

Abstract
Membrane peptidases are a multi-functional group of ectoenzymes that have been implicated in the control of growth and differentiation of many cellular systems. Here, using aminopeptidase N/CD13 as an example, Dagmar Riemann and colleagues discuss the role of cell–cell contact in peptidase regulation and the influence of peptidases on cellular functions.
Keywords: CD10, CD13, CD26, Aminopeptidase, Cell–cell contact, Peptidase regulation, Leukemia typing, Myeloid marker, Ectoenzyme
Membrane peptidases are a group of ubiquitously occurring ectoenzymes with a broad functional repertoire. In protein metabolism, their functional importance is well documented, especially in peptide degradation and amino acid scavenging in the brush border membranes of renal and intestinal microvilli. However, they also perform more subtle tasks. Not only do they cleave peptide mediators, resulting in activation or inactivation, but they also function as receptors and as molecules participating in adhesion or in signal transduction (reviewed in Ref. 1). Cell-surface peptidases might have a key role in the control of growth and differentiation of many cellular systems by modulating the activity of peptide factors, as well as by regulating their access to adjacent cells. Whether the enzymatic activity is necessary for all of these different functions remains to be determined.
In hematopoiesis, the expression of surface peptidases is a characteristic of several distinct developmental stages. Molecules such as CD10/neutral endopeptidase 24.11 [common acute lymphoblastic leukemia antigen (CALLA)] and CD13/aminopeptidase N (APN) have been used for years in the characterization and typing of leukemia or lymphoma cells. The sequencing of cDNA clones for these antigens revealed their identity as well-known peptidases that occur ubiquitously in the organism and function in a much broader field than hematopoiesis2, 3. In normal bone marrow, the expression of CD10 is tightly regulated and restricted to lymphocyte precursors and mature granulocytes. CD13 is expressed on stem cells and during most developmental stages of myeloid cells4. Also, cells committed to the earliest stages of B- or T-cell differentiation5, 6 are CD13 positive but become negative upon maturation. Lymphocytes of peripheral blood, spleen or tonsils are CD13 negative.
These earlier observations formed the basis for regarding CD13 as a myelomonocytic marker. However, CD13 mRNA has been detected in leukemic T-cell lines such as HUT78 despite a lack of CD13 surface expression7. Furthermore, induction of CD13 expression has been reported after culture of pediatric acute lymphoblastic leukemia (ALL) cells for 2 to 3 days, a phenomenon that could be inhibited by cycloheximide8. ALL cells are frequently reported to express CD13 and it has been suggested that aberrant CD13 expression, also designated ‘myeloid coexpression’ or lineage infidelity, reflects neoplastic transformation. Otherwise, CD13-expressing lymphoid cells could be a repetition of phenotypes seen on minor cell populations. Thus, cases of pediatric B-lineage ALL with CD13 expression do not appear to have a poorer prognosis than ‘typical’ leukemia cases, although some studies have suggested so for adult patients in this category (reviewed in Ref. 9). Lymphoproliferative diseases presumed to arise from a more differentiated cell, such as chronic lymphocytic leukemia and lymphomas, might similarly exhibit CD13 expression, associated with a diffuse pattern of bone marrow infiltration10.
CD13/aminopeptidase N as an ectoenzyme
What kind of enzyme is CD13? Some of the characteristic features of the enzyme (reviewed in Ref. 11) are summarized in Box 1 . Like CD10, CD26/dipeptidylpeptidase IV and aminopeptidase A (APA), CD13 is an exopeptidase functioning as an ectoenzyme, that is, it displays its activity extracellularly (Fig. 1 ). The HELAH motif of the Zn2+-binding active site reveals the metallopeptidase nature of CD13 (superfamily of gluzincins12). The catalytic activity is closely related to that of the bacterial enzyme thermolysin13, the only enzyme in this family crystallized so far. CD13 is anchored to the cell membrane by a transmembrane helical region near the N-terminus, with only a small region of the N-terminus (8–10 amino acids) protruding into the cytoplasm. In most species, CD13 exists as a homodimer, dimerization occurring in the endoplasmic reticulum prior to the Golgi-associated complex glycosylation14.
Box 1. Box 1. Gene structure, occurrence and enzymology of CD13.
-
•
Human gene localization and size: 15q25 - q26; 35 kb
-
•
mRNA 3.5 kb; comprising 20 exons
-
•
Occurrence: widespread, main sources being liver, brush borders of kidney, small intestine and placenta. In the myelomonocytic lineage, CD13 is found on precursors, monocytes, basophils, eosinophils and neutrophils. Peripheral blood and tonsillar lymphocytes are negative.
-
•
Action: metalloexopeptidase that removes unsubstituted, N-terminal amino acids with neutral side chains (Ala>Phe>Leu>Gly) from peptides and amide or arylamide derivatives of amino acids. The peptide bond preceding proline is resistant to CD13.
-
•
Routine substrate: Ala-pNA at pH 7.0.
-
•
Inhibitors: o-phenanthroline, EDTA, amastatin, actinonin, probestin, bestatin.
-
•
Natural substrates: vasoactive peptides (lysyl-bradykinin, angiotensin III), neuropeptide hormones (leu- and met-enkephalin, neurokinin A, somatostatin), cytokines and immunomodulating peptides (interleukin 8, tuftsin, thymopentin).
Fig. 1.
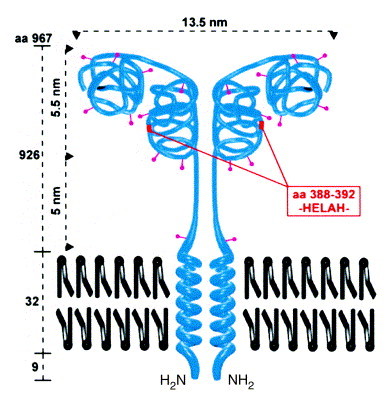
Structure of CD13. The type II membrane protein is found as a dimer of two non-covalently associated subunits with a relative molecular mass of 160 kDa each. CD13 has ten N-glycosylation sites (∼400 carbohydrate residues comprising approximately 40% of the molecular mass). Analogous to aminopeptidase A (APA)71, a two-domain structure can be hypothesized with a substrate-binding site in the C-terminal and the active center in the N-terminal domain. The dimensions of the molecule are taken from Noren et al.11
CD13 function
The functional role of CD13 varies depending on its location. Often, other membrane peptidases such as CD10 or CD26 co-localize with CD13 and seem to cooperate in peptide degradation. In the intestinal brush border, CD13 as well as CD10 or CD26 are involved in the terminal degradation of small peptides and in amino acid scavenging15 (in this location 6–8% of the protein is CD13). In synaptic membranes, CD13 and CD10 (enkephalinase) inactivate endorphins and enkephalins16. Furthermore, like CD26 (17, 18), CD13 has been considered an auxiliary adhesion molecule. When melanoma cells form colonies, the majority of CD13 molecules relocate to sites of cell–cell contact; in those cells CD13 seems to be tightly associated with extracellular matrix (ECM) components19. Whereas CD26 is involved in the adherence of metastatic breast and prostate carcinoma cells to lung endothelium20, CD13 mediates migration of different human metastatic tumor cell lines through matrigel19, 21. Recently, CD13 was shown to be the major receptor for the transmissible gastroenteritis virus (TGEV) (Ref. 22) that causes a severe gastroenteritis in piglets, and for the human coronavirus 229E (Ref. 23) that causes upper respiratory infections. TGEV binds to a region between residues 717 and 813 that lies within the C-terminal domain22; the peptidase activity of CD13 appears not to be involved in virus binding, which is in accordance with the position of the active site located in the N-terminal domain (see Fig. 1).
CD13 regulation
Because there is no zymogen pro-form, the activity of CD13 is modulated either by regulation of its synthesis or degradation, or by an endogenous inhibitor. Interestingly, natural peptides such as bradykinin or substance P inhibit the enzyme in micromolar concentrations24, but as yet there are no reports of regulatory circuits involving these two peptides that otherwise are good substrates for CD10. The first insights into CD13 expression suggest that it is regulated by a variety of different mechanisms. In addition to developmentally regulated expression, preliminary studies suggest a varying expression of CD13 during cell-growth and differentiation. For example, maturation and differentiation of monocytes is accompanied by increasing CD13 expression25. Furthermore, T-cell-derived cytokines such as interleukin 4 (IL-4) and interferon γ (IFN-γ) can upregulate CD13 mRNA and protein as well as enzyme activity in monocytes/macrophages, endothelial cells and renal tubular epithelial cells26, 27, with a maximum achieved after 2–3 days. In a variety of cells, the levels of CD13 protein and enzyme activity correlate with the abundance of CD13 transcripts.
Different promoters control transcription in myeloid cells and fibroblasts compared with epithelial cells28, 29. In epithelial cells, transcription is controlled by a classical promoter containing a TATA box upstream from the translation initiation codon. By contrast, myeloid cell and fibroblast transcripts originate from multiple sites clustered in an upstream exon located 8 kb from the exon containing the initiator codon. The two promoter regions differ with respect to transcription factor binding sites, but the encoded proteins are believed to be structurally identical. Myb and Ets are transacting factors for the myeloid promoter and cooperate with the transcription factors CBP/p300 and c-Maf (30, 31).
With respect to hematopoietic cells, CD13 has long been considered to be specific for the myeloid lineage. However, CD13+ T cells have been found outside the peripheral blood in various conditions, for example in the synovial fluid of patients with different forms of arthritis32, in the pericardial fluid of patients undergoing thoracic surgery for heart valve replacement33, or as tumor-infiltrating lymphocytes, especially of renal cell carcinoma34. In highly purified B- and T-cell preparations neither mitogens nor cytokines, such as IL-4 or IFN-γ, promote expression of CD13 on lymphocytes35.
Cell–cell contact as a paradigm of peptidase regulation
In experiments designed to study the regulation of the lymphocytic expression of CD13, direct contact of lymphocytes with various cells expressing CD13 (for example, fibroblast-like synoviocytes, HUVEC, epithelial cells and monocytes/macrophages) has been shown to induce mRNA synthesis, as well as surface expression of catalytically active CD13 (Ref. 35). The time course of CD13 expression on lymphocytes is rapid and differs from the well-described temporal characteristics of an increase in CD13 expression on monocytes and renal tubular epithelial cells elicited by IL-4. Exposure of tonsillar T and B cells to synoviocytes for as little as 1 h is sufficient to induce the expression of CD13 on the lymphocytes, and peak levels of CD13 mRNA expression persist for up to 16 h. Mitogens enhance lymphocytic CD13 expression when co-cultured with different adherent cells. Direct cell–cell contact between lymphocytes and adherent cells is required for CD13 induction on lymphocytes in vitro. Neither soluble APN from human kidney nor ECM proteins can induce lymphocytic CD13 expression. Furthermore, recent data point to a role for CD13 on adherent cells in the induction of CD13 on lymphocytes; cell–cell contact with CD13+ cells such as the transformed endothelial cell line ECV304 does not result in lymphocytic CD13 expression. Similarly, ECV304 cells transfected with an expression vector for soluble APN (enzyme lacking its membrane anchor) are insufficient, whereas ECV cells transfected with a CD13 expression vector and sorted for CD13+ cells can induce CD13 expression on lymphocytes36. Peripheral blood lymphocytes allowed to migrate into a three-dimensional collagen lattice with synoviocytes, are another good example of cell–cell contact induced expression of lymphocytic CD13 (Fig. 2 ).
Fig. 2.
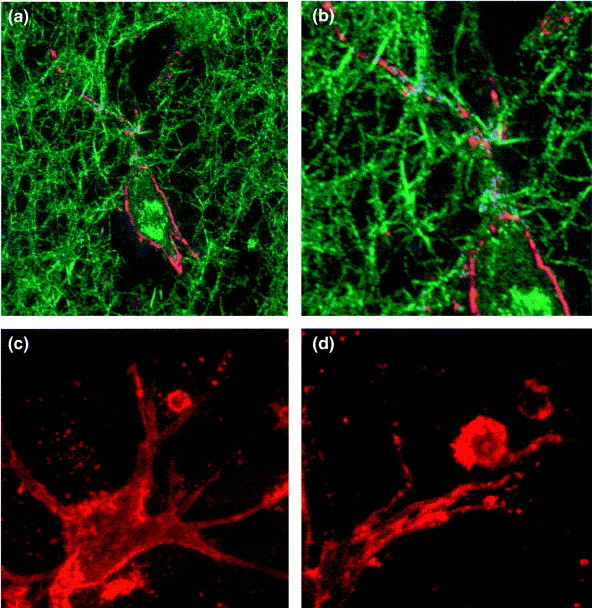
Confocal laser scanning micrographs of CD13 surface expression on fibroblast-like synoviocytes and on lymphocytes in a three-dimensional collagen matrix. (a) Confocal reflection contrast in combination with immunofluorescence staining is used to show surface expression of CD13 on synoviocytes (red) as well as reflection of collagen (type I) fibers (green). (b) Part of (a) in higher magnification. Points of close contact between collagen fibers (reflection mode) and CD13 surface staining on the synoviocyte membrane (immunofluorescence) appear in blue. (c) and (d) Immunofluorescence localization of CD13 on synoviocytes and on T cells. Peripheral blood lymphocytes were allowed to migrate into a three-dimensional collagen lattice and to adhere to synoviocytes within 3 h at 37°C. Cells were subsequently stained for CD13 (red) and fixed. Magnification for (a), ×1000; (b), ×1700; (c), ×1000; (d), ×1900.
Regulation induced by cell–cell contact has been observed for several other peptidases in co-cultivation assays using differing types of cells and studied in very different contexts (Table 1 ). The compiled results not only show induction of peptidases, but also downregulation; for example, CD13 expression on early B-lineage ALL cells is decreased after contact with bone marrow stromal cells37. Stromal-cell-mediated suppression of CD13 on B cells requires direct contact, is due to inhibition of gene transcription and is reversed by removing cells from stroma. Lymphocytes are not only cells that are responsive to signals from stromal cells, but can themselves induce upregulation of proteolytic activity in monocytes and fibroblasts45. Furthermore, direct cell–cell contact has not been found as an inevitable precondition in all cases; for example, bone marrow stromal cells positive for APA (BP-1/6C3) rapidly and selectively induce APA expression on B-cell precursors coincident with their growth. As a soluble factor, IL-7 can partially mimic the effect, inducing both APA expression as well as proliferation of B-cell precursors40.
Table 1.
Regulation of peptidases during cocultivation of human leukocytes with cells of different origina
| Cell-cell | |||||||
|---|---|---|---|---|---|---|---|
| Enzyme | Cells involved |
Time | contact | ||||
| Enzyme | location | Stimulatorsb | Responders | Effect | required | required | Refs |
| Aminopeptidase N/ | Plasma membrane | Bone marrow stromal | B-cell precursors | ↓ | Days | Yes | 37 |
| CD13 | cells | ||||||
| Synoviocytes, | B and T cells | ↑ | min-h | Yes | 35 | ||
| monocytes, HUVEC | |||||||
| Neutral | Plasma membrane | Bone marrow stromal | B-cell precursors | ↑ | Days | Yes | 38, 39 |
| endopeptidase | cells | ||||||
| 24.11/CD10 | |||||||
| Aminopeptidase A | Plasma membrane | Bone marrow stromal | B-cell precursors | ↑ | Days | No? | 40 |
| cells | |||||||
| Angiotensin | Plasma membrane | T cells | Monocytes | ↑ | Days | ? | 41 |
| converting enzyme/ | |||||||
| CD143 | |||||||
| Leucine | Cytosol | T cells | Monocytes | ↑ | Days | ? | 41 |
| aminopeptidase | |||||||
| Tissue factor/CD142 | Plasma membrane | HUVEC | Monocytes, | ↑ | min-h | ? | 42, 43 |
| THP-1, U937 | |||||||
| Interstitial collagenase | Secreted | PBL, Jurkat, HUT78 | THP-1 | ↑ | Days | Yes | 44 |
| (MMP-1) | |||||||
| Gelatinase B (MMP-9) | Secreted | Synovial T cell clones | THP-1, fibroblasts | ↑ | Days | Yes | 45 |
| PBL, Jurkat | THP-1 | ↑ | Days | ? | 46 | ||
Abbreviations: MMP, matrix metalloproteinase; PBL, peripheral blood lymphocytes.
‘Stimulators’ are those cells named that provide signals for the ‘responder’ cells to produce the respective enzymes.
In view of the reported possibility of direct protein transfer between co-cultured cells (reported for lysosomal enzymes47) it is important to note that, in most cases, induction of transcription of mRNA has been shown as proof of de novo synthesis of the induced enzymes. In some of the examples, induction across species barriers was possible [examples include CD13 (Ref. 37) and leucine aminopeptidase41], whereas other systems required autologous cells (as in the case of angiotensin converting enzyme41). Cell–cell contact can also upregulate membrane peptidases outside the hematopoietic system. As one example, CD13 expression in microglia is upregulated by culture of microglia on astrocytes48. In addition, ECM structures might be involved in cell–cell-contact-dependent regulation of proteases, as there are proteases that are obviously regulated by contact with ECM structures alone, such as tissue factor induced in the myeloid cell line THP-1 after culture on fibronectin49.
The expression of membrane peptidases on bone marrow stromal cells seems to be an important feature for the maturation of B-cell precursors and the regulation of their membrane peptidases. Cell–cell contact with stromal cells expressing CD13 causes a downregulation of CD13 on maturing B cells37; stromal cells expressing APA induce APA expression on B-cell precursors40. Similarly, maturing B cells grow in a microenvironment where CD10 is expressed both on stromal cells and on the precursor cells in close contact with the stromal layer39. Furthermore, stromal membrane peptidases in thymus seem of significance for the maturation of lymphocytic precursors, but only a few cases have been reported so far. Thymic stromal cells that bind double-positive (CD4+CD8+) thymocytes exhibit strong activity of CD13 and CD10 (Ref. 50), and in fetal thymus organ cultures a delayed thymocyte maturation has been observed after inhibition of CD10 (Ref. 51) and after inhibition of aminopeptidases with bestatin52.
At points of contact between stroma and lymphocytic precursors, local concentrations of specific peptides crucial for the differentiation or proliferation, or both, of lymphocytes have to be considered. Until now, however, no physiological peptide substrates of membrane peptidases such as CD13, CD10 or APA have been clearly implicated in the regulation of growth or maturation of lymphatic precursors. It has been proposed that a point of convergence between adhesion and peptidase substrates could be cytoplasmic tyrosine kinase focal adhesion kinase (p125FAK), which is activated and autophosphorylated both by ligation of integrins as well as by peptidase substrates that act through G-protein-coupled receptors (such as bombesin-related peptide substrates for CD10)53. Focal adhesion kinase can associate with Src or PI 3-kinase, or both, leading to activation of several signaling pathways. Many integrin-triggered reactions, for example activation of protein tyrosine kinases, such as pp60src, pp125fak and pp72syk are shared with signals generated by more traditional agonist receptors, such as those for growth factors and cytokines (reviewed in Ref. 54). Receptors for growth factors or cytokines could partition into complexes assembled by integrins, and in these complexes they might become activated and signal more efficiently.
At present it remains rather speculative to consider the possible triggering reactions for peptidase inductions during cell–cell contact. Interestingly, in some cases plasma membrane preparations from stimulator cells can substitute for intact cells, regardless of whether the stimulated peptidase is a membrane ectoenzyme or a secreted enzyme (see Table 1). In addition, fixation of stimulators does not often abrogate peptidase induction35, 44, 45. Thus, triggering molecules should be expressed on the cell surface; however, the reports to date only hint at the molecules possibly involved, such as fibronectin-integrin49, CD40–CD40L46 or E-selectin-sialyl Lewis x interactions43.
Possible role for CD13 in various types of inflammation
In inflammatory diseases such as arthritis, infiltrating lymphocytes and resident tissue cells are in close proximity, allowing the cells to communicate not only by secretion of soluble mediators but also by direct cell–cell contact. The T-cell-synoviocyte interaction has been reported to result in T-cell survival55, increased levels of tumor necrosis factor α (TNF-α), IFN-γ and IL-6 in co-culture supernatants, as well as in the upregulation of CD54 and CD106 on synovial fibroblasts56. Tonsillar B cells cultured over confluent monolayers of synoviocytes secrete high levels of immunoglobulins and differentiate into plasma cells after 8–12 days57. CD13, CD10 and CD26 were shown to be co-localized on the cell surface of synovial cells, with identical profiles in cells derived from rheumatoid and osteoarthritic joints58. It has been claimed that the fibroblastic synovial cells are a potent source of peptide-degrading activities in the joint: with (1) CD10 responsible for metabolism of bradykinin; (2) CD13 and CD10 acting together as Leu-enkephalin degrading enzymes; and (3) CD10 and CD26 inactivating substance P (Ref. 58). The finding that anti-CD13 monoclonal antibodies (mAbs) can inhibit the increased IL-6 production observed after co-culture of blood monocytes and synoviocytes59 points to a more extended role of CD13 in cell–cell contacts within the inflamed joint. Together with membrane peptidases on synoviocytes, we propose an important role for lymphocytic peptidases in joint inflammatory processes. In addition, we suggest that cell–cell contact between lymphocytes and synoviocytes acts as a regulatory event in their expression.
The observation that contact with different stromal cells induces CD13 expression on lymphocytes can explain the occurrence of CD13+ lymphocytes in various locations, which might be of physiological relevance. In the immune system, ectopeptidases can exert such diverse functions as signal transduction across the cell membrane, and regulation of the concentration of bioactive peptides and of cellular adhesion. However, in comparison with CD26, which is much better characterized with respect to its possible involvement in immune functions (reviewed in 60, 61), there are only very few experiments proving such functions for CD13. For example, CD13 has been implicated in the trimming on the cell surface of peptides that protrude out of major histocompatibility complex (MHC) class II molecules62. Pool sequencing experiments of natural HLA-DR,-DQ and -DP ligands revealed a highly dominant proline at position 2 (Ref. 63), supporting a role for CD13-like activity in antigen processing. T cells with a bright expression of CD26 have been found to preferentially migrate through a monolayer of endothelial cells on collagen gels64. Similarly, a CD13-mediated adhesion to cells and ECM structures could be a common mechanism shared by various motile cells. Furthermore, lymphocytic expression of CD13 represents an increased cellular potential for inactivation of inflammatory mediators, as shown for the degradation of enkephalins by rat splenocytes65. Hence, CD13 could play a specific role in the control of the communication between the neuroendocrine and the immune system. CD13 or CD26, either tested separately or in combination, were not able to degrade rIL-1α, rIL-2, recombinant granulocyte colony-stimulating factor (rG-CSF) or natural IL-2 despite the presence of susceptible N-terminal peptide sequences66. Nevertheless, recent studies show that chemokines are substrates for CD26 (67, 72, 73, 74). Processing of the CC chemokine RANTES by CD26 changes specificity of receptor and of target cells67, 72. With respect to this finding, the one report describing degradation as well as inactivation of the CXC chemokine IL-8 by CD13 (Ref. 68) acquires special importance. Cell–cell contact is a potent stimulus for chemokine secretion69. Hence, the question arises: are there common mechanisms in cell–cell contact that cause secretion of IL-8 and induction of its inactivator CD13? Furthermore, chemokines, similar to neuropeptides, signal with G-protein-coupled receptors, with the involvement of the aforementioned focal adhesion kinase (see as example Ref. 70), leading to the potential for common regulations in the immuno-neuroendocrine axis.
Concluding remarks
The dual regulatory aspects of membrane peptidases – being regulated by cell–cell contacts themselves as well as influencing cellular functions – are the key points of the facts and speculations presented here. An increased understanding of the function of individual CD13+ cells in controlling the level of peptide growth factors and their effects on cellular neighbours might throw light on specific peptide-mediated intercellular interactions in complex tissues. Future work will be aimed at defining the regulatory signals of CD13 induction and at the identification of natural peptidase ligands such as substrates and effectors.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (SFB 387). We are especially grateful to M. Danielsen (The Panum Institute, Copenhagen) and M.O. Lively (Wake Forest University, School of Medicine, Winston-Salem, NC, USA) for many helpful discussions. For help with laser scanning microscopy we are grateful to B. Hause (Institute of Plant Biochemistry) and H. Aurich (Institute of Physiological Chemistry, Martin Luther University). The art work of M. Pahl for the CD13 structure is gratefully acknowledged.
References
- 1.Shipp M.A., Look A.T. Blood. 1993;82:1052–1070. [PubMed] [Google Scholar]
- 2.Look A.T., Ashmun R.A., Shapiro L.H., Peiper S.C. J. Clin. Invest. 1989;83:1299–1307. doi: 10.1172/JCI114015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Letarte M., Vera S., Tran R. J. Exp. Med. 1988;168:1247–1253. doi: 10.1084/jem.168.4.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Drexler H.G. Leukemia. 1987;1:697–705. [PubMed] [Google Scholar]
- 5.Syrjälä M., Ruutu T., Jansson S.E. Br. J. Haematol. 1994;88:679–684. doi: 10.1111/j.1365-2141.1994.tb05104.x. [DOI] [PubMed] [Google Scholar]
- 6.Spits H., Lanier L.L., Phillips J.H. Blood. 1995;85:2654–2670. [PubMed] [Google Scholar]
- 7.Lendeckel U., Wex T., Kähne T., Frank K., Reinhold D., Ansorge S. Cell. Immunol. 1994;153:214–226. doi: 10.1006/cimm.1994.1019. [DOI] [PubMed] [Google Scholar]
- 8.Hara J., Kawa-Ha K., Yumura-Yagi K. Leukemia. 1991;5:19–25. [PubMed] [Google Scholar]
- 9.Ryan D.H. Clin. Chim. Acta. 1992;206:9–23. doi: 10.1016/0009-8981(92)90004-a. [DOI] [PubMed] [Google Scholar]
- 10.Pinto A., Zagonel V., Carbone A. Leuk. Lymphoma. 1992;6:209–218. [Google Scholar]
- 11.Noren, O., Sjöström, H. and Olsen, J. (1997) in Cell Surface Peptidases in Health and Disease (Kenny, A.J. and Boustead, C.M., eds), pp. 175–191, BIOS Scientific Publishers
- 12.Hooper N.M. FEBS Lett. 1994;354:1–6. doi: 10.1016/0014-5793(94)01079-x. [DOI] [PubMed] [Google Scholar]
- 13.Luciani N., Marie-Claire M., Ruffet E., Beaumont A., Roques B.P., Fournie-Zaluski M-C. Biochemistry. 1998;37:686–692. doi: 10.1021/bi971705p. [DOI] [PubMed] [Google Scholar]
- 14.Danielsen E.M. Biochemistry. 1994;33:1599–1605. doi: 10.1021/bi00172a041. [DOI] [PubMed] [Google Scholar]
- 15.Turner, A.J., Hooper, N.M. and Kenny, A.J. (1987) in Mammalian Ectoenzymes (Kenny, A.J. and Turner A.J., eds), p. 211, Elsevier Science
- 16.Matsas R., Stephenson S.L., Hryszko J., Turner A.J., Kenny A.J. Biochem. J. 1985;231:445–449. doi: 10.1042/bj2310445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Löster K., Zeilinger K., Schuppan D., Reutter W. Biochem. Biophys. Res. Commun. 1995;217:341–348. doi: 10.1006/bbrc.1995.2782. [DOI] [PubMed] [Google Scholar]
- 18.Piazza G.A., Callanan H.M., Mowery J., Hixson D.C. Biochem. J. 1989;262:327–334. doi: 10.1042/bj2620327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menrad A., Speicher D., Wacker J., Herlyn M. Cancer Res. 1993;53:1450–1455. [PubMed] [Google Scholar]
- 20.Johnson R.C., Zhu D., Augustin-Voss H.G., Pauli B.U. J. Cell Biol. 1993;121:1423–1432. doi: 10.1083/jcb.121.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saiki I., Fujii H., Yoneda J. Int. J. Cancer. 1993;54:137–143. doi: 10.1002/ijc.2910540122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delmas B., Gelfi J., L'Haridon R. Nature. 1992;357:417–420. doi: 10.1038/357417a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yeager C.L., Ashmun R.A., Williams R.K. Nature. 1992;357:420–422. doi: 10.1038/357420a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Y., Wellner D., Scheinberg D.A. Biochem. Biophys. Res. Commun. 1995;208:664–674. doi: 10.1006/bbrc.1995.1390. [DOI] [PubMed] [Google Scholar]
- 25.Laouar A., Wietzerbin J., Bauvois B. Int. Immunol. 1993;5:965–973. doi: 10.1093/intimm/5.8.965. [DOI] [PubMed] [Google Scholar]
- 26.van Hal P.T., Hopstaken-Broos J.P., Prins A. J. Immunol. 1994;153:2718–2728. [PubMed] [Google Scholar]
- 27.Riemann D., Kehlen A., Langner J. Clin. Exp. Immunol. 1995;100:277–283. doi: 10.1111/j.1365-2249.1995.tb03665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shapiro L.H., Ashmun R.A., Roberts W.M., Look A.T. J. Biol. Chem. 1991;266:11999–12007. [PubMed] [Google Scholar]
- 29.Olsen J., Laustsen L., Kärnström U., Sjöström H., Noren O. J. Biol. Chem. 1991;266:18089–18096. [PubMed] [Google Scholar]
- 30.Yang C., Shapiro L.H., Rivera M., Kumar A., Brindle P.K. Mol. Cell. Biol. 1998;18:2218–2229. doi: 10.1128/mcb.18.4.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hedge S.P., Kumar A., Kurschner C., Shapiro L.H. Mol. Cell. Biol. 1998;18:2729–2737. doi: 10.1128/mcb.18.5.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riemann D., Schwachula A., Hentschel M., Langner J. Immunobiology. 1993;187:24–35. doi: 10.1016/S0171-2985(11)80243-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Riemann D., Wollert H.G., Menschikowski J., Mittenzwei S., Langner J. Int. Arch. Allergy Immunol. 1994;104:48–56. doi: 10.1159/000236708. [DOI] [PubMed] [Google Scholar]
- 34.Riemann D., Göhring B., Langner J. Immunol. Lett. 1994;42:19–23. doi: 10.1016/0165-2478(94)90029-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riemann D., Kehlen A., Thiele K., Löhn M., Langner J. J. Immunol. 1997;158:3425–3432. [PubMed] [Google Scholar]
- 36.Thiele K., Riemann D., Kehlen A., Löhn M., Vogel L.K., Langner J. Adv. Exp. Med. Biol. 1997;421:81–84. doi: 10.1007/978-1-4757-9613-1_11. [DOI] [PubMed] [Google Scholar]
- 37.Saito M., Kumagai M., Okazaki T. Leukemia. 1995;9:1508–1516. [PubMed] [Google Scholar]
- 38.Salles G., Chen C.Y., Reinherz E.L., Shipp M.A. Blood. 1992;80:2021–2029. [PubMed] [Google Scholar]
- 39.Ishii E., Greaves A., Grunberger T., Freedman M.H., Letarte M. Leukemia. 1995;9:175–184. [PubMed] [Google Scholar]
- 40.Welch P.A., Burrows P.D., Namen A., Gillis S., Cooper M.D. Int. Immunol. 1990;2:697–705. doi: 10.1093/intimm/2.8.697. [DOI] [PubMed] [Google Scholar]
- 41.Rohrbach M.S., Conrad A.K. Clin. Exp. Immunol. 1991;83:510–515. doi: 10.1111/j.1365-2249.1991.tb05670.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Collins P.W., Noble K.E., Reittie J.R., Hoffbrand A.V., Pasi K.J., Yong K.L. Br. J. Haematol. 1995;91:963–970. doi: 10.1111/j.1365-2141.1995.tb05420.x. [DOI] [PubMed] [Google Scholar]
- 43.Lo S.K., Cheung A., Zheng Q., Silverstein R.L. J. Immunol. 1995;154:4768–4777. [PubMed] [Google Scholar]
- 44.Lacraz S., Isler P., Vey E., Welgus H.G., Dayer J-M. J. Biol. Chem. 1994;269:22027–22033. [PubMed] [Google Scholar]
- 45.Miltenburg A.M., Lacraz S., Welgus H.G., Dayer J-M. J. Immunol. 1995;154:2655–2667. [PubMed] [Google Scholar]
- 46.Malik N., Greenfield B.W., Wahl A.F., Kiener P.A. J. Immunol. 1996;156:3952–3960. [PubMed] [Google Scholar]
- 47.Olsen I., Abraham D., Shelton I., Bou-Gharios G., Muir H., Winchester B. Biochim. Biophys. Acta. 1988;968:312–322. doi: 10.1016/0167-4889(88)90022-5. [DOI] [PubMed] [Google Scholar]
- 48.Lucius R., Sievers J., Mentlein R. J. Neurochem. 1995;64:1841–1847. doi: 10.1046/j.1471-4159.1995.64041841.x. [DOI] [PubMed] [Google Scholar]
- 49.McGilvray I.D., Lu Z., Bitar R., Dackiw A.P.B., Davreux C.J., Rotstein O.D. J. Biol. Chem. 1997;272:10287–10294. doi: 10.1074/jbc.272.15.10287. [DOI] [PubMed] [Google Scholar]
- 50.Small M., Kaiser M., Tse W., Heimfeld S., Blumberg S. Eur. J. Immunol. 1996;26:961–964. doi: 10.1002/eji.1830260438. [DOI] [PubMed] [Google Scholar]
- 51.Guerin S., Mari B., Maulon L., Belhacene N., Marguet D., Auberger P. FASEB J. 1997;11:376–381. doi: 10.1096/fasebj.11.5.9141505. [DOI] [PubMed] [Google Scholar]
- 52.Appasamy P.M., Kenniston T.W., Amoscato A.A. Cell. Immunol. 1997;177:1–8. doi: 10.1006/cimm.1997.1099. [DOI] [PubMed] [Google Scholar]
- 53.Zachary I., Rozengurt E. Cell. 1992;71:891–894. doi: 10.1016/0092-8674(92)90385-p. [DOI] [PubMed] [Google Scholar]
- 54.Shattil S.J., Ginsberg M.H. J. Clin. Invest. 1997;100:1–5. doi: 10.1172/JCI119500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scott S., Pandolfi F., Kurnick J.T. J. Exp. Med. 1990;172:1873–1876. doi: 10.1084/jem.172.6.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bombara M.P., Webb D.L., Conrad P. J. Leukocyte Biol. 1993;54:399–406. doi: 10.1002/jlb.54.5.399. [DOI] [PubMed] [Google Scholar]
- 57.Dechanet J., Merville P., Durand I., Banchereau J., Miossec P. J. Clin. Invest. 1995;95:456–463. doi: 10.1172/JCI117685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bathon J.M., Proud D., Mizutani S., Ward P.E. J. Clin. Invest. 1992;90:981–991. doi: 10.1172/JCI115975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chomarat P., Rissoan M.C., Pin J.J., Banchereau J., Miossec P. J. Immunol. 1995;155:3645–3652. [PubMed] [Google Scholar]
- 60.Morimoto C., Schlossman S.F. Immunol. Rev. 1998;161:55–70. doi: 10.1111/j.1600-065x.1998.tb01571.x. [DOI] [PubMed] [Google Scholar]
- 61.von Bonin A., Hühn J., Fleischer B. Immunol. Rev. 1998;161:43–53. doi: 10.1111/j.1600-065x.1998.tb01570.x. [DOI] [PubMed] [Google Scholar]
- 62.Larsen S.L., Pedersen L.O., Buus S., Stryhn A. J. Exp. Med. 1996;184:183–189. doi: 10.1084/jem.184.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Falk K., Rötzschke O., Stevanovic S., Jung G., Rammensee H-G. Immunogenetics. 1994;39:230–242. doi: 10.1007/BF00188785. [DOI] [PubMed] [Google Scholar]
- 64.Masuyama J., Berman J.S., Cruikshank W.W., Morimoto C., Center D.M. J. Immunol. 1992;148:1367–1374. [PubMed] [Google Scholar]
- 65.Amoscato A.A., Spiess R.R., Sansoni S.B., Herberman R.B., Chamber W.H. Brain Behav. Immun. 1993;7:176–187. doi: 10.1006/brbi.1993.1019. [DOI] [PubMed] [Google Scholar]
- 66.Hoffmann T., Faust J., Neubert K., Ansorge S. FEBS Lett. 1993;336:61–64. doi: 10.1016/0014-5793(93)81609-4. [DOI] [PubMed] [Google Scholar]
- 67.Oravecz T., Pall M., Roderiquez G. J. Exp. Med. 1997;186:1865–1872. doi: 10.1084/jem.186.11.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kanayama N., Kajiwara Y., Goto J. J. Leukocyte Biol. 1995;57:129–134. doi: 10.1002/jlb.57.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smith R.E., Hogaboam C.M., Strieter R.M., Lukacs N.W., Kunkel S.L. J. Leukocyte Biol. 1997;62:612–619. doi: 10.1002/jlb.62.5.612. [DOI] [PubMed] [Google Scholar]
- 70.Bacon K.B., Szabo M.C., Yssel H., Bolen J.B., Schall T.J. J. Exp. Med. 1996;184:873–882. doi: 10.1084/jem.184.3.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hesp J.R., Hooper N.M. Biochemistry. 1997;36:3000–3007. doi: 10.1021/bi962401q. [DOI] [PubMed] [Google Scholar]
- 72.Proost P., De Meester I., Schols D. J. Biol. Chem. 1998;273:7222–7227. doi: 10.1074/jbc.273.13.7222. [DOI] [PubMed] [Google Scholar]
- 73.Van Coillie E., Proost P., Van Aelst I. Biochemistry. 1998;37:12672–12680. doi: 10.1021/bi980497d. [DOI] [PubMed] [Google Scholar]
- 74.Proost P., Struyf S., Schols D. FEBS Lett. 1998;432:73–76. doi: 10.1016/s0014-5793(98)00830-8. [DOI] [PubMed] [Google Scholar]