

Abstract
Studies of radiation interaction with tumor cells often focus on apoptosis as an end point; however, clinically relevant doses of radiation also promote autophagy and senescence. Moreover, functional p53 has frequently been implicated in contributing to radiation sensitivity through the facilitation of apoptosis. To address the involvement of apoptosis, autophagy, senescence and p53 status in the response to radiation, the current studies utilized isogenic H460 non-small cell lung cancer cells that were either p53-wild type (H460wt) or null (H460crp53). As anticipated, radiosensitivity was higher in the H460wt cells than in the H460crp53 cell line; however, this differential radiation sensitivity did not appear to be a consequence of apoptosis. Furthermore, radiosensitivity did not appear to be reduced in association with the promotion of autophagy, as autophagy was markedly higher in the H460wt cells. Despite radiosensitization by chloroquine in the H460wt cells, the radiation-induced autophagy proved to be essentially nonprotective, as inhibition of autophagy via 3-methyl adenine (3-MA), bafilomycin A1 or ATG5 silencing failed to alter radiation sensitivity or promote apoptosis in either the H460wt or H460crp53 cells. Radiosensitivity appeared to be most closely associated with senescence, which occurred earlier and to a greater extent in the H460wt cells. This finding is consistent with the in-depth proteomics analysis on the secretomes from the H460wt and H460crp53 cells (with or without radiation exposure) that showed no significant association with radioresistance-related proteins, whereas several senescence-associated secretory phenotype (SASP) factors were upregulated in H460wt cells relative to H460crp53 cells. Taken together, these findings indicate that senescence, rather than apoptosis, plays a central role in determination of radiosensitivity; furthermore, autophagy is likely to have minimal influence on radiosensitivity under conditions where autophagy takes the nonprotective form.
INTRODUCTION
Lung cancer is one of the leading causes of cancer-related deaths worldwide (1). Along with immunotherapy, chemoradiation is a primary treatment for advanced, unresectable disease; however, resistance to the currently used therapeutics often results in low cure rates and treatment failure (2, 3). The molecular basis for both radiation and chemotherapeutic drug sensitivity and resistance remains to be fully understood, making it difficult to develop therapeutic strategies that circumvent resistance mechanisms and improve survival outcomes in non-small cell lung cancer (NSCLC).
Generally, the goal of therapy is to induce cell death through mechanisms such as apoptosis. However, recently published studies have demonstrated that autophagy is virtually always present in tumor cells undergoing radiation-induced stress (4–6). Autophagy acts as a defensive cellular response that is triggered by unfavorable environmental conditions such as nutrient deprivation and hypoxia (7, 8). In response to these stresses, autophagy mediates the degradation of cellular organelles such as mitochondria and endoplasmic reticulum to generate energy and necessary metabolic precursors to prolong cell survival (9).
Hypoxia- and starvation-induced autophagy represent largely cytoprotective responses (10), whereas the role(s) of chemotherapy- and radiation-induced autophagy are less clear (11). The cytoprotective function of autophagy is generally thought to reflect efforts by the tumor cell to survive, in large part by preventing the cell from undergoing apoptosis (12, 13). However, there are also extensive examples of studies where inhibition of autophagy fails to sensitize the tumor cells to the initiating stress which we have termed “nonprotective autophagy” (14–18). The relevance and potential clinical importance of the non-protective form of autophagy relates to efforts to sensitize malignancies to therapy through autophagy inhibition. That is, unless the autophagy induced in the clinic is cytoprotective in function, there is unlikely to be a therapeutic advantage to its inhibition (19).
The cytoprotective form of autophagy is often considered a mechanism of drug and radiation resistance in tumors (20, 21). However, despite extensive evidence that pharmacological inhibitors of autophagy, such as chloroquine (CQ) and 3-methyladenine (3-MA), or genetic inhibition of autophagy can increase tumor cell sensitivity to these therapies (21), it has not been unequivocally established that autophagy uniformly reduces radiosensitivity.
In addition to autophagy, it has become apparent in recent years that tumor cells can also respond to radiation by entering a state of growth arrest with characteristics of senescence (16, 22, 23). Autophagy and senescence often occur in parallel (24) and there is evidence that autophagy can accelerate the induction of senescence (25), although we and others have shown that the two responses can also be dissociated (26, 27).
Finally, several reports have shown that proteomics studies can be very useful in discerning cellular phenotypes (28–30). Protein secretion can affect the tumor microenvironment and secreted proteins reach the blood stream where they can serve as potential biomarkers for personalized cancer treatment (31–35). Consequently, we characterized protein secretion (i.e., secretome) of tumor cellular response to radiation using high-performance liquid chromatography mass spectrometry (LC-MS/MS)-based proteomics.
MATERIALS AND METHODS
Antibodies and Reagents
The following primary antibodies were used: SQSTM1/p62 (BD Biosciences, San Jose, CA), ATG5, LC3B and GAPDH (Cell Signaling Technology® Inc., Danvers, MA) and TP53 (BD Biosciences). Secondary antibodies used were: horseradish peroxidase (HRP)-conjugated secondary antibodies (Cell Signaling Technology); TRITC-conjugated secondary antibodies or FITC-conjugated secondary antibodies (Invitrogen™, Carlsbad, CA). Hoechst 33258 was purchased from Thermo Fisher Scientific™, Inc. (Rockford, IL).
Cell Lines
H460 cells were obtained from ATCC® (NCI-H460; Gaithersburg, MD). p53 knockout H460 cells (H460crp53) were generated as described elsewhere (36). Briefly, cells were co-transfected (3 × 106 cells in 10-cm dish) with 1 μg CRISPR-Cas9 plasmid targeting the p53 loci and 1 μg of a homology-directed repair plasmid for p53 (both from Santa Cruz Biotechnology® Inc., Santa Cruz, CA). Cells were transfected using PolyJet™ reagent (SignaGen Laboratories, Rockville, MD) following the manufacturer’s guidelines. After 72 h, cells were exposed to 2.5 μg/ml puromycin with daily media exchanges to replenish the selection agent. After all cells transfected with 1 μg of a control CRISPR/Cas9 plasmid (Santa Cruz Biotechnology, Inc.) were killed (~96 h), the cells were allowed to recover and grow as individual colonies, which were then selected and examined for expression of p53 by Western blotting. shATG5 H460 cells were generated as described below. Mission shRNA bacterial stocks for ATG5 were purchased from Sigma-Aldrich (St. Louis, MO). Lentiviruses were produced in HEK 293TN cells co-transfected using EndoFectin™ Lenti Transfection Reagent (GeneCopoeia, Rockville, MD) with a packaging mixture of psPAX2 and pMD2.G constructs (Addgene, Cambridge, MA). Media containing the viruses was used to infect the H460 cells.
Cell Culture and Treatment
H460wt and H460crp53 cells were cultured in DMEM supplemented with 10% (v/v) fetal bovine serum (Thermo Fisher Scientific), 100 u/ml penicillin G sodium (Invitrogen) and 100 μg/ml streptomycin sulfate (Invitrogen). Cells were incubated at 37°C under humidified 5% CO2. Cell seeding occurred at day 0 followed by irradiation at day 1 using a 137Cs irradiator. Media was replaced every other day.
Growth Inhibition and Clonogenic Survival
Temporal studies of cell growth inhibition were based on cell viability by trypan blue exclusion. Cells were stained with 0.4% trypan blue (Sigma-Aldrich) and immediately counted using a hemocytometer. For clonogenic survival assays, cells were plated in six-well plates at a density of 200–300 cells per well. Where indicated, cells were pretreated with CQ (0, 5 or 10 μM) or 3-MA (0 or 1 mM) for 3 h, followed by exposure to 6 Gy of radiation. After 11-days of incubation postirradiation, the cell colonies were washed with phosphate buffered saline (PBS; Life Technologies, Grand Island, NY) and then fixed with 100% methanol, stained with 0.1% crystal violet (Sigma-Aldrich) and counted.
Determination of Cell Apoptosis
Apoptosis was monitored by annexin V-FITC/propidium iodide (PI) staining (Annexin V-FITC Apoptosis Detection Kit; BD Biosciences) according to the manufacturer’s instructions. Samples were analyzed by BD FACSCanto™ II and BD FACSDiva™ software at Virginia Commonwealth University Flow Cytometry Core Facility, as described elsewhere (37).
Determination and Quantification of Acidic Vesicles with Acridine Orange Staining
Cells were seeded in six-well plates and incubated overnight. After irradiation alone or in the presence of the indicated autophagy inhibitor, cells were stained with 1 μg/ml acridine orange at 37°C for 15 min and then washed with PBS. Cells were observed under an inverted fluorescence microscope (Olympus, Tokyo, Japan). For quantification of autophagic vesicles (AVOs), cells were trypsinized, harvested and washed with PBS. Pellet fractions were resuspended in PBS and analyzed by BD FACSCanto II and BD FACSDiva software. All experimental procedures were performed with cells protected from light.
Western Blotting and Immunofluorescence Staining
Western blotting was performed as described elsewhere (37). Densitometric analysis of the Western blots was performed using ImageJ2 software (NIH, Bethesda, MD). All protein band densities were normalized for their corresponding GAPDH loading control band densities. For immunofluorescence staining, after irradiation for the indicated time periods, cells were fixed with 100% methanol for 15 min, permeabilized with 0.1% Triton™ X-100 for 15 min and then blocked with 5% bovine serum albumin. Cells were incubated with primary antibody (1:100) overnight, and then exposed to FITC- or TRITC-conjugated secondary antibody for 2 h at room temperature. Finally, nuclei were stained using Hoeschst 33258. Immunofluorescence was detected by inverted fluorescence microscopy (Olympus, Tokyo, Japan).
Beta-Galactosidase and C12FDG Staining
Beta-galactosidase (β-gal) staining to monitor senescence was performed as described elsewhere by Dimri et al. (38). To quantify β-gal-positive senescent cells, after irradiation, cells were treated with bafilomycin (100 nM; Sigma-Aldrich) for 1 h to achieve lysosomal alkalization, followed by staining with C12FDG (10 μM; Thermo Fisher Scientific) for 2 h at 37°C. After incubation, cells were collected and analyzed by flow cytometry (39).
Reactive Oxygen Species (ROS) Staining
Cells were seeded in six-well plates and incubated overnight. After irradiation alone or in the presence of the indicated autophagy inhibitor, cells were stained with 10 μM 2′,7′-dichlorofluorescin diacetate (Sigma-Aldrich) at 37°C for 20 min and then washed with PBS. For quantification of ROS generation, cells were trypsinized, harvested and washed with PBS. Pellet fractions were resuspended in PBS and analyzed using BD FACSCanto II and BD FACSDiva software. All experimental procedures were performed with cells protected from light.
Cell Cycle Analysis
Cells were seeded in six-well plates and incubated overnight. After irradiation, cells were trypsinized, harvested and washed with PBS on the indicated days. Cells were fixed in 70% ethanol for 15 min, and made permeable utilizing 0.1% (v/v) Triton X-100 in PBS for 15 min prior to adding the staining solution [50 μg/ml propidium iodide (Sigma-Aldrich), 0.2 mg/ml ribonuclease A (Sigma-Aldrich)]. Cells were then incubated with the staining solution for 1 h and pellets were harvested. Pellet fractions were resuspended in PBS and analyzed using BD FACSCanto II and BD FACSDiva software. All experimental procedures were performed with cells protected from light.
H460 Secretome Collection
H460wt and H460crp53 cells were seeded with 10 ml DMEM in 12 × 10 cm dishes at a density of 100,000 cells per ml for all plates except the untreated control plates (20,000 cells per ml). Cells were allowed to attach and grow to ~85% confluency over 60 h. At ~85% confluency, the 10% FBS culture media was aspirated and the cells were washed twice with 3 ml of PBS 1× (pH 7.4; Gibco®, Grand Island, NY) and then once with 3 ml of serum-free DMEM. A final 10-ml volume of serum-free DMEM was added to all plates. A set of 3 × p53+/+ and 3 3 p53−/− plates were then immediately exposed to 6 Gy, after which all plates were incubated at 37°C and 5% CO2. The serum-free media samples containing secreted proteins (secretomes) were collected into 15-ml tubes after a 72 h incubation period and stored at −80°C until processing.
Proteomics Sample Preparation
The collected secretome samples were removed from −80°C incubation, thawed at room temperature and centrifuged at 2,500 rpm for 5 min at 8°C to remove cellular debris. The supernatant was then collected and concentrated in 3-kDa MWCO filters by centrifugation at 5,000g for 30 min at 8°C in 5-ml aliquots until a final volume of ~500 μl of concentrated secretome remained in the filter. The concentrated secretome was washed three times with 500 μl of Tris HCl buffer, pH 8.1, via centrifugation at 5,000g for 30 min at 8°C and transferred into 500-μl, 10-kDa MWCO filters. Concentrated secretome in the filters was then made up to 300 μl with Tris-HCl pH 8.1 buffer and transferred into 1.5-ml centrifuge tubes. Total protein concentration was determined at 280 nm using a BioTek Synergy H1 fitted with a Take3 Plate (BioTek® Instruments Inc., Winooski, VT). The entire 300 μl of each secretome sample was pipetted into new 10-kDa MWCO filters and processed for proteomics analysis using the FASP method (34). Briefly, each secretome sample in the 10-kDa filter was centrifuged at 15,000g for 10 min, and 50 μl of 50 mM dithiothreitol (DTT) was then added to each sample (to give a final concentration of 25 mM DTT) and incubated for 45 mins at 56°C. Reduced samples were then alkylated with 100 μl of 80 mM iodoacetamide (IAA; 40 mM final concentration) at room temperature for 30 min in the dark. After reduction and alkylation, samples were centrifuged at 15,000g for 10 min and subsequently rinsed with 350 μl of Tris-HCl, pH 8.1. Trypsin digestion was performed with 5 μl, of 1 μg/μl Trypsin Gold (Promega) solution. Samples were incubated overnight at 37°C and then digestion terminated with 200 μl of 50 mM acetic acid (Fluka™ Analytical, Morris Plains, NJ). Tryptic peptides from the FASP procedure were desalted with C18 StageTips (Thermo Fisher Scientific) per the manufacturer’s instructions and then reconstituted in 20 μl of 2% acetonitrile and 0.1% formic acid for nanoLC-MS/MS analysis
LC-MS/MS Analysis
The LC-MS/MS system consists of an Eksigent® nLC 415 (AB SCIEX™, Framingham, MA) coupled to a Q-Exactive™ (Thermo Fisher Scientific) mass spectrometer fitted with the Nanospray-Flex™ ionization source. The reverse phase analytical column (75 μm i.d., 10 μm emitter, PicoFrit®; New Objective Inc., Woburn, MA) was custom packed to a length of 20 cm with Magic AQ C18 5 μm and 200Å material at 1,000 psi with a pressure bomb (Next Advance Inc., Troy NY). A 2-μl sample of desalted peptide sample was loaded onto the analytical column and eluted at a flow rate of 300 nl/min using the following linear gradient: 5% B (0–2 min), 35% B (85 min), 75% B (95–100 min), 5% B (105 min) and held for 15 min until the run finished at 120 min where mobile phase A = 2% acetonitrile, 0.1% formic acid and mobile phase B = 98% acetonitrile, 0.1% formic acid. The electrospray voltage was set to +1.80 kV and the Q-Exactive inlet temperature and S-lens setting were maintained at 250°C and 62 V, respectively. Full scan (400–1,600 m/z) resolution was set at 70,000 FWHM with an AGC target of 3 × 106. MS/MS was set to a resolution of 17,500 with an AGC target of 2 × 104 at 120 ms maximum inject time and selection of the top 12 ions at a 30-s dynamic exclusion. HCD voltage was maintained at 27 NCE throughout.
LC-MS/MS Protein Identification, Quantification and Data Analysis
Q-Exactive proteomics raw files were processed in MaxQuant version 1.5.8.3 with the Andromeda search algorithm and the Uniprot Human proteome database; up to two missed cleavages, mass accuracies: MS = 5 ppm, MS/MS = 0.02 Da; fixed modifications: carbamidomethyl (C), variable modifications: acetyl (N-terminus) and methionine oxidation (M), and a false discovery rate (FDR) of 1%. The LFQ algorithm in MaxQuant was used for quantifying proteins with at least two peptides shared between samples. Perseus version 1.5.8.8 and JMP Pro 13 statistical software were used for statistical analysis. Gene Ontology (GO) annotation enrichment analysis was performed using the David Bioinformatics Resource version 6.8 (40, 41).
Statistical Analysis
Unless otherwise indicated, data are shown as mean ± SD from three separate experiments. One-way ANOVAs followed by the Tukey’s post hoc test and two-tailed t tests were used to assess statistical differences between groups with the GraphPad Prism 5.0 software; P < 0.05 was considered statistically significant.
RESULTS
Radiation Sensitivity in p53 Wild-Type and p53 Knockout H460 Cells
The involvement of p53 in sensitivity to radiation has been somewhat inconclusive (42, 43). It has long been thought that p53 is required for radiation-induced apoptosis, suggesting, as a general principle, that p53 wild-type H460 cells should have greater radiosensitivity than cells lacking functional p53. We addressed this question using H460wt and H460crp53 non-small cell lung cancer cells. Radiosensitivity was evaluated by clonogenic survival, where the cells were exposed to a range of doses and then allowed to form colonies for 10 days. Closely adhering to the paradigm in the literature, the H460wt cells were more sensitive to radiation (IC50 ~ 2.69 ± 0.21 Gy) than the H460crp53 cells (IC50 ~ 4.16 ± 0.19 Gy), as shown in Fig. 1A. Complementary studies of cell viability over a period of 7 days, where radiation was observed to induce a time-dependent growth inhibition, generated data that was consistent with the clonogenic survival assay whereby the H460wt cells were more radiosensitive than H460crp53 cells based on comparative rates of growth after 6 Gy irradiation (Fig. 1B). The growth of nonirradiated H460wt and H460crp53 cells was essentially identical.
FIG. 1.
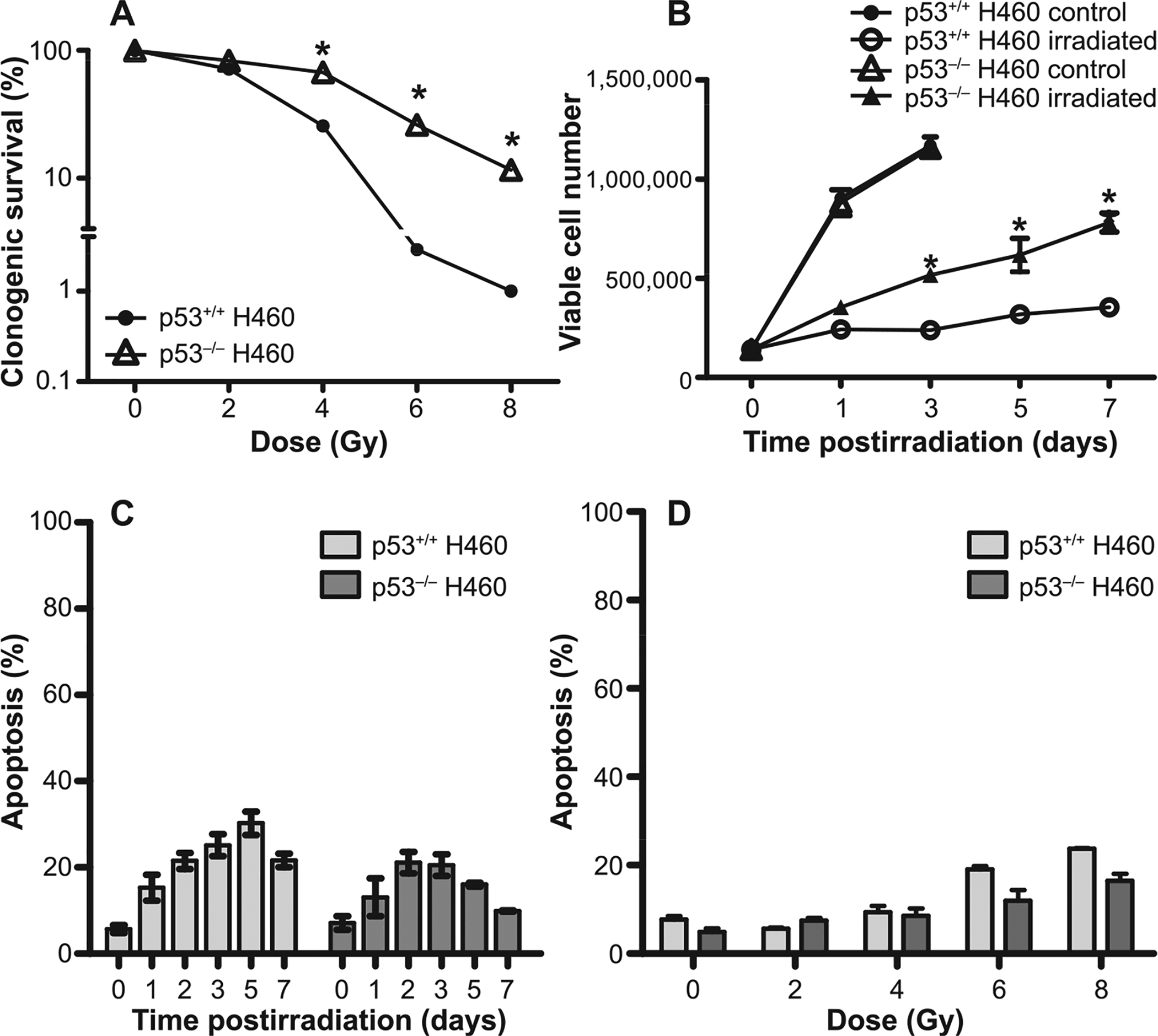
Sensitivity to ionizing radiation in H460wt and H460crp53 cells. Panel A: Clonogenic survival assay. H460wt and H460crp53 cells were exposed to the indicated doses and incubated for 11 days prior to determination of colony formation. Panel B: Cell viability. Cells were exposed to 6 Gy, incubated with fresh media for the indicated number of days and viable cell number determined by trypan blue exclusion. Panel C: Apoptosis time course. After 6 Gy irradiation, apoptotic cells were identified by annexin V/PI staining and flow cytometry at the indicated days. Panel D: Apoptosis dose response. After irradiation at the indicated doses, cells were incubated with fresh media for two days and apoptotic cells were identified by annexin V/PI staining and flow cytometry. Results are from three independent experiments. *P < 0.05, irradiated H460wt vs. H460crp53 cells.
Assuming the differences in radiation sensitivity were likely consequences of extensive apoptosis in the H460wt cells, apoptosis was assessed by annexin V/PI staining. Unexpectedly, however, as Fig. 1C indicates, the extent of radiation-induced apoptosis was low and essentially identical in both cell lines, falling between 15 and 25% within 72 h after 6 Gy irradiation. These observations were confirmed by dose-response study of radiation-induced apoptosis (Fig. 1D). Consequently, apoptosis does not appear to account for the observed differences in radiosensitivity in the two cell lines.
Radiation-Induced Autophagy is Significantly Elevated in the H460wt Cells
There is extensive evidence in the literature that inhibition of autophagy can increase tumor cell sensitivity to different forms of therapy, including radiation (44–47). Consequently, one possible explanation for the differences observed in radiosensitivity could have been the preferential promotion of cytoprotective autophagy in the H460 p53 null cells (48). To investigate this possibility, the extent of radiation-induced autophagy was determined in both cell lines. An initial screening involved acridine orange staining of cells exposed to 6 Gy after 72 h of incubation (Fig. 2A). While acridine orange is not specific for early autophagosome formation, it serves to screen for later stages in the autophagy degradation process that is characterized by the formation of acidic vacuoles (49). Acridine orange staining, quantified by flow cytometry, demonstrated a dose-dependent increase in the formation of acidic vesicular organelles (AVOs) (50) in both H460wt and H460crp53 cells (Fig. 2B). AVO formation was significantly higher in the H460wt cells. While indicative of a more pronounced autophagic response in the H460wt cells, this difference appeared to be inconsistent with the greater radiation sensitivity in the H460wt cells.
FIG. 2.
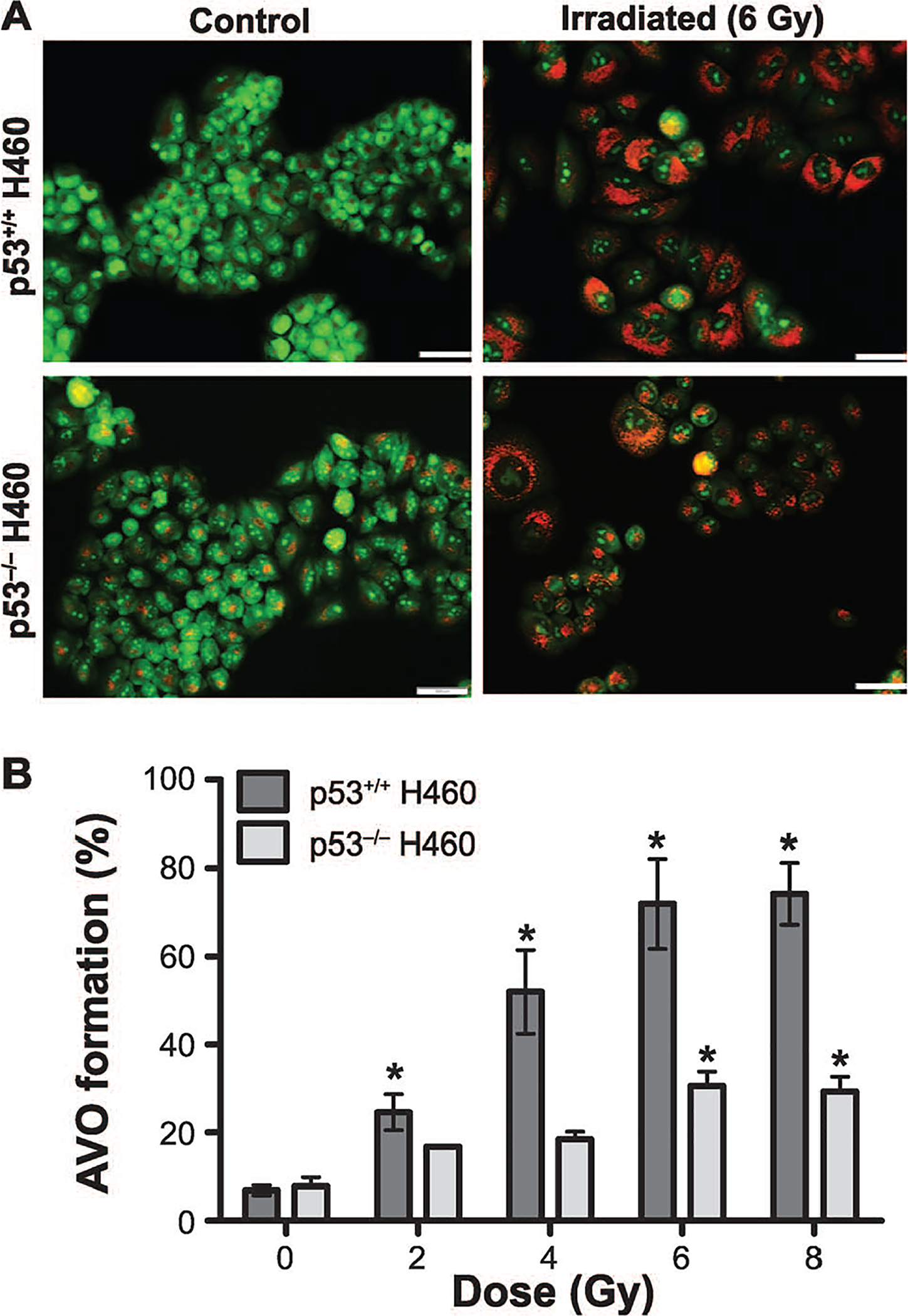
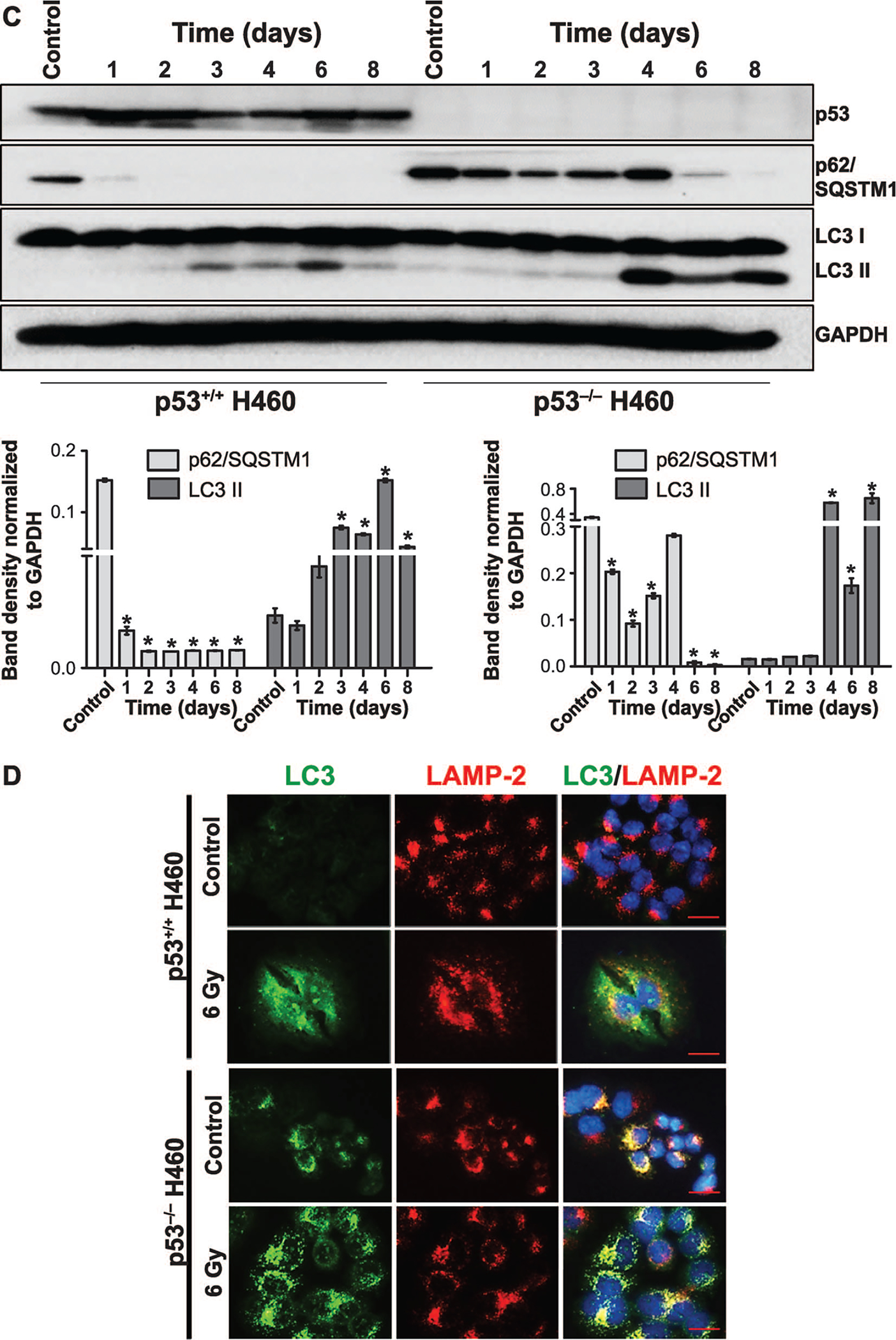
Radiation-induced autophagy in H460wt and H460crp53 cells. Panel A: Acridine orange staining. Three days after 6 Gy irradiation, cells were stained with acridine orange; images were taken at identical magnification (scale bar = 200 μm). Panel B: Quantification of acridine orange staining. Autophagy was quantified based on acridine orange staining as measured by flow cytometry. Panel C: Western blotting for levels of relevant proteins. The status of p53 in both H460 cell lines was confirmed by Western blotting. Autophagy was assessed based on p62/SQSTM1 degradation and the conversion of LC3 I to LC3 II. The bar graph in each panel indicates the relative band intensity generated from densitometric scans of three independent experiments in arbitrary densitometric units. Panel D: Co-localization of LC3 and LAMP. Fluorescence microscopy showing LC3 and LAMP2 co-localization in response to 6 Gy radiation. Imaging was performed 3 days after irradiation (scale bar = 20 μm, n = 2). Unless stated otherwise, data were from three independent experiments. *P < 0.05, control vs. irradiated cells.
To further compare the extent of autophagy and to assess whether radiation-induced autophagy is going to completion (i.e., autophagic flux), degradation of p62/SQSTM1 was evaluated using Western blotting in both cell lines. Figure 2C indicates that while autophagic flux is clearly occurring in both cell lines, autophagy goes to completion much earlier (day 2) in the H460wt cells, whereas p62/SQSTM1 degradation is markedly delayed (day 6) in the H460crp53 cells, largely complementing the finding that autophagy is more extensive in the p53 wt cells. Radiation also promoted lipidation of LC3B (conversion from LC3 I to LC3 II) in both cell lines, but occurred somewhat earlier in the H460wt cells than in the H460crp53 cells, as demonstrated in Fig. 2C (quantification is presented in the bar graphs below the figure). Figure 2D demonstrates the merging of LC3 with the lysosomal marker, LAMP, 72 h (3 days) postirradiation, further confirming radiation activated autophagic flux in both H460wt and H460crp53 cells, which is consistent with the data indicating degradation of p62/SQSTM1 in both cell lines. Collectively, these results suggest autophagy is unlikely to be serving a cytoprotective function since cells that undergo more extensive autophagy (the H460wt cells) in response to radiation are clearly more radiosensitive than the H460crp53 cells.
Investigating the Nature of Autophagy in H460wt and H460crp53 Cells
Our data clearly suggest that, despite radiation promoting more extensive autophagy in the H460wt cells than in the H460crp53 cells, autophagy was not conferring resistance to radiation, indicating that autophagy was not serving a cytoprotective function. An alternative explanation for these observations could have been that autophagy would have a cytotoxic function in the p53 wt cells (51) and a cytoprotective function in the H460crp53 cells. To determine the nature of the radiation-induced autophagy, the lysosomal acidification that is necessary for the progression of autophagy was blocked using chloroquine. Chloroquine has been reported to increase radiation-induced growth inhibition in several cell lines including colon cancer, breast cancer and liver cancer (14, 52). Both H460 cell lines were pretreated with CQ (10 μM) for 3 h followed by exposure to multiple doses (0–8 Gy) and co-incubated for 72 h (3 days). Acridine orange staining confirmed that CQ treatment results in lysosomal alkalization (a yellow rather than orange coloration), which was accompanied by accumulation of p62/SQSTM1 and LC3B II in both the H460wt and the H460crp53 cell lines (Fig. 3A and B) (quantification is presented in the bar graphs below the figure). Figure 3C shows that CQ sensitized the H460wt cells to radiation, which would be indicative of a cytoprotective function, while failing to influence radiation sensitivity in the H460crp53 cells, indicative of a nonprotective function. Consequently, although the studies using CQ are suggestive of a cytoprotective function of radiation-induced autophagy in p53 wt cells, this does not coincide with the more pronounced radiosensitivity observed in these cells.
FIG. 3.
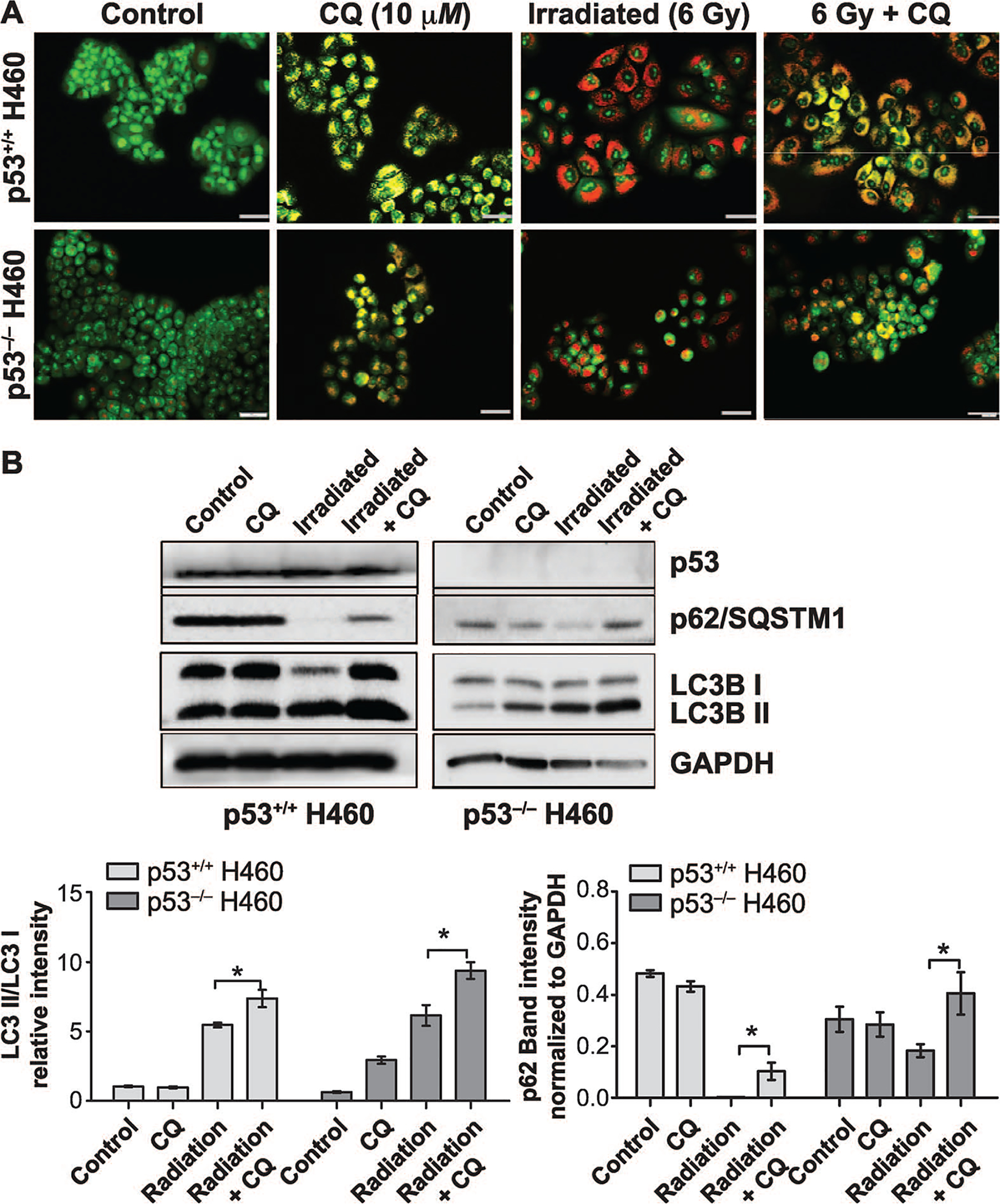
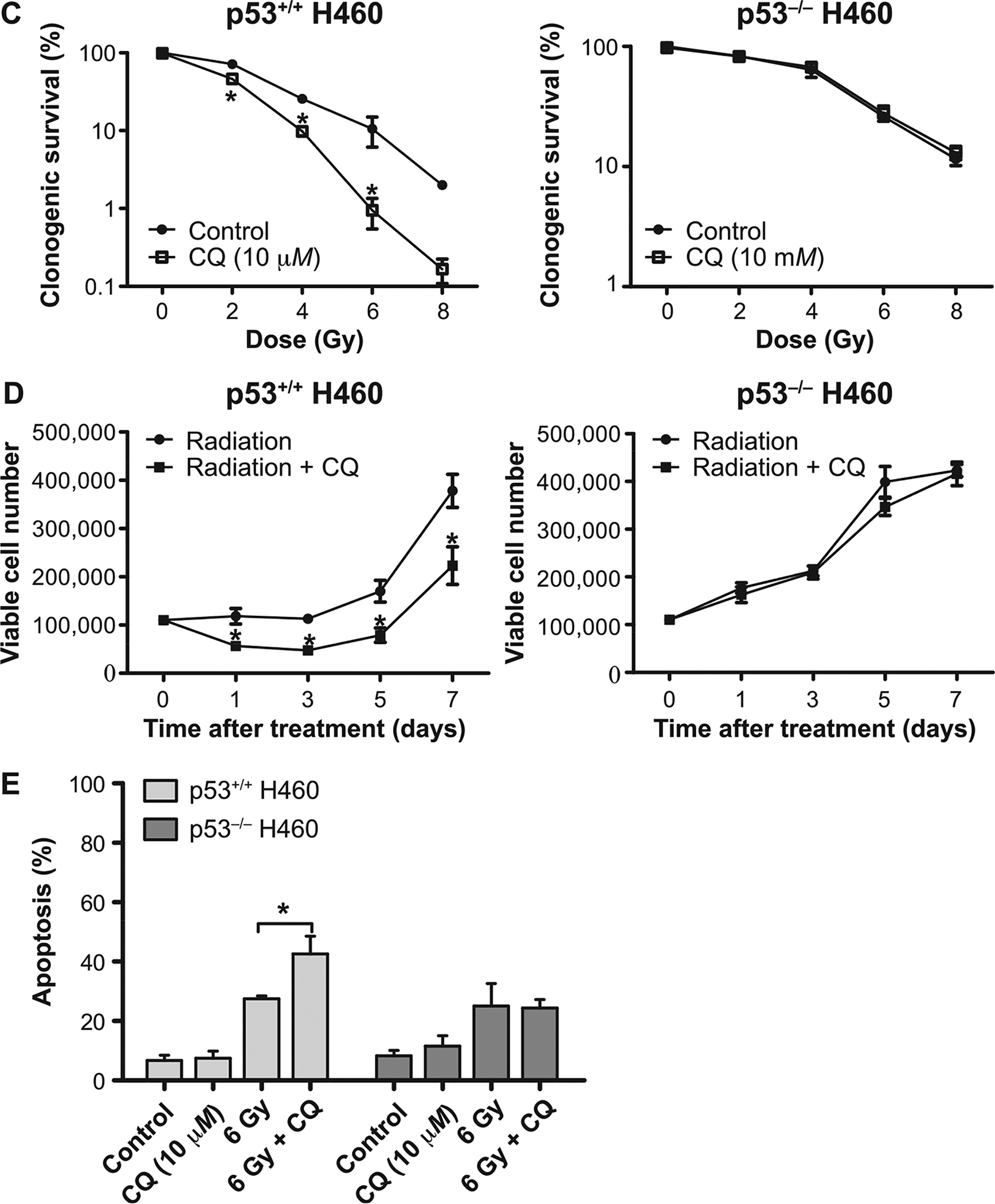
Influence of chloroquine (CQ) on radiation sensitivity in H460wt and H460crp53 cells. Panel A: Inhibition of autophagy by CQ. Fluorescence microscopy showing acridine orange-stained vacuoles induced by 6 Gy radiation alone or with CQ (10 μM) treatment (scale bar = 200 μm). Panel B: Inhibition of autophagy by CQ. Western blot showing autophagy blockade by CQ (10 μM) based on levels of p62/SQSTMQ and LC3 II. Cells were pretreated with CQ for 3 h prior to irradiation and protein was isolated after 3 days. The bar graph in each panel indicates the relative band intensity generated from densitometric scans of three independent experiments in arbitrary densitometric units. Panel C: Influence of autophagy inhibition on radiation sensitivity. Clonogenic survival assay indicating that CQ (10 μM) increased radiation-induced cell growth inhibition in H460wt cells, but not in H460crp53 cells. Panel D: Influence of autophagy inhibition on radiosensitivity. Cell viability assay indicating that CQ increased sensitivity of H460wt cells to radiation (6 Gy), but not H460crp53 cells. Panel E: Influence of autophagy inhibition on radiation-induced apoptosis. Annexin V/PI staining indicating that CQ (10 μM) increased radiation-induced apoptosis (after 2 days) in H460wt cells, but not in H460crp53 cells. Results were from three independent experiments. *P < 0.05, irradiated only cells vs. irradiated and CQ-treated cells.
As shown in Fig. 3D, a temporal analysis of cell viability in both cell lines was performed, confirming that the combination of 6 Gy irradiation and CQ enhanced growth inhibition in the H460wt cells, but not in the H460crp53 cells. Furthermore, annexin V/PI staining showed CQ resulted in a significant (albeit modest) increase in radiation-induced apoptosis (from ~24–37%) in the H460wt cells, but did not alter apoptosis in the H460crp53 cells (Fig. 3E). Taken together, CQ inhibition of radiation-induced autophagy suggests that autophagy provides a modest (albeit statistically significant) survival advantage to cells with functional p53, which is again inconsistent with the more pronounced radiosensitivity evident in the H460wt cells.
Pharmacological and Genetic Inhibition of Autophagy Fails to Support the Protective Function of Autophagy in the H460wt Cells
We had considered two possible (and closely related) explanations for the observation that the extensive autophagy in the H460wt cells did not confer reduced radiation sensitivity compared to the H460crp53 cells with low levels of autophagy. One is that the radiation-induced autophagy failed to interfere with apoptosis. However, as shown in Fig. 3E, pharmacological inhibition of autophagy by CQ did, in fact, enhance the extent of radiation-induced apoptosis in the H460wt cells. Another possibility is that the autophagy was not actually cytoprotective and that the sensitization observed in the H460wt cells might have been due to off-target effects of the chloroquine, as has been proposed by the Thorburn laboratory (53). In this context, published studies have shown that chloroquine increases intracellular ROS production, and may ultimately result in mitochondrial dysfunction and increased apoptosis (54, 55). As shown in Supplementary Fig. S1 (http://dx.doi.org/10.1667/RR15099.1.S1), an analysis of ROS generation was performed in response to CQ and another autophagy inhibitor, 3-MA, a class III PI3K inhibitor that has been widely used to prevent the initiation of autophagy (56), both alone and in combination with 6 Gy irradiation (studies using 3-MA are also shown in Fig. 4). We observed a significant increase in ROS production in the H460wt cells pretreated with CQ, an effect that was not apparent in cells pretreated with 3-MA. This could explain the radiosensitization observed with CQ.
FIG. 4.
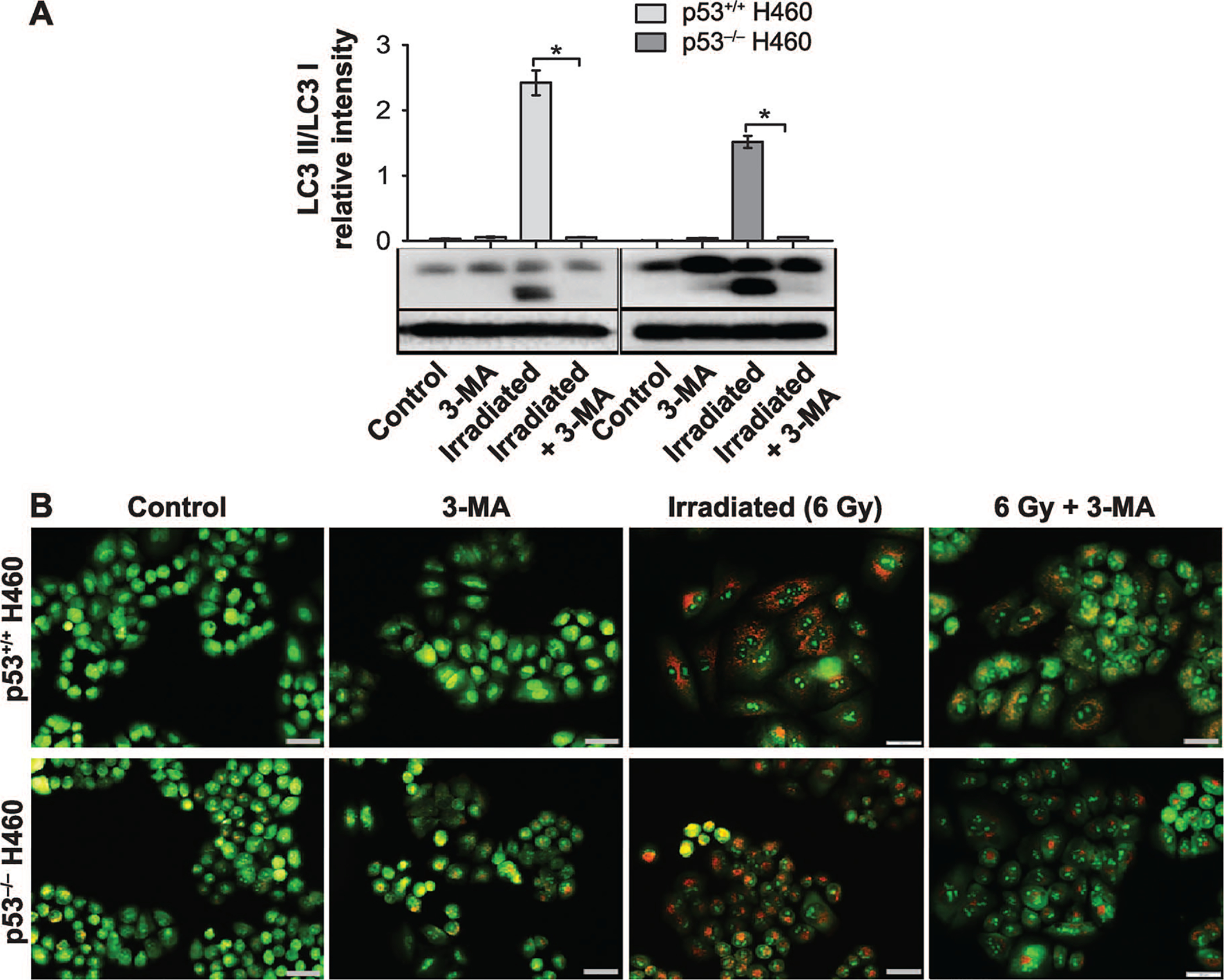
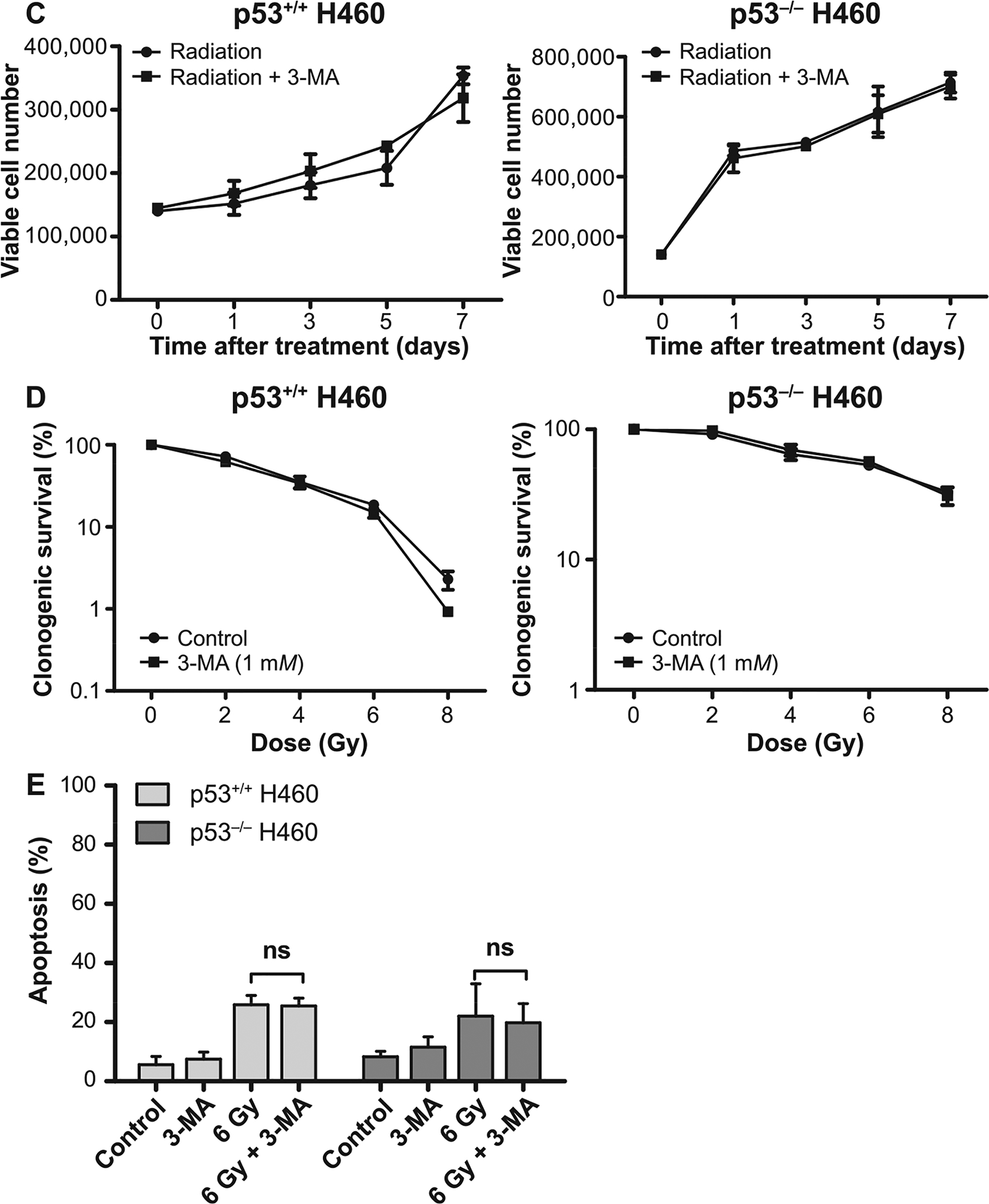
3-MA fails to alter radiation sensitivity in H460wt or H460crp53 cells. Panel A: Inhibition of autophagy by 3-MA. Western blot showing autophagy blockade by 3-MA (1 mM) based on levels of LC3 II. Cells were pretreated with 3-MA for 3 h prior to irradiation and protein was collected 3 days postirradiation. The bar graph in each panel indicates the relative band intensity generated from densitometric scans of two independent experiments in arbitrary densitometric units. Panel B: Inhibition of autophagy by 3-MA. Cells were pretreated with 3-MA for 3 h prior to irradiation and cells were stained with acridine orange 3 days postirradiation (scale bar = 200 μm). Panel C: Influence of 3-MA on radiation sensitivity. Cell viability assay indicating that 3-MA has no effect on radiosensitivity in either H460wt or H460crp53 cells. Cells were pretreated with 3-MA for 3 h followed by radiation exposure. Panel D: Influence of 3-MA on radiation sensitivity. Clonogenic survival assay indicating that 3-MA has no effect on radiosensitivity in either cell line. Cells were pretreated with 3-MA for 3 h followed by an additional 11 days postirradiation. Panel E: Influence of 3-MA on radiation-induced apoptosis. Annexin V/PI staining showing that 3-MA has no effect on radiation-induced apoptosis in either cell line. Cells were pretreated with 3-MA for 3 h prior to irradiation and apoptosis was assessed after 2 days. Unless stated otherwise, data were from three independent experiments. *P < 0.05, irradiated only cells vs. irradiated and 3-MA-treated cells.
Given that ROS production was not increased by 3-MA in either the H460wt or H460crp53 cells, (Supplementary Fig. S1; http://dx.doi.org/10.1667/RR15099.1.S1), the effects of 3-MA on radiosensitivity were investigated in both cell lines. Figure 4A and B confirms that the 3-MA was effective in inhibiting autophagy in both the H460wt and the H460crp53 cells based upon the decreased LC3 conversion and reduced formation of acridine orange-stained acidic vacuoles. Furthermore, consistent with our premise that the radiation-induced autophagy in the H460wt cells was not actually cytoprotective, pretreatment with 3-MA had no effect on colony formation or growth inhibition (Fig. 4C and D) induced by exposure to 6 Gy in either the H460wt or the H460crp53 cells. Furthermore, as would be expected based on the lack of sensitization, 3-MA failed to increase radiation-induced apoptosis in either cells line (Fig. 4E), supporting the tentative conclusion that the radiation-induced autophagy was, in fact, nonprotective in both cell lines.
To confirm these latter findings using 3-MA, short hairpin RNA was used to knock down Atg5, an autophagy regulatory gene, in both the H460wt and the H460crp53 cell lines. As shown in Fig. 5A, the status of Atg5 in two cell lines was verified by Western blotting. Atg5 knockdown decreased radiation-induced LC3B lipidation and prevented p62/SQSTM1 degradation in both the H460wt and the H460crp53 cell lines, indicating that shAtg5 effectively inhibited autophagy (Fig. 5A) (quantification is presented in the bar graphs accompanying the figure). Here, genetic interference with autophagy yielded similar results to the outcome of autophagy inhibition with 3-MA, where autophagy-compromised cells were not sensitized to radiation (Fig. 5B and C). Moreover, knockdown of Atg5 in both cell lines again failed to increase the extent of radiation-induced apoptosis (Fig. 5D). Finally, to confirm these findings when late stage autophagy is inhibited, we used bafilomycin A1, which disrupts autophagic flux by inhibiting V-ATPase-dependent decrease in lysosomal pH, and thus, autophagosome-lysosome fusion (57). Bafilomycin A1 interfered with the completion of radiation-induced autophagy in H460wt cells based on failure of lysosomal acidification and upon p62 accumulation (Supplementary Fig. S2A and B, respectively; http://dx.doi.org/10.1667/RR15099.1.S1). Again, in agreement with the previous observations, bafilomycin A1 had no impact on radiation-induced growth inhibition (Supplementary Fig. S2C). These results further confirm that CQ effects in H460wt was likely nonspecific, that late-stage autophagy inhibition does not interfere with radiosensitivity in this cell model and that autophagy plays a non-protective role in H460 cells regardless of p53 status.
FIG. 5.
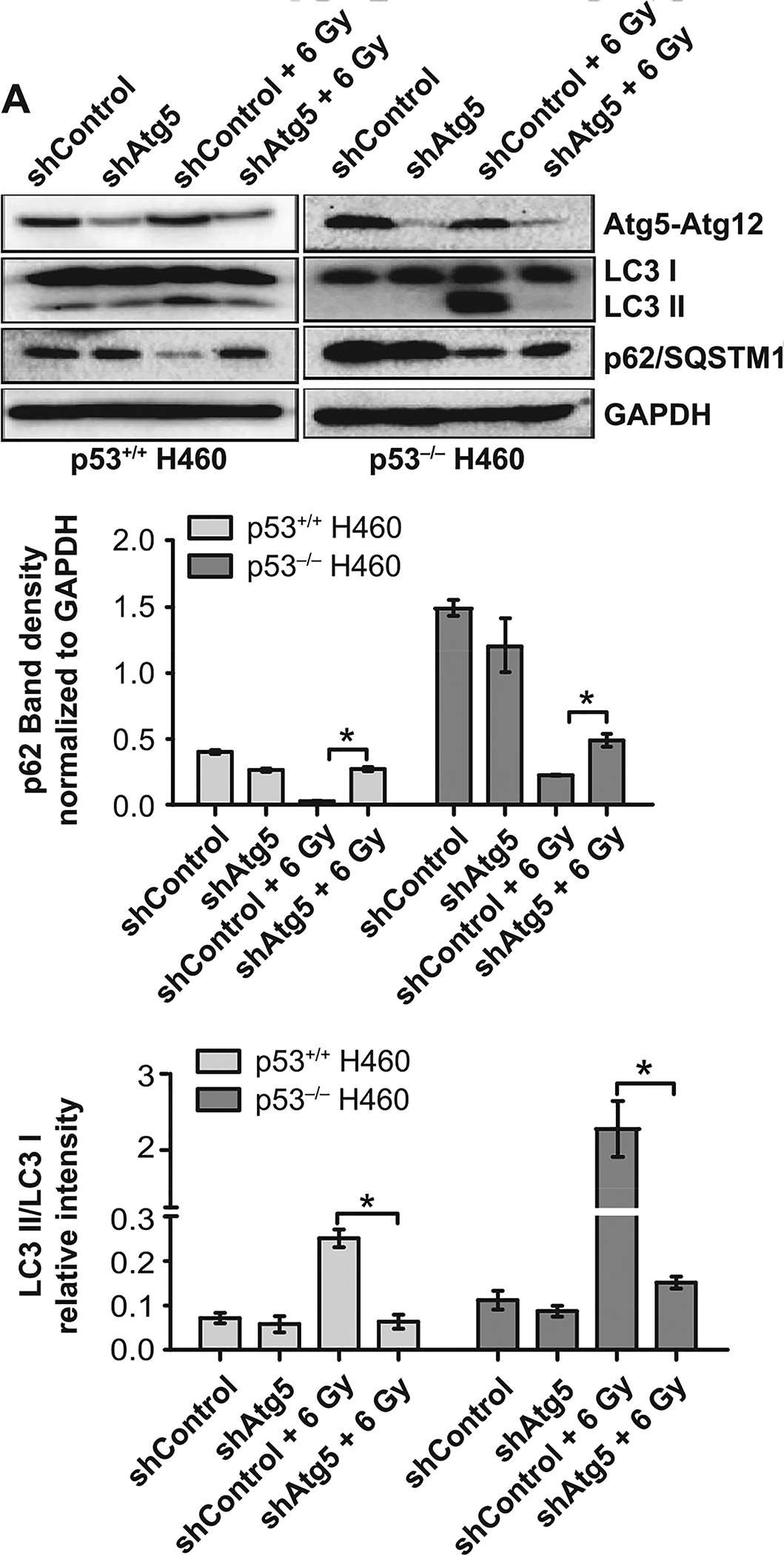
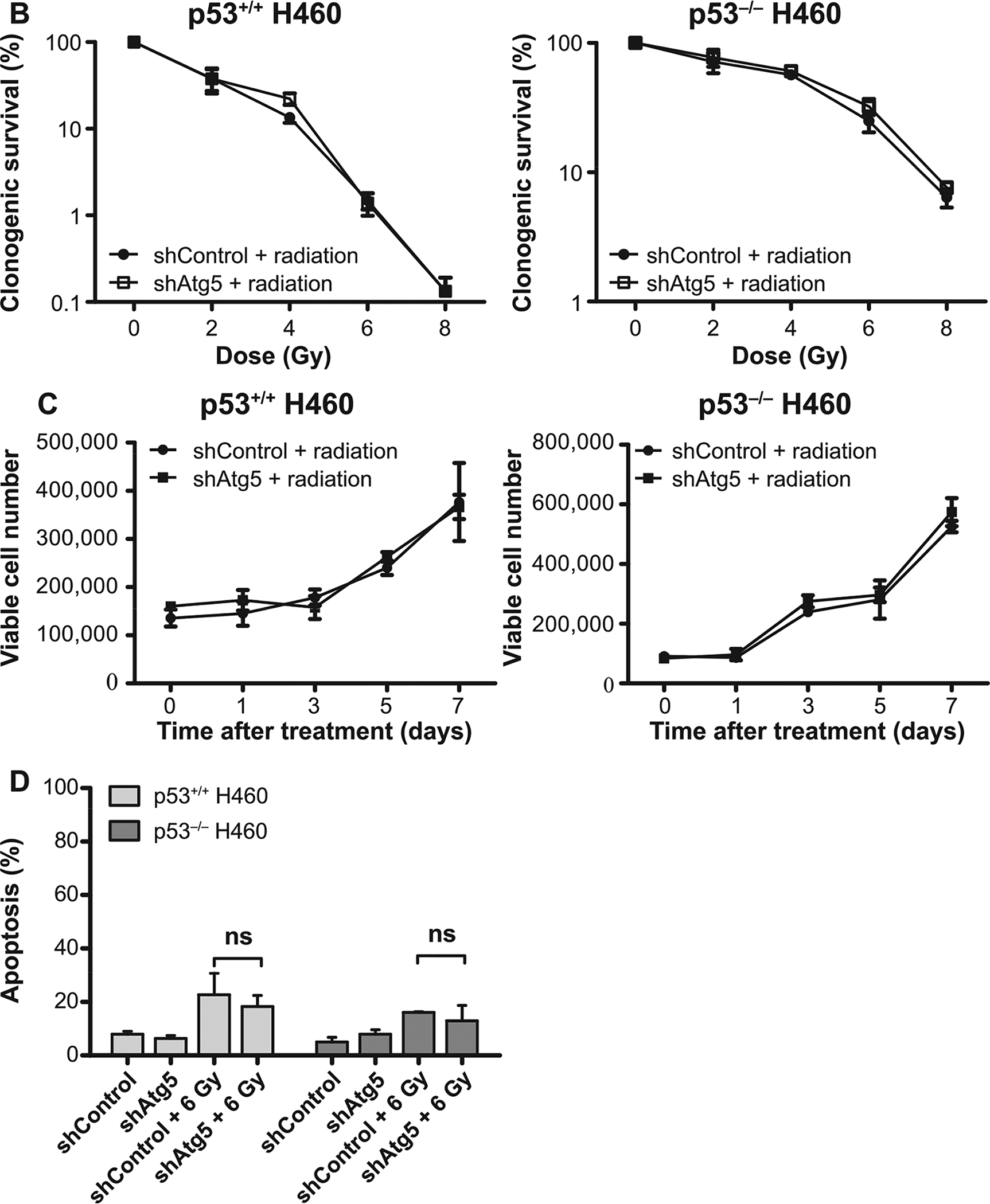
Atg5 knockdown fails to alter radiation sensitivity in H460wt or H460crp53 cells. Panel A: ATG5 knockdown. Cells were collected 3 days postirradiation. Western blot showing ATG5 knockdown in H460wt and H460crp53 cell lines; inhibition of autophagy in shAtg5 H460wt and shAtg5 H460crp53 cell lines is indicated by reduced conversion of LC3 I to LC3 II and interference with degradation of p62/SQSTM1. The bar graph in each panel indicates the relative band intensity generated from densitometric scans of two independent experiments in arbitrary densitometric units. Panel B: Lack of radiation sensitization by autophagy inhibition. Clonogenic survival assay showing that Atg5 knockdown has no effect on radiosensitivity in either cell line. Cells were irradiated at the indicated doses and incubated for 11 days prior to assessment of colony formation. Panel C: Lack of radiation sensitization by autophagy inhibition. Temporal viability assay indicating that Atg5 knockdown has no effect on radiosensitivity in both H460wt and H460crp53 cells. Cells were 6 Gy irradiated. Panel D: Autophagy inhibition does not increase the extent of radiation-induced apoptosis. Annexin V/PI staining indicating that radiation-induced apoptosis was unaltered after Atg5 knockdown in both H460wt and H460crp53 cell lines. Unless stated otherwise, data are from three independent experiments. *P < 0.05, irradiated shControl cells vs. irradiated shAtg5 cells.
Radiation Sensitivity in H460 Cells may be Linked to Senescence Rather than Autophagy or Apoptosis
In previously published work, we and others have demonstrated that senescence and autophagy are primary responses to radiation in a variety of experimental tumor cell models (16, 58). Therefore, we investigated the possibility that differential induction of senescence might influence sensitivity/resistance to radiation in the H460 cells, particularly since the cells’ response to radiation appeared to be primarily, though not exclusively, through growth inhibition rather than cell death (Fig. 1B and C). Widel et al. have shown that induction of senescence differed in p53+/+ and p53−/− HCT116 cells (59), where the p53-deficient cells showed a significant reduction in the expression of senescence markers compared to the p53-proficients cells. Our early published studies of senescence induced by doxorubicin in breast tumor cells also demonstrated that senescence occurred preferentially in tumor cells with wild-type p53 (23). Similarly, β-galactosidase staining showed that the H460wt cells undergo an early and widespread senescence, while the H460crp53 cells show delayed and modest senescence after irradiation (Fig. 6A). Microscopically, irradiated H460crp53 cells demonstrated less staining and largely maintained their original cell morphology compared to the H460wt cells, which showed significantly higher staining and extended, senescent-like morphology. Consistent with the β-galactosidase staining results, flow cytometry showed that senescence was much more extensive and occurred much earlier (after 3 days) in irradiated H460wt cells, but only increased modestly after day 7 in irradiated H460crp53 cells (Fig. 6B). Moreover, levels of p21, which is often considered a collateral marker of senescence (60), were increased in H460wt cells after day 3 of treatment, whereas the level of p21 appeared to be essentially unaltered (though initially much higher) in H460crp53 cells (Fig. 6C). Finally, to determine whether the nature of growth arrest differed substantially in the H460wt and H460crp53 cells, cell cycle distribution was monitored over a period of 11 days postirradiation. Interestingly and perhaps unexpectedly, both cell lines showed a similar response profile, undergoing an initial G2/M growth arrest that was subsequently resolved (Supplementary Figure S3; http://dx.doi.org/10.1667/RR15099.1.S1).
FIG. 6.
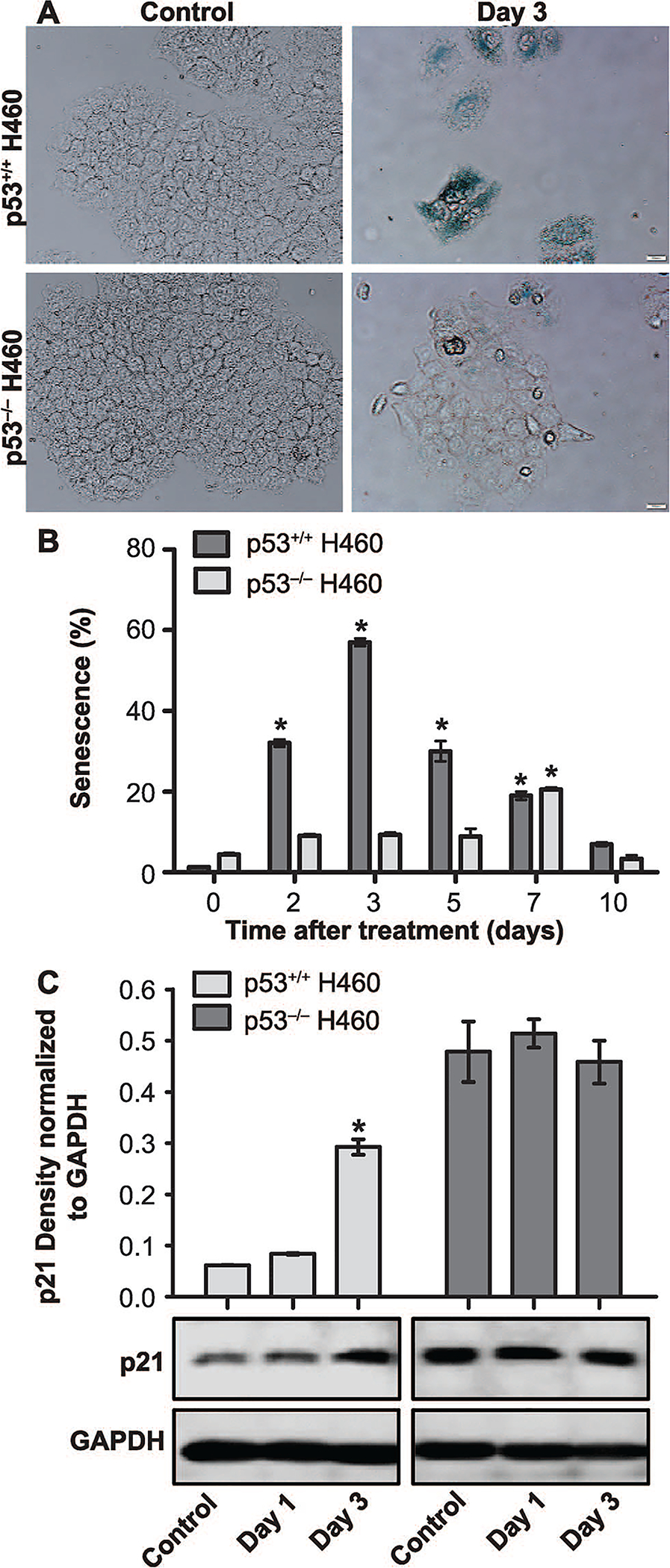
Radiation-induced senescence in H460wt and H460crp53 cells. Panel A: Beta-galactosidase staining and cell morphology. Beta-galactosidase staining indicating the induction of senescence by exposure (6 Gy) in both cell lines (scale bar = 20 μm). Panel B: Quantification of senescence. C12FDG staining and flow cytometry to quantify the extent of senescence in H460wt cells and H460crp53 cells. Panel C: Influence of radiation on levels of p21 associated with senescence. Western blot showing increased p21 induced by radiation in the H460wt cells but not the H460crp53 cells. Unless stated otherwise, data are from three independent experiments. *P < 0.05, control vs. irradiated group.
Secretome Analysis of Irradiated H460 Cells
In an effort to identify protein signatures that could differentiate the interplay between autophagy, senescence, radiation sensitivity and resistance, we used label-free quantitative proteomics (LFQ) to analyze the secretome of irradiated and nonirradiated control H460wt and H460crp53 cells. Based on the results of our cell viability analysis, clonogenic survival and senescence assays in which key differences were observed between H460wt and H460crp53 cells at day 3 (Figs. 1A–B and 6A–B), we collected conditioned media (secretomes) at 72 h from the four experimental groups (i.e., irradiated H460wt and H460crp53 cells and nonirradiated H460wt and H460crp53 cells). We identified a total of 2,092 secreted proteins (Supplementary Tables; http://dx.doi.org/10.1667/RR15099.1.S2), of which 1,595 were quantified based on their presence in ≥2 replicates of at least one out of the four experimental conditions. Functional enrichment analysis was performed on the 1,595 quantified proteins. Enriched cellular components (GOCC) in the quantified secretome proteins included extracellular exosome (~3.24-fold, P value = 1.04 × 10−275), cytoplasm (~1.67-fold, P value = 1.07 × 10−66), extracellular space (~1.73-fold, P value = 1.72 × 10−14), cytosol (~2.39-fold, P value = 1.26 × 10−140) and membrane (~2.26-fold, P value = 6.89 × 10−70) (Supplementary Fig. S4A and Tables). The molecular functions (GOMF) most enriched in this same dataset include protein binding (~1.37-fold, P value 3.77 × 10−61), poly(A) RNA binding (~3.91-fold, P value = 1.33 × 10−158) and chaperone binding (~3.61-fold, P value = 1.64 × 10−7), which is one of the more important processes in autophagy (Supplementary Fig. S4B; http://dx.doi.org/10.1667/RR15099.1.S1 and Tables; http://dx.doi.org/10.1667/RR15099.1.S2). Translation initiation (~7.22-fold, P value = 1.64 × 10−62) and cell-cell adhesion (~4.99-fold, P value = 1.28 × 10−58) were some of the biological processes (GOBP) enriched in the dataset (Supplementary Fig. S4C and Tables).
Quantitative statistical analysis revealed differential secretion of 29 proteins among the untreated H460wt and H460crp53 cells and 112 proteins between the exposed H460wt and H460crp53 cells (Supplementary Fig. S4D–E; http://dx.doi.org/10.1667/RR15099.1.S1). Within the untreated group, 10 proteins were upregulated and 19 proteins were downregulated in the H460wt, while 19 were upregulated in the H460crp53 secretome (Supplementary Fig. S4D). Within the exposed group, 28 proteins were upregulated and 74 proteins were downregulated in H460wt vs. H460crp53 secretome (Supplementary Fig. S4E).
Secretion of Radioresistance-Associated Proteins is Higher in H460crp53 Cells
Proteins associated with radiation resistance within the 1,595 proteins dataset were reported in Table 1 (61–65). In untreated control cells, none of the identified resistance-associated proteins displayed significant differences in the secretome between H460wt and H460crp53 cells, whereas three proteins showed significant differences between H460wt and H460crp53 cells after irradiation (Fig. 7A–B, Table 1). Where a significant difference (≥2-fold) was observed, as with RAC1, KLC4 and VIM (shown in red), levels of the radioresistance-associated proteins were observed to be higher in the H460crp53 cells (Fig. 7B), which is consistent with the lower radiosensitivity seen in these cells. Secretion of PLOD3 was observed to be significantly higher in H460wt cells after irradiation, albeit by <2-fold.
TABLE 1.
Resistance-Associated Proteins and Senescence-Associated Secretory Phenotype (SASP) Factors in the Secretome of H460wt and H460crp53 Cells
| Control | Irradiated | |||||||
|---|---|---|---|---|---|---|---|---|
| Protein accession | Protein name | Gene name | N: Sequence coverage (%) | N: Mol. weight (kDa) | −Log10 (P value) | Difference | −Log10 (P value) | Difference |
| Resistance-associated proteins | ||||||||
| P08670 | Vimentin | VIM | 46.8 | 53.651 | 0.2595 | 0.5584 | 3.1221 | −1.5103 |
| P63000 | Ras-related C3 botulinum toxin substrate 1 | RAC1 | 43.2 | 21.45 | 0.0769 | 0.1594 | 3.1066 | −1.2153 |
| Q9NSK0 | Kinesin light chain 4 | KLC4 | 8.4 | 62.506 | 0.1875 | −0.3751 | 2.1090 | −2.1474 |
| O60568 | Procollagen-lysine,2-oxoglutarate 5-dioxygenase 3 | PLOD3 | 32 | 84.784 | 0.1275 | −0.1237 | 1.7999 | 0.7549 |
| P36952 | Serpin B5 | SERPINB5 | 22.7 | 42.1 | 0.0163 | 0.0218 | 1.5157 | −0.5508 |
| P49327 | Fatty acid synthase | FASN | 40.3 | 273.42 | 0.5180 | −0.9924 | 1.4559 | −0.4168 |
| P27695 | DNA-(apurinic or apyrimidinic site) lyase | APEX1 | 46.2 | 35.554 | 0.6547 | −0.9087 | 1.0257 | −0.4148 |
| P08758 | Annexin A5 | ANXA5 | 23.1 | 35.936 | 0.4295 | 0.3975 | 1.0061 | 1.0789 |
| P31947 | 14-3-3 protein sigma | SFN | 48.8 | 27.774 | 0.6745 | −0.8959 | 0.9709 | −0.9161 |
| P15531 | Nucleoside diphosphate kinase A | NME1 | 65.1 | 17.149 | 0.0049 | 0.0068 | 0.5840 | −0.1710 |
| P62491 | Ras-related protein Rab-11A | RAB11A | 29.8 | 24.488 | 0.2692 | −0.3305 | 0.5442 | −0.1878 |
| Q5JPE7 | Nodal modulator 2 | NOMO2 | 6.2 | 139.38 | 0.1650 | −0.1895 | 0.5106 | 0.4930 |
| P31930 | Cytochrome b-c1 complex subunit 1, mitochondrial | UQCRC1 | 5.4 | 52.645 | 0.3140 | −0.7046 | 0.4073 | 0.5895 |
| P30040 | Endoplasmic reticulum resident protein 29 | ERP29 | 30.7 | 28.993 | 0.4358 | 1.6002 | 0.0920 | 0.0590 |
| P62937 | Peptidyl-prolyl cis-trans isomerase A | PPIA | 67.3 | 18.012 | 0.1148 | 0.5008 | 0.0495 | −0.0518 |
| Senescence-associated secretory proteins | ||||||||
| P09341 | Growth-regulated alpha protein | CXCL1 | 45.8 | 11.301 | 4.1403 | 5.3441 | 5.3041 | 3.7295 |
| P05231 | Interleukin-6 | IL6 | 42.9 | 21.494 | 0.6601 | 1.2458 | 3.6702 | 3.7453 |
| P42830 | C-X-C motif chemokine 5 | CXCL5 | 43 | 11.972 | 0.0768 | 0.3904 | 3.4760 | 3.1293 |
| P55268 | Laminin subunit beta-2 | LAMB2 | 26.1 | 195.98 | 1.9894 | 1.2477 | 3.3122 | 0.6565 |
| P03956 | Interstitial collagenase | MMP1 | 11.1 | 54.006 | 0.5693 | 0.5751 | 3.2670 | 1.0677 |
| O15230 | Laminin subunit alpha-5 | LAMA5 | 28 | 399.73 | 0.7360 | 0.5856 | 2.8928 | 1.0147 |
| P17936 | Insulin-like growth factor-binding protein 3 | IGFBP3 | 53.2 | 28.98 | 0.4682 | −0.7370 | 2.6325 | 1.4437 |
| P22692 | Insulin-like growth factor-binding protein 4 | IGFBP4 | 34.9 | 27.934 | 0.5261 | 1.8182 | 2.5945 | 1.0343 |
| P19876 | C-X-C motif chemokine 3 | CXCL3 | 34.6 | 11.342 | 0.0594 | 0.0906 | 2.5362 | 4.2024 |
| P07942 | Laminin subunit beta-1 | LAMB1 | 34.1 | 200.48 | 1.8644 | 1.3326 | 2.3580 | 0.6935 |
| P25391 | Laminin subunit alpha-1 | LAMA1 | 32.6 | 337.08 | 0.7519 | 1.2152 | 2.1027 | 0.7547 |
| P16035 | Metalloproteinase inhibitor 2 | TIMP2 | 18.6 | 24.399 | 0.7339 | −0.9250 | 2.0269 | 0.9077 |
| P24592 | Insulin-like growth factor-binding protein 6 | IGFBP6 | 15.8 | 25.322 | 1.0006 | −1.9892 | 1.5490 | 0.7319 |
| Q16270 | Insulin-like growth factor-binding protein 7 | IGFBP7 | 24.8 | 29.13 | 0.6870 | −1.0294 | 1.4286 | 0.1357 |
| P03950 | Angiogenin | ANG | 19.7 | 16.55 | 0.2687 | −0.3723 | 1.2610 | −2.0546 |
| P01033 | Metalloproteinase inhibitor 1 | TIMP1 | 52.7 | 23.171 | 0.7765 | 2.9735 | 1.2018 | 0.7942 |
| P07858 | Cathepsin B | CTSB | 36 | 37.821 | 0.6240 | 2.1588 | 1.1950 | 0.6746 |
| P02751 | Fibronectin | FN1 | 42.7 | 262.62 | 1.0938 | 1.3290 | 1.0122 | 0.7676 |
| P11047 | Laminin subunit gamma-1 | LAMC1 | 48 | 177.6 | 1.1392 | 0.6569 | 0.7737 | 0.4710 |
| P18065 | Insulin-like growth factor-binding protein 2 | IGFBP2 | 46.8 | 34.814 | 0.1856 | −0.4689 | 0.7302 | 0.4093 |
| P14174 | Macrophage migration inhibitory factor | MIF | 11.3 | 12.476 | 0.0951 | 0.2367 | 0.3994 | 0.3085 |
FIG. 7.
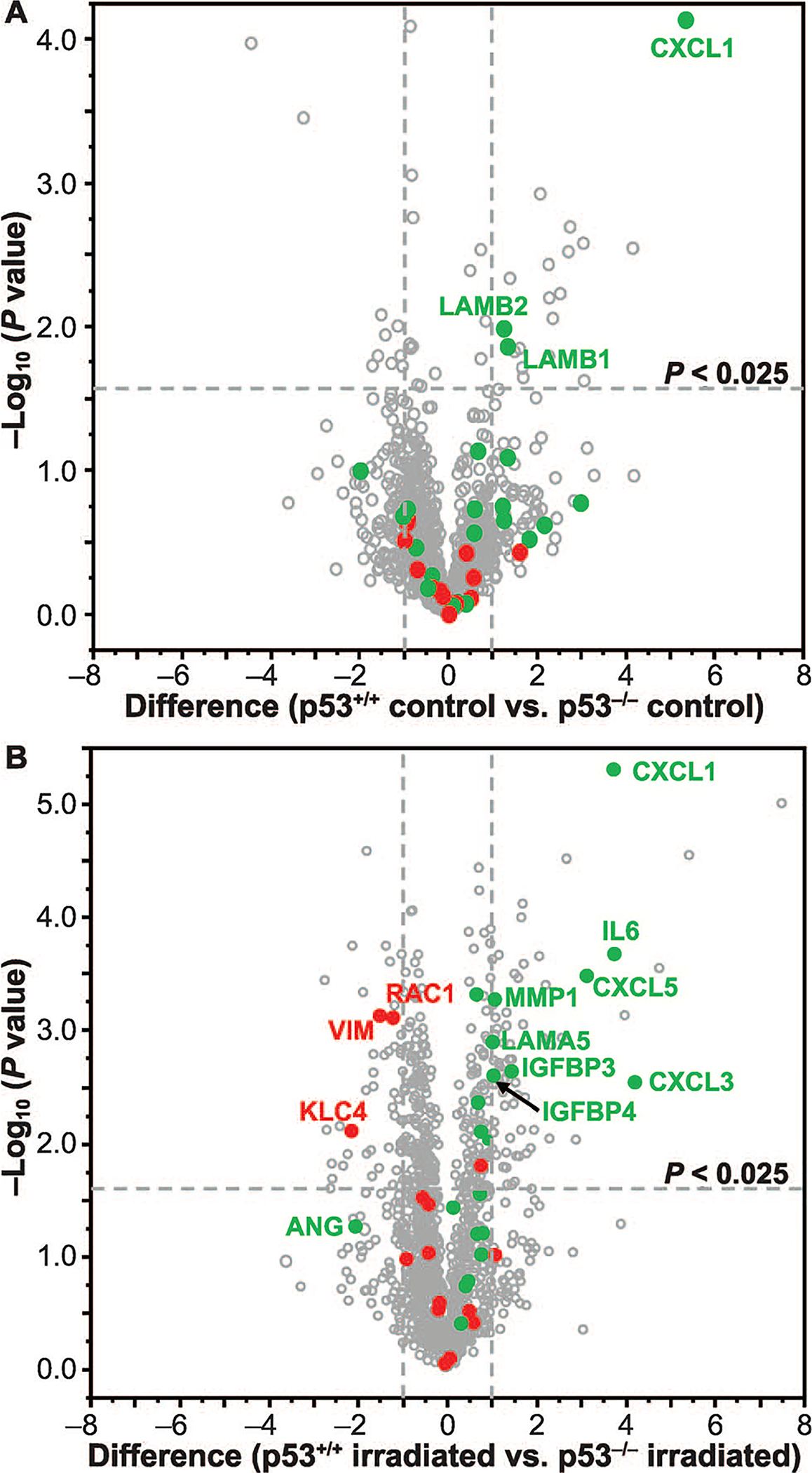
Differential protein secretion in H460wt and H460crp53 non-small cell lung cancer cells. Volcano plots of −log10 P value vs. difference in log2 LFQ intensity showing secretion of resistance-associated proteins (shown in red) and senescence-associated secretory proteins (shown in green) in nonirradiated controls (panel A) and irradiated H460 cells (panel B).
Secretion of SASP Factors is Significantly Higher in H460wt cells
Given the increased senescence in the H460wt cells, we investigated the dataset for the senescence-associated secretory phenotype (SASP) factors. Table 1 shows a list of 21 SASP factors, from Coppe et al. (66), which were identified in this study. A total of 14 proteins, representing 66.67% of identified SASP factors, were differentially regulated (P < 0.05) in the secretome of irradiated H460wt cells relative to the H460crp53 secretome (Fig. 7B and Table 1). Importantly, eight of the 14 proteins significantly upregulated in the irradiated H460wt cell secretome, including CXCL1, CXCL3, CXCL5, IL6, MMP1, LAMA5, IGFBP3 and IGFBP4 (shown in green), exhibited marked differences (≥2-fold change; P < 0.025). Additionally, LAMB1, LAMB2 and CXCL1 showed significant differences in the untreated control cells with higher levels in the H460crp53 secretome (Fig. 7A). In contrast, only ANG exhibited greater secretion (<2-fold) in irradiated H460crp53 cells compared to H460wt cells (Fig. 7B).
DISCUSSION
Ionizing radiation triggers a spectrum of responses in tumor cells including apoptosis, necrosis, autophagy and senescence (67, 68); however, the role of each of these responses in tumor cell radiation sensitivity or resistance has not been fully elucidated.
The current work was designed to evaluate the contributions of apoptosis, autophagy and senescence to radiation sensitivity in H460 non-small cell lung cancer cells that are either wild-type or deficient in p53, given that p53 status has often been considered central to the radiation response in tumor cells (69). We found that radiation sensitivity in H460 cells appears to be unrelated to the extent of apoptosis, which is low and essentially identical in both cell lines, consistent with several previously published studies (70–72). Importantly, in the context of apoptosis, the findings reported in the literature have reiterated the limited induction of apoptosis in response to radiation, suggesting it is neither the sole, nor the predominant response (23, 73).
Autophagy is an established response to cellular stress that can be considered a “first responder” to both cancer chemotherapy and radiation (16, 74, 75). There has been a tendency in the literature toward focusing on the cytoprotective function of autophagy within the context of autophagy inhibition as a potential therapeutic strategy for chemosensitization and radiosensitization. Early studies reported by the Rodemann group, as well as others, identified cytoprotective autophagy in response to radiation in a number of experimental tumor cell models (47, 76–78), such as NSCLC, breast cancer and glioblastoma. However, in addition to the cytoprotective form of autophagy, there is accumulating evidence suggesting that autophagy can contribute to or mediate drug cytotoxicity (79, 80), as well as a non-protective role, wherein autophagy inhibition has no demonstrable effect on chemosensitivity or radiosensitivity (15–19, 81), although the terminology (nonprotective autophagy) has yet to become widely adopted.
Radiation induces autophagy in both cell lines, but more extensively and earlier in the p53 wild-type cells; however, autophagy does not confer radiation resistance, since the H460wt cells demonstrated greater radiation sensitivity than that in the p53 deficient H460 cells as opposed to the lesser radiation sensitivity that might have been expected with cytoprotective autophagy. To evaluate whether the autophagy induced by radiation was cytoprotective or non-protective (or possibly cytotoxic), we inhibited autophagy both pharmacologically and genetically by exposing the H460 cells to CQ, 3-MA and bafilomycin A1 and by knockdown of the autophagy regulatory gene Atg5. Unexpectedly, only CQ treatment increased the radiosensitivity of the H460wt cells, whereas 3-MA, bafilomycin A1 and genetic autophagy inhibition showed no effect on radiosensitivity in either cell line, raising further reservations about the utilization of chloroquine as an autophagic inhibitor in the experimental discernment of the nature of autophagy. This finding supports the studies by Maycotte et al., which suggested that chemosensitization by CQ could occur independently of autophagy inhibition (53).
Our current studies as well as the work of other laboratories have demonstrated an increase in intracellular ROS generation with chloroquine treatment, an effect that is not evident upon treatment with 3-MA, in response to radiation and chemotherapy (55). The combination of CQ and 6 Gy exposure showed an increase in ROS production compared to radiation alone or in combination with 3-MA; moreover, the extent of ROS generation was greater in p53-wild-type compared to p53-null H460 cells, adding further complexity to p53-mediated effects. Additionally, other laboratories have shown chloroquine may induce apoptosis via p53-dependent pathways leading to increased apoptosis, as well as accentuating mitochondrial fragmentation and dysfunction in already damaged mitochondria, an effect evident in irradiated cells (54, 82, 83). Furthermore, when autophagy is inhibited utilizing bafilomycin A1, another late-stage autophagy inhibitor, there is no effect on radiosensitivity in H460wt cells. Overall, excluding the effect of chloroquine leads to the conclusion that the autophagy induced by radiation is nonprotective in the H460 cells, regardless of p53 status. Consequently, we confirm the existence of nonprotective autophagy, in support of previously published findings both in our laboratory and the work of others (15–18, 81, 84).
In support of the findings that suggest that autophagy does not represent a resistance mechanism in H460wt cells, MS-based secretome analysis revealed no significant enrichment of resistance-associated proteins in the H460wt cells. On the contrary, where significant differences existed in resistance-associated protein secretion between H460wt and H460crp53 cells, levels were consistently higher in H460crp53 cells, further supporting the assertion that radiation-induced autophagy in H460wt cells does not confer resistance. In a published study of radiation-resistant H460 cells by Yun et al., eight (including four novel) proteins, namely FASN, VIM, GRP78, UQCRC1, PAI-2, NOMO2, KLC4 and PLOD3, were shown to differ significantly between radiosensitive and radioresistant H460 cells (62). The current study identified six of these proteins, i.e., FASN, VIM, UQCRC1, NOMO2, KLC4 and PLOD3 (Table 1), with the results of our quantitative analysis indicating that two proteins (KLC4 and VIM), which are also associated with radioresistance in esophageal cancer, demonstrated a significant increase in the irradiated H460crp53 secretome compared to the H460wt secretome (85).
Our findings add to the complexity and uncertainty of the function of p53 in radiation sensitivity. In published studies of Hep3B2.1–7 hepatocellular cancer cells that lack p53 expression, it was found that these cells less radiosensitive than HepG2 cells with functional p53, albeit due to an attenuation in apoptosis induction (86). Similarly, Cheng et al. reported that induction of p53 expression in H1299 lung cancer cells resulted in enhanced radiosensitivity, again due to increased apoptosis induction (87). In the current work, H460 cells with functional p53 are also more radiosensitive than their p53-null counterparts; however, p53 status had no significant effect on apoptosis levels in response to radiation. Instead, our studies suggest that a primary response to radiation in both cell lines is a (transient) senescence, which may be the basis for differences in radiosensitivity as suggested by earlier work from our group relating to the involvement of p53 in chemotherapy-induced senescence (23). These conclusions concur with previous work performed in our laboratory, that cellular senescence is a fundamental response to radiotherapy in multiple glioblastoma multiforme cell lines (88).
In further support of these conclusions, studies by Efimova et al. demonstrated an increase in senescence both in vitro and in vivo in response to radiotherapy when combined with a PARP inhibitor to block repair of double-strand breaks induced by exposure to 6 Gy (73). Similar studies from our laboratory using HCT116 colon carcinoma cells showed a transient induction of senescence followed by proliferative recovery in response to radiation, which is further supported by the transient growth arrest observed for both H460 isogenic cell lines (16). Moreover, together with the results of our β-galactosidase staining and morphological observations showing evidence of increased senescence in H460wt cells, the MS-based secretome study revealed a significant upregulation of 7 out of 21 identified SASP factors in the secretome of irradiated H460wt cells, whereas only one of these proteins demonstrated increased secretion in irradiated H460crp53 compared to H460wt cells. Taken together, these results suggest that senescence, and not autophagy, may be responsible for the differential molecular signatures and radiosensitivity observed between the isogenic H460 cell lines.
Clinical trials currently in progress that putatively inhibit autophagy using chloroquine or hydroxychloroquine are based, in part, on the premise that autophagy represents a mechanism of treatment resistance (89, 90). In the current work, we used a model of radiation-induced autophagy in the isogenic H460 NSCLC cell line to compare radiation sensitivity, with the premise that tumor cells in which autophagy is cytoprotective would be expected to be significantly less sensitive to radiation than tumor cells in which the autophagy did not exhibit the cytoprotective function. However, despite modest sensitization to radiation of the p53-wild-type H460 cells with chloroquine, the radiation-induced autophagy proved to be nonprotective in both cell lines.
It should be emphasized that these conclusions focus solely on the cell autonomous effects of autophagy inhibition. The potential utility of autophagy inhibition as a therapeutic strategy becomes even more complex when one further considers the cell non-autonomous impact of autophagy inhibition (81, 91). Therefore, it is critical to emphasize, as previously stated (92), that an observation of a chemotherapeutic drug or radiation promoting autophagy in a tumor cell does not, of itself, predict the capacity of the autophagy to play a cytoprotective function and/or promote resistance; consequently, inhibition of the induced autophagy does not obligatorily represent a useful therapeutic approach for chemosensitization or radiosensitization.
These observations indicate that enhanced radiation sensitivity in cells expressing wild-type p53 is not obligatorily a consequence of differential apoptosis but may be related to the extent of senescence. However, a caveat to these findings is that it is currently not technically feasible to conclusively demonstrate that senescence is directly responsible for the observed differences in radiosensitivity since, unlike the case of autophagy, there are no pharmacological or genetic approaches available to selectively block senescence.
The current studies also indicate that radiation-induced autophagy is not necessarily cytoprotective, but may also be nonprotective; consequently, extensive autophagy does not necessarily confer radiation resistance or, conversely, contribute to radiation-induced cell killing. Of necessity also considering the possibility of additional outcomes such as mitotic catastrophe, necrosis and necroptosis, these studies indicate that the response of a tumor cell to radiation is likely to include apoptosis, autophagy and senescence; furthermore, the extent to which each of these factors contribute to radiation sensitivity or resistance in a particular malignancy cannot be predicted and is likely to vary depending on the genetic background of the tumor (or tumor cell lines) being studied.
Supplementary Material
Fig. S1. Effect of autophagy inhibition on ROS generation in response to radiation in H460wt and H460crp53 cells.
Fig. S2. Influence of Bafilomycin A1 (Baf A1) on radiation sensitivity in H460wt cells.
Fig. S3. Radiation induces G2/M phase arrest in H460wt and H460crp53 cells.
Fig. S4. Functional and statistical analysis of H460 cell secretome.
ACKNOWLEDGMENTS
Work in Dr. Gewirtz’s laboratory was supported by the Office of the Assistant Secretary of Defense for Health Affairs through the Breast Cancer Research Program (grant no. W81XWH-14-1-0088 to DAG) and a Massey Center Support Grant (no. P30 CA016059). Santiago Lima was supported by the National Institutes of Health (grant no. K22 CA187314). We gratefully acknowledge financial support (JX) from the China Scholarship Council. Services and products in support of the research project were generated by the VCU Massey Cancer Center Flow Cytometry Shared Resource, supported in part, with funding from NIHNCI Cancer Center Support Grant no. P30 CA016059.
Footnotes
Editor’s note. The online version of this article (DOI: 10.1667/RR15099.1) contains supplementary information that is available to all authorized users.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018; 68:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Lin JJ, Shaw AT. Resisting resistance: targeted therapies in lung cancer. Trends Cancer 2016; 2:350–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim ES. Chemotherapy resistance in lung cancer. Adv Exp Med Biol 2016; 893:189–209. [DOI] [PubMed] [Google Scholar]
- 4.Maycotte P, Thorburn A. Autophagy and cancer therapy. Cancer Biol Ther 2011; 11:127–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amaravadi RK, Thompson CB. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin Cancer Res 2007; 13:7271–9. [DOI] [PubMed] [Google Scholar]
- 6.Gewirtz DA. The autophagic response to radiation: relevance for radiation sensitization in cancer therapy. Radiat Res 2014; 182:363–7. [DOI] [PubMed] [Google Scholar]
- 7.Hu YL, DeLay M, Jahangiri A, Molinaro AM, Rose SD, Carbonell WS, et al. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res 2012; 72:1773–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sato K, Tsuchihara K, Fujii S, Sugiyama M, Goya T, Atomi Y, et al. Autophagy is activated in colorectal cancer cells and contributes to the tolerance to nutrient deprivation. Cancer Res 2007; 67:9677–84. [DOI] [PubMed] [Google Scholar]
- 9.Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ 2005; 12 Suppl 2:1509–18. [DOI] [PubMed] [Google Scholar]
- 10.Mazure NM, Pouyssegur J. Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol 2010; 22:177–80. [DOI] [PubMed] [Google Scholar]
- 11.Gewirtz DA. The four faces of autophagy: implications for cancer therapy. Cancer Res 2014; 74:647–51. [DOI] [PubMed] [Google Scholar]
- 12.Zhou Y, Sun K, Ma Y, Yang H, Zhang Y, Kong X, et al. Autophagy inhibits chemotherapy-induced apoptosis through downregulating Bad and Bim in hepatocellular carcinoma cells. Sci Rep 2014; 4:5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007; 8:741–52. [DOI] [PubMed] [Google Scholar]
- 14.Bristol ML, Emery SM, Maycotte P, Thorburn A, Chakradeo S, Gewirtz DA. Autophagy inhibition for chemosensitization and radiosensitization in cancer: do the preclinical data support this therapeutic strategy? J Pharmacol Exp Ther 2013; 344:544–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chakradeo S, Sharma K, Alhaddad A, Bakhshwin D, Le N, Harada H, et al. Yet another function of p53–the switch that determines whether radiation-induced autophagy will be cytoprotective or nonprotective: implications for autophagy inhibition as a therapeutic strategy. Mol Pharmacol 2015; 87:803–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alotaibi M, Sharma K, Saleh T, Povirk LF, Hendrickson EA, Gewirtz DA. Radiosensitization by PARP inhibition in dna repair proficient and deficient tumor cells: proliferative recovery in senescent cells. Radiat Res 2016; 185:229–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saleh T, Cuttino L, Gewirtz DA. Autophagy is not uniformly cytoprotective: a personalized medicine approach for autophagy inhibition as a therapeutic strategy in non-small cell lung cancer. Biochim Biophys Acta 2016; 1860:2130–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eng CH, Wang Z, Tkach D, Toral-Barza L, Ugwonali S, Liu S, et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc Natl Acad Sci U S A 2016; 113:182–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gewirtz DA. The challenge of developing autophagy inhibition as a therapeutic strategy. Cancer Res 2016; 76:5610–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ko A, Kanehisa A, Martins I, Senovilla L, Chargari C, Dugue D, et al. Autophagy inhibition radiosensitizes in vitro, yet reduces radioresponses in vivo due to deficient immunogenic signalling. Cell Death Differ 2014; 21:92–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levy JM, Thorburn A. Targeting autophagy during cancer therapy to improve clinical outcomes. Pharmacol Ther 2011; 131:130–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suzuki K, Mori I, Nakayama Y, Miyakoda M, Kodama S, Watanabe M. Radiation-induced senescence-like growth arrest requires TP53 function but not telomere shortening. Radiat Res 2001; 155:248–53. [DOI] [PubMed] [Google Scholar]
- 23.Jones KR, Elmore LW, Jackson-Cook C, Demasters G, Povirk LF, Holt SE, et al. p53-Dependent accelerated senescence induced by ionizing radiation in breast tumour cells. Int J Radiat Biol 2005; 81:445–58. [DOI] [PubMed] [Google Scholar]
- 24.Gewirtz DA. Autophagy and senescence in cancer therapy. J Cell Physiol 2014; 229:6–9. [DOI] [PubMed] [Google Scholar]
- 25.Huang YH, Yang PM, Chuah QY, Lee YJ, Hsieh YF, Peng CW, et al. Autophagy promotes radiation-induced senescence but inhibits bystander effects in human breast cancer cells. Autophagy 2014; 10:1212–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gewirtz DA. Autophagy and senescence: a partnership in search of definition. Autophagy 2013; 9:808–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goehe RW, Di X, Sharma K, Bristol ML, Henderson SC, Valerie K, et al. The autophagy-senescence connection in chemotherapy: must tumor cells (self) eat before they sleep? J Pharmacol Exp Ther 2012; 343:763–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan C, Kumar C, Bohl S, Klingmueller U, Mann M. Comparative proteomic phenotyping of cell lines and primary cells to assess preservation of cell type-specific functions. Mol Cell Proteomics 2009; 8:443–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Becker L, Liu NC, Averill MM, Yuan W, Pamir N, Peng Y, et al. Unique proteomic signatures distinguish macrophages and dendritic cells. PloS One 2012; 7:e33297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flor AC, Wolfgeher D, Wu D, Kron SJ. A signature of enhanced lipid metabolism, lipid peroxidation and aldehyde stress in therapy-induced senescence. Cell Death Discov 2017; 3:17075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perez-Soler R Individualized therapy in non-small-cell lung cancer: future versus current clinical practice. Oncogene 2009; 28:S38–45. [DOI] [PubMed] [Google Scholar]
- 32.Kraya AA, Piao S, Xu X, Zhang G, Herlyn M, Gimotty P, et al. Identification of secreted proteins that reflect autophagy dynamics within tumor cells. Autophagy 2015; 11:60–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chenau J, Michelland S, de Fraipont F, Josserand V, Coll JL, Favrot MC, et al. The cell line secretome, a suitable tool for investigating proteins released in vivo by tumors: application to the study of p53-modulated proteins secreted in lung cancer cells. J Proteome Res 2009; 8:4579–91. [DOI] [PubMed] [Google Scholar]
- 34.Hu R, Huffman KE, Chu M, Zhang Y, Minna JD, Yu Y. Quantitative secretomic analysis identifies extracellular protein factors that modulate the metastatic phenotype of non-small cell lung cancer. J Proteome Res 2016; 15:477–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO 2011; 30:4701–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lima S, Takabe K, Newton J, Saurabh K, Young MM, Leopoldino AM, et al. TP53 is required for BECN1- and ATG5-dependent cell death induced by sphingosine kinase 1 inhibition. Autophagy 2018:1–16; Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma K, Goehe RW, Di X, Hicks MA 2nd, Torti SV, Torti FM, et al. A novel cytostatic form of autophagy in sensitization of non-small cell lung cancer cells to radiation by vitamin D and the vitamin D analog, EB 1089. Autophagy 2014; 10:2346–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 1995; 92:9363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc 2009; 4:1798–806. [DOI] [PubMed] [Google Scholar]
- 40.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44–57. [DOI] [PubMed] [Google Scholar]
- 42.Cuddihy AR, Bristow RG. The p53 protein family and radiation sensitivity: Yes or no? Cancer Metastasis Rev 2004; 23:237–57. [DOI] [PubMed] [Google Scholar]
- 43.Gudkov AV, Komarova EA. The role of p53 in determining sensitivity to radiotherapy. Nature Rev Cancer 2003; 3:117–29. [DOI] [PubMed] [Google Scholar]
- 44.Cerniglia GJ, Karar J, Tyagi S, Christofidou-Solomidou M, Rengan R, Koumenis C, et al. Inhibition of autophagy as a strategy to augment radiosensitization by the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVPBEZ235. Mol Pharmacol 2012; 82:1230–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Y, Li X, Guo L, Wu X, He C, Zhang S, et al. Combining radiation with autophagy inhibition enhances suppression of tumor growth and angiogenesis in esophageal cancer. Mol Med Rep 2015; 12:1645–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu Z, Yan Y, Zeng S, Qian L, Dai S, Xiao L, et al. Reducing autophagy and inducing G1 phase arrest by aloperine enhances radio-sensitivity in lung cancer cells. Oncol Rep 2017. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 47.Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res 2008; 68:1485–94. [DOI] [PubMed] [Google Scholar]
- 48.Abedin MJ, Wang D, McDonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ 2007; 14:500–10. [DOI] [PubMed] [Google Scholar]
- 49.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12:1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sasaki K, Tsuno NH, Sunami E, Tsurita G, Kawai K, Okaji Y, et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer 2010; 10:370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ 2008; 15:171–82. [DOI] [PubMed] [Google Scholar]
- 52.Schonewolf CA, Mehta M, Schiff D, Wu H, Haffty BG, Karantza V, et al. Autophagy inhibition by chloroquine sensitizes HT-29 colorectal cancer cells to concurrent chemoradiation. World J Gastrointest Oncol 2014; 6:74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maycotte P, Aryal S, Cummings CT, Thorburn J, Morgan MJ, Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy 2012; 8:200–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qu X, Sheng J, Shen L, Su J, Xu Y, Xie Q, et al. Autophagy inhibitor chloroquine increases sensitivity to cisplatin in QBC939 cholangiocarcinoma cells by mitochondrial ROS. PloS One 2017; 12:e0173712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen P, Luo X, Nie P, Wu B, Xu W, Shi X, et al. CQ synergistically sensitizes human colorectal cancer cells to SN-38/CPT-11 through lysosomal and mitochondrial apoptotic pathway via p53-ROS cross-talk. Free Radic Biol Med 2017; 104:280–97. [DOI] [PubMed] [Google Scholar]
- 56.Palmeira dos Santos C, Pereira GJ, Barbosa CM, Jurkiewicz A, Smaili SS, Bincoletto C. Comparative study of autophagy inhibition by 3MA and CQ on Cytarabine induced death of leukaemia cells. J Cancer Res Clin Oncol 2014; 140:909–20. [DOI] [PubMed] [Google Scholar]
- 57.Mauvezin C, Neufeld TP. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 2015; 11:1437–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Frame FM, Savoie H, Bryden F, Giuntini F, Mann VM, Simms MS, et al. Mechanisms of growth inhibition of primary prostate epithelial cells following gamma irradiation or photodynamic therapy include senescence, necrosis, and autophagy, but not apoptosis. Cancer Med 2016; 5:61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Widel M, Lalik A, Krzywon A, Poleszczuk J, Fujarewicz K, Rzeszowska-Wolny J. The different radiation response and radiation-induced bystander effects in colorectal carcinoma cells differing in p53 status. Mutat Res 2015;778:61–70. [DOI] [PubMed] [Google Scholar]
- 60.Fang L, Igarashi M, Leung J, Sugrue MM, Lee SW, Aaronson SA. p21Waf1/Cip1/Sdi1 induces permanent growth arrest with markers of replicative senescence in human tumor cells lacking functional p53. Oncogene 1999; 18:2789–97. [DOI] [PubMed] [Google Scholar]
- 61.Feng XP, Yi H, Li MY, Li XH, Yi B, Zhang PF, et al. Identification of biomarkers for predicting nasopharyngeal carcinoma response to radiotherapy by proteomics. Cancer Res 2010; 70:3450–62. [DOI] [PubMed] [Google Scholar]
- 62.Yun HS, Baek JH, Yim JH, Um HD, Park JK, Song JY, et al. Radiotherapy diagnostic biomarkers in radioresistant human H460 lung cancer stem-like cells. Cancer Biol Ther 2016; 17:208–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Allal AS, Kahne T, Reverdin AK, Lippert H, Schlegel W, Reymond MA. Radioresistance-related proteins in rectal cancer. Proteomics 2004; 4:2261–9. [DOI] [PubMed] [Google Scholar]
- 64.Lacombe J, Azria D, Mange A, Solassol J. Proteomic approaches to identify biomarkers predictive of radiotherapy outcomes. Expert Rev Proteomics 2013; 10:33–42. [DOI] [PubMed] [Google Scholar]
- 65.Skvortsov S, Debbage P, Cho WC, Lukas P, Skvortsova I. Putative biomarkers and therapeutic targets associated with radiation resistance. Expert Rev Proteomics 2014; 11:207–14. [DOI] [PubMed] [Google Scholar]
- 66.Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 2010; 5:99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Okada H, Mak TW. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat Rev Cancer 2004; 4:592–603. [DOI] [PubMed] [Google Scholar]
- 68.Maier P, Hartmann L, Wenz F, Herskind C. Cellular pathways in response to ionizing radiation and their targetability for tumor radiosensitization. Int J Mol Sci 2016; 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fei P, El-Deiry WS. P53 and radiation responses. Oncogene 2003; 22:5774–83. [DOI] [PubMed] [Google Scholar]
- 70.Wang F, Tang J, Li P, Si S, Yu H, Yang X, et al. Chloroquine enhances the radiosensitivity of bladder cancer cells by inhibiting autophagy and activating apoptosis. Cell Physiol Biochem 2018; 45:54–66. [DOI] [PubMed] [Google Scholar]
- 71.Luo H, Yount C, Lang H, Yang A, Riemer EC, Lyons K, et al. Activation of p53 with Nutlin-3a radiosensitizes lung cancer cells via enhancing radiation-induced premature senescence. Lung Cancer 2013; 81:167–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shafaee Z, Schmidt H, Du W, Posner M, Weichselbaum R. Cyclopamine increases the cytotoxic effects of paclitaxel and radiation but not cisplatin and gemcitabine in Hedgehog expressing pancreatic cancer cells. Cancer Chemother Pharmacol 2006; 58:765–70. [DOI] [PubMed] [Google Scholar]
- 73.Efimova EV, Mauceri HJ, Golden DW, Labay E, Bindokas VP, Darga TE, et al. Poly(ADP-ribose) polymerase inhibitor induces accelerated senescence in irradiated breast cancer cells and tumors. Cancer Res 2010; 70:6277–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Garbar C, Mascaux C, Giustiniani J, Merrouche Y, Bensussan A. Chemotherapy treatment induces an increase of autophagy in the luminal breast cancer cell MCF7, but not in the triple-negative MDA-MB231. Sci Rep 2017; 7:7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E, et al. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res 2001; 61:439–44. [PubMed] [Google Scholar]
- 76.Ito H, Daido S, Kanzawa T, Kondo S, Kondo Y. Radiation-induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. International J Oncology 2005; 26:1401–10. [PubMed] [Google Scholar]
- 77.Lomonaco SL, Finniss S, Xiang C, Decarvalho A, Umansky F, Kalkanis SN, et al. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int J Cancer 2009; 125:717–22. [DOI] [PubMed] [Google Scholar]
- 78.Chaachouay H, Ohneseit P, Toulany M, Kehlbach R, Multhoff G, Rodemann HP. Autophagy contributes to resistance of tumor cells to ionizing radiation. Radiother Oncol 2011; 99:287–92. [DOI] [PubMed] [Google Scholar]
- 79.Shimizu S, Yoshida T, Tsujioka M, Arakawa S. Autophagic cell death and cancer. Int J Mol Sci 2014; 15:3145–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sharma K, Le N, Alotaibi M, Gewirtz DA. Cytotoxic autophagy in cancer therapy. Int J Mol Sci 2014; 15:10034–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011; 334:1573–7. [DOI] [PubMed] [Google Scholar]
- 82.Chaanine AH, Gordon RE, Nonnenmacher M, Kohlbrenner E, Benard L, Hajjar RJ. High-dose chloroquine is metabolically cardiotoxic by inducing lysosomes and mitochondria dysfunction in a rat model of pressure overload hypertrophy. Physiol Rep 2015; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim EL, Wustenberg R, Rubsam A, Schmitz-Salue C, Warnecke G, Bucker EM, et al. Chloroquine activates the p53 pathway and induces apoptosis in human glioma cells. Neuro Oncol 2010; 12:389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gewirtz DA. An autophagic switch in the response of tumor cells to radiation and chemotherapy. Biochem Pharmacol 2014; 90:208–11. [DOI] [PubMed] [Google Scholar]
- 85.Su H, Jin X, Zhang X, Zhao L, Lin B, Li L, et al. FH535 increases the radiosensitivity and reverses epithelial-to-mesenchymal transition of radioresistant esophageal cancer cell line KYSE-150R. J Transl Med 2015; 13:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gomes AR, Abrantes AM, Brito AF, Laranjo M, Casalta-Lopes JE, Goncalves AC, et al. Influence of P53 on the radiotherapy response of hepatocellular carcinoma. Clin Mol Hepatol 2015; 21:257–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cheng G, Kong D, Hou X, Liang B, He M, Liang N, et al. The tumor suppressor, p53, contributes to radiosensitivity of lung cancer cells by regulating autophagy and apoptosis. Cancer Biother Radiopharm 2013; 28:153–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Quick QA, Gewirtz DA. An accelerated senescence response to radiation in wild-type p53 glioblastoma multiforme cells. J Neurosurg 2006; 105:111–8. [DOI] [PubMed] [Google Scholar]
- 89.Rangwala R, Leone R, Chang YC, Fecher LA, Schuchter LM, Kramer A, et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014; 10:1369–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014; 10:1359–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gewirtz DA, Tyutyunyk-Massey L, Landry JW. The potentially conflicting cell autonomous and cell non-autonomous functions of autophagy in mediating tumor response to cancer therapy. Biochem Pharmacol 2018; 153:46–50. [DOI] [PubMed] [Google Scholar]
- 92.Gewirtz DA. When cytoprotective autophagy isn’t… and even when it is. Autophagy 2014; 10:391–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Effect of autophagy inhibition on ROS generation in response to radiation in H460wt and H460crp53 cells.
Fig. S2. Influence of Bafilomycin A1 (Baf A1) on radiation sensitivity in H460wt cells.
Fig. S3. Radiation induces G2/M phase arrest in H460wt and H460crp53 cells.
Fig. S4. Functional and statistical analysis of H460 cell secretome.