp/+ mice, indicating that, like social behavioral deficits, the phenotype of increased c-fos+ cells in response to social cues in the PFC and amygdala of Pten cHet mice displays differential responsiveness to Mtor or Ctnnb1 mutations.
Figure 4.

Neuronal hyperactivity in Pten conditional heterozygous mice is corrected in Pten and Mtor double conditional heterozygous mice. (A) Schema showing the procedure of social exposure. (B) Representative images of c-fos staining in the prelimbic cortex (PrL) and basolateral amygdala (BLA) of socially exposed mice. Scale bar: 100 μm. (C and D) Quantification of c-fos+ neurons in the PrL(C) and BLA (D) of socially exposed mice. One-way ANOVA and Tukey’s post hoc tests were used. *P < 0.05. n.s. indicates no significant difference. N = 7 animals per genotype. F(3, 24) = 8.25, P = 0.0006 (C); F(3, 24) = 7.462, P = 0.0011 (D).
Discussion
Macrocephaly and brain overgrowth are replicated findings in a subset of individuals with ASD. Studies have suggested that brain size is positively correlated with the severity of behavioral phenotypes in individuals with ASD [Courchesne, Carper, & Akshoomoff, 2003; Sacco et al., 2007]. In addition to macrocephaly, altered functional connectivity and activity in response to social cues are also well-replicated findings in individuals with ASD [Ashwin, Baron-Cohen, Wheelwright, O’Riordan, & Bullmore, 2007; Bookheimer, Wang, Scott, Sigman, & Dapretto, 2008; Corbett et al., 2009; Dalton et al., 2005; Hadjikhani, Joseph, Snyder, & Tager-Flusberg, 2007; Monk et al., 2010; Weng et al., 2011]. In fact, among the ASD risk genes, a large fraction of them are directly involved in regulating synaptic function, suggesting that altered synaptic connectivity might be a proximal cause of ASD in some cases [Bourgeron, 2015; Delorme et al., 2013]. Studies from mouse models of ASD risk genes reveal that abnormal structural and functional connectivity can cause ASD-relevant circuit dysfunction and behavioral phenotypes [e.g., Huang et al., 2016; Peça et al., 2011; Rothwell et al., 2014]. On the other hand, studies have reported that transient early brain overgrowth is sufficient to cause ASD-relevant behavioral phenotypes (e.g., [Belinson et al., 2016; Fang et al., 2014]. Here, we showed that Pten cHet brain overgrowth can be rescued by a heterozygous mutation in Ctnnb1, but not Mtor; however, the increased potential excitatory connectivity (measured by vGluT1 puncta) and social behavioral deficits can be rescued by a heterozygous mutation in Mtor, but not Ctnnb1. This suggests that brain overgrowth and social behavioral deficits may be dissociable phenotypes in mature animals, and that altered connectivity may be a substrate of social behavioral deficits in a mouse model of Pten haploinsufficiency. Our findings here are consistent with previous studies showing mutations in Pten result in altered synaptic connectivity, which can be rescued by reducing mTOR activity and signaling [Huang et al., 2016; Kwon et al., 2006; Zhou et al., 2009].
In this study, we have used heterozygous mutations to suppress, but not abolish, the expression of β-Catenin and mTOR. This is because full knockout of Ctnnb1 or Mtor leads to severe cell death and differentiation phenotypes that would make interpretation of effects on the more subtle Pten cHet brain overgrowth and social behavioral phenotypes difficult. Although we found that heterozygous deletion of Mtor does not rescue the brain overgrowth shown in Pten cHet mice, this is likely due to gene dosage effects of Mtor. In fact, our previous study showed that heterozygous deletion of Mtor alone does not alter brain mass [Chen et al., 2015]. Given that conditional homozygous mutation of Mtor has been shown to reduce brain size [Ka et al., 2014], and that mTOR acts downstream of Pten, we predict that conditional homozygous mutation of Mtor in Pten heterozygous mice would result in brain undergrowth. Therefore, our results indicate that mTOR does not modify Pten-dependent brain overgrowth when only one copy of Mtor is deleted, but will likely modify this phenotype upon homozygous deletion of Mtor. Likewise, our results reveal that heterozygous deletion of Ctnnb1 does not rescue connectivity phenotypes and social behavioral deficits, but this could also be due to dosage. Studies have shown that knockout of Ctnnb1 leads to decreased dendritic arborization [Gao, Arlotta, Macklis, & Chen, 2007], and expression of stabilized β-catenin can increase dendritic complexity and the number of dendritic spines [Durak et al., 2016]. Thus, we suspect that knockout of Ctnnb1 in Pten heterozygous mice might modify the connectivity phenotype, although this would require an approach that bypasses the role of β-catenin in regulating cell proliferation and neurogenesis [Chen et al., 2015; Chenn & Walsh, 2002; Groszer et al., 2001], such as knockout of Ctnnb1 in postmitotic neurons of Pten heterozygous mice.
Our previous study showed evidence of hyperconnectivity from mPFC projections to the BLA in germline Pten heterozygous mice [Huang et al., 2016]. In the present study, we did not find evidence of increased vGluT1 or vGAT puncta in the BLA of Pten cHet mice (Emx1-Cre+; Ptenloxp/+). This suggests that the increased synaptic boutons from mPFC projections to the BLA shown in germline Pten heterozygous mice may be due to non-cell autonomous function of Pten. For example, the increased connectivity in the BLA may require deletion of Pten in neurons other than those in the Emx1-Cre-expressing lineage, or in microglia and astrocytes, which have been shown to play roles in sculpting synapse development and function [Allen & Eroglu, 2017; Paolicelli et al., 2011; Weinhard et al., 2018]. In the present study, we generated conditional heterozygosity for Pten using Emx1-Cre, which is primarily restricted in expression to cortical pyramidal neurons and some glia cells [Gorski et al., 2002]. Nevertheless, we found evidence of increased excitatory connectivity in the mPFC of Pten cHet mice, and that normalizing this phenotype by reduction of mTOR rescues the social behavioral deficit.
In the present study, we examined Pten cHet mice, which exhibit brain overgrowth, elevated c-Fos reactivity in response to social cues and vGluT1 puncta in the mPFC, and social behavioral deficits. We found that macroscale brain overgrowth and social behavioral deficits are dissociable phenotypes in Pten cHet mice. We also found that the increased neuronal activity in response to social cues and vGluT1 puncta in the mPFC coincide with social behavioral deficits in Pten cHet mice. While the contribution of transient early brain overgrowth caused by Pten haploinsufficiency to these phenotypes remains to be explored, our results are consistent with the possibility that mTOR-mediated microscale neuronal growth and connectivity is likely a key substrate for social behavioral deficits in Pten haploinsufficient mice.
Acknowledgments
We thank members in the Page laboratory (Amy Clipperton-Allen, Julien Sejourne, Ori Cohen, Aya Zucca, and Jenna Levy) for helpful feedback on this manuscript. This research was supported by gift funds from Ms. Nancy Lurie Marks, National Institute of Health (NIH) Grant R01MH105610, the Fraternal Order of Eagles, The American Honda and Children’s Healthcare Charity Inc., and an anonymous donor.
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest to declare.
References
- Adolphs R (2010). What does the amygdala contribute to social cognition? Annals of the New York Academy of Sciences, 1191, 42–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen NJ, & Eroglu C (2017). Cell biology of astrocyte-synapse interactions. Neuron, 96, 697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral DG, Schumann CM, & Nordahl CW (2008). Neuroanatomy of autism. Trends in Neurosciences, 31, 137–145. [DOI] [PubMed] [Google Scholar]
- Ashwin C, Baron-Cohen S, Wheelwright S, O’Riordan M, & Bullmore ET (2007). Differential activation of the amygdala and the “social brain” during fearful face-processing in Asperger Syndrome. Neuropsychologia, 45, 2–14. [DOI] [PubMed] [Google Scholar]
- Belinson H, Nakatani J, Babineau BA, Birnbaum RY,Ellegood J, Bershteyn M, … Wynshaw-Boris A (2016). Prenatal β-catenin/Brn2/Tbr2 transcriptional cascade regulates adult social and stereotypic behaviors. Molecular Psychiatry, 21, 1417–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bookheimer SY, Wang AT, Scott A, Sigman M, & Dapretto M (2008). Frontal contributions to face processing differences in autism: evidence from fMRI of inverted face processing. Journal of the International Neuropsychological Society, 14, 922–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeron T (2015). From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nature Reviews Neuroscience, 16, 551–563. [DOI] [PubMed] [Google Scholar]
- Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, … Kemler R (2001). Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development, 128, 1253–1264. [DOI] [PubMed] [Google Scholar]
- Butler MG, Dasouki MJ, Zhou X-P, Talebizadeh Z,Brown M, Takahashi TN, … Eng C (2005). Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. Journal of Medical Genetics, 42, 318–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Cai G, Chaste P, Nygren G, Goldsmith J,Reichert J, … Betancur C (2007). Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. American Journal of Medical Genetics, Part B, 144B, 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Huang W-C, Séjourné J, Clipperton-Allen AE, & Page DT (2015). Pten mutations alter brain growth trajectory and allocation of cell types through elevated β-catenin signaling. The Journal of Neuroscience, 35, 10252–10267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenn A, & Walsh CA (2002). Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science, 297, 365–369. [DOI] [PubMed] [Google Scholar]
- Clipperton-Allen AE, & Page DT (2014). Pten haploinsufficient mice show broad brain overgrowth but selective impairments in autism-relevant behavioral tests. Human Molecular Genetics, 23, 3490–3505. [DOI] [PubMed] [Google Scholar]
- Corbett BA, Carmean V, Ravizza S, Wendelken C,Henry ML, Carter C, & Rivera SM (2009). A functional and structural study of emotion and face processing in children with autism. Psychiatry Research, 173, 196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchesne E, Carper R, & Akshoomoff N (2003). Evidence of brain overgrowth in the first year of life in autism. JAMA, 290, 337–344. [DOI] [PubMed] [Google Scholar]
- Dalton KM, Nacewicz BM, Johnstone T, Schaefer HS,Gernsbacher MA, Goldsmith HH, … Davidson RJ (2005). Gaze fixation and the neural circuitry of face processing in autism. Nature Neuroscience, 8, 519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorme R, Ey E, Toro R, Leboyer M, Gillberg C, & Bourgeron T (2013). Progress toward treatments for synaptic defects in autism. Nature Medicine, 19, 685–694. [DOI] [PubMed] [Google Scholar]
- Donovan APA, & Basson MA (2017). The neuroanatomy of autism – a developmental perspective. Journal of Anatomy, 230, 4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durak O, Gao F, Kaeser-Woo YJ, Rueda R, Martorell AJ, Nott A, … Tsai L-H (2016). Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nature Neuroscience, 19, 1477–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang W-Q, Chen W-W, Jiang L, Liu K, Yung W-H, Fu AKY, & Ip NY (2014). Overproduction of upper-layer neurons in the neocortex leads to autism-like features in mice. Cell Reports, 9, 1635–1643. [DOI] [PubMed] [Google Scholar]
- Fidler DJ, Bailey JN, & Smalley SL (2000). Macrocephaly in autism and other pervasive developmental disorders. Developmental Medicine and Child Neurology, 42, 737–740. [DOI] [PubMed] [Google Scholar]
- Frazier TW, Embacher R, Tilot AK, Koenig K, Mester J, & Eng C (2015). Molecular and phenotypic abnormalities in individuals with germline heterozygous PTEN mutations and autism. Molecular Psychiatry, 20, 1132–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Arlotta P, Macklis JD, & Chen J (2007). Conditional knock-out of beta-catenin in postnatal-born dentate gyrus granule neurons results in dendritic malformation. Journal of Neuroscience, 27, 14317–14325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JLR, & Jones KR (2002). Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. Journal of Neuroscience, 22, 6309–6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann T (2013). The role of medial prefrontal cortex in early social cognition. Frontiers in Human Neuroscience, 7, 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groszer M, Erickson R, Scripture-Adams DD, Lesche R,Trumpp A, Zack JA, … Wu H (2001). Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science, 294, 2186–2189. [DOI] [PubMed] [Google Scholar]
- Hadjikhani N, Joseph RM, Snyder J, & Tager-Flusberg H(2007). Abnormal activation of the social brain during face perception in autism. Human Brain Mapping, 28, 441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N, & Sonenberg N (2004). Upstream and downstream of mTOR. Genes & Development, 18, 1926–1945. [DOI] [PubMed] [Google Scholar]
- Huang W-C, Chen Y, & Page DT (2016). Hyperconnectivity of prefrontal cortex to amygdala projections in a mouse model of macrocephaly/autism syndrome. Nature Communications, 7, 13421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ka M, Condorelli G, Woodgett JR, & Kim W-Y (2014). mTOR regulates brain morphogenesis by mediating GSK3 signaling. Development, 141, 4076–4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanner L (1943). Autistic disturbances of affective contact. TheNervous Child, 2, 217–250. [PubMed] [Google Scholar]
- Kwon C-H, Luikart BW, Powell CM, Zhou J, Matheny SA, Zhang W, … Parada LF (2006). Pten regulates neuronal arborization and social interaction in mice. Neuron, 50, 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lainhart JE, Piven J, Wzorek M, Landa R, Santangelo SL, Coon H, & Folstein SE (1997). Macrocephaly in children and adults with autism. Journal of the American Academy of Child and Adolescent Psychiatry, 36, 282–290. [DOI] [PubMed] [Google Scholar]
- Lyall K, Croen L, Daniels J, Fallin MD, Ladd-Acosta C,Lee BK, … Newschaffer C (2017). The changing epidemiology of autism spectrum disorders. Annual Review of Public Health, 38, 81–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride KL, Varga EA, Pastore MT, Prior TW, Manickam K,Atkin JF, & Herman GE (2010). Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Research, 3, 137–141. [DOI] [PubMed] [Google Scholar]
- Monk CS, Weng S-J, Wiggins JL, Kurapati N,Louro HMC, Carrasco M, … Lord C (2010). Neural circuitry of emotional face processing in autism spectrum disorders. Journal of Psychiatry & Neuroscience, 35, 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page DT, Kuti OJ, Prestia C, & Sur M (2009). Haploinsufficiency for Pten and Serotonin transporter cooperatively influences brain size and social behavior. Proceedings of the National Academy of Sciences of the United States of America, 106, 1989–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, … Gross CT (2011). Synaptic pruning by microglia is necessary for normal brain development. Science, 333, 1456–1458. [DOI] [PubMed] [Google Scholar]
- Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, … Feng G (2011). Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature, 472, 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persad A, Venkateswaran G, Hao L, Garcia ME, Yoon J, Sidhu J, & Persad S (2016). Active β-catenin is regulated by the PTEN/PI3 kinase pathway: A role for protein phosphatasePP2A. Genes & Cancer, 7, 368–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Escudero I, Oliver MD, Andrés-Pons A, Molina M, Cid VJ, & Pulido R (2011). A comprehensive functional analysis of PTEN mutations: Implications in tumor-and autism-related syndromes. Human Molecular Genetics, 20, 4132–4142. [DOI] [PubMed] [Google Scholar]
- Rothwell PE, Fuccillo MV, Maxeiner S, Hayton SJ,Gokce O, Lim BK, … Südhof TC (2014). Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell, 158, 198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco R, Militerni R, Frolli A, Bravaccio C, Gritti A, Elia M,… Persico AM (2007). Clinical, morphological, and biochemical correlates of head circumference in autism. Biological Psychiatry, 62, 1038–1047. [DOI] [PubMed] [Google Scholar]
- Varga EA, Pastore M, Prior T, Herman GE, & McBride KL(2009). The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genetics in Medicine, 11, 111–117. [DOI] [PubMed] [Google Scholar]
- Weinhard L, di Bartolomei G, Bolasco G, Machado P,Schieber NL, Neniskyte U, … Gross CT (2018). Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nature Communications, 9, 1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng S-J, Carrasco M, Swartz JR, Wiggins JL, Kurapati N, Liberzon I, … Monk CS (2011). Neural activation to emotional faces in adolescents with autism spectrum disorders. Journal of Child Psychology and Psychiatry, 52, 296–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, & Yu D (2010). PI(3)king apart PTEN’s role in cancer.Clinical Cancer Research, 16, 4325–4330. [DOI] [PubMed] [Google Scholar]
- Zhou J, Blundell J, Ogawa S, Kwon C-H, Zhang W, Sinton C, … Parada LF (2009). Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. Journal of Neuroscience, 29, 1773–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]