

Abstract
A growth in recreational drug use will lead to a rise in delayed posthypoxic leukoencephalopathy cases. Physicians may inadvertently misdiagnose this rare condition as a primary psychiatric disorder by not maintaining a broad differential diagnosis.
Keywords: drug overdose, hypoxic brain injury in adults, leukoencephalopathy, MRI
A growth in recreational drug use will lead to a rise in delayed posthypoxic leukoencephalopathy cases. Physicians may inadvertently misdiagnose this rare condition as a primary psychiatric disorder by not maintaining a broad differential diagnosis.
1. INTRODUCTION
Delayed posthypoxic leukoencephalopathy (DPHL) is a rare sequela of hypoxic brain injury that has been described in literature since the 1970s.1 It occurs following several different causes of cerebral hypo‐oxygenation including predominantly carbon monoxide (CO) poisoning, but also chemotherapy agents, recreational drug overdose, and myocardial infarction.2, 3 DPHL is characterized by its clinical presentation and radiological findings, including a lucid interval for a few weeks following an inciting hypoxic event with a resultant acute encephalopathic presentation.1, 4 Brain imaging is characteristic for central white matter demyelination without necrosis or chronic inflammatory changes, which is best seen on Magnetic resonance imaging (MRI) with T2/FLAIR with high‐intensity signaling. The cerebral cortex, putamen, thalamus, hippocampus, brainstem, and cerebellum are usually spared.3, 5, 6, 7
It is important to exclude other possible diagnoses that may mimic DPHL. Acute toxic leukoencephalopathy also symmetrically affects periventricular white matter but does not exhibit a period of recovery.3, 8 Another similar condition, delayed spongiform leukoencephalopathy (DSL), is caused by heroin inhalation (“chasing‐the‐dragon toxicity”) and may be related to the exposure of toxins in the vapor.1 That condition is characterized neuropathologically by spongiosis with massive astrocytosis without evidence of hypoxic lesions typically found in DPHL.9 Additionally, DSL is primarily associated with involvement of infratentorial structures such as the brainstem and especially the cerebellar white matter.3, 10
The pathophysiology and the relative rarity of this condition remain unclear.3 There has been speculation based on two cases that found an association with decreased arylsulfatase A activity (27%–50% of normal) from peripheral leukocytes, also known as arylsulfatase A pseudodeficiency.6, 7 It is thought that a relative deficiency would lead to accumulation of toxic byproducts from the hypoxic damage preventing remyelination. However, at least six subsequent case reports, five previously and the case reported here, did not find such association.4
2. CASE DESCRIPTION
The patient is a 28‐year‐old female with history of drug abuse previously on suboxone 5 months prior and migraine headaches who overdosed on intravenous recreational drugs. She has a family history of bipolar disorder in a paternal aunt but had no psychiatric disorders herself.
While out of state, the patient was initially found unconscious for an unknown duration in the field by EMS with agonal breathing and acute hypoxic respiratory failure to an unknown degree requiring a nonrebreather mask. She had some positive response to naloxone via intranasal and intravenous administration with the patient less obtunded. Upon arrival at the Emergency Department, she became more alert but was confused and combative. Initial noncontrast head Computed Tomography (CT) was unremarkable for acute changes, as well as repeat imaging 24 hours later. Initial urine toxicology screening was positive for benzodiazepines but negative for opiates. Serum alcohol level was negative. A boyfriend reported intravenous heroin use immediately prior to this presentation, and the patient was noted to have track marks on her right upper extremity. The patient also used benzodiazepine immediately prior to the inciting event for anxiety, although no prescriptions were in the state's reporting system for controlled substances. On the second day of her hospitalization, the patient required elective intubation due to status epilepticus from suspected benzodiazepine withdrawal. She was uneventfully extubated the following day. She was alert and oriented ×3 with no focal neurologic deficits and left against medical advice on day 3 of the hospitalization.
Five days following the initial hospitalization in the same state as the initial inciting event, the patient was noted by her family to have retrograde amnesia and an inability to care for herself due to confusion. Upon admission to a different hospital, the patient was noted to be only oriented to self and unable to provide history regarding her recent hospitalization. Urine drug screen was negative. Liver enzymes were elevated on admission (ALT 384 μ/L, AST 227 μ/L) due to Hepatitis C. She had an electroencephalogram (EEG) which showed frequent epileptiform activity in the occipital region without electrographic seizures. The patient was started on topiramate 50 mg BID and levetiracetam 500 mg BID for seizure prophylaxis. Imaging with head CT without contrast showed low attenuation of the caudate heads and lentiform nucleus, suggestive of hypoxic injury with clinical encephalopathy (Figure 1). MRI of the brain was unremarkable during this admission (Figure 2). Due to labile emotions, she was started on 0.25mg q8h lorazepam as needed. After 2 days of hospitalization, the patient was discharged, as she was alert and oriented ×3 without confusion and with a nonfocal neurologic examination.
Figure 1.
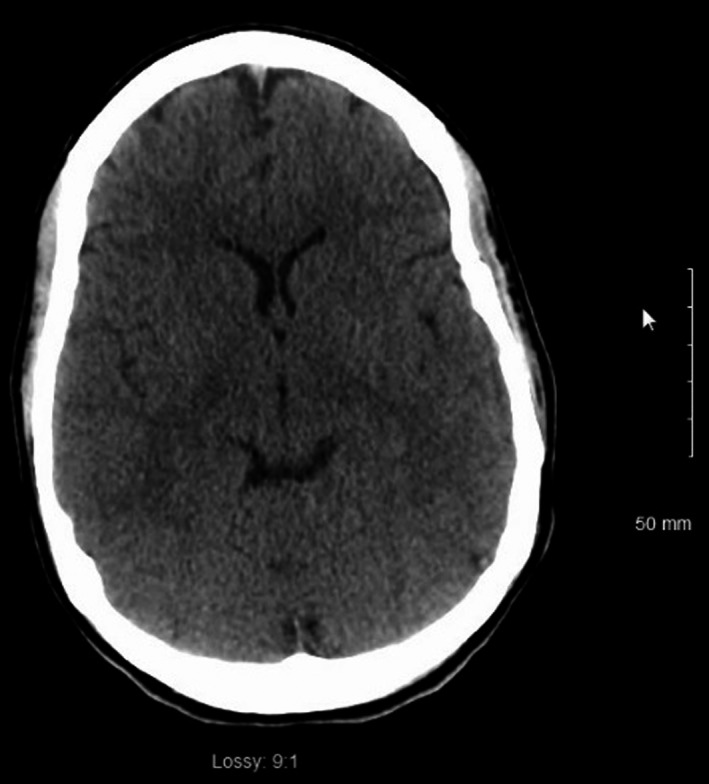
CT head 5 d postanoxic brain injury with low attenuation in bilateral caudate heads and lentiform nuclei
Figure 2.
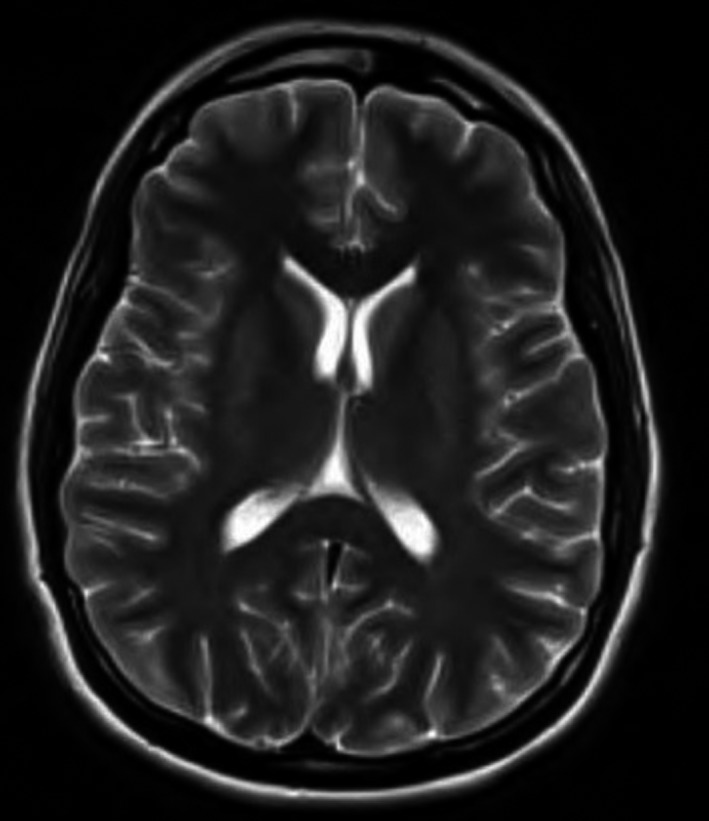
MRI brain 5 d postanoxic brain injury with no significant abnormalities
Four weeks after the initial event, the patient presented to our hospital due to acute psychosis of 2 days' duration, including confusion, agitation, lack of sleep, flat affect, and hallucinations. Family reported that she was doing well at home following discharge from the second hospitalization, except for short‐term memory loss. Upon admission, the patient was alert and oriented to self only with a nonfocal neurologic examination. Montreal Cognitive Assessment (MOCA) administered on admission showed significant cognitive impairment with a score of 16/30. Urine drug screen during this hospitalization was negative. Liver enzymes were mildly elevated. Head CT without contrast revealed bilateral symmetric areas of abnormal low attenuation in the basal ganglia involving the caudate heads and anterior aspect of the putamen due to anoxic brain injury (Figure 3). Brain MRI with contrast demonstrated a hyperintense T2 signal in the basal ganglia with mild white matter changes in bilateral frontal paraventricular areas (Figure 4). Infectious Disease was consulted, and the workup was negative, including HIV, CMV, HSV, Lyme disease, syphilis, and acute EBV. No cerebral spinal fluid analysis was done due to patient's clinical condition and parent's wishes to monitor clinically with low to no index of suspicion for infectious etiology, as no fevers, leukocytosis, or meningeal signs were present. Arylsulfatase A level in leukocytes was within normal limits. The patient had multiple EEG's which failed to show any significant seizure activity, including during the patient's delusional episodes.
Figure 3.
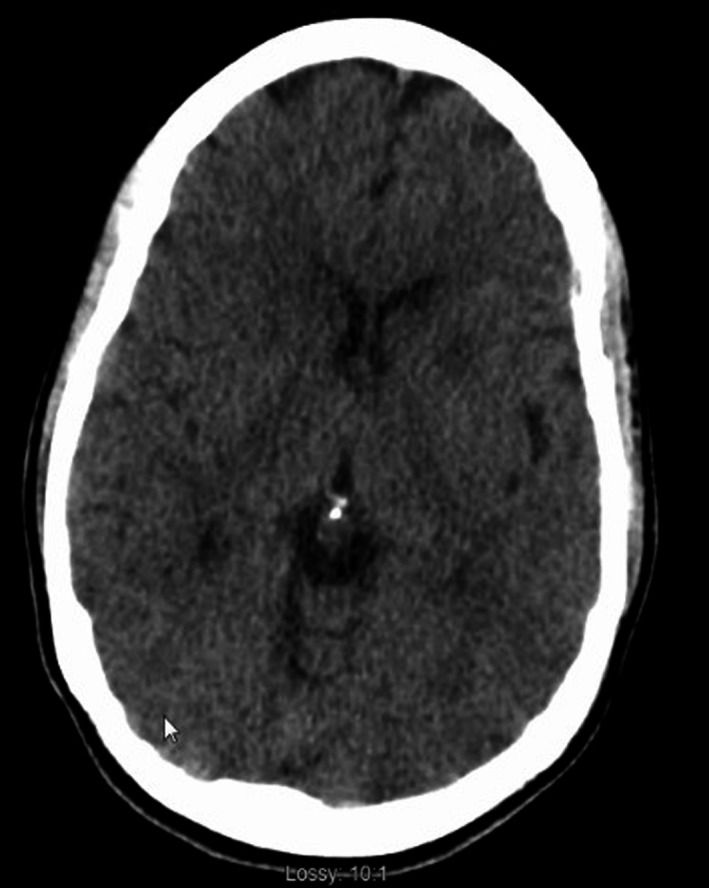
CT head one month postanoxic brain injury with low attenuation in bilateral basal ganglia
Figure 4.
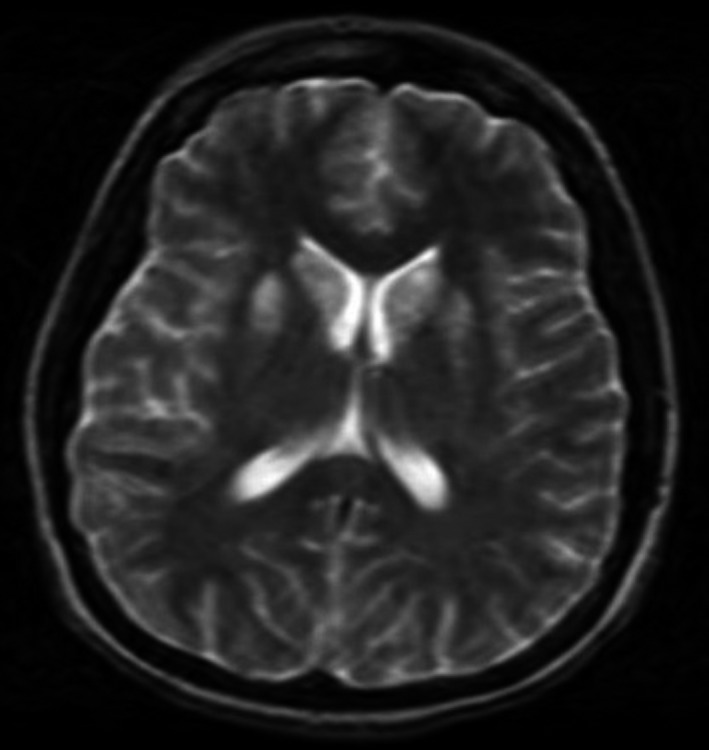
MRI brain 1 mo postanoxic brain injury with symmetric abnormal hyperintense T2 signal abnormality involving the bilateral caudate nuclei, anterior lentiform nuclei, and globus pallidus, compatible with hypoxic injury
Psychiatry was consulted, who declared there was no underlying primary psychiatric condition. Her home topiramate 50 mg BID and levetiracetam 500 mg BID were discontinued due to concern of worsening her clinical features. She was started on olanzapine 5 mg q6h, trazodone 100 mg nightly, and scheduled lorazepam 2 mg q6h. However, she had minimal response to this regimen with frequent agitation and delusional outbreaks. She was transitioned to quetiapine (50 mg, 50 mg, and 100 mg) TID with 50 mg PRN and lorazepam 2 mg q6h PRN with good response, which were continued as outpatient. Carbamazepine 200 mg BID was started during the admission for seizures and continued at discharge. Propranolol 10 mg TID was initiated and continued upon discharge for tachycardia due to agitation. The patient was alert and oriented to person, place, and time without hallucinations or paranoia upon discharge home. She was calm, relaxed and more interactive. She had improved sleep, with seven hours per night.
3. DISCUSSION
The patient's clinical course is consistent with DPHL,2, 3, 5 as she had an inciting event with acute encephalopathy and a lucid period for approximately 3 weeks when she was doing well at home. She subsequently had return of acute encephalopathy with psychosis 4 weeks after the initial event. There is some limitation though, as most patients return to baseline prior to the acute encephalopathy, with some patients returning to their work environments.3, 5 This patient had not returned to baseline but did significantly improve after the inciting event, with short‐term memory loss as the major symptom. By the end of the third admission, the patient had significantly improved in her behavioral symptoms. The majority of patients have reversibility of the disease manifestations in one year following the acute event with some residual cognitive dysfunction.1, 2, 3, 5, 11 Twelve months after discharge, follow‐up with the mother and father was completed to determine the progression of disease. The patient had mostly recovered with respect to her presentation compared to baseline health. At that time, she had a part‐time job, was obtaining her driver's license and was able to independently perform her activities of daily living. She did have minimal persistent short‐term memory loss with an infrequent labile mood. The patient's overall course of disease is consistent with DPHL.
The patient had corresponding characteristic imaging with CT and MRI of the brain revealing basal ganglia demyelination, which is only seen in 46% of DPHL patients.3, 5, 6, 11 Even though her initial imaging was relatively unremarkable, we confirmed with our radiology department that imaging can be delayed with respect to symptoms. The final extent of the disease process to the brain can be seen at approximately three months after the inciting event, which we unfortunately do not have in our records.
This patient had a normal arylsulfatase A level, which does not exclude DPHL as several case studies have failed to find an association. It is suggested that the arylsulfatase A level may be pseudodeficient for predisposition of DPHL. There are no data available for the prevalence of pseudodeficiency, and many patients have reported normal levels.1, 2, 6, 11 There needs to be more research to determine if there is a causal relationship of arylsulfatase A for predisposition of DPHL, as well as other potential markers of predisposition.
It is known that opioids are the largest inciting recreational drug.3 This patient has a remarkable case as her urine drug screen was only positive for benzodiazepines, which have rarely been reported as a causative agent for DPHL.3 This patient's disease process will facilitate physician awareness of DPHL due to recreational drug overdose solely from benzodiazepines. It is possible that other recreational drugs were present in the patient's system but not recognizable on completed drug testing, and thus, we could possibly have an incomplete picture of the inciting cause.
This case report is unique as the available literature does not discuss specific medication options or guidelines.1, 3, 5, 11 Currently, there exists only symptomatic treatment. This patient required a trial and error approach of medications, along with the knowledge of a family history of bipolar disorder with good response to quetiapine as a preliminary guide. Our determination of antipsychotic (quetiapine), benzodiazepine (lorazepam), anticonvulsant (carbamazepine), and beta‐blocker (propranolol) as a combination treatment as an effective regimen for the patient can help other physicians achieve management with symptomatic treatment for DPHL. It is possible that the medications were not the only factor in the improvement in her acute psychosis symptoms, as she was admitted for 17 days and the symptoms may have also improved over time through a natural course.
4. CONCLUSION
Delayed posthypoxic leukoencephalopathy is a rare but important sequela of hypoxic brain injury, which has been in the literature for several decades but rarely discussed. It is important to continue to discuss and bring awareness of DPHL in the primary care forum as it can be easily misdiagnosed as a primary psychiatric condition. When DPHL is due to recreational drug use, it is most commonly due to opioids but may be due to other drugs including benzodiazepines. Other conditions, including primary psychiatric and infections conditions, need to be excluded. Predisposition for DPHL may be due to pseudodeficiency of arylsulfatase A levels, but this was not seen in our patient. Effective treatment for DPHL for our patient required a multidisciplinary approach with both medication and strong social support. We found that using a multimodal treatment regimen of both antipsychotic and mood stabilizers was effective in managing our patient's symptoms. Limitations inherent to studying and managing a rare condition is unavoidable. More research is needed to understand the pathophysiology and potential causes of DPHL in order to provide effective treatments to these patients.
CONFLICT OF INTEREST
The authors have no conflicts of interest, financial disclosures, or ethical conflicts to declare.
AUTHOR CONTRIBUTIONS
Dr Kara Kaplan: collected the data, analyzed the results, and wrote the article; Dr Ariful Alam: supported in the article writing, analyzed the results, and presented criticism to the paper; Dr Braydon Dymm: contributed to research and diagnosis, and writing of the article; Dr Hisham Valiuddin: contributed to the interpretation of the results, presented criticism to the paper; and Dr Sangeetha Nanthabalan: collaborated in data collection and diagnosis, analyzed the results, and supervised the work.
CONSENT FOR PUBLICATION
Written informed consent was obtained from the patient's parents for publication of this case report and any accompanying images.
Kaplan K, Alam A, Dymm B, Valiuddin H, Nanthabalan S. Rare anoxic brain injury sequela of delayed posthypoxic leukoencephalopathy due to recreational drug overdose with benzodiazepines. Clin Case Rep. 2020;8:635–639. 10.1002/ccr3.2705
REFERENCES
- 1. Shprecher D, Mehta L. The syndrome of delayed post‐hypoxic leukoencephalopathy. NeuroRehabilitation. 2010;26(1):65‐72. [PMC free article] [PubMed] [Google Scholar]
- 2. Jang SH, Kwon HG. Injury of ascending reticular activating system associated with delayed post‐hypoxic leukoencephalopathy: a case report. BMC Neurol. 2017;17:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aljarallah S, Al‐Hussain F. Acute fatal posthypoxic leukoencephalopathy following benzodiazepine overdose: a case report and review of the literature. BMC Neurol. 2015;15:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shprecher DR, Flanigan KM, Smith AG, Smith SM, Schenkenberg T, Steffens J. Clinical and diagnostic features of delayed hypoxic leukoencephalopathy. J Neuropsychiatry Clin Neurosci. 2008;20(4):473‐477. [DOI] [PubMed] [Google Scholar]
- 5. Geraldo AF, Silva C, Neutal D, Neto LL, Albuquerque L. Delayed leukoencephalopathy after acute carbon monoxide intoxication. J Radiol Case Rep. 2014;8(5):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gottfried JA, Mayer SA, Shungu DC, Chang Y, Duyn JH. Delayed posthypoxic demyelination. Association with arylsulfatase A deficiency and lactic acidosis on proton MR spectroscopy. Neurology. 1997;49(5):1400‐1404. [DOI] [PubMed] [Google Scholar]
- 7. Weinberger LM, Schmidley JW, Schafer IA, et al. Delayed postanoxic demyelination and arylsulfatase‐A pseudodeficiency. Neurology. 1994;44:152‐154. [DOI] [PubMed] [Google Scholar]
- 8. McKinney AM, Kieffer SA, Paylor RT, SantaCruz KS, Kendi A, Lucato L. Acute toxic leukoencephalopathy: potential for reversibility clinically and on MRI with diffusion‐weighted and FLAIR imaging. AJR Am J Roentgenol. 2009;193(1):192‐206. [DOI] [PubMed] [Google Scholar]
- 9. Rizzuto N, Morbin M, Ferrari S, et al. Delayed spongiform leukoencephalopathy after heroin abuse. Acta Neuropathol. 1997;94(1):87‐90. [DOI] [PubMed] [Google Scholar]
- 10. Keogh CF, Andrews GT, Spacey SD, Forkheim KE, Graeb DA. Neuroimaging features of heroin inhalation toxicity: “chasing the dragon”. AJR Am J Roentgenol. 2003;180:847‐850. [DOI] [PubMed] [Google Scholar]
- 11. Molloy S, Soh C, Williams TL. Reversible delayed posthypoxic leukoencephalopathy. Am J Neuroradiol. 2006;27:1763‐1765. [PMC free article] [PubMed] [Google Scholar]