

Abstract
Type 1 and advanced type 2 diabetes treatment involves daily injections or continuous infusion of exogenous insulin aimed at regulating blood glucose levels in the normoglycemic range. However, current options for insulin therapy are limited by the risk of hypoglycemia and are associated with suboptimal glycemic control outcomes. Therefore, a range of glucose-responsive components that can undergo changes in conformation or show alterations in intermolecular binding capability in response to glucose stimulation have been studied for ultimate integration into closed-loop insulin delivery or “smart insulin” systems. Here, we present an overview of the evolution and recent progress in the development of molecular approaches for glucose-responsive insulin delivery systems, a rapidly growing subfield of precision medicine. We focus on three central glucose-responsive moieties, including glucose oxidase, phenylboronic acid, and glucose-binding molecules. Future opportunities and challenges regarding translation are also discussed.
Keywords: drug delivery, diabetes treatment, insulin delivery, glucose-responsive, device
Graphical Abstract
The evolution and recent progress of the mechanism, formulation and delivery route for glucose-responsive insulin delivery are summarized. The perspectives and challenges for clinical translation are also discussed. This review aims to provide fundamentals for designing desirable and applicable glucose-responsive insulin delivery systems in the future.

In healthy individuals, blood glucose is tightly controlled by endogenous insulin secreted by β-cells, which can sense the fluctuation of blood glucose level and release insulin accordingly. Increasing blood glucose level results in increased insulin secretion from the β-cells, while low blood glucose level inhibits further insulin secretion.[1] Due to the short half-life of insulin,[2] the amount of insulin in the blood is therefore accurately and synchronously controlled. However, for insulin-dependent diabetes, including type 1 and advanced type 2 diabetes, daily administration of exogenous insulin remains central to treatment.[3] In this setting, the primary challenge of insulin therapy involves the precise administration of adequate insulin doses over the course of the day, including at meal times and overnight to achieve tight glucose control while simultaneously avoiding the episodes of severe hypoglycemia.[4] Unfortunately, due to the risk of insulin-induced hypoglycemia, the potential of insulin therapy for glycemic control is not realized in many individuals with diabetes.
Therefore, a therapeutic system that can mimic the endogenous “smart” and glucose-dependent insulin release profile is critical for achieving satisfactory diabetes management while avoiding hypoglycemia. Such a system would essentially offer precision medicine at the individual level by offering a treatment approach that would adapt to an individuals’ glucose level in real time, thereby achieving high treatment efficacy while reducing undesirable side effects.[5] The direct integration of glucose-responsive components into insulin molecules or insulin delivery systems offers one solution to these challenges by mimicking the dynamic and bio-responsive insulin secretion from pancreatic β-cells.[6] Therefore, multidisciplinary approaches have been exploited to render insulin glucose-responsive by adjusting its pharmacokinetics and pharmacodynamics.[5b, 7] To date, the predominant glucose-sensing moieties for constructing chemically-synthetic glucose-responsive insulin delivery systems utilize glucose oxidase (GOx),[8] phenylboronic acid (PBA),[9] and glucose-binding molecules.[10] With each of these molecular tools, researchers have created numerous modified insulin molecules as well as delivery formulations and devices, including hydrogels (bulk or micro/nanosized), membranes, nanovesicles, microneedle array patches, liposomes, and cells (Figure 1).[7j, 11] The ability of these formulations to release insulin is triggered by glucose-induced binding-capability change, swelling/contraction, dissolution, pore-size alternation, and degradation. However, the construction of smart insulin systems capable of blood glucose regulation with the avoidance of hypoglycemia remains elusive.[12] It is likely that such systems will rely on the future development of novel glucose-sensing moieties and/or integration with a suitable device.[13] In this perspective, we review the evolution of main strategies in this field of research focusing on the significant points of recent progress.
Figure 1.

Schematic of the mechanism, formulation, and delivery route for glucose-responsive insulin delivery.
1. Glucose oxidase-based systems
There is a large family of glucose-responsive components that are triggered to release insulin by changes in the physiological environment signaled via products of the glucose-oxidation reaction catalyzed by GOx. GOx is a flavin-containing glycoprotein that catalyzes glucose to produce hydrogen peroxide (H2O2) and gluconic acid in the presence of oxygen, as shown in Figure 1.[14] Due to its high selectivity towards glucose, this catalytic enzyme is predominately utilized in glucose-sensing systems that respond to the alteration of local pH, H2O2 concentrations, and O2 levels induced by GOx-catalyzed oxidation of glucose.
1.1. pH-responsive systems
The decrease in pH associated with the conversion of glucose into gluconic acid has been widely exploited to trigger conformational or structural changes for subsequent controlled drug release.[15] Examples of such strategies include the pH-triggered swelling/contraction of hydrogels and membranes,[16] degradation of hydrogels or particles,[17] “on-off” of membranes or particles with “gate”,[18] and biomimetic membrane fusion[8d]. Each approach is discussed in greater detail below.
1.1.1. Swelling/contraction of gels and membranes
pH-sensitive hydrogels are usually categorized as acidic hydrogels or basic hydrogels, both of which undergo a significant volume change in response to pH fluctuations in the local environment.[19] Basic hydrogels are ionized and hence swell at lower pH levels, while acidic hydrogels lose a charge and thus contract in the same conditions.[20] Multiple research teams have leveraged these relationships to construct insulin delivery systems and release encapsulated insulin in a glucose concentration-dependent manner.
Ishihara et al. immobilized GOx onto a crosslinked polymeric membrane incorporated with polymer prepared from N,N-diethylaminoethyl methacrylate and 2-hydroxypropyl methacrylate.[8c] In this study, higher glucose concentrations increased the permeability of insulin and promoted its subsequent release. Then, hydroxyethyl acrylate, N,N-diethylaminoethyl methacrylate and 4-trimethylsilylstyrene were co-polymerized and used to prepare a polymer capsule for loading both insulin and GOx.[21] Insulin release rate was positively correlated with glucose concentrations, and the glucose-triggered swelling and contraction of the gel were shown to be reversible. Meanwhile, Horbett and co-workers reported insulin delivery systems consisting of immobilized GOx in pH-responsive polymeric membranes and hydrogels.[22] Peppas and co-workers also developed GOx-containing poly(methacrylic acid-g-ethylene glycol) hydrogels with glucose sensitivity.[8a, 23] In physiological pH ranges, the hydrogel swelled to approximately 20 times compared to that at the dry state. Upon exposure to glucose, the pH of the solution rapidly decreased, causing the hydrogel to collapse and forcing out insulin solution. Gu et al. prepared a glucose-responsive microgel composed of an acid-responsive chitosan matrix, GOx/catalase (CAT) nanocapsules, and recombinant human insulin. In vivo results demonstrated prolonged normoglycemia in streptozotocin (STZ)-induced type 1 diabetic mice as compared with native insulin.[24] The inhibitory effect of H2O2 on the reaction was overcome by co-immobilization of CAT with GOx to convert H2O2 to O2, which increased the availability of the reactants and enhanced the swelling rate.[25] An alternate approach utilized MnO2[26] or cerium oxide nanoparticles (CeNPs) (Figure 2a–b)[27] to compensate for oxygen consumption by glucose oxidation. CeNPs exhibited enhanced thermo-stability compared with CAT, as well as appreciable H2O2-scavenging ability (Figure 2c–d). This hybrid membrane was integrated into an implantable device for glucose-sensing and controlled insulin release (Figure 2e–g).[28] Following the development of glucose-sensitive swelling hydrogels, many studies were conducted to optimize self-regulated glucose-sensitive systems combining GOx with pH-responsive components, including membranes[26, 28–29] and hydrogels.[30] Recently, Chu et al. provided an example of combining techniques mentioned above and fabricated a wireless, compact implantable microdevice for closed-loop delivery of insulin in diabetic rats.[31] This device demonstrated the ability to regulate blood glucose levels under in vivo conditions.
Figure 2.
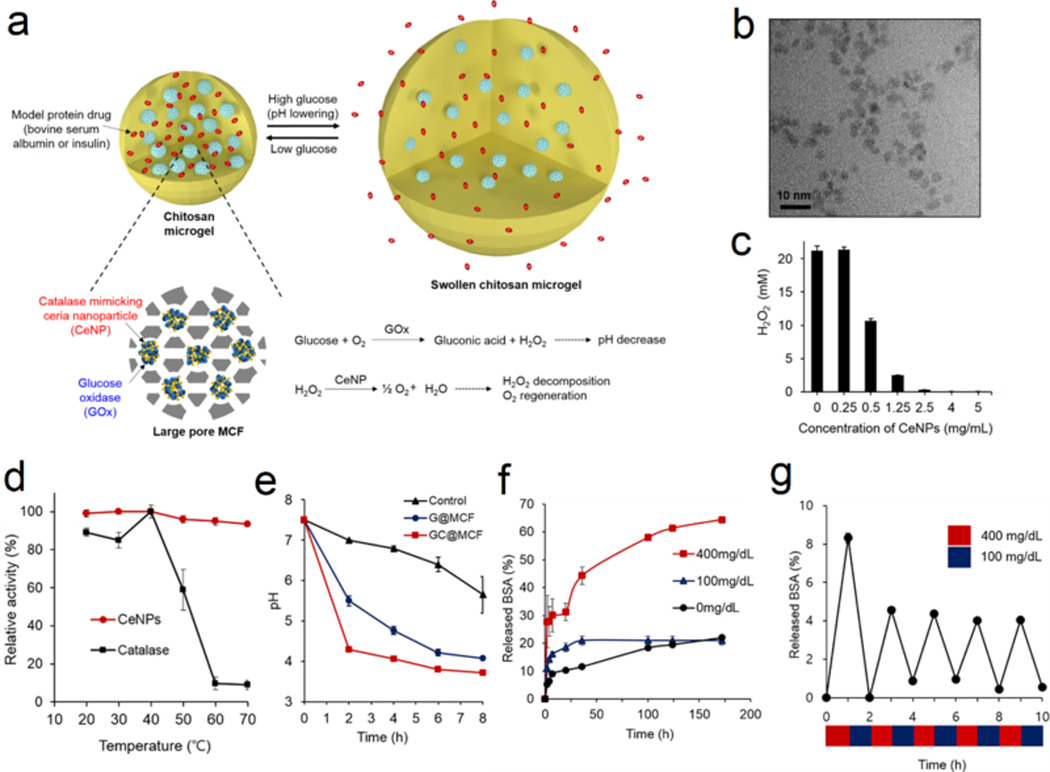
Glucose-responsive platforms integrated with H2O2-scavenging CeNPs for insulin delivery. (a) Schematic of the glucose-responsive chitosan microgels loaded with GOx and CeNPs. (b) TEM images of CeNPs. (c) H2O2 consuming assay via CeNPs incubation. (d) The enhanced stability of CeNPs compared with catalase. (e) Change of pH of different solution over time in 400 mg/mL glucose solution. (f) Glucose-triggered BSA release at different glucose concentrations. (g) Pulsatile BSA release from microgel via alternating glucose concentration between 100 and 400 mg/dL. All data with error bars were presented as mean ± SD (n=3). Reprinted with permission.[27] Copyright 2017, American Chemical Society.
1.1.2. Degradation of hydrogels or particles
Another strategy of achieving pH-responsive release is to encapsulate insulin inside an acid-disintegrable formulation, such as hydrogels, liposomes, and polymeric vesicles. Li et al. developed a pH-responsive peptide and used it to form injectable glucose-responsive delivery hydrogel through encapsulation of GOx, CAT, and insulin.[17a] With the same acidification mechanism, Lim et al. reported the use of pH-sensitive biomimetic peptide coacervate droplets as an insulin reservoir. However, these systems suffer from premature leakage of insulin in the absence of glucose, an issue that requires further optimization.[32]
Compared with bulk hydrogels, microgels or polymeric vesicles generally exhibit an elevated response rate to an external stimulus because of their decreased size and increased specific surface area.[33] Wu et al. fabricated a positively-charged acid-disintegrable microgel with covalently-immobilized GOx and CAT, in which insulin was encapsulated through electrostatic interactions.[34] The acidification triggered by the GOx/CAT cascade enzymatic reactions cleaved the crosslinking moieties and reduced the negative charge of the insulin, leading to insulin release at high glucose levels. Gu et al. prepared nanoparticles coated with chitosan to be positively-charged or alginate to be negatively-charged. The mixing of these two oppositely-charged particles formed an injectable hydrogel (Figure 3a–c).[17b] A dramatic difference in insulin release rate was observed when the gel was exposed to 100 and 400 mg/dL glucose solutions (Figure 3d). In vivo studies showed that type 1 diabetic mice treated with this gel had normoglycemia for more than 10 days after a single injection (Figure 3e).
Figure 3.
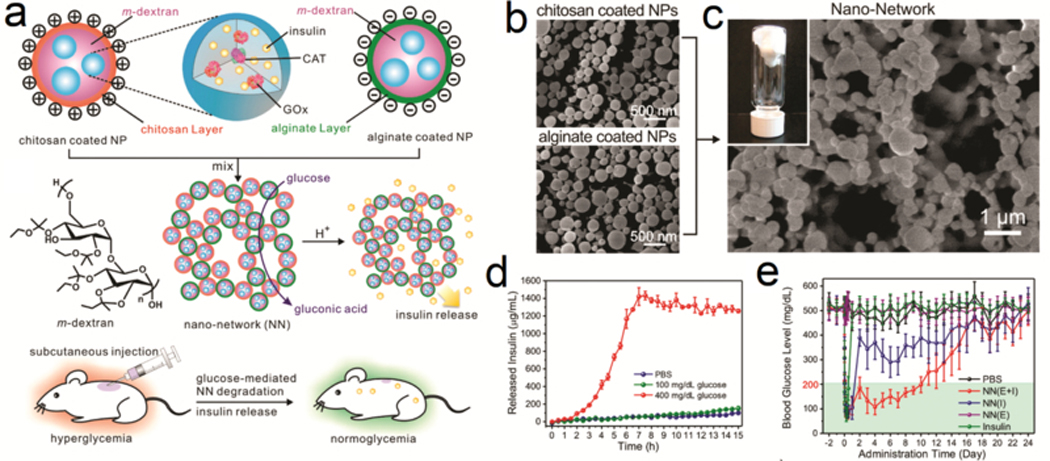
Glucose-responsive microgel network. (a) Schematic of the preparation of microgel-network via mixing positively-charged chitosan-coated NPs and negatively-charged alginate NPs. (b-c) Representative SEM images of NPs and the nano-network. Inset was the representative image of gel-like nano-network. (d) Glucose-concentration dependent insulin release from the gel. Data were presented as mean ± SD (n=2). (e) The blood glucose regulation performance of the nano-network in type 1 diabetic mouse model. The data were presented as mean ± SD (n=8). Reprinted with permission.[17b] Copyright 2013, American Chemical Society.
Compared to microgels, polymeric vesicles have an even smaller size and can be readily prepared from amphiphilic copolymers. Tai et al. reported an acid-sensitive amphiphilic polymer poly(ethylene glycol)-block-poly(Ser-Ketal) prepared from a water-soluble poly(ethylene glycol)-block-poly(serine).[35] GOx/CAT/insulin was co-loaded into these polymeric vesicles in a single step. In the presence of glucose, the conversion of glucose to gluconic acid led to the hydrolysis of the insulin-encapsulated system and converted the copolymer from amphiphilic to hydrophilic, therefore destabilizing the vesicle structure and promoting insulin release.[35]
While both microgel and polymeric vesicles are prepared from synthetic materials, liposomes have the distinct advantage of high biocompatibility and biodegradability. Kim et al. reported the utilization of pH-sensitive liposomes for in vitro glucose-responsive insulin delivery.[36] This was further evaluated in in vivo studies by Jain et al..[37] Compared to a non-glucose responsive formulation, the glucose-responsive formulation showed an enhanced response rate to reduce blood glucose levels back to normal range after a glucose challenge. Xia et al. used GOx-modified erythrocytes as an insulin carrier (designated GOx-INS-ERs).[38] A single injection of this formulation resulted in normoglycemia in diabetic rats within one hour. In addition, normoglycemia was maintained normally for up to 9 days.
In recent years, metal-organic frameworks (MOFs) have emerged as a novel platform for drug delivery.[39] Among the large family of MOFs, zeolitic imidazolate frameworks-8 (ZIF-8), prepared from Zn2+ ions and 2-methylimidazolate ligands as bridging units, has become popular in biomedical applications due to high biocompatibility and degradability in acidic conditions. Willner and co-workers validated ZIF-8 for GOx-based insulin delivery strategy (Figure 4a–c).[40] With the addition of glucose (50 mM), an obvious morphology change of these enzyme-loaded nanosized MOFs was observed via scanning microscopy within one hour (Figure 4c), which led to a glucose-triggered insulin release (Figure 4d–e). In addition, insulin release is only observed in a glucose solution, indicating its tendency to selectively release insulin in the presence of glucose (Figure 4f).
Figure 4.

(a) Schematic of the ZIF-8 NMOFs loaded with GOx and insulin for pH-triggered insulin release. (b) Confocal microscopy image of NMOFs loaded with FITC-labeled insulin (green, I) and coumarin-modified GOx (blue, II). The brightfield image (III) and the overlap of (I, II, and III) were also presented (IV). (c) Representative SEM image of original NMOFs loaded with insulin and GOx (I). These NMOFs were subsequently treated with glucose (50 mM) for one hour (II) or PBS for two days (III). (d) Representative fluorescence spectra of FITC-insulin solution when the NMOFs were exposed to a series of glucose concentrations (a to e, 0, 1, 5, 10, and 50 mM). (e) FITC-insulin release from NMOFs at various glucose concentrations (a to e, 0, 5, 10, 20 and 50 mM). (f) The fluorescence spectra of FITC-insulin in solution after NMOFs were treated with glucose (50 mM, a), galactose (50 mM, b), β-lactose (50 mM, c), sucrose (50 mM, d), and a pure buffer solution (e). Reprinted with permission.[40] Copyright 2018, American Chemical Society.
1.1.3. “On-off” of membranes or particles with “gate”
Ensuring the stability of drug molecules, protecting them from digestion, and spatiotemporally controlling the released dose of a drug are necessary considerations in the design of high-efficacy drug delivery systems with minimal side effects.[41] Since the 1980s, microcapsules with the capacity to respond to external stimuli have been developed widely.[42] Such smart delivery platforms usually share a similar structure, which includes an inner core and a stimuli-responsive outer shell. The former provides a protected space for drug loading, while the latter can be designed as the “gate” to control drug release. Chu et al. reported a microcapsule that could reversibly respond to glucose.[18b] They utilized the plasma-grafted poly(acrylic acid) as the pH-responsive gates docked in the membrane pores, where the immobilized GOx enzymes acted as glucose sensors and catalyzer that induced pH changes. Since the binding of PBA to glucose is pH dependent, Wang and co-workers utilized this “gating” strategy for fabricating an ultrasound-assisted glucose-responsive insulin release device.[43] Aznar et al. presented mesoporous silica nanoparticles gated with enzymes.[18a, 44] The mesoporous material was capped with an active cyclodextrin-modified-glucose oxidase (CD-GOx) through an inclusion complex formed between the cyclodextrins and the propylbenzimidazole group on the solid support. The catalytic activity of CD-GOx induced the protonation of benzimidazole, which resulted in dethreading of the inclusion complex and release of the drug cargo.
Although their use is less frequent among GOx-based glucose-responsive delivery systems, inorganic particles that respond to environmental change can also be used as the “gate”. Xu et al. adopted ZnO quantum dots as the pH-responsive switch and applied it to cap the nanopores of mesoporous bioactive glasses via electrostatic interaction to seal GOx/CAT and insulin inside the pores.[45] ZnO QDs were dissolved when pH dropped, leading to the disassembly of the mesoporous bioactive glasses and the delivery of insulin.
1.1.4. Membrane fusion
The physiologic insulin secretion from β-cells is mediated by the vesicle-membrane fusion between intracellular insulin-encapsulated granules and plasma membranes.[46] Recently, Gu and co-workers reported an artificial β-cell superstructure for glucose-triggered membrane fusion-mediated insulin delivery (Figure 5a).[8d] In this “vesicles-in-vesicle” system, the small inner liposomes were loaded with insulin solution to mimic the insulin granules in β-cells, while GOx and CAT were loaded into the internal space of the large outer liposomes, which was further functionalized with glucose transporter 2 and proton channel gramicidin A. Under a normoglycemic condition, the system was maintained at near physiological pH, so polyethylene glycol-conjugated cytosine-rich DNA (PEG-CDNA) and guanine-rich DNA (GDNA–CH) anchored on inner liposomes were linked together to shield the peptide K from recognizing peptide E on the inner surface of outer liposomes. Under hyperglycemic conditions, the pH in the system decreased significantly, leading to the detachment of PEG-CDNA and the deshielding of peptide K, which subsequently recognized the peptide E and induced the membrane fusion and insulin release (Figure 5b). Both glucose-dependent and pulsatile insulin release were validated with this system (Figure 5c–d). The capacity for prolonged blood glucose regulation with negligible hypoglycemia was also confirmed in studies of type 1 diabetic mice (Figure 5e).
Figure 5.
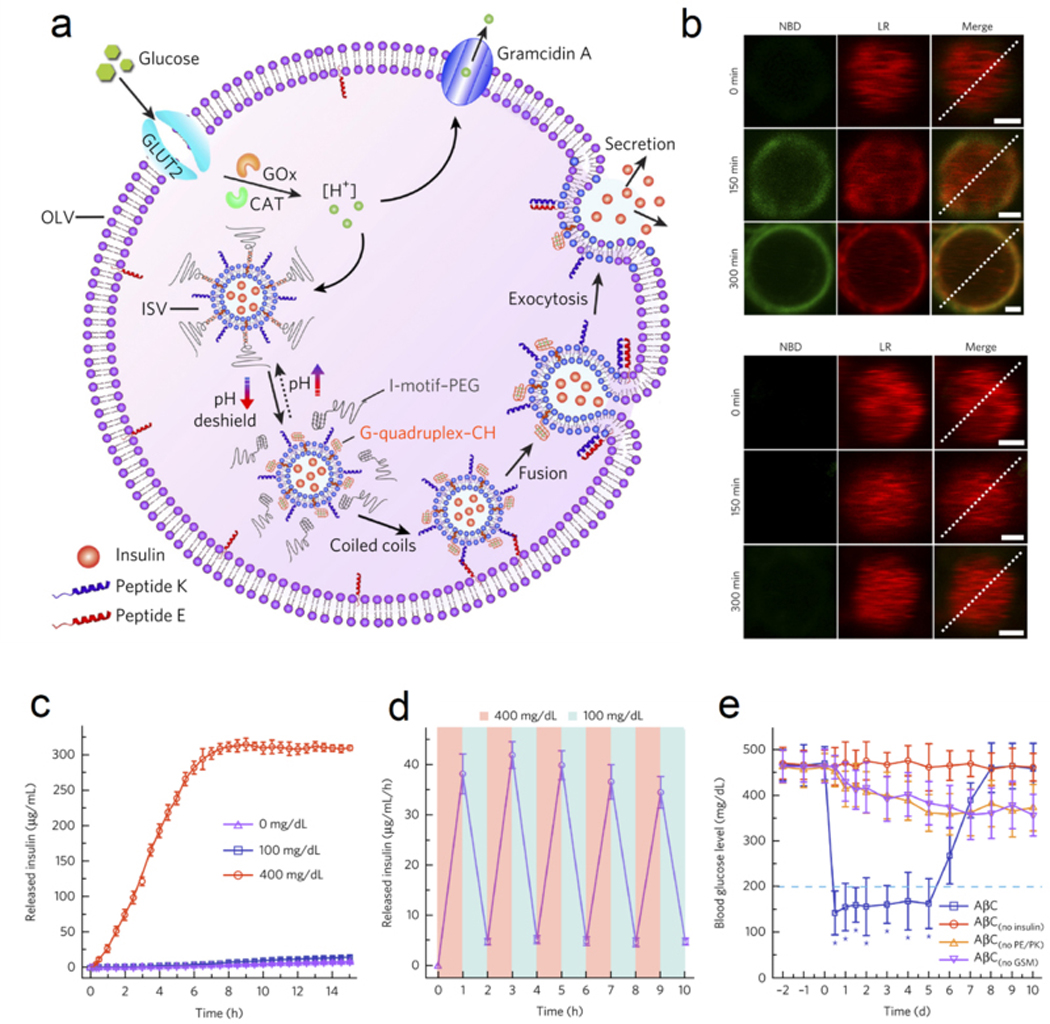
The design of membrane-fusion-mediated glucose responsive insulin release. (a) Schematic of the biochemical procedure of AβCs for glucose-triggered insulin release. (b) Upper, high glucose concentration (400 mg/mL) triggered membrane fusion; Lower, low glucose (100 mg/mL) failed to trigger membrane fusion. (c) Glucose-responsive insulin release from AβCs. Data were presented as mean ± SD (n=3). (d) Pulsatile insulin release by switching glucose concentration between 100 and 400 mg/dL. Data were presented as mean ± SD (n=3). (e) The glucose regulation ability of AβCs in type 1 diabetic mice. Data were presented as mean ± SD (n=5). *P < 0.001. Reproduced with permission.[8d] Copyright 2017, Springer Nature.
1.2. Hypoxia-responsive microneedle array patch
The GOx-catalyzed glucose-oxidation reaction produces H2O2 and gluconic acid, simultaneously consuming oxygen to generate an in-situ hypoxic microenvironment. Gu and co-workers developed hypoxia-sensitive material for achieving rapid and glucose-responsive insulin delivery using a microneedle array patch (Figure 6).[8b] The water-soluble hyaluronic acid was converted to amphiphilic structures via functionalization of hydrophobic 2-nitroimidazole groups, which could be reduced to hydrophilic 2-aminoimidazoles under bio-reductive and hypoxic conditions (Figure 6a), resulting in its dissociation and release of the encapsulated insulin (Figure 6b). The researchers prepared vesicles loaded with GOx and insulin (GRV(E+I)) and loaded them into the tips of the microneedles (Figure 6c–d). As a comparison, they also prepared GRV(1/2E+I) with half the amount of GOx and GRV(I) without GOx. In vivo treatment in type 1 diabetic mice confirmed the ability of microneedle patch loaded with GRV(E+I) to regulate blood glucose in a normal range with limited hypoglycemia in healthy mice (Figure 6e–g), while the microneedle patch loaded with GRV(1/2E+I) or GRV(I) showed negligible treatment efficacy. This work demonstrated the first prototype of the smart insulin patch combining the painless microneedle patch with a glucose-responsive component,[47] an approach that has potential to improve health outcomes and quality of life for people with diabetes in future translational work.[48]
Figure 6.

The design and results of hypoxia-responsive microneedle patch for insulin delivery. (a) Schematic of the glucose-responsive microneedle patch for insulin delivery. (b) Insulin release from nanoparticles was promoted with increased glucose concentration. The data were presented as mean ± SD (n=3). (c-d) Representative SEM (c) and fluorescence (d) images of microneedle patch loaded with FITC-labeled insulin. (e) Insertion of microneedle patch into skins of mice. (f) Blood glucose regulation of the microneedle patch was validated in type 1 diabetic mouse model. (g) Plasma insulin levels of mice treated with various microneedle patches. Data were presented as mean ± SD (n=5). *P < 0.05 for treatment efficacy of GRV(E+I) loaded microneedle array patch compared with GRV(1/2E+I) or GRV(E+I) loaded microneedle array patch. Reproduced with permission.[8b] Copyright 2015, National Academy of Sciences.
1.3. H2O2-responsive systems
In addition to scavenging the H2O2 generated from the GOx catalytic reaction, H2O2 itself can also be used as a trigger for insulin release.[49] For example, Hu et al. utilized a transcutaneous microneedle array patch strategy to directly exploit the generated H2O2 to oxidize and hydrolyze the grafted phenylboronic esters of an amphiphilic copolymer.[50] Upon exposure to generated H2O2, the scaffold became water-soluble, leading to the disassembly of polymeric vesicles and insulin release.[47a] A second H2O2-responsive polymeric vesicle system self-assembled from a poly(phenylboronic acid pinacol ester)-containing triblock copolymer was also used for a smart insulin delivery patch design.[51] In addition, Xu et al. integrated microneedles with insulin-loaded and H2O2-responsive mesoporous silica nanoparticles.[52] Wang et al. upgraded the microneedle into a core-shell structure by loading GOx and insulin at the core of the device with a surrounding shell embedded with CAT that serves as an active filter to scavenge excessive H2O2 to mitigate the risk of inflammation caused by H2O2.[47b, c]
2. Glucose-binding molecule (particle)-based systems
Naturally-occurring glucose-binding molecules such as lectin,[53] enzymes that bind glucose as a substrate such as GOx,[54] and synthetic polymer-imprinted particles[55] all have been exploited to prepare glucose-responsive insulin delivery systems. Recently, the glucose-binding abilities of glucose transporter[10c] and aptamer[10d] have also emerged in the realm of glucose-sensing and insulin delivery research.
The most well-studied molecule in this respect is Concanavalin A (Con A), a protein of the lectin family that can reversibly and specifically bind to glucose and mannose with a high affinity.[7a] In the presence of divalent cations, four Con A molecules associate with each other to form a tetramer assembly, which can bind to four glucose molecules and function as a crosslinker of macromolecules with pendant saccharide moieties.[7a] The gel is the most prominent formulation that has been used in the exploration of the reversible binding between glucose and Con A. In the presence of glucose, the competitive binding of free glucose to Con A impairs the binding between Con A and saccharide, leading to a decrease in the crosslinking density of the gel. In another approach, glucose can induce the detachment of saccharide-modified insulin from Con A-immobilized gels.
2.1. Responsive hydrogel crosslinked by Con A
Inspired by the findings that Con A could form a precipitate as a polymer with pendant glucose moieties,[56] Nakamae and co-workers reported a hydrogel prepared from glucosyloxyethy1 methacrylate (GEMA) and N,N’-methylene-bis-acrylamide (MBAAm) via copolymerization in the presence of Con A. The gel expanded in the presence of high glucose concentrations.[57] The same strategy was further explored to prepare glucose-sensing gels by synthesizing glucose-grafted polymers from allyl glucose.[58] Park and co-workers also developed a sol-gel phase-reversible hydrogel from the copolymer synthesized from 1-vinyl-2-pyrrolidinone and allyl glucose.[56] The addition of glucose turned the gel into a solution which could return to a gel via dialysis in water. Because the leakage of Con A from hydrogel was noted to impair the reversible nature of the swelling, Con A was immobilized onto polymer networks.[59] These Con A-modified polymers were further used with native or modified dextrin for constructing glucose-responsive delivery systems.[60]
2.2. Irresponsive gel immobilized with Con A
Another type of glucose-responsive insulin release utilizing Con A was constructed by conjugating insulin to one or more glucose molecules, which can then tether insulin to the Con A-constituted hydrogels. The pioneering work in this field was published in Science.[10b] In their study, a maltose-insulin conjugate was chemically synthesized and evaluated on a column filled with Con A-coupled sepharose 4B. Insulin release peak with increased intensity was observed in response to a stepwise increase in glucose concentrations. Following this work, insulin was conjugated to various glucose-like molecules, such as oligosaccharides maltose, maltotriose, mannotriose, and mannotetrose,[61] p-aminophenyl-α-D-glucopyranoside,[62] and glycosyl poly(ethylene glycol).[63] Despite alternation in chemical structure, glucose-modified insulin molecules showed comparable activity to native insulin in regulating the blood glucose levels in a rodent model.[62a, 64] Because micro-sized hydrogel formulations can improve the glucose response rate and increase feasibility of administration, microsized hydrogel prepared from Con A and chitosan/or dextrin,[65] nanosized hydrogel prepared from poly(NIPAM),[66] polymersomes prepared from diblock polymer grafted with glucose for glucose breathing,[67] and co-assembly from Con A and amylopectin[68] have also been studied.
2.3. Endogenous lectin-targeted systems
Recently, a glucose-responsive insulin analog formulation was developed for mitigating hypoglycemia by targeting the mannose receptor (MR) C-type 1, a lectin that is primarily expressed on hepatic sinusoidal endothelial cells, specific macrophages, and dendritic cells.[69] After conjugating carbohydrate moieties to insulin, an insulin analog named MK-2640 was able to recognize the lectin structures.[10a, 70] After binding to the MR, MK-2640 can be internalized and degraded in lysosomes. Because of the modification, the binding affinity of MK-2640 toward the insulin receptor decreased, but it acquired a Kd of 3.4 nM toward MR. In addition, its binding to MR was inhibited by glucose with an IC50 of 8 mM when the MK-2640 was set as 4 nM. The in vivo clearance rate via the MR path was modulated by blood glucose levels. In healthy dogs, when the blood glucose was lowered stepwise from 280 to 80 mg/dL, the MR-mediated clearance rate of MK-2640 increased progressively, leading to a ∼30% reduction of its availability for binding to the insulin receptor. Despite extensive and exciting preliminary studies,[10a] the clinical trial of MK-2640 initiated in 2014 failed.[71] In this clinical study, a nonsignificant decrease (6%) in the clearance rate was observed under hyperglycemic conditions compared to that under normoglycemic conditions, which indicated a great challenge for further clinical development.
2.4. Glucose transporter-mediated insulin delivery
Recently, Gu and co-workers conceived of the concept to use the glucose transporter (Glut) to achieve glucose-responsive insulin delivery.[10c] In this work, each insulin molecule was equipped with two glucosamine molecules to obtain the glucose-conjugated insulin (Glc-insulin), which was endowed with the ability to bind to the Gluts on red blood cells . After mixing Glc-insulin with freshly-obtained red blood cells overnight, the Glc-insulin was loaded onto the plasma membranes of the red blood cells. Glc-insulin release from red blood cells was promoted with the stepwise increase in glucose concentrations in in vitro studies. In a chemically-induced type 1 diabetic mouse model, Glc-insulin-loaded red blood cells showed a prolonged blood glucose regulation ability as compared with free insulin. Importantly, a plasma insulin spike associated with glucose peak was observed in response to a glucose challenge.
In addition, Gu group has further exploited glucose transporter inhibitor as a Glut-targeting agent (Figure 7a).[72] They prepared an insulin analog (designated i-insulin) with insulin conjugated to a Glut inhibitor called Glut-i2. Under a normoglycemic state, i-insulin could bind to Glut on living cells and establish an i-insulin reservoir. Upon a glucose challenge, the elevated glucose level triggered the dissociation of i-insulin-Glut complex and the release of i-insulin, which subsequently bound to the insulin receptors to trigger the blood glucose clearance. In addition, the dissociation of i-insulin-Glut also released free Glut, which also promoted glucose transport into cells. Under a hypoglycemic state, enhanced binding of i-insulin to Glut could help reduce the glucose transport activity of Glut, therefore mitigating the risk of hypoglycemia. In type 1 diabetic mice, i-insulin showed prolonged blood glucose regulation ability compared with native insulin. If the second injection of i-insulin was given after the blood glucose was normalized, negligible hypoglycemia and extended normoglycemia were observed (Figure 7b–c). In addition, mitigated hypoglycemia of i-insulin treatment in healthy mice was observed as compared with native insulin (Figure 7d).
Figure 7.
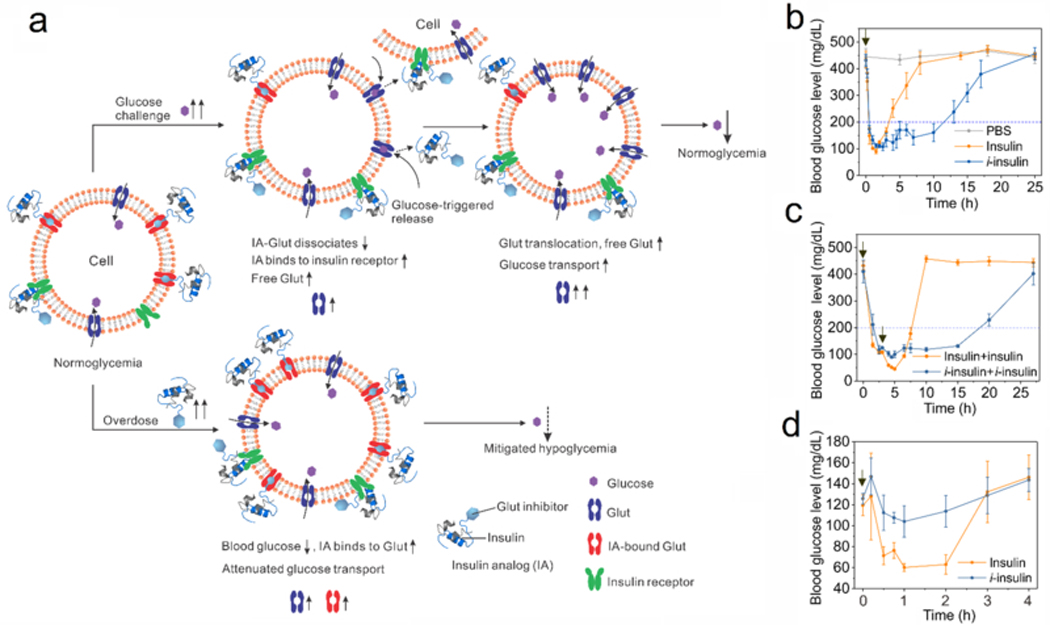
Glucose-responsive modulation of glucose transport activity for mitigating hypoglycemia. (a) Schematic of glucose-responsive glucose transport activity using an insulin analog (IA). Under a normoglycemia state, insulin analog binds to glucose transporter (Glut) and insulin receptor for blood glucose regulation. When blood glucose level increases because of carbohydrate administration, insulin analog is released into plasma or interstitial liquid because of glucose-triggered dissociation of IA-Glut complex. Subsequently, insulin analog binds to insulin receptor for reducing the blood glucose level. Meanwhile, more glucose accessible and free Glut is available for taking glucose into cells. Upon excess insulin administration, the formation of IA-Glut complex reduces the active Glut, therefore attenuating the glucose transport and mitigating the risk of hypoglycemia. (b) The blood glucose levels of diabetic mice administrated with i-insulin. Insulin was used as a control. The data were presented as mean ± SD (n=5). The black arrow indicated the treatment. (c) The blood glucose levels of diabetic mice receiving two sequential treatments at 0 h and 3 h. The time of treatment was indicated by black arrows. The data were presented as mean ± SD (n=5). (d) The blood glucose levels of healthy mice receiving treatment of i-insulin and native insulin. Black arrows indicated the time of treatment. The data were presented as mean ± SD (n=5). Reproduced with permission.[72] Copyright 2019, National Academy of Sciences.
2.5. Other glucose-binding molecules
A significant challenge for glucose-binding molecules is the selective binding of different carbohydrates.[73] Davis and co-workers have conducted numerous studies in this area of research (Figure 8). They have developed various cyclic structures for recognizing monosaccharide,[74] disaccharide,[74a, 75] polysaccharide,[76] O-linked β-N-acetylglucosamine,[77] and β-glucosyl[78]. Recently, they synthesized a cyclic compound with high affinity and selectivity toward glucose, offering significant progress towards future clinical applications.[10e]
Figure 8.
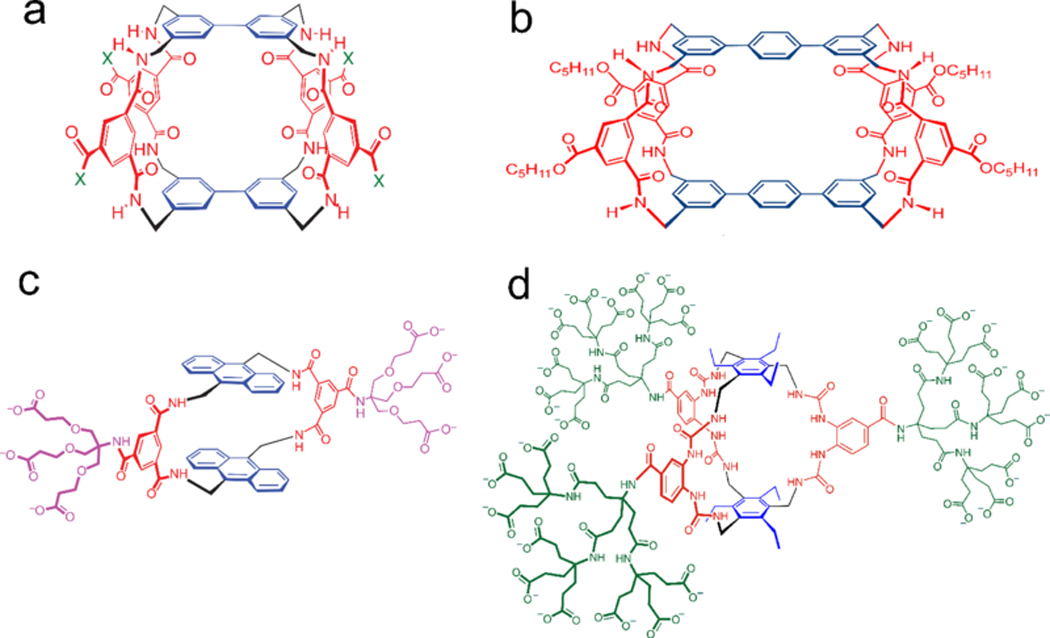
The structure of saccharide-binding molecules. (A) The chemical structure of a Tricyclic polyamide receptor for carbohydrate. X represents substituents. Reproduced with permission.[74a] Copyright 2004, John Wiley and Sons. (B) The chemical structure of a disaccharide-recognizing molecule. Reproduced with permission.[75a] Copyright 2002, John Wiley and Sons. (C) The chemical structure of a monocyclic molecule for recognizing glucose. Reproduced with permission.[74b] Copyright 2012, Springer Nature. (D) The chemical structure of a synthetic receptor for glucose with high affinity. Reproduced with permission.[10e] Copyright 2018, Springer Nature.
3. Phenylboronic acid-based systems
Compared with GOx and glucose-binding molecules, phenylboronic acid (PBA) is a synthetic compound with high stability and durability in the physiological environment, making these molecules reliable glucose sensors for glucose-responsive insulin delivery device design. Kuivila et al. observed and demonstrated the formation of an ester from aromatic boronic acids and a series of diol-containing compounds.[79] It was subsequently found that a cis-1, 2- or 1, 3-diol compound favored the formation of esters with PBA.[79] This complexation was reversible, allowing the disruption of the network built on phenylboronic acid-diol complex via adding glucose afterwards.[15b, 80]
In aqueous milieu, PBA exists as both an uncharged trigonal planar state and an anionic tetrahedral form in dynamically balance as shown below:
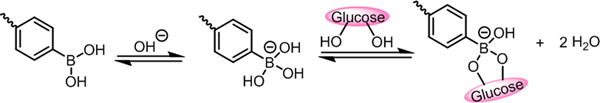
Glucose in the solution can reversibly and dynamically bind to the anionic tetrahedral structure, shifting the equilibrium toward the anionic form of PBA with enhanced hydrophilicity and increased negative charge density.[81]
Several strategies, including glucose-triggered swelling, dissociation, and competitive replacement, have been applied for the construction of glucose-sensitive platforms. Bulk hydrogels, micro-/nanogels, and self-assembled micelles can be prepared from molecules with a PBA moiety or boronate ester. Elevated glucose levels in the surrounding solution disintegrate the phenylboronic-diol bond, leading to the swelling of the carrier and the subsequent release of insulin. Alternatively, insulin can be conjugated to a diol-containing compound (or PBA) and subsequently used in combination with a PBA-containing gel (or a diol-containing matrix). In these design, the competitive binding between glucose and the diol units for PBA lead to glucose-triggered insulin release from carriers.[82]
3.1. Bulk hydrogels
As a three-dimensional structure containing a considerable amount of water, a hydrogel is supported and maintained via the chemically or physically crosslinked three-dimensional polymeric scaffold.[83] Thus, the incorporation of PBA into the polymeric network can endow the hydrogel with a glucose-responsive performance at the desired environment. The addition of glucose shifts the equilibrium toward the formation of boronate ester, inducing altered interactions among the polymer chains or between the polymer chains and water molecules.[84] The increased hydrophilicity or negative charge density results in an increased distance between the polymer chains, driven by steric or electrostatic repulsion respectively, ultimately leading to the release of the encapsulated drug.
In 1991, Kitano et al. proposed the concept of a glucose-responsive gel with potential application for insulin delivery. The hydrogel was prepared from poly(vinyl alcohol) (PVA) and poly(N-vinyl-2-pyrrolidone) with pendant phenylboronic acid moieties in basic aqueous solution.[85] The subsequent addition of glucose led to a continuous decrease in the viscosity in the gel.[86] Following this work, N,N-dimethylaminopropyl acrylamide was incorporated into the polymer structure, leading to enhanced water solubility at physiological pH and the formation of a stable hydrogel with PVA at pH 7.4. A model drug, myoglobin, was shown to be released promptly upon the addition of glucose (1000 mg/dL).[87] In addition, the amino group in this polymer was assumed to stabilize the phenylboronic acid-diol complex.[88] A glucose-responsive hydrogel for insulin delivery was also later prepared by Kataoka.[89] The bulk gel was prepared from N-isopropylacrylamide (NIPAAm) and 3-acrylamidophenylboronic acid (AAPBA) with MBAAm as the crosslinker. A pulsatile glucose-responsive insulin release was observed at 28°C and pH 9.[89] Several approaches were further developed to improve the glucose-responsive behavior at the physiological environment (pH 7.4 and 37 °C).
One approach is the introduction of adjacent functional groups onto PBA molecules, which renders the anionic tetrahedral geometry more favorable and produces a new phenylboronic acid with a pKa near 7.4. Therefore, Kataoka and co-workers introduced 4-(1,6-dioxo-2,5-diaza-7-oxamyl) phenylboronic acid (DDOPBA) (pKa=7.8) into poly(N-isopropylmethacrylamide) (poly(NIPMAA)), which had a lower critical solution temperature (LCST) higher than poly(N-isopropylacrylamide).[90] The glucose-responsive swelling of this gel occurred over time at 20 °C, 25 °C, and 30 °C, along with an increased ratio of DDOPBA. Following this work, 4-(2-acrylamidoethylcarbamoyl)-3-fluorophenylboronic acid (AmECFPBA) was found to possess a pKa around 7.2.[9a] An optimized concentration of AmECFPBA (7.5 mol%) was incorporated into PNIPMAA to prepare a cylinder gel. When the glucose concentration was cycled between 100 and 200 mg/dL at 37 °C and pH 7.4, a dehydrated skin layer was formed at lower glucose concentrations, suppressing the further penetration of either glucose or insulin through the gel.[9a] A pulsatile insulin release pattern was achieved when varying the glucose concentration between 100 mg/dL and 300 mg/dL, showing a 10-fold difference of insulin release rate that was achieved across the range of glucose concentrations.[9a]
In 2017, this hydrogel formulation was combined with a catheter in a novel device (Figure 9a–c).[9b] The pores perpendicular to the long axis on the catheter served as the channel for communication between the internal insulin solution and the glucose concentration in the outer interstitial fluid. Meanwhile, the gel covering the pores functioned as the glucose-responsive “gate“ for tuning the rate of insulin diffusion through the pores. After implanting this device under the skin of type 1 diabetic mice, blood glucose levels decreased from around 500 mg/dL to 300 mg/dL, when the type 1 diabetic mice were normally fed. A glucose tolerance test was also performed on overnight-fasted diabetic mice. A serum human insulin spike associated with the blood glucose peak was observed, suggesting the in vivo blood glucose-triggered insulin release (Figure 9d–e). In addition, a glucose-responsive transdermal patch for insulin delivery was also developed.[91] Three generally used monomers, NIPAAm, AmECFPBA, and MBAAm, were employed to form a crosslinked hydrogel inside the microneedle template via radical polymerization. The microneedle array patch was able to penetrate through the epidermal layer of the skin, offering communication channels between insulin solution and subcutaneous tissue.
Figure 9.

Boronate gel-based glucose-responsive insulin delivery. (a) The mechanism for glucose-responsive insulin release. A skin at the surface of the gel formed at low glucose level, inhibiting the insulin release; at high glucose levels, the skin was hydrated and allowed the insulin permeation. (b) The gel was loaded into a silicone catheter with the “skin” layer as the gate. (c) Schematic of the glucose-responsive insulin release. (d) Glucose tolerance test toward type 1 diabetic mice. *P < 0.05, **P < 0.01. (e) An in vivo insulin release into plasma associated with glucose peak was observed. ##P < 0.01 versus 0 min. n=6 to 12. Reproduced with permission.[9b] Copyright 2017, AAAS.
Another approach to improve the glucose-responsive performance in the physiological environment is to introduce new components into polymer structures. For example, a hydrogel composed of both crosslinked and grafted poly(N-isopropylacrylamide-co-3-acrylamidophenylboronic acid) [poly(NIPAM-co-AAPBA)] exhibited glucose-triggered volume expansion at 37 °C and pH 8.8.[92] Miyahara and co-workers introduced water-soluble N-acryloyl-l-alanine (CIPAAm) into a polymer network to increase the overall hydrophilicity. The introduction of CIPAAm maintained the sharpness of the volume phase transition of the polymeric network yet achieved a 163% increase of the volume upon the addition of glucose (500 mg/dL) at 37 °C and pH 7.4. [93] In another formulation, maleimide-glucosamine (MAGA) and AAPBA were copolymerized to give poly(AAPBA-r-MAGA) with various ratios.[94] When the ratio of AAPBA to MAGA in the prepared polymer was 2.7: 1, a 3-fold enhanced insulin release rate was observed when the glucose concentration increased from 100 to 300 mg/dL.
3.2. Micro-/nanogels
Microgels for use in this realm are typically prepared directly using precipitation or emulsion polymerization methods.[95] Alternatively, they can be generated from the self-assembly of macromolecule chains driven by intra- or intermolecular PBA-diol complexation or hydrophobic interactions. Like bulk gels, the binding of free glucose to PBA weakens the inter- or intramolecular forces of attraction, leading to the swelling or dissociation of the microgel or nanogel and thus triggering insulin release.
Zhou and co-workers employed a post-modification method to prepare monodisperse glucose-sensitive poly(NIPAAm-PBA) microgels.[95b] In their study, 3-aminophenylboronic acid (APBA) was grafted onto the poly(N-isopropylacrylamide-co-acrylic acid) backbone of a crosslinked microgel prepared via precipitation polymerization.[95b] In the presence of glucose (10 mM), the size of the 10% APBA microgel doubled from 80 nm to 160 nm at 25 °C and pH 9. Similarly, another group reported a direct polymerization method to prepare PBA-containing microgels (165–185 nm measured at 40 °C) using precipitation polymerization.[95a] The swelling ratios were tested at three different pH (pH=7.5, 8.5, 9.5) in Tris buffer (2 mM), with the evident expansion of microgel observed only at pH 8.5 and 9.5. Meanwhile, other methods have also explored the synthesis of core-shell or shell-only nanogels, but the glucose-responsive temperature remained lower than 37 °C.[96]
Besides the poly(NIPAAm)-based microgels, Jin et al. prepared nanogels from amphiphilic random glycopolymers rich in inter- or intramolecular complexation among the PBA and glycols.[94] Insulin was encapsulated into these nanoparticles, after which the insulin release rate was observed while changing the glucose concentration from 0 to 300 mg/dL at 37 °C and pH 7.4. Since the pKa of AAPBA was around 8.5, its optimal glucose-responsive performance was observed at pH 8.5. The researchers proposed that the swelling of the nanoparticles, as a result of competitive binding with glucose, gave the system the capacity to respond to glucose in the physiological environment. Recently, a complexation-driven self-assembly micellular platform was also prepared.[97] When the surrounding glucose concentration was increased stepwise from 200 mg/dL to 500 mg/dL, the micelles expanded in size; when the glucose concentration was increased to 1000 mg/dL, the micelles completely disintegrated. In addition, a 3-fold enhancement of insulin release rate was observed when the glucose solution was increased from 100 mg/dL to 200 mg/dL.
3.3. Self-assembled micelles
The introduction of phenylboronic acid into the hydrophobic chain has also been used to generate an amphiphilic copolymer with glucose-responsive behavior. A micelle system prepared from poly(ethylene glycol)-block-poly(acrylic acid-co-acryl amidophenylboronic acid) achieved glucose-responsive insulin release in the physiological range of glucose exposure.[82a] A subsequent study revealed that the coordination between the carboxylate anion and phenylboronic acid helped reduce the pKa of phenylboronic acid, thereby increasing its responsiveness to glucose at lower pH.[98] Meanwhile, Yang[99] and Yuan[100] prepared a glucose-responsive micellular carrier by introducing a phenylborate ester monomer, 2-phenylboronic esters-1,3-dioxane-5-ethyl methacrylate, into an amphiphilic polymer chain. This copolymer self-assembled into spherical micelles with core-shell structures at pH 7.4. In the presence of high glucose concentrations, glucose competed with diols to bind to PBA, thereby switching the polymer from amphiphilicity to hydrophilicity. In a hyperglycemic glucose solution (400 mg/dL), the enhanced release rate of FITC-labeled insulin was observed.[99e] A more detailed study of the factors that affected the glucose-responsive performance or insulin loading capacity, such as the molecular weight and the insulin-encapsulating method, was further explored.[99a]
3.4. Liposome formulation for oral administration
Oral administration of insulin has remained a goal since its discovery.[101] However, insulin is vulnerable to destruction by the enzymes in the gastrointestinal (GI) tract, and it is challenging for insulin to cross the intestinal epithelium.[102] Recently, Gu and co-workers have developed an oral insulin formulation for postprandial glycemic regulation (Figure 10). In this system, insulin was loaded into the neonatal Fc receptor (FcRn)-targeted liposomes, which was further equipped with a glucose-responsive hyaluronic acid-PBA shell[103]. After the liposomes were orally administrated, the HA shell detached from liposomes in response to increased intestinal glucose levels associated with ingestion of a meal. Subsequently, the binding of exposed Fc to FcRn expressed on epithelial cells in the small intestine facilitated the transepithelial transport of insulin-liposomes. In vitro studies confirmed a glucose-triggered zeta-potential increase after glucose exposure, and in vivo blood-glucose regulation was confirmed after the insulin was administrated orally.
Figure 10.
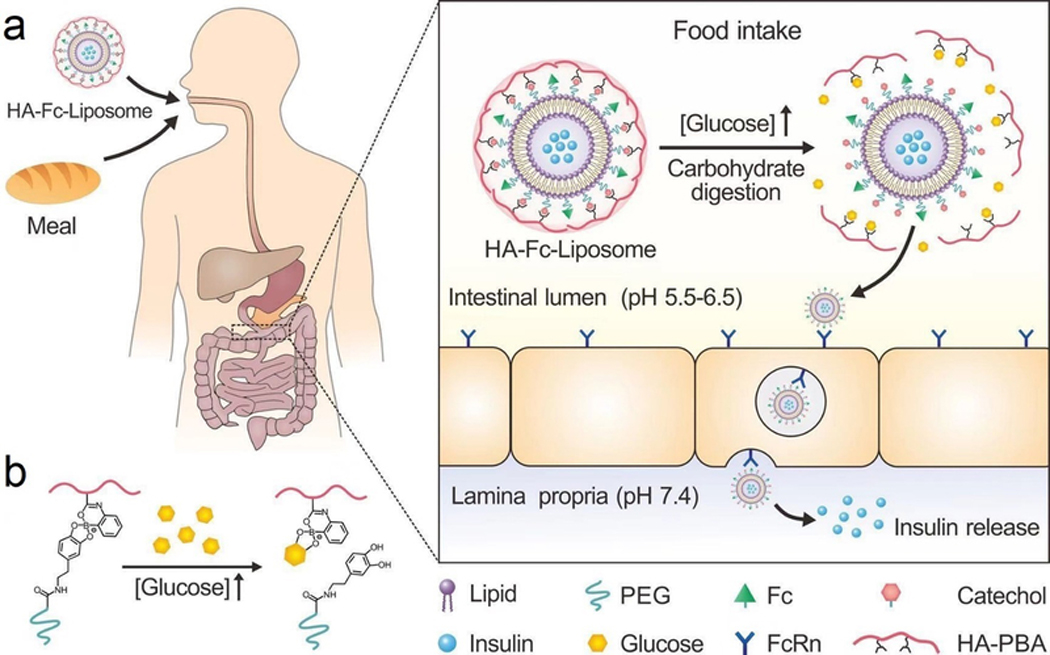
Schematic of an oral formulation for glucose-responsive insulin delivery. (a) Insulin was loaded into the liposomes coated with Fc, which facilitates the FcRn-mediated insulin absorption through the intestine. (b) Schematic of the glucose-responsive detachment of HA shell. HA, hyaluronic acid. Reproduced with permission.[103] Copyright 2018, Springer Nature.
3.5. PBA- or diol-immobilized carrier
A distinct approach to achieve glucose-responsive insulin release involves directly engineering the insulin structure to explore the reversible and glucose-competitive binding between PBA and diols. Typically, insulin is first either modified with PBA or diols and subsequently loaded into the corresponding diol- or PBA-immobilized carriers.
Tokano and co-workers found that diol-containing compounds were able to form covalent complexes with PBA-containing gel beans made from m-methacrylamidophenylboronic acid, acrylamide, and MBAAm.[104] Among these compounds, gluconic acid showed higher binding affinity than glucose; therefore, it was subsequently used for conjugation to give a novel insulin conjugate (designated G-insulin). By exploiting the specific recognition between gluconic acid with PBA, G-insulin was loaded into the PBA-gel beans. A pulsatile insulin release profile corresponding to alterations in glucose concentrations was confirmed at pH 8.5.[105] The researchers later improved this system by applying amine-containing PBA gel-beans, which was able to form complex with G-insulin at physiological pH.[106] Following this work, a gel prepared from NIPAAm, AAPBA, and MBAAm was synthesized to improve the glucose-responsive insulin release. At pH 9 and 28 °C, an on-off G-insulin release from the gel was observed by switching the glucose concentration.
Alternatively, the conjugation of insulin to PBA has also been studied by other groups.[107] Hoeg-jensen et al. conjugated insulin to several sulfonamide PBAs to leverage their pKa of around 7.4.[108] The dissociation constant (Kd) between glucose and obtained modified insulin was measured to be in the range of 15–31 mM. Glucamine-derived resin was also used for evaluating glucose-responsiveness. After loading insulin and zinc ions onto the resin, a steep and instant insulin release was observed in response to the glucose challenges (5, 25, 50 mM).
3.6. Endogenous albumin carrier
The improved water solubility of PBA associated with the formation of its boronate ester with glucose has also been explored as a potential trigger for insulin-PBA release from the carrier. Therefore, researchers have explored switching the interaction between the aliphatic phenylboronic acid and the circulating serum albumin as an additional glucose-responsive strategy for insulin delivery.[109] In this study, insulin was modified with various aliphatic phenylboronic acids showing different pKa. Under hyperglycemic conditions, the hydrophobic PBA was converted to a hydrophilic boronate ester with glucose, leading to weakened hydrophobic interactions between the aliphatic chain and albumin, thereby triggering the insulin release. This modified insulin not only retained similar long-acting activity as insulin-detemir but also showed the capacity to regulate its activity dynamically in response to fluctuating blood glucose levels.
3.7. Charge-reversal polymeric carrier for insulin delivery
Recently, Gu and coworkers developed a glucose-responsive charge-reversal polymeric carrier for insulin (Figure 11). In their study, a synthetic polymer with pendant amino groups and FPBA groups was prepared. In the absence of glucose, the polymer was positively charged, making it suitable for loading the negatively-charged insulin with a high loading efficiency and capability. Under a normoglycemic state, the polymer was still positively charged, and release insulin slowly; under a hyperglycemic state, the positive charge was significantly compromised and even reversed for immediate insulin release (Figure 11a). In vitro studies confirmed its glucose-binding ability and glucose-stimulated insulin release (Figure 11b–c). Further studies in both diabetic mice and minipigs validated the in vivo glucose-triggered insulin release from the complex into blood (Figure 11d–i).
Figure 11.
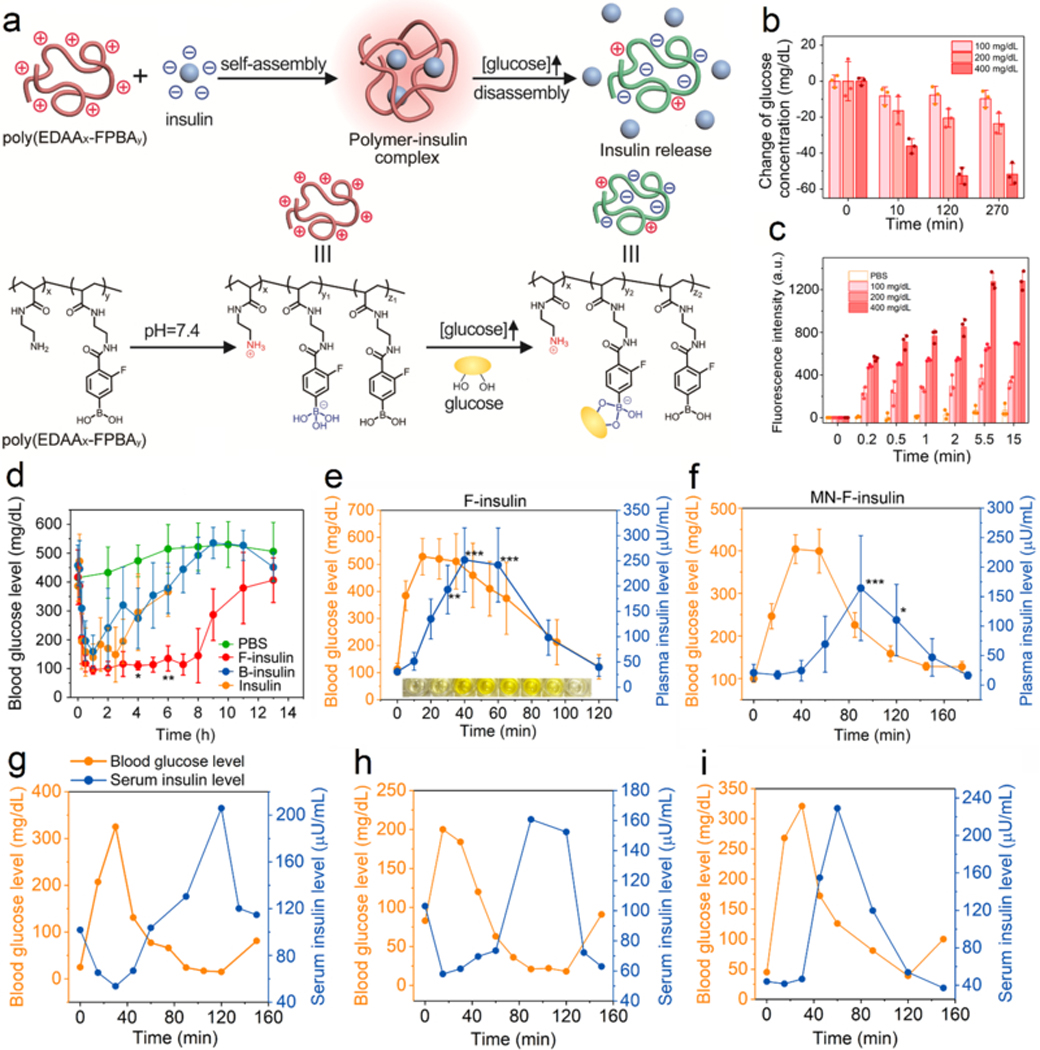
Glucose-responsive insulin release from the complex prepared from insulin and a charge-reversal cationic polymer. (a) Schematic of glucose-triggered charge-reversal and subsequent insulin release. (b) Glucose-binding ability of polymer. (c) Glucose-triggered insulin release. (d) Blood glucose regulation ability of the prepared complex in a type 1 diabetic mouse model. n=5. *P < 0.05, **P < 0.01. (e) Diabetic mice receiving treatment were further administrated with glucose. Both blood glucose levels and plasma insulin levels were monitored. n=4 to 5. *P < 0.05, **P < 0.01, ***P < 0.001. (f) Diabetic mice receiving treatment of microneedle patches loaded with complex were further administrated with glucose. Both blood glucose levels and plasma insulin levels were monitored. n=4. *P < 0.05, **P < 0.01, ***P < 0.001. (g-i) Diabetic minipigs receiving complex treatment were administrated with dextrose solution intravenously. Both blood glucose levels and serum insulin levels were measured. Each Figure indicated one diabetic minipig. Reproduced with permission. Copyright 2017, AAAS.
4. Discussion
To date, glucose-responsive systems stimulated by GOx, PBA or glucose-binding molecules have been validated at physiologically relevant conditions and in animal studies. Meanwhile, an increasing number of other systems with in vivo glucose-responsive performance have also been demonstrated. Looking ahead to human translation, these systems have the potential to offer a fully personalized approach to diabetes therapy, as a single glucose-responsive insulin system could adapt to different individuals’ specific glucose levels over the day and night to optimize glycemic control. As precision medicine expands to diabetes care, including significant technologic efforts to deliver insulin in a glucose-responsive way, this work offers further options for individualizing insulin delivery for the avoidance of hyper- and hypoglycemia.[110]
However, to date, MK-2640, a glycosylated insulin, is the only glucose-responsive insulin to be tested in a human clinical trial. Despite promising results in diabetic dog and minipig models,[70, 111] the clinical trial of this insulin analog was not successful. As pointed out by the researchers involved in the clinical study, incomplete understanding of quantitative differences across species might complicate the prediction of the clinical outcomes of glucose-responsive insulins when translated to humans. The lack of other clinical trial data at this time points to several major issues with the translation of currently available glucose-responsive insulin delivery systems; for example, more investigation is required to evaluate toxicity and efficacy.[7c, 112]
First, the carriers employed to achieve glucose-responsive insulin delivery raise challenges relating to biocompatibility issues. For example, GOx catalyzes the oxidation of glucose to gluconic acid and H2O2, the latter of which can cause inflammation or even severe tissue damage.[113] In addition, GOx may cause a host immune response;[114] this is also a problem that has been encountered by Con A.[115] The toxicity of PBA for human remains unclear; therefore, a thorough evaluation of its biosafety must be accomplished prior to translational studies in humans. Although these considerations pose significant challenges for translational studies in the future, several existing efforts have already produced promising strategies to mitigate these risks. For example, the transdermal microneedle array patch can deliver insulin in a glucose-responsive manner while retaining the materials associated with devices/formulations outside the body.[8b, 48b, 116] Moreover, glucose-modified insulin that can target endogenous glucose-binding proteins may potentially eliminate the use of additional carriers.[10a, c] Finally, an orally-delivered glucose-responsive insulin system may offer a way to circumvent major biocompatibility issues.[103]
Second, the in vivo reliability of these systems warrants further study. It is critical for researchers in this field to carry out in vivo studies, as the physiological environment may be entirely different from the in vitro buffer solutions used for experiments. In addition, animal studies provide significantly more information on burst release or other release profiles, biocompatibility issues, glucose-response rates, the risk for hypoglycemia, and other important factors that may affect considerations for clinical translation. For example, the dynamic insulin release from carriers into blood circulation can be evaluated in mice via a glucose tolerance test, in addition to clamp technology.[10c] These in vivo studies will also provide information regarding how the components of body fluid can affect the activity of enzymes or proteins, as well as insights into complicated in vivo interactions that affect glucose-responsive performance.
Third, specific goals for the design of glucose-responsive insulin delivery should be clearly specified. An ideal glucose-responsive insulin delivery system or insulin analog is ultimately one that can entirely replace the role of β-cells. However, it may be more realistic to focus on engineering a glucose-responsive insulin delivery system to replace specific aspects of β-cell functionality. For example, glucose-responsive basal insulin could offer overnight and fasting blood glucose regulation with reduced risk of hypoglycemia, while other formulations of glucose-responsive insulin can be designed as meal-time insulin for post-prandial glucose control. In the future, these different insulin systems could also be incorporated into insulin pumps. In combination with the recent advances in the field, these diverse goals for future work will facilitate the optimization of current systems, the development of novel designs, and the ultimate clinical translation of glucose-responsive insulin delivery systems.
Acknowledgements
This work was supported by the grants from NIH (grant No. R01 DK112939 01A1), JDRF (3-SRA-2015–117-Q-R, 1-PNF-2019–674-S-B), National Science Foundation (grant no. 1708620), American Diabetes Association (grant no. 1–15-ACE-21).
Biography

Jinqiang Wang earned his Ph.D. degree from Zhejiang University in 2013, under the guidance of Prof. Youqing Shen in the Department of Chemical Engineering. He is currently a postdoctoral associate working with Prof. Zhen Gu in the bioengineering department at University of California, Los Angeles. His research interests include development of new materials and systems for both cancer drug and insulin delivery.

Zejun Wang obtained her Bachelor Degree in Chemistry in 2013 from Hunan University and her Ph.D. degree in Inorganic Chemistry at Shanghai Institute of Applied Physics, Chinese Academy of Sciences, in 2018. In the same year, she joined Prof. Zhen Gu’s research group at University of California, Los Angeles as a postdoctoral research fellow. Her research interests mainly focus on the design and synthesis of analytical probes, biomedical devices and biomimetic materials.

Zhen Gu is a Professor in the Department of Bioengineering and the California NanoSystems Institute at University of California, Los Angeles (UCLA). Dr. Gu received his B.S. degree in Chemistry and M.S. degree in Polymer Chemistry and Physics from Nanjing University. In 2010, he obtained Ph.D. degree at UCLA, under the guidance of Dr. Yi Tang in the Department of Chemical and Biomolecular Engineering. He was a Postdoctoral Associate working with Dr. Robert Langer at MIT and Harvard Medical School during 2010 to 2012. Before he moved to UCLA in 2018, he had been appointed as a Jackson Family Distinguished Professor in the Joint Department of Biomedical Engineering at the University of North Carolina at Chapel Hill and North Carolina State University. His group studies controlled drug delivery, bio-inspired materials and nanobiotechnology, especially for cancer and diabetes treatment.
Footnotes
Conflict of Interests
Prof. Z. Gu is the co-founder of Zenomics Inc. Dr. J. Yu is the chief scientific officer of Zenomics Inc.
Contributor Information
Jinqiang Wang, Department of Bioengineering, University of California, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, CA 90095, USA.
Zejun Wang, Department of Bioengineering, University of California, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, CA 90095, USA.
Jicheng Yu, Zenomics Inc., Durham, NC 27709, USA.
Anna R. Kahkoska, Department of Medicine, University of North Carolina School of Medicine, Chapel Hill, NC 27599, USA;
John B. Buse, Department of Medicine, University of North Carolina School of Medicine, Chapel Hill, NC 27599, USA
Zhen Gu, Department of Bioengineering, University of California, Los Angeles, CA 90095, USA; California NanoSystems Institute, University of California, Los Angeles, CA 90095, USA; Zenomics Inc., Durham, NC 27709, USA; Jonsson Comprehensive Cancer Center, University of California, Los Angeles, CA 90095, USA;; Center for Minimally Invasive Therapeutics, University of California, Los Angeles, CA 90095, USA.
References
- [1].Koeslag JH, Saunders PT, Terblanche E, J. Physiol.-London 2003, 549, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Matthews DR, Rudenski AS, Burnett MA, Darling P, Turner RC, Clin. Endocrinol. (Oxf.) 1985, 23, 71. [DOI] [PubMed] [Google Scholar]
- [3].a) Yu J, Zhang Y, Bomba H, Gu Z, Bioeng. Transl. Med. 2016, 1, 323; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Berenson DF, Weiss AR, Wan ZL, Weiss MA, Ann. N. Y. Acad. Sci. 2011, 1243, E40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Ohkubo Y, Kishikawa H, Araki E, Miyata T, Isami S, Motoyoshi S, Kojima Y, Furuyoshi N, Shichiri M, Diabetes Res. Clin. Pr. 1995, 28, 103; [DOI] [PubMed] [Google Scholar]; b) Yang J, Cao Z, Control J. Release 2017, 263, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Traverso G, Langer R, Sci. Transl. Med. 2015, 7, 289ed6; [DOI] [PubMed] [Google Scholar]; b) Sun WJ, Hu QY, Ji WY, Wright G, Gu Z, Physiol. Rev. 2017, 97, 189; [Google Scholar]; c) Collins FS, Varmus H, Engl N. J. Med. 2015, 372, 793; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Gu Z, Bioeng. Transl. Med. 2016, 1, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Cryer P, Hypoglycemia in diabetes: pathophysiology, prevalence, and prevention, American Diabetes Association, 2016; [Google Scholar]; b) Bakh NA, Cortinas AB, Weiss MA, Langer RS, Anderson DG, Gu Z, Dutta S, Strano MS, Nat. Chem. 2017, 9, 937. [DOI] [PubMed] [Google Scholar]
- [7].a) VandenBerg MA, Webber MJ, Adv. Funct. Mater. 2019, 1801466; [DOI] [PubMed] [Google Scholar]; b) Rodbard D, Diabetes Technol. Ther. 2016, 18 Suppl 2, S3; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Langer R, Science 1990, 249, 1527; [DOI] [PubMed] [Google Scholar]; d) Chen Z, Wang Z, Gu Z, Acc. Chem. Res. 2019, 52, 1255; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Mo R, Jiang T, Di J, Tai W, Gu Z, Chem. Soc. Rev. 2014, 43, 3595; [DOI] [PubMed] [Google Scholar]; f) Yu J, Zhang Y, Yan J, Kahkoska AR, Gu Z, Int. J. Pharm. 2018, 544, 350; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Lu Y, Sun W, Gu Z, Control J. Release 2014, 194, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Zhang YQ, Yu JC, Shen QD, Gu Z, Prog. Chem. 2015, 27, 11; [Google Scholar]; i) DiSanto RM, Subramanian V, Gu Z, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2015, 7, 548; [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Di J, Yu JC, Ye YQ, Ranson D, Jindal A, Gu Z, Cell Mol. Bioeng. 2015, 8, 445. [Google Scholar]
- [8].a) Hassan CM, Doyle FJ, Peppas NA, Macromolecules 1997, 30, 6166; [Google Scholar]; b) Yu J, Zhang Y, Ye Y, DiSanto R, Sun W, Ranson D, Ligler FS, Buse JB, Gu Z, Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 8260; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ishihara K, Kobayashi M, Ishimaru N, Shinohara I, Polym. J. 1984, 16, 625; [Google Scholar]; d) Chen Z, Wang J, Sun W, Archibong E, Kahkoska AR, Zhang X, Lu Y, Ligler FS, Buse JB, Gu Z, Nat. Chem. Biol. 2018, 14, 86; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Yu J, Zhang Y, Hu X, Wright G, Gu Z, Ann. Biomed. Eng. 2016, 44, 1931; [DOI] [PubMed] [Google Scholar]; f) Yu J, Qian C, Zhang Y, Cui Z, Zhu Y, Shen Q, Ligler FS, Buse JB, Gu Z, Nano Lett. 2017, 17, 733. [DOI] [PubMed] [Google Scholar]
- [9].a) Matsumoto A, Ishii T, Nishida J, Matsumoto H, Kataoka K, Miyahara Y, Angew. Chem. Int. Ed. Engl. 2012, 51, 2124; [DOI] [PubMed] [Google Scholar]; b) Matsumoto A, Tanaka M, Matsumoto H, Ochi K, Moro-Oka Y, Kuwata H, Yamada H, Shirakawa I, Miyazawa T, Ishii H, Kataoka K, Ogawa Y, Miyahara Y, Suganami T, Sci. Adv. 2017, 3, eaaq0723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Yang R, Wu M, Lin S, Nargund RP, Li X, Kelly T, Yan L, Dai G, Qian Y, Dallas-Yang Q, Fischer PA, Cui Y, Shen X, Huo P, Feng DD, Erion MD, Kelley DE, Mu J, JCI Insight 2018, 3, e97476; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Brownlee M, Cerami A, Science 1979, 206, 1190; [DOI] [PubMed] [Google Scholar]; c) Wang C, Ye Y, Sun W, Yu J, Wang J, Lawrence DS, Buse JB, Gu Z, Adv. Mater. 2017, 29, 1606617; [DOI] [PubMed] [Google Scholar]; d) Nakatsuka N, Yang KA, Abendroth JM, Cheung KM, Xu X, Yang H, Zhao C, Zhu B, Rim YS, Yang Y, Weiss PS, Stojanovic MN, Andrews AM, Science 2018, 362, 319; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Tromans RA, Carter TS, Chabanne L, Crump MP, Li H, Matlock JV, Orchard MG, Davis AP, Nat. Chem. 2019, 11, 52. [DOI] [PubMed] [Google Scholar]
- [11].Zhang Y, Yu JC, Shen Q, Gu Z, Prog. Chem. 2015, 27, 11. [Google Scholar]
- [12].Wagner AM, Gran MP, Peppas NA, Acta Pharm. Sin. B 2018, 8, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Farmer TG, Edgar TF, Peppas NA, J. Pharm. Pharmacol. 2008, 60, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Wilson R, Turner APF, Biosens. Bioelectron. 1992, 7, 165; [Google Scholar]; b) Huggett ASG, Nixon DA, Lancet 1957, 2, 368. [DOI] [PubMed] [Google Scholar]
- [15].a) Kost J, Langer R, Trends Biotechnol. 1992, 10, 127; [DOI] [PubMed] [Google Scholar]; b) Wu Q, Wang L, Yu H, Wang J, Chen Z, Chem. Rev. 2011, 111, 7855; [DOI] [PubMed] [Google Scholar]; c) Kost J, Langer R, Adv. Drug Deliver. Rev. 2012, 64, 327; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kost J, Langer R, Adv. Drug Deliver. Rev. 2001, 46, 125. [DOI] [PubMed] [Google Scholar]
- [16].a) Caldorera-Moore M, Peppas NA, Adv. Drug Deliver. Rev. 2009, 61, 1391; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Morishita M, Lowman AM, Takayama K, Nagai T, Peppas NA, Journal Of Control. Release 2002, 81, 25; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kim B, Peppas NA, Macromolecules 2002, 35, 9545. [Google Scholar]
- [17].a) Li X, Fu M, Wu J, Zhang C, Deng X, Dhinakar A, Huang W, Qian H, Ge L, Acta Biomater. 2017, 51, 294; [DOI] [PubMed] [Google Scholar]; b) Gu Z, Aimetti AA, Wang Q, Dang T, Zhang Y, Veiseh O, Cheng H, Langer RS, Anderson DG, ACS Nano 2013, 7, 4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Aznar E, Villalonga R, Gimenez C, Sancenon F, Marcos MD, Martinez-Manez R, Diez P, Pingarron JM, Amoros P, Chem. Commun. 2013, 49, 6391; [DOI] [PubMed] [Google Scholar]; b) Chu L, Li Y, Zhu J, Wang H, Liang Y, Control J. Release 2004, 97, 43. [DOI] [PubMed] [Google Scholar]
- [19].a) Slaughter BV, Khurshid SS, Fisher OZ, Khademhosseini A, Peppas NA, Adv. Mater. 2009, 21, 3307; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Qiu Y, Park K, Adv. Drug Deliver. Rev. 2012, 64, 49. [Google Scholar]
- [20].a) Peppas NA, Hilt JZ, Khademhosseini A, Langer R, Adv. Mater. 2006, 18, 1345; [Google Scholar]; b) Brannonpeppas L, Peppas NA, Chem. Eng. Sci. 1991, 46, 715. [Google Scholar]
- [21].Ishihara K, Matsui K, Polymer Sci. J. C Polymer Lett. 1986, 24, 413. [Google Scholar]
- [22].a) Kost J, Horbett TA, Ratner BD, Singh M, J. Biomed. Mater. Res. 1985, 19, 1117; [DOI] [PubMed] [Google Scholar]; b) Albin G, Horbett TA, Ratner BD, Control J. Release 1985, 2, 153; [Google Scholar]; c) Albin GW, Horbett TA, Miller SR, Ricker NL, Control J. Release 1987, 6, 267. [Google Scholar]
- [23].Parker RS, Doyle FJ 3rd, Peppas NA, IEEE Trans. Biomed. Eng. 1999, 46, 148. [DOI] [PubMed] [Google Scholar]
- [24].Gu Z, Dang T, Ma M, Tang B, Cheng H, Jiang S, Dong Y, Zhang Y, Anderson DG, ACS Nano 2013, 7, 6758. [DOI] [PubMed] [Google Scholar]
- [25].Podual K, Doyle FJ, Peppas NA, Control J. Release 2000, 67, 9. [DOI] [PubMed] [Google Scholar]
- [26].Gordijo CR, Shuhendler AJ, Wu X, Adv. Funct. Mater. 2010, 20, 1404. [Google Scholar]
- [27].Kim MY, Kim J, ACS Biomater. Sci. Eng. 2017, 3, 572. [DOI] [PubMed] [Google Scholar]
- [28].Gordijo CR, Koulajian K, Shuhendler AJ, Bonifacio LD, Huang H, Chiang S, Ozin GA, Giacca A, Wu X, Adv. Funct. Mater. 2011, 21, 73. [Google Scholar]
- [29].a) Iwata H, Matsuda T, Membrane Sci J. 1988, 38, 185; [Google Scholar]; b) Kurian P, Kasibhatla B, Daum J, Burns CA, Moosa M, Rosenthal KS, Kennedy JP, Biomaterials 2003, 24, 3493. [DOI] [PubMed] [Google Scholar]
- [30].a) Traitel T, Cohen Y, Kost J, Biomaterials 2000, 21, 1679; [DOI] [PubMed] [Google Scholar]; b) Zhang K, Wu X, Control J. Release 2002, 80, 169; [DOI] [PubMed] [Google Scholar]; c) Wu W, Mitra N, Yan EC, Zhou S, ACS Nano 2010, 4, 4831; [DOI] [PubMed] [Google Scholar]; d) Farahani BV, Ghasemzaheh H, Afraz S, RSC Adv. 2016, 6, 26590. [Google Scholar]
- [31].Chu MK, Chen J, Gordijo CR, Chiang S, Ivovic A, Koulajian K, Giacca A, Wu XY, Sun Y, Lab Chip 2012, 12, 2533. [DOI] [PubMed] [Google Scholar]
- [32].Lim ZW, Ping Y, Miserez A, Bioconjug. Chem. 2018, 29, 2176. [DOI] [PubMed] [Google Scholar]
- [33].Kawaguchi H, Polym. Int. 2014, 63, 925. [Google Scholar]
- [34].Wu Y, Hu H, Hu J, Liu S, Macromol. Rapid. Comm. 2012, 33, 1852. [DOI] [PubMed] [Google Scholar]
- [35].Tai W, Mo R, Di J, Subramanian V, Gu X, Buse JB, Gu Z, Biomacromolecules 2014, 15, 3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chong-Kook K, Eun-Bin I, Soo-Jeong L, Yu-Kyoung O, Seok-Kyu H, Int. J. Pharm. 1994, 101, 191. [Google Scholar]
- [37].Jain SK, Amit KC, Chalasani KB, Jain AK, Chourasia MK, Jain A, Jain NK, J. Drug. Deliv. Sci. Tec. 2007, 17, 399. [Google Scholar]
- [38].Xia D, He H, Wang Y, Wang K, Zuo H, Gu H, Xu P, Hu Y, Acta Biomater. 2018, 69, 301. [DOI] [PubMed] [Google Scholar]
- [39].Zhou HC, Long JR, Yaghi OM, Chem. Rev. 2012, 112, 673. [DOI] [PubMed] [Google Scholar]
- [40].Chen W, Luo GF, Vázquez-González M, Cazelles R, Sohn YS, Nechushtai R, Mandel Y, Willner I, ACS Nano 2018, 12, 7538. [DOI] [PubMed] [Google Scholar]
- [41].Fenton OS, Olafson KN, Pillai PS, Mitchell MJ, Langer R, Adv. Mater. 2018, 30, 1705328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Okahata Y, Lim H, Nakamura G, Hachiya S, J. Am. Chem. Soc. 1983, 105, 4855. [Google Scholar]
- [43].Diez P, Esteban-Fernandez de Avila B, Ramirez-Herrera DE, Villalonga R, Wang J, Nanoscale 2017, 9, 14307. [DOI] [PubMed] [Google Scholar]
- [44].Oroval M, Diez P, Aznar E, Coll C, Marcos MD, Sancenon F, Villalonga R, Martinez-Manez R, Chem.-Eur. J. 2017, 23, 1353. [DOI] [PubMed] [Google Scholar]
- [45].Xu B, Cao Q, Zhang Y, Yu W, Zhu J, Liu D, Jiang G, ACS Biomater. Sci. Eng. 2018, 4, 2473. [DOI] [PubMed] [Google Scholar]
- [46].a) Rorsman P, Renstrom E, Diabetologia 2003, 46, 1029; [DOI] [PubMed] [Google Scholar]; b) Gao ZF, Reavey-Cantwell J, Young RA, Jegier P, Wolf BA, J. Biol. Chem. 2000, 275, 36079. [DOI] [PubMed] [Google Scholar]
- [47].a) Hu X, Yu J, Qian C, Lu Y, Kahkoska AR, Xie Z, Jing X, Buse JB, Gu Z, ACS Nano 2017, 11, 613; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang J, Ye Y, Yu J, Kahkoska AR, Zhang X, Wang C, Sun W, Corder RD, Chen Z, Khan SA, Buse JB, Gu Z, ACS Nano 2018, 12, 2466; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang Y, Wang J, Yu J, Wen D, Kahkoska AR, Lu Y, Zhang X, Buse JB, Gu Z, Small 2018, 14, 1704181; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhang Y, Yu J, Kahkoska AR, Wang J, Buse JB, Gu Z, Adv. Drug Deliver. Rev. 2018, 10.1016/0378-5173(94)90214-3; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Ye Y, Yu J, Wen D, Kahkoska AR, Gu Z, Adv. Drug Deliver. Rev. 2018, 127, 106; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Chen G, Yu J, Gu Z, Diabetes Sci J. Technol. 2018, 13, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].a) Lu Y, Aimetti AA, Langer R, Gu Z, Nat. Rev. Mater. 2017, 2, 16075; [Google Scholar]; b) Veiseh O, Langer R, Nature 2015, 524, 39. [DOI] [PubMed] [Google Scholar]
- [49].Uchiyama T, Kiritoshi Y, Watanabe J, Ishihara K, Biomaterials 2003, 24, 5183. [DOI] [PubMed] [Google Scholar]
- [50].Hu XL, Zhang YG, Xie ZG, Jing XB, Bellotti A, Gu Z, Biomacromolecules 2017, 18, 649. [DOI] [PubMed] [Google Scholar]
- [51].Tong Z, Zhou J, Zhong J, Tang Q, Lei Z, Luo H, Ma P, Liu X, ACS Appl. Mater. Interfaces 2018, 10, 20014. [DOI] [PubMed] [Google Scholar]
- [52].Xu B, Jiang GH, Yu WJ, Liu D, Zhou J, Zhang Y, Sun SQ, Liu Y, J. Mater. Chem. B 2017, 5, 8200. [DOI] [PubMed] [Google Scholar]
- [53].a) Goldstein IJ, Hayes CE, Adv. Carbohydr. Chem. Biochem, 1978, 35, 127; [DOI] [PubMed] [Google Scholar]; b) Gabor F, Bogner E, Weissenboeck A, Wirth M, Adv. Drug Deliv. Rev. 2004, 56, 459. [DOI] [PubMed] [Google Scholar]
- [54].a) Chen M, Huang C, He C, Zhu W, Xu Y, Lu Y, Chem. Commun. 2012, 48, 9522; [DOI] [PubMed] [Google Scholar]; b) Ye T, Bai X, Jiang X, Wu Q, Chen S, Qu A, Huang J, Shen J, Wu W, Polym. Chem. 2016, 7, 2847. [Google Scholar]
- [55].a) Parmpi P, Kofinas P, Biomaterials 2004, 25, 1969; [DOI] [PubMed] [Google Scholar]; b) Chen G, Guan Z, Chen C, Fu L, Sundaresan V, Arnold FH, Nat. Biotechnol. 1997, 15, 354; [DOI] [PubMed] [Google Scholar]; c) Seong H, Lee HB, Park K, J. Biomater. Sci. Polym. Ed. 2002, 13, 637. [DOI] [PubMed] [Google Scholar]
- [56].Morris JE, Hoffman AS, Fisher RR, Biotechnol. Bioeng. 1993, 41, 991. [DOI] [PubMed] [Google Scholar]
- [57].Miyata T, Jikihara A, Nakamae K, Hoffman AS, Macromol. Chem. Physic. 1996, 197, 1135. [Google Scholar]
- [58].a) Obaidat AA, Park K, Pharm. Res. 1996, 13, 989; [DOI] [PubMed] [Google Scholar]; b) Obaidat AA, Park K, Biomaterials 1997, 18, 801. [DOI] [PubMed] [Google Scholar]
- [59].a) Tanna S, Taylor MJ, Adams G, J. Pharm. Pharmacol. 1999, 51, 1093; [DOI] [PubMed] [Google Scholar]; b) Miyata T, Jikihara A, Nakamae K, Hoffman AS, J. Biomat. Sci.-Polym. E. 2004, 15, 1085. [DOI] [PubMed] [Google Scholar]
- [60].a) Taylor MJ, Tanna S, Sahota TS, Drug. Dev. Ind. Pharm. 2008, 34, 73; [DOI] [PubMed] [Google Scholar]; b) Adams GG, Cui Y, Mitchell JH, Taylor MJ, Rheol. Acta 2006, 45, 611; [Google Scholar]; c) Tanna S, Sahota TS, Sawicka K, Taylor MJ, Biomaterials 2006, 27, 4498; [DOI] [PubMed] [Google Scholar]; d) Taylor MJ, Tanna S, Sahota TS, Voermans B, Eur. J. Pharm. Biopharm. 2006, 62, 94; [DOI] [PubMed] [Google Scholar]; e) Zhang R, Tang M, Bowyer A, Eisenthal R, Hubble J, React. Funct. Polym. 2006, 66, 757; [Google Scholar]; f) Taylor MJ, Tanna S, Sahota TS, Pharm. Dev. Technol. 2010, 15, 80. [DOI] [PubMed] [Google Scholar]
- [61].Brownlee M, Cerami A, Diabetes 1983, 32, 499. [DOI] [PubMed] [Google Scholar]
- [62].a) Seminoff LA, Gleeson JM, Zheng J, Olsen GB, Holmberg D, Mohammad SF, Wilson D, Kim SW, Int. J. Pharm. 1989, 54, 251; [Google Scholar]; b) Seminoff LA, Olsen GB, Kim SW, Int. J. Pharm. 1989, 54, 241. [Google Scholar]
- [63].Liu F, Song S, Mix D, Baudys M, Kim SW, Bioconjug. Chem. 1997, 8, 664. [DOI] [PubMed] [Google Scholar]
- [64].Veiseh O, Tang B, Whitehead KA, Anderson DG, Langer R, Nat. Rev. Drug Discov. 2015, 14, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].a) Yin R, Han J, Zhang J, Nie J, Colloids Surf. B Biointerfaces 2010, 76, 483; [DOI] [PubMed] [Google Scholar]; b) Yin R, Tong Z, Yang D, Nie J, Carbohydr. Polym. 2012, 89, 117; [DOI] [PubMed] [Google Scholar]; c) Yin R, Wang K, Du S, Chen L, Nie J, Zhang W, Carbohydr. Polym. 2014, 103, 369. [DOI] [PubMed] [Google Scholar]
- [66].Ye T, Yan S, Hu Y, Ding L, Wu W, Polym. Chem. 2014, 5, 186. [Google Scholar]
- [67].Xiao Y, Sun H, Du J, J. Am. Chem. Soc. 2017, 139, 7640. [DOI] [PubMed] [Google Scholar]
- [68].Chang RR, Li M, Ge SJ, Yang J, Sun QJ, Xiong L, Ind. Crop Prod. 2018, 112, 98. [Google Scholar]
- [69].Figdor CG, van Kooyk Y, Adema GJ, Nat. Rev. Immunol. 2002, 2, 77. [DOI] [PubMed] [Google Scholar]
- [70].Kaarsholm NC, Lin S, Yan L, Kelly T, van Heek M, Mu J, Wu M, Dai G, Cui Y, Zhu Y, Carballo-Jane E, Reddy V, Zafian P, Huo P, Shi S, Antochshuk V, Ogawa A, Liu F, Souza SC, Seghezzi W, Duffy JL, Erion M, Nargund RP, Kelley DE, Diabetes 2018, 67, 299. [DOI] [PubMed] [Google Scholar]
- [71].Krug AW, Visser SAG, Tsai K, Kandala B, Fancourt C, Thornton B, Morrow L, Kaarsholm NC, Bernstein HS, Stoch SA, Crutchlow M, Kelley DE, Iwamoto M, Clin. Pharmacol. Ther. 2019, 105, 417. [DOI] [PubMed] [Google Scholar]
- [72].Wang J, Yu J, Zhang Y, Kahkoska AR, Wang Z, Fang J, Whitelegge JP, Li S, Buse JB, Gu Z, Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Davis AP, Wareham RS, Angew. Chem. Int. Ed. Engl. 1999, 38, 2978. [PubMed] [Google Scholar]
- [74].a) Klein E, Crump MP, Davis AP, Angew. Chem. Int. Ed. Engl. 2004, 44, 298; [DOI] [PubMed] [Google Scholar]; b) Ke C, Destecroix H, Crump MP, Davis AP, Nat. Chem. 2012, 4, 718. [DOI] [PubMed] [Google Scholar]
- [75].a) Lecollinet G, Dominey AP, Velasco T, Davis AP, Angew. Chem. Int. Ed. Engl. 2002, 114, 4093; [DOI] [PubMed] [Google Scholar]; b) Klein E, Ferrand Y, Auty EK, Davis AP, Chem. Commun. 2007, 2390; [DOI] [PubMed] [Google Scholar]; c) Ferrand Y, Crump MP, Davis AP, Science 2007, 318, 619; [DOI] [PubMed] [Google Scholar]; d) Sookcharoenpinyo B, Klein E, Ferrand Y, Walker DB, Brotherhood PR, Ke C, Crump MP, Davis AP, Angew. Chem. Int. Ed. Engl. 2012, 51, 4586. [DOI] [PubMed] [Google Scholar]
- [76].Mooibroek TJ, Casas-Solvas JM, Harniman RL, Renney CM, Carter TS, Crump MP, Davis AP, Nat. Chem. 2016, 8, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ferrand Y, Klein E, Barwell NP, Crump MP, Jimenez-Barbero J, Vicent C, Boons GJ, Ingale S, Davis AP, Angew. Chem. Int. Ed. Engl. 2009, 48, 1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Barwell NP, Crump MP, Davis AP, Angew. Chem. Int. Ed. Engl. 2009, 48, 7673. [DOI] [PubMed] [Google Scholar]
- [79].Kuivila HG, Keough AH, Soboczenski EJ, J. Org. Chem. 1954, 19, 780. [Google Scholar]
- [80].Yetisen AK, Jiang N, Fallahi A, Montelongo Y, Ruiz-Esparza GU, Tamayol A, Zhang YS, Mahmood I, Yang S-A, Kim KS, Butt H, Khademhosseini A, Yun S-H, Adv. Mater. 2017, 29, 1606380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Zhao L, Huang QW, Liu Y, Wang Q, Wang LY, Xiao SS, Bi F, Ding JX, Materials 2017, 10, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].a) Wang B, Ma R, Liu G, Li Y, Liu X, An Y, Shi L, Langmuir 2009, 25, 12522; [DOI] [PubMed] [Google Scholar]; b) Yesilyurt V, Webber MJ, Appel EA, Godwin C, Langer R, Anderson DG, Adv. Mater. 2016, 28, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].a) Kumarasamy D, Ghosh MK, Giri TK, Polymer-Based Responsive Hydrogel for Drug Delivery, 2018; [Google Scholar]; b) Sharifzadeh G, Hosseinkhani H, Adv. Healthc. Mater. 2017, 6, 1700801. [DOI] [PubMed] [Google Scholar]
- [84].Cambre JN, Sumerlin BS, Polymer 2011, 52, 4631. [Google Scholar]
- [85].Kitano S, Kataoka K, Koyama Y, Okano T, Sakurai Y, Makromol. Chem., Rapid Commun. 1991, 12, 227. [Google Scholar]
- [86].Kitano S, Koyama Y, Kataoka K, Okano T, Sakurai Y, Control J. Release 1992, 19, 162. [Google Scholar]
- [87].Kitano S, Hisamitsu I, Koyama Y, Kataoka K, Okano T, Sakurai Y, Polymer Adv. Tech. 1991, 2, 261. [Google Scholar]
- [88].Hisamitsu I, Kataoka K, Okano T, Sakurai Y, Pharm. Res. 1997, 14, 289. [DOI] [PubMed] [Google Scholar]
- [89].Kataoka K, Miyazaki H, Bunya M, Okano T, Sakurai Y, J. Am. Chem. Soc. 1998, 120, 12694. [Google Scholar]
- [90].Matsumoto A, Ikeda S, Harada A, Kataoka K, Biomacromolecules 2003, 4, 1410. [DOI] [PubMed] [Google Scholar]
- [91].Chen S, Matsumoto H, Moro-oka Y, Tanaka M, Miyahara Y, Suganami T, Matsumoto A, Adv. Funct. Mater. 2018, 29, 1807369. [Google Scholar]
- [92].Zhang S, Chu L, Xu D, Zhang J, Ju X, Xie R, Polym. Adv. Technol. 2008, 19, 937. [Google Scholar]
- [93].Matsumoto A, Yamamoto K, Yoshida R, Kataoka K, Aoyagi T, Miyahara Y, Chem. Commun. 2010, 46, 2203. [DOI] [PubMed] [Google Scholar]
- [94].Jin X, Zhang X, Wu Z, Teng D, Zhang X, Wang Y, Wang Z, Li C, Biomacromolecules 2009, 10, 1337. [DOI] [PubMed] [Google Scholar]
- [95].a) Lapeyre V, Gosse I, Chevreux S, Ravaine V, Biomacromolecules 2006, 7, 3356; [DOI] [PubMed] [Google Scholar]; b) Zhang Y, Guan Y, Zhou S, Biomacromolecules 2006, 7, 3196. [DOI] [PubMed] [Google Scholar]
- [96].a) Zhang Y, Guan Y, Zhou S, Biomacromolecules 2007, 8, 3842; [DOI] [PubMed] [Google Scholar]; b) Lapeyre V, Ancla C, Catargi B, Ravaine V, Colloid Interface Sci J. 2008, 327, 316. [DOI] [PubMed] [Google Scholar]
- [97].Gaballa H, Theato P, Biomacromolecules 2019, 20, 871. [DOI] [PubMed] [Google Scholar]
- [98].Wang B, Ma R, Liu G, Liu X, Gao Y, Shen J, An Y, Shi L, Macromol. Rapid. Commun. 2010, 31, 1628. [DOI] [PubMed] [Google Scholar]
- [99].a) Yao Y, Zhao L, Yang J, Yang J, Biomacromolecules 2012, 13, 1837; [DOI] [PubMed] [Google Scholar]; b) Zhang G, Zhang X, Shen H, Yang J, Yang J, RSC Adv. 2014, 4, 49964; [Google Scholar]; c) Zhao L, Zhang X, Yao Y, Yu C, Yang J, Macromol. Chem. Phys. 2014, 215, 1609; [Google Scholar]; d) Zhang Y, Zhao W, Yang J, Hammouda B, Yang J, Cheng G, Eur. Polym. J. 2016, 83, 173; [Google Scholar]; e) Yao Y, Wang X, Tan T, Yang J, Soft Matter 2011, 7, 7948. [Google Scholar]
- [100].Yuan W, Li L, Zou H, RSC Adv. 2015, 5, 80264. [Google Scholar]
- [101].Carino GP, Mathiowitz E, Adv. Drug Deliv. Rev. 1999, 35, 249. [DOI] [PubMed] [Google Scholar]
- [102].a) Pridgen EM, Alexis F, Kuo T, Levy-Nissenbaum E, Karnik R, Blumberg RS, Langer R, Farokhzad OC, Sci. Transl. Med. 2013, 5, 213ra167; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Banerjee A, Ibsen K, Brown T, Chen R, Agatemor C, Mitragotri S, Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 7296; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mukhopadhyay P, Mishra R, Rana D, Kundu PP, Prog. Polym. Sci. 2012, 37, 1457. [Google Scholar]
- [103].Yu J, Zhang Y, Wang J, Wen D, Kahkoska AR, Buse JB, Gu Z, Nano Res. 2018, 10.1007/s12274-018-2264-9. [DOI] [Google Scholar]
- [104].Shiino D, Murata Y, Kataoka K, Koyama Y, Yokoyama M, Okano T, Sakurai Y, Biomaterials 1994, 15, 121. [DOI] [PubMed] [Google Scholar]
- [105].Shiino D, Kataoka K, Koyama Y, Yokoyama M, Okano T, Sakurai Y, J. Intel. Mat. Syst. Str. 1994, 5, 311. [DOI] [PubMed] [Google Scholar]
- [106].Shiino D, Murata Y, Kubo A, Kim YJ, Kataoka K, Koyama Y, Kikuchi A, Yokoyama M, Sakurai Y, Okano T, Control J. Release 1995, 37, 269. [Google Scholar]
- [107].a) Langer RS, Anderson DG, Gu Z, Aimetti AA, 2012, WO 2013123491 A1; [Google Scholar]; b) Hoeg-Jensen T, Havelund S, Nielsen PK, Markussen J, J. Am. Chem. Soc. 2005, 127, 6158. [DOI] [PubMed] [Google Scholar]
- [108].Hoeg-Jensen T, Ridderberg S, Havelund S, Schaffer L, Balschmidt P, Jonassen I, Vedso P, Olesen PH, Markussen J, J. Pept. Sci. 2005, 11, 339. [DOI] [PubMed] [Google Scholar]
- [109].Chou DH, Webber MJ, Tang B, Lin AB, Thapa LS, Deng D, Truong JV, Cortinas AB, Langer R, Anderson DG, Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Boughton CK, Hovorka R, Sci. Transl. Med. 2019, 11, eaaw4949. [DOI] [PubMed] [Google Scholar]
- [111].Moore MC, Kelley DE, Camacho RC, Zafian P, Ye T, Lin SN, Kaarsholm NC, Nargund R, Kelly TM, Van Heek M, Previs SF, Moyes C, Smith MS, Farmer B, Williams P, Cherrington AD, Diabetes 2018, 67, 1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Peppas NA, Khademhosseini A, Nature 2016, 540, 335. [DOI] [PubMed] [Google Scholar]
- [113].Saravanakumar G, Kim J, Kim WJ, Adv. Sci. 2017, 4, 1600124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Broom WA, Coulthard CE, Gurd MR, Sharpe ME, Brit. J. Pharm. Chemoth. 1946, 1, 225. [PMC free article] [PubMed] [Google Scholar]
- [115].Sumner JB, Howell SF, Bacteriol J. 1936, 32, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Ye YQ, Yu JC, Wang C, Nguyen NY, Walker GM, Buse JB, Gu Z, Adv. Mater. 2016, 28, 3115. [DOI] [PMC free article] [PubMed] [Google Scholar]