

A previously unidentified group of Sp7-dependent CD24a+ stem cells, called MDPSCs, were found to promote dental pulp regeneration.
Abstract
Dental pulp is critical to maintain the vitality of a tooth. Regeneration of pulpo-dentinal complex is of great interest to treat pulpitis and pulp necrosis. In this study, through three-dimensional spheroid culture, a group of unique multipotent stem cells were identified from mouse dental papilla called multipotent dental pulp regenerative stem cells (MDPSCs). MDPSCs exhibited enhanced osteogenic/odontogenic differentiation capabilities and could form regenerative dentin and neurovascular-like structures that mimicked the native teeth in vivo. Further analysis revealed that CD24a was the bona fide marker for MDPSCs, and their expansion was highly dependent on the expression of a key transcriptional factor, Sp7. Last, CD24a+ cells could be detected in primary dental papilla in mice and human, suggesting that MDPSCs resided in their native niches. Together, our study has identified a previously unidentified group of multipotent pulp regenerative stem cells with defined molecular markers for the potential treatment of pulpitis and pulp necrosis.
INTRODUCTION
Dental pulp plays a central role in regulating the critical vitality of a tooth, including induction of dentin formation, maintaining nutritional supply, and providing the sensational neural network that responds to external stimuli (1). Exposure of dental pulp to environmental bacterial infection due to untreated caries, trauma, and multiple restorations would cause pulpitis and pulp necrosis, the most common types of oral diseases. Clinical management of these diseases are notoriously difficult, and patients usually end up receiving root canal treatment as the standard of care, which replaces the dental pulp with filling materials and results in a permanently devitalized tooth (2). Therefore, functional regeneration of dental pulp is of great interest in both translational medicine and basic research.
Over the past decade, multiple stem cells of mesenchyme origin have been identified and characterized from various tooth tissues, including stem cells from apical papilla (SCAP) (3), stem cells from human exfoliated deciduous teeth (SHED) (4), dental follicle stem cells (5), periodontal ligament stem cells (6), dental pulp stem cells (DPSCs) (7), and dental papilla cells (DPCs) (8). Since discovery, these cells have been extensively studied about their differentiation and regeneration capabilities (9). However, the regenerative potential of these cells for dental pulp is highly variable. For DPSCs, initial studies indicated that these cells retained typical features of mesenchymal stem cells, which could be induced to differentiate into adipose and chondrogenic tissues (7). When mixed with a hydroxyapatite/tricalcium phosphate (HA/TCP ceramic powder) carrier, DPSCs could be induced to form dentin-like structures with an interface layer of odontoblast-like cells (7). It was also reported that DPSCs seeded with a poly-d,l-lactide/glycolide could form a continuous layer of regenerative dentin in vivo (10). More recently, cultured DPSC aggregates were shown to regenerate vascular-containing dental pulp in human patients, although regeneration of dentin-like structures seemed to be limited (11). In addition, DPCs and SHED cells were also shown to be able to regenerate pulp-like structures in vivo (8, 10). However, as mesenchymal stem cells are famously heterogeneous, only a small fraction of single colonies from SHEDs contained dental pulp regenerative capability (4), and multiple different subgroups within primary DPCs from the developing teeth were also documented in a recent study of single-cell transcriptome analysis (12). Therefore, it is still unclear that dental pulp regeneration capability from tooth stem cells was from a single multipotent stem cell population or from several populations with distinct differentiation potentials.
Meanwhile, although several types of stem cells have been isolated from teeth, one often overlooked fact is that these cells were all maintained and expanded through a traditional two-dimensional (2D) adherent culture system, which is notoriously known as inadequate in supporting long-term expansion of stem cells, including mesenchymal stromal cells (MSCs) (13). On the other hand, 3D spheroid culture has increasingly become widely accepted as a better choice that offers a higher degree of biological and physiological relevance, which leads to the discovery and characterization of many tissue stem cells, such as mammary gland stem cells (14), intestinal stem cells (15), and neural progenitor cells (16). More recently, this system was further adopted to use in organoid cultures to mimic organogenesis and disease pathogenesis processes in human (17). However, the use of spheroid culture in supporting self-renewal of tooth-derived stem cells was rather limited. Thus, it is important to develop a 3D spheroid culture system for tooth-derived stem cells, which may enable us to further dissect their lineage commitment and characterize subset cell groups with true tissue regenerative potentials.
In this study, we used a stem cell–supportive spheroid 3D culture system in DPCs and identified a previously undocumented multipotent population of stem cells that could form DPC spheres and maintained their self-renewal state over multiple passages. DPC spheres were positive for pluripotency markers such as Oct4, Sox2, and Nanog and exhibited enhanced osteogenic and odontogenic differentiation capabilities in vitro. When transplanted in vivo, these cells could further develop to form regenerative dentin and neurovascular-like structures that mimicked the native teeth. Therefore, we name these cells as multipotent dental pulp regenerative stem cells (MDPSCs). Last, using transcriptome analysis and flow cytometry–mediated cell sorting, MDPSCs were identified as a group of CD24+ cells, of which the self-renewal was highly dependent on Sp7 expression, a key transcription factor (TF) in bone formation and tooth development (18, 19). Last, we provided evidence that the presence of MDPSCs could also be detected in primary tissues from dental papilla in mice and human patients. Together, our data indicated that a previously unidentified group of pulpo-dentinal regenerative CD24+ stem cells were present in dental papilla and expandable in 3D culture, which would have high therapeutic potential in the treatment of pulpitis and pulp necrosis.
RESULTS
Cells from mouse dental papilla could initiate sphere formation in 3D culture
To test sphere formation in DPCs, we reasoned that a stem cell supportive 3D culture system would be required. Muse cells (multilineage differentiating stress enduring cells) are a group of multipotent stem cells identified from 3D culture of bone marrow stromal cells that could be differentiated into multiple lineages in vitro and in vivo (20). Although it is still highly controversial due to the lack of in vivo identities (21), the isolation (21) and application of muse cells in animal disease models are well documented from multiple laboratories (22–24). These evidences indicated that the 3D culture system used in muse cells identification might be stromal stem cell supportive, which makes it attractive to use in our primary DPC culture to test whether there are previous undocumented stem cells in dental papilla. We modified our culture method to incorporate the 3D culture system, where primary DPCs at passage 2 (DPCs P2; ~day 6) would be switched to a 3D suspension culture system (Fig. 1A). Unexpectedly, when primary DPCs were cultured in 3D for about 7 days, spontaneous formation of spheres could be readily noticed, which had relatively smooth surface appearance (Fig. 1B) and were of variable diameters ranging from 30 to 100 μm (fig. S1A). In addition, DPC spheres could be passaged over time through an adherence-suspension cycle for at least twice without proliferation loss (fig. S1, B and C). To exclude the possibility that DPC spheres were derived from cell aggregates rather than single cells, primary DPCs were first labeled with lentivirus of two different colors [mCherry and enhanced green fluorescent protein (EGFP)], respectively, before seeded in 3D culture (fig. S2A). DPC spheres seemed to develop from single starting cells (fig. S2B) and continuously grew over time (Fig. 1C). Moreover, DPC spheres were positive for proliferation and pluripotency markers such as Ki67, Oct4, Sox2, and Nanog, as confirmed by both immunostaining and reverse transcription polymerase chain reaction (RT-PCR) (Fig. 1, D and E). In comparison, only a minor fraction of primary DPCs P2 was positively stained with these pluripotency and proliferation markers (fig. S3). Together, these data suggested that a group of sphere-forming cells existed in mouse dental papilla, which were likely multipotent stem cells.
Fig. 1. A group of multipotent sphere-forming stem cells existed in dental papilla.
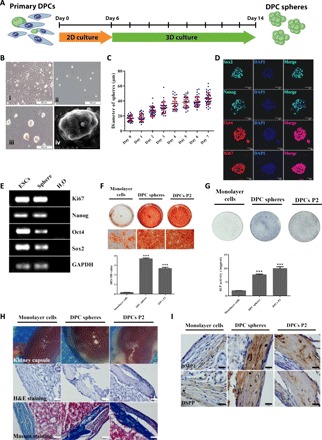
(A) Schematic diagram of the 3D culture assay. DPCs were isolated from tooth germs of neonatal pups and cultured in standard DPC medium for about 1 week before further seeded in MC culture medium to initiate sphere formation. Spheres and control monolayer cells were collected at day 8 for further characterization. (B) Representative images for the typical morphology of primary mouse DPCs P2 (i) and DPC spheres on days 0 (ii) and 7 (iii). Scale bars, 200 μm. Scanning electron microscopy morphology of DPC spheres (iv). Scale bar, 20 μm. (C) 3D culture supported continuous growth of DPC spheres. The growth and diameter of DPC sphere were monitored over time in 3D culture system. (D) DPC spheres were positive for pluripotency and proliferation markers including Oct4, Sox2, Nanog, and Ki67. DPC spheres were collected at day 8 and immunostained with antibodies against Oct4, Sox2, Nanog, and Ki67. DAPI (4′,6-diamidino-2-phenylindole) was used for nuclear staining. Scale bars, 50 μm. (E) Expression of Oct4, Sox2, Nanog, and Ki67 in DPC spheres were also confirmed by RT-PCR. Complementary DNAs from mESCs (mouse embryonic stem cells) were used as the positive control and H2O as the negative control. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (F) Alizarin Red S (ARS) staining and quantification indicated DPC sphere–derived cells retained differentiation capabilities compared with 2D monolayer cultures. Monolayer cells, DPC sphere–derived cells, and DPCs at P2 were incubated in osteoinductive medium (OIM) for 10 days, respectively. Sphere-derived cells exhibited enhanced mineralization from monolayer cultures. Error bar represents two independent experiments with triplicates. ***P < 0.001 (Student’s t test). OD, optical density. (G) ALP staining and quantification indicated DPC sphere–derived cells retained differentiation capabilities compared with 2D monolayer cultures. Cells were incubated in OIM for 7 days. Error bar represents two independent experiments with triplicates. ***P < 0.001. (H) Strongly mineralized structures were formed by DPC sphere–derived cells when transplanted in renal capsules. Monolayer cells, DPC sphere–derived cells, and DPCs P2 were implanted into renal capsule of C57BL/6 mice together with Matrigel. Samples were harvested after 4 weeks. The hematoxylin and eosin (H&E) and Masson staining were used to show the mineralized structures. Scale bars, 50 μm. (I) Immunohistochemical analysis of odontogenic markers (DMP1 and DSPP) indicated that DPC sphere–derived cells retained strong potential for odontogenic differentiation. Scale bars, 20 μm.
DPC spheres exhibited enhanced osteogenic/odontogenic differentiation capability in vitro and in vivo
To characterize the differentiation capacity of DPC spheres, cells derived from DPC spheres were cultured in osteoinduction medium (OIM) for 10 and 7 days, respectively, before Alizarin Red S (ARS) and alkaline phosphatase (ALP) staining. Cells from traditional monolayer 2D culture (the same culture time as 3D), which usually showed substantial loss of differentiation potentials, and DPCs P2, which were the starting parental cells, were used as the negative and positive controls, respectively. Both ARS and ALP staining demonstrated that cells from DPC spheres had retained enhanced mineralization capability and higher ALP activity than monolayer cells, similar to DPCs P2, which was also confirmed by quantitative analysis (Fig. 1, F and G). DPC spheres exhibited enhanced differentiation capability than adult DPSCs as well (fig. S4). These data indicated that cells from DPC spheres retained robust osteogenic differentiation capability in 3D culture.
To explore the developmental potential of DPC spheres in vivo, cells from the same three groups were combined with Matrigel before transplanted into renal capsule of C57BL/6 mice for 4 weeks. Hematoxylin and eosin (H&E) and Masson staining suggested that mineralized structures formed in both DPC spheres and DPCs P2 groups, but not in monolayer group, where only collagen fibers were noticeable (Fig. 1H). Similar results were also found when cells matrix was mixed with HA/TCP ceramic powder to enhance induction (fig. S5). Immunohistochemical analysis of odontogenic marker genes dentin matrix protein 1 (DMP1) and dentin sialophosphoprotein (DSPP) further confirmed that the newly formed mineralized tissues were from cells of odontoblast-like lineage (Fig. 1I). Together, these data suggested that cells from DPC spheres retained robust odontogenic capabilities in vivo.
DPC spheres enabled regeneration of pulpo-dentinal complex-like tissues in vivo
As shown above, DPC spheres were derived from dental papilla and retained robust osteogenic/odontogenic capabilities, we sought to examine whether cells from DPC spheres would enable regeneration of more complex tissues, such as the pulpo-dentinal complex. To mimic the dental root environment and stimulate tissue regeneration, subcutaneous transplantation of a swine-treated dentin matrix (TDM) model was chosen, as TDM was too large and hard for traditional transplantation under kidney capsule. Cells from DPC spheres, primary DPCs P2, and monolayer cultures were mixed with Matrigel and inserted into TDM before subcutaneously transplanted into immunocompromised mice for 4 weeks (Fig. 2A). Regenerated pulpo-dentinal complex-like structures were observed only in the DPC spheres group, including microvessel formation (marked with black arrows) as indicated by H&E staining, which resembled the natural pulp tissue (Fig. 2B). In addition, a layer of polarized cells lining along the newly formed dentin-like tissue (marked with white arrows) was similar to the odontoblasts along the pulp-dentin interface in the native teeth (Fig. 2B) (10, 18). These observations did not seem to be an isolated event as similar findings were repetitively recorded in a separate independent experiment (fig. S6). To further characterize newly formed tissues by DPC spheres, immunostaining of dentin-related markers (DMP1 and DSPP), vascularization-related markers [vascular endothelial growth factor (VEGF) and CD31], and neural markers [glial fibrillary acidic protein (GFAP), S100, and NF200] were performed. In all cases, robust immunofluorescent signals were detected in DPC spheres group, similar to the native teeth (Fig. 2, C to E, and fig. S7). These data suggested that cells from DPC spheres could enable efficient regeneration of pulpo-dentinal complex-like tissues in vivo, while DPCs P2 and monolayer cells could not.
Fig. 2. DPC spheres enabled functional regeneration of the pulpo-dentinal complex-like tissue in vivo.
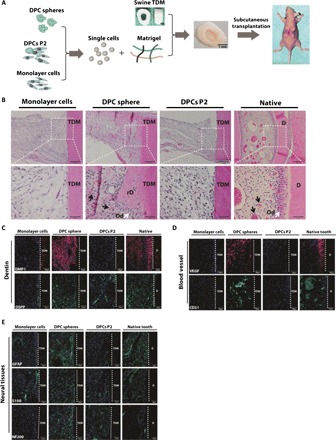
(A) Schematic diagram of TDM transplantation in nude mice. Single-cell suspension was prepared from DPC spheres, monolayer cells, and DPCs P2 before mixed with Matrigel. Cell-matrix mixtures were then inserted into swine TDM before subcutaneously transplanted into recipient nude mice for 4 weeks. Photo credit: Hong Chen, Sichuan University. (B) A pulpo-dentinal complex-like structure was formed only in the DPC spheres group. Tissues were harvested about 4 weeks after transplantation, and H&E staining was used to evaluate the regenerated tissue from each cell group. (C) Regenerated tissue from DPC spheres was positive for dentin-specific markers DMP1 and DSPP. rD, regenerated dentin; Od, odontoblast-like cells. Scale bars (from top to bottom), 100 μm (first lane), 20 μm (second lane), and 50 μm (the rest of the lane). (D) Regenerated tissue from DPC spheres was positive for blood vessel–specific markers VEGF and CD31. (E) Regenerated tissue from DPC spheres was positive for neural tissue–specific markers GFAP, S100, and NF200.
Transcriptome analysis identified signature gene network in DPC spheres
To investigate the potential molecular mechanism that leads to functional differences between monolayer cells, DPC spheres, and primary DPCs, transcriptome analysis was performed in these three groups of cells. Principal components analysis indicated that DPC spheres were significantly different from monolayer cells and DPCs P2 (Fig. 3A), which was consistent with the observed phenotype in vitro and in vivo. Significantly, differentially expressed genes were also identified in these cells and clustered into four groups (Fig. 3B): Group I represented genes highly enriched in DPC spheres, which was the most interesting group to us; group II represented genes highly expressed in monolayer cells; group III represented genes highly expressed in both monolayer cells and DPC P2; and group IV represented genes highly expressed only in DPCs P2. Gene ontology (GO) analysis revealed a significant enrichment of ossification-associated genes in DPC spheres, and the top 20 significantly enriched GO biological processes in DPC spheres indicated that up-regulated genes in spheres were associated with ossification and odontogenesis (Fig. 3C). Meanwhile, signaling pathway analysis indicated that Wnt and phosphatidylinositol 3-kinase pathway genes were also enriched in DPC spheres (Fig. 3D). To identify the potential putative markers for DPC spheres, we compared our transcriptome analysis with surface protein–encoding genes from multiple sources [Cell Surface Protein Atlas, http://wlab.ethz.ch/cspa/; GO database, http://geneontology.org (25); dental gene database, http://bite-it.helsinki.fi; and CD (cluster of determination) marker handbook, BD Biosciences]. Top 15 candidate surface markers were listed (Fig. 3E). Among these genes, Cd24a, Bambi, and Lgr6 were particularly interesting to us. Bambi, BMP (bone morphogenetic protein) and actin membrane-bound inhibitor, had a high fragments per kilobase million in DPC spheres and top fold changes when compared with DPCs P2 and monolayer cells. Lgr6 is a member of a group of unique Wnt signaling G protein–coupled receptor mediators, which is well documented for its role in both tissue (26) and cancer stem cells (27). Cd24a is also previously documented to take part in various processes of tissue and tumorigenic stem cells (28, 29). Therefore, these three genes were chosen as candidates for further validation. To confirm the expression level of three surface marker candidate genes, RT–quantitative PCR (qPCR) analysis was performed, and the results were consistent with the sequencing data, suggesting that the dataset obtained from transcriptome analysis accurately reflected gene expression differences between three cell groups (Fig. 3F). In summary, through transcriptome analysis, we have identified genes that were specifically expressed in DPC spheres, and three surface markers—Bambi, Lgr6, and Cd24a—were chosen for further functional validation.
Fig. 3. Transcriptome analysis identified the enriched genes in DPC spheres.
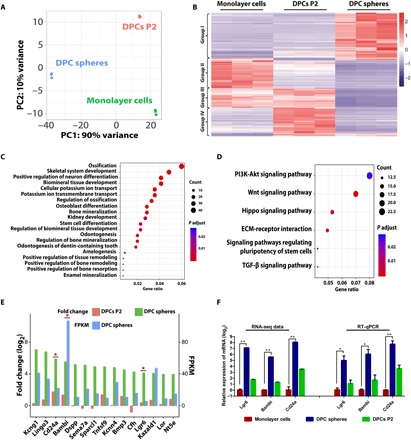
(A) Principal components (PC) analysis of DPC spheres indicated that these cells were significantly different from monolayer cells and DPCs P2. (B) Heat map of the differentially expressed genes in DPC spheres. RNA sequencing (RNA-seq) analysis of DPC spheres, monolayer cells, and DPCs P2 identified significantly differentially expressed genes in these populations. Cutoff line: Fold change > 2 and adjusted P value of <0.01. (C) GO analysis indicated a significant enrichment of ossification associated genes in DPC spheres. Top 20 significantly enriched GO biological processes in DPC spheres were shown. (D) Signaling pathway analysis indicated that top 2 significantly enriched signaling pathway were Wnt and phosphatidylinositol 3-kinase (PI3K) pathway in DPC spheres. TGF-β, transforming growth factor–β. (E) A group of surface markers were identified to be enriched in DPC spheres. Top 15 candidate surface markers were listed by comparing differential gene expression profile between DPC spheres and monolayer cells and overlapping with surface protein database (fold change ≥ 2). Candidates chosen for further validation were labeled with a star. FPKM, fragments per kilobase million. (F) Confirmation of surface marker candidate genes by RT-qPCR. RT-qPCR analysis was performed for selected candidate marker genes enriched in DPC spheres, including Lgr6, Bambi, and Cd24a. Error bar represents triplicate independent samples. *P < 0.05 and **P < 0.01 (Student’s t test). GAPDH was used as the house keeping control.
CD24a was the bona fide surface marker for sphere-initiating cells
To characterize the potential candidate surface markers, cells from monolayer culture, DPCs P2, and DPC spheres were immunostained with respective antibodies to evaluate whether the protein levels were consistent with our transcriptome analysis. Notably, CD24a protein expression was indeed only enriched in cells from DPC spheres (Fig. 4A), while no differential expression of BAMBI and LGR6 was observed (fig. S8), which was also confirmed by quantitative Western blotting (Fig. 4, B and C). Flow cytometry analysis indicated that cells from DPC spheres had a high percentage of strong CD24a+ cells (CD24a++, 72.9%) (Fig. 4D). This group of cells was also present in DPCs P2 at a much lower rate (9.32%) and gradually lost over time in monolayer culture (0.179%), which matched with the functional phenotypic difference of these cells.
Fig. 4. CD24a was the bona fide surface marker for sphere-initiating cells.
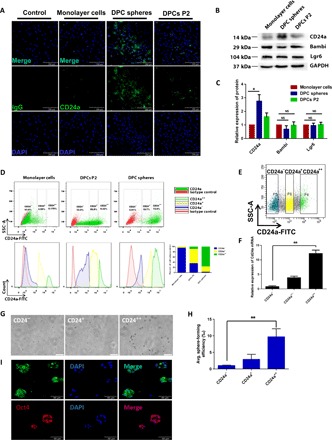
(A) Cells from DPC spheres were confirmed to have an enriched population of CD24a+ cells. Cells from each group were immunostained anti-mouse CD24a antibody before taking images. (B and C) Western blot and quantitative analysis of protein expression for CD24a, Bambi, and Lgr6 in DPCs P2, monolayer cells, and DPC spheres. Error bar represents data from two independent experiments. *P < 0.01 (Student’s t test). NS, not significant. (D) Cells from DPC spheres had a high percentage of CD24a++ cells. Single-cell suspension from monolayer cells, DPC spheres, and DPCs P2 was prepared and immunostained with anti-CD24a antibody labeled with fluorescein isothiocyanate (FITC). From top to bottom, scatter diagram and histogram were showed, respectively. Sphere-derived cells were CD24++ (72.9%), CD24+ (19.7%), and CD24− (2.91%). monolayer cells were CD24++ (0.179%), CD24+ (4.99%), and CD24− (93.8%). DPCs P2 were CD24++ (9.32%), CD24+ (50.8%), and CD24− (33%) as shown in the bar graph. Rat normal immunoglobulin G (IgG) was used as the negative control. SSC-A, side scatter area. (E) CD24a+ cells were collected through fluorescence-activated cell sorting (FACS). Three subpopulations of primary DPCs were sorted by FACS based on their expression of CD24a. (F) CD24a expression in the FACS-sorted cells was further confirmed by RT-qPCR. Error bar represents data from triplicate wells. (G) Only CD24a++ exhibited strong sphere-forming capability. Sorted cells were 3D cultured in MC medium for 7 days before taking images. Scale bars, 200 μm. (H) CD24a++ cells showed significantly higher sphere formation efficiency (17.04 ± 3.87‰) than CD24− cells (1.76 ± 0.24‰). **P < 0.01 (Student’s t test). (I) Spheres formed by sorted CD24a++ cells were positive for pluripotency markers Sox2 and Oct4. Spheres were collected at day 8 and immunostained with antibodies against Sox2 and Oct4. DAPI was used for nuclear staining.
To further validate that CD24a+ cells were indeed sphere-initiating cells, three subpopulations of primary DPCs were sorted by fluorescence-activated cell sorting (FACS) based on their expression of CD24a (Fig. 4E), and expression of CD24a was also confirmed in sorted cells by RT-qPCR (Fig. 4F). Sorted CD24a++, CD24a+, and CD24a− cells were then cultured in 3D system for 7 days, and only CD24a++ cells exhibited strong sphere-forming capability but not CD24a+ and CD24a− cells (Fig. 4, G and H). Moreover, spheres formed by sorted CD24a++ cells were also positive for pluripotency markers, such as Sox2 and Oct4 (Fig. 4I). Therefore, CD24a was indeed the bona fide surface marker that could be used to isolate these sphere-initiating cells, and we named these cells as MDPSCs.
Sp7 was the key TF to drive self-renewal of MDPSCs in vitro
To investigate the molecular mechanisms regulating self-renewal of MDPSCs, top 15 candidate TFs that were significantly enriched in DPC spheres were identified from the transcriptome profile (Fig. 5A). Among them, three TFs—Dlx3, Rcor2, and Sp7—were further chosen for downstream characterization based on their reported functions and relative expression abundance in DPC spheres. Expression of these three TFs was first confirmed by RT-qPCR (Fig. 5B), and two short hairpin RNAs (shRNAs) targeting each TF were designed and cloned into the pLKO-puro lentiviral vector. Knockdown efficiency of these shRNAs was confirmed by transducing primary DPCs with shRNA lentiviruses and RT-qPCR analysis of target genes. All three TFs could be effectively knocked down by both shRNAs (Fig. 5C). Primary DPCs with shRNAs were then seeded in 3D culture system, and sphere formation capability was quantified at day 7. Knockdown of Sp7, but not Dlx3 and Rcor2, strongly abolished DPC sphere formation (Fig. 5D). Quantitative analysis revealed that both the sphere size and quantity were significantly reduced in Sp7 shRNA knockdown samples (Fig. 5, E and F). To exclude the possibility that this phenomenon was strain specific, we further confirmed the results in a separate mouse strain (BALB/c). DPC spheres could also be induced in primary DPCs from Balb/c, and knockdown of Sp7 abolished sphere formation in these cells (fig. S9). Together, these data indicated that Sp7 was the key TF to drive self-renewal of MDPSCs in 3D culture.
Fig. 5. Sp7 was the key TF to drive self-renewal of MDPSCs in vitro.
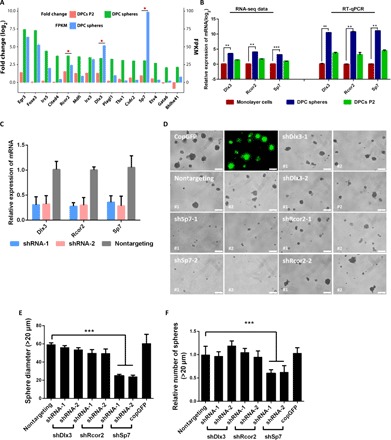
(A) A group of TFs that may be crucial for MDPSCs self-renewal were identified in DPC sphere–enriched genes. Top 15 candidate TFs were listed by comparing differential gene expression profile between DPC spheres versus monolayer cells and DPCs P2 (fold change ≥ 2). Candidates chosen for further validation were labeled with a star. (B) Expression of several enriched TFs were further confirmed by RT-qPCR. Error bar represents triplicate assays. Gapdh was used as the house keeping gene. (C) Efficient knockdown of targeted TFs by shRNAs. shRNAs targeting Dlx3, Rcor2, and Sp7 were cloned into the pLKO-puro lentiviral vector. Cell were transduced with lentivirus and harvested at day 3 for RNA extraction. RT-qPCR was then used to evaluate the knockdown efficiency of each shRNA. **P < 0.01 and ***P < 0.001 (Student’s t test). Gapdh was used as the housekeeping control. (D) Knockdown of Sp7 resulted in significant decrease of sphere formation from primary DPCs. Representative images for sphere formation in shRNAs knockdown cells were shown. Lenti-copGFP was used to monitor transduction efficiency. Nontargeting shRNA was used as the negative control. Scale bars, 100 μm. (E) Spheres from shSp7 group exhibited significant smaller diameter. Error bar represents data from three independent experiments with triplicate wells. ***P < 0.001 [one-way analysis of variance (ANOVA)]. (F) The overall number of spheres formed in shSp7-transduced cells was also significantly lower. Error bar represents data from three independent experiments with triplicate wells. ***P < 0.001 (one-way ANOVA).
MDPSCs were present in dental papilla from mice and human patients
To establish whether CD24a+ MDPSCs existed in primary tissue from dental papilla, immunofluorescence and immunohistochemical staining were performed for tooth germ sections from day 1 postnatal mouse pups. A group of CD24a+ cells were identified to be mainly present in the mesenchymal tissue underlying inner enamel epithelium layer (Fig. 6, A and B). CD24a+ cells were also present in primary human dental papilla isolated from the extracted third molar germ of an 11-year-old female patient receiving orthodontic treatment (Fig. 6C). RT-PCR also detected the expression in CD24a in human samples (Fig. 6D). Flow cytometry sorting indicated that cells from human dental papilla indeed had a substantial group of CD24+ cells (9.37%), and the expression of CD24 was also confirmed in sorted cells by RT-qPCR (Fig. 6E). Therefore, we concluded that CD24a+ MDPSCs were likely present in primary dental papilla from both mice and human patients.
Fig. 6. CD24a+ cells were present in both mouse tooth germs and human dental papilla tissues.
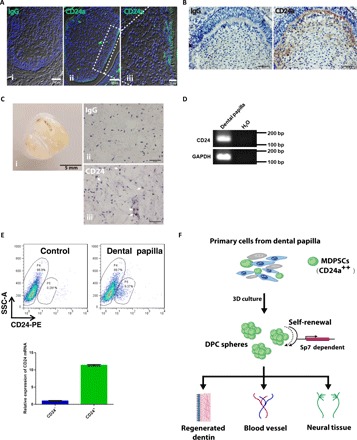
(A) CD24a+ cells were present in the developing mouse tooth germ. Immunofluorescence staining was performed for tooth germ sections from day 1 postnatal mice. Scale bars, 50 μm (i and ii) and 20 μm (iii). (B) Immunohistochemical analysis of the developing mouse tooth germ confirmed the presence of CD24a+ cells. Scale bars, 50 μm. (C) CD24a+ cells were also present in primary samples from human dental papilla. Representative CD24a+ cells were pointed out with white arrowheads. Scale bars, 50 μm (ii and iii). Photo credit: Hong Chen, Sichuan University. (D) The expression of CD24a in human DPCs was confirmed by RT-PCR. H2O was used as the negative control. bp, base pair. (E) Human DPCs had a subpopulation of CD24a+ cells (9.37%). Single-cell suspension from human DPCs was prepared and immunostained with anti-CD24a antibody labeled with PE (phycoerythrin). PBS was used as the blank control. CD24a expression in the FACS-sorted cells was further confirmed by RT-qPCR. Error bar represents data from triplicate wells. (F) Model image for MDPSCs. MDPSCs were defined as a group of CD24a+ cells present in dental papilla, which can be expanded through a 3D culture system. Self-renewal of MDPSCs seemed to be Sp7 dependent. When transplanted in vivo, MDPSCs could give rise to regenerated dentin, blood vessel, and neural tissues, which indicated that these cells had therapeutic potential for dental pulp regeneration.
DISCUSSION
In this study, we identified a previously undocumented group of multipotent stem cells from dental papilla, which could initiate DPC sphere formation in a stem cell supportive 3D culture environment and expressed typical pluripotent markers such as Oct4, Sox2, and Nanog. Typically, these cells could be passaged and clonally expanded over time through an adherence-suspension cycle for at least two to three cycles (2 to 3 weeks) without obvious loss of proliferation and self-renewal capacity. In addition, they exhibited enhanced osteogenic/odontogenic differentiation capabilities in vitro and in vivo and were capable of regenerating functional pulpo-dentinal complex-like tissues. We further provided evidence that CD24a was the bona fide surface marker that could be used to isolate these cells, and their self-renewal in 3D culture was highly dependent on Sp7 expression, a key TF for bone formation and tooth development. Last, these cells were also found to be present in primary dental papilla tissues from both mice and human. We therefore named these cells as MDPSCs (Fig. 6F).
Although several tooth stem cell types have been identified and characterized over the past decade, regeneration of functional pulpo-dentinal complex remains a challenge. The key challenge is to rebuild vascularized pulp-like tissue accompanying with highly organized histological structure resembling the native pulp, including microvessel in the central region and dentin deposition by newly differentiated odontoblasts in the peripheral existing dentinal wall (10, 30). Several types of tooth stem cells—such as SHED (31), SCAP (10), DFCs (dental follicle cells) (DFCs) (32), and DPSCs (7, 11)—have been applied in dental pulp regeneration [some even in clinics (11, 33)], but the regeneration efficiency is variable, partially due to the lack of a stem cell marker that can distinguish pluripotency of tooth MSCs versus degenerated cells over long-term 2D culture. In addition, since MSCs are notoriously heterogeneous (34), it is rather difficult to determine whether the observed tissue regenerative potential was from a single subpopulation of stem cells or from several different ones. In this regard, the discovery and characterization of MDPSCs not only offer an alternative cell source for highly efficient regeneration of pulpo-dentinal complex-like tissues but also provide an important piece of evidence that a single group of CD24a+ cells would be able to drive the regeneration process.
CD24a, also known as the heat-stable antigen, has been previously reported as one of key stem cell markers for several normal and pathogenic cell lineages including mammary gland cells, pancreatic cancer cells, and hepatocellular carcinoma cells (2, 28–30). During development, progenitor cells and metabolically active cells have higher CD24a expression than terminally differentiated cells, which reveals the close relationship between CD24a and cellular pluripotency and its strong correlation with self-renewal state (29, 35, 36). In tooth stem cells, only one publication briefly mentioned that a small percentage of SCAP cells was CD24a+, and its presence was decreased upon osteodifferentiation (37). Therefore, our study provides the report that CD24a+ cells may represent a unique group of dental pulp regenerative stem cells, which could be expanded through 3D culture. It is also noteworthy to point out that although CD24a+ cells could be identified in primary human dental papilla, further work is warranted to investigate whether these cells can be expanded through the same 3D culture system and whether these cells also retain dental pulp regenerative potential as their counterparts in mice. In addition, as the sphere-forming efficiency of sorted mouse CD24a++ cells was only ~10‰, two potential explanation could be raised: (i) The 3D culture system was not supportive enough, which requires further optimization of the medium formulation, and (ii) additional marker may be needed to further enrich the MDPSCs with high regenerative potential. Therefore, lineage tracing with CD24a would also be required to dissect the contribution of MDPSCs in tissue development and repair under physiological conditions. However, as CD24a was expressed in early mouse embryos (38), a tamoxifen-inducible cyclic adenosine 5′-monophosphate response element (CRE) strategy would be needed to control CRE expression and label CD24a+ cells specifically in dental papilla.
Together, we have identified a unique group of CD24a+ stem cells in dental papilla, which exhibited enhanced osteogenic/odontogenic differentiation capabilities and could regenerate pulpo-dentinal complex-like tissues in vivo. Our study not only sheds light on the hidden treasure of underappreciated subpopulations in tooth mesenchymal tissues but also provides an excellent alternative cell source for potential translational use in clinical management of pulpitis and pulp necrosis.
MATERIALS AND METHODS
Cell culture
Isolation and culture of primary DPCs
The dental papilla was separated from maxillary and mandibular molar tooth germ of neonatal mouse and digested in a solution containing type I collagenase (3 mg/ml) (catalog no. C0130; Sigma-Aldrich, St. Louis, MO) for 25 min at 37°C. Single-cell suspensions and dental papilla tissue were cultured in EpiCM (epithelial cell medium) basal medium supplemented with 2% fetal bovine serum (FBS) (catalog no. 0010, STEMCELL Technologies, USA), 1% cell growth supplement [EpiCGS (epithelial cell growth supplement), catalog no. 4152, STEMCELL Technologies, USA], and 1% penicillin/streptomycin solution (catalog no. 0503, STEMCELL Technologies, USA). DPCs P2 were used for this study. All cells were grown in a humidified incubator (Thermo Fisher Scientific, USA) at 37°C with 5% CO2, and the medium was refreshed every 3 days.
DPC sphere formation assay
Primary DPCs P2 were seeded onto six-well ultralow attachment plates (catalog no. 3471, Corning, USA) at a density of 1 × 104 cells/cm2 in basal EpiCM medium supplemented with methylcellulose (MC) (catalog no. M03134, STEMCELL Technologies, USA). Cell medium and MC were thoroughly mixed at a 3:2 (v/v) ratio by gentle pipetting to minimize cell aggregation (MC culture hereafter). To ensure nutrient supply, fresh medium (1 of 10 of the initial volume) was gently added to the culture every 2 days. The size and number of spheres were recorded after 7 days. For secondary and tertiary sphere formation, the spheres were collected, dissociated into single cells using 0.25% trypsin/EDTA (catalog no. SM-2003-C, Millipore, USA), and transferred to MC culture as described above at a density of 2.5 × 103 cells/cm2 in 24-well ultralow attachment plates (catalog no. 3473, Corning, USA). To prove that the spheres were derived from single cells rather than aggregation, primary DPCs were marked with lenti-mCherry and lenti-EGFP, respectively, before cultured in 3D under same culture condition described above.
ARS and ALP assays
To detect osteogenic differentiation capability of DPC spheres, sphere-derived cells were cultured in OIM [α–minimum essential medium supplemented with 10% FBS, 5 mM l-glycerophosphate, 100 nM dexamethasone, and ascorbic acid (50 μg/ml)]. Calcium deposition in the extracellular matrix was determined by ARS staining (pH 4.3; catalog no. A5533, Sigma-Aldrich, USA) at day 10 after differentiation. Mineralized bone nodules were destained with 10% cetylpyridinium chloride in double distilled water, and the calcium concentration was determined by absorbance measurements at 405 nm. ALP activity was detected according to the manufacturer’s recommendations using an ALP color development kit (catalog no. C3206, Beyotime, China) and an ALP activity kit (catalog no. A059-2, Jiancheng, China) and normalized with total protein loadings. The absorbance of each well was determined by measurements at 520 nm.
In vivo transplantation
Renal capsule transplantation
To characterize the osteogenic/odontogenic differentiation capabilities of monolayer cells, sphere-derived cells and DPCs P2 in vivo cells were mixed with Matrigel (catalog no. 354234, BD Biosciences, USA) at a density of 1.0 × 106 cells/10 μl, with or without HA/TCP ceramic powder (20 mg per sample). Cell-Matrigel pellets (a total of approximately 3.3 × 105 cells in 3 μl of pellet) were then transplanted into the left renal capsule of each 8-week-old C57BL/6 male mouse (Dashuo Experimental Animal Co. Ltd., Chengdu, China). Eighteen mice were randomly designated into six groups: monolayer cells mixed with or without HA/TCP group, sphere-derived cells with or without HA/TCP group, and DPCs P2 with or without HA/TCP group. All animal experimental procedures used were approved by the Institutional Animal Care and Use Committee of Sichuan University. All animals were maintained under standardized conditions with the temperature of 21°C and a 12-hour light cycle and had free access to food and water. The grafts were obtained at 4 weeks after transplantation, which were further fixed with 4% paraformaldehyde, decalcified with buffered 10% EDTA for 1 week, and then embedded in paraffin for further experiments.
Subcutaneous transplantation in nude mice
To prepare TDM for cell implants, incisor teeth were extracted from the swine mandible. Periodontal ligament tissues were carefully scraped away along with partial removal of outer cementum, inner dental pulp tissue, and predentin. High-speed air turbine handpiece was used to cut the root canal into pieces of 2 to 3 mm in length. Next, the root canal pieces were concussed for three times (5 to 6 min each time) by an ultrasonic cleaner before further exposed to a series of decreasing EDTA solution (from 17 to 5%) for 5 min and washed with deionized water for 5 min (three times). Last, the prepared TDM was in DPC culture medium with penicillin (50 U/ml) and streptomycin (50 mg/ml) at 4°C. Then, monolayer cells, sphere-derived cells, and DPCs P2 were combined with Matrigel (1.0 × 106 cells/10 μl) and seeded into the treated root canals, where the end near the skin of TDM was sealed with radiopaque calcium hydroxide composition (Dycal, catalog no. 10800, DENSPLY, USA) to prevent cell leakage and then subcutaneously implanted the root in immunocompromised nude mice for 4 weeks.
Cytological and histological staining
Cellular immunofluorescence
Cells were washed twice with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde at room temperature for 30 min. Fixed cells were permeabilized with 0.5% Triton X-100 for 15 min (permeabilization was not performed for cell surface antigens). Cells were then blocked in 5% bovine serum albumin in PBS containing 0.1% Triton X-100 for 1 hour at room temperature. Cells were stained with primary antibodies overnight at 4°C and then washed three times with PBS. Secondary antibodies were stained for 30 min at room temperature and then washed three times with PBS before 4′,6-diamidino-2-phenylindole staining (catalog no. D1306, Thermo Fisher Scientific, USA) for 5 min. Cells were lastly examined under a fluorescence confocal microscope (Olympus FV1000, Japan).
H&E and Masson staining
The samples were decalcified in 10% EDTA (pH 7.0) for 1 month and embedded in paraffin. Samples were sectioned at 5 μm thickness. Paraffin tissue sections were deparaffinized in xylene and rehydrated through graded ethanol solutions. Then, the sections were stained with H&E or Masson’s trichrome (catalog no. BA-4079, Baso, China) according to the manufacturer’s instructions.
Immunofluorescence for tissue sections
The preparation of tissue sections was the same as H&E staining. Paraffin tissue sections were deparaffinized in xylene and rehydrated through graded ethanol solutions. As for frozen sections, they were rehydrated through PBS. Endogenous peroxidases were blocked using 3% hydrogen peroxide. For antigen retrieval, the paraffin sections were processed by 0.1% trypsin solution (catalog. no. X1020, Solarbio, USA) for 30 min at 37°C, while the frozen sections were processed by 1× quick antigen retrieval solution for 5 min at room temperature (catalog no. KGIHC005, KeyGen Biotech, China). Then, the sections were blocked by 10% normal goat serum for 30 min to 1 hour at 37°C and incubated with primary antibodies overnight at 4°C. Subsequent steps were performed the same as cellular immunofluorescence described above.
Immunohistochemical analysis of tissue sections
Briefly, the experimental procedures were the same with tissue sections immunofluorescence until incubating with secondary antibodies. Then, the sections were stained using a 3,3-diaminobenzidine kit. Subsequent steps were performed according to the manufacturer’s recommendations, and the immune reactions were visualized under a light microscope (Olympus BX43F, JEOL, Tokyo, Japan).
The primary and secondary antibodies and their dilutions used in this study were as follows: rabbit anti-Sox2 (1:200; ab97959, Abcam, MA, USA), rabbit anti-Nanog (1:200; ab80892, Abcam, MA, USA), rabbit anti-OCT4 (1:200; D121072, Sangon Biotech, Shanghai, China), rabbit anti-Ki67 (1:200; ab15580, Abcam, MA, USA), rabbit anti-DMP1 (1:200; 3844-100, BioVision, CA, USA), rabbit anti-DSPP (1:200; 508413, Zenbio, Chengdu, China), rabbit anti-CD31 (1:200; NB100-2284, Novus Bio, CO, USA), rabbit anti-VEGF (1:200; ab46154, Abcam, MA, USA), rabbit anti-GFAP (1:200; ab7260, Abcam, MA, USA), rabbit anti-S100 (1:200; ab868, Abcam, MA, USA), mouse anti-NF200 (1:200; ab82259, Abcam, MA, USA), rat anti-CD24 (1:200; ab64064, Abcam, MA, USA), rabbit anti-Lgr6 (1:200; ab126747, Abcam, MA, USA), rabbit anti-BAMBI/NMA (non-metastatic gene A) (1:200; ab203070, Abcam, MA, USA), mouse anti-CD24 (1:200; NB100-77903, Novus Bio, CO, USA), Alexa Fluor 488 goat anti-mouse (1:400; A11001, Invitrogen, Eugene, OR, USA), Alexa Fluor 488 goat anti-rabbit (1:400; A11008, Invitrogen, Eugene, OR, USA), Alexa Fluor 555 goat anti-rabbit (1:400; A21428, Invitrogen, Eugene, OR, USA), Alexa Fluor 488 donkey anti-rat (1:400; A21208, Invitrogen, Eugene, OR, USA), goat anti-rabbit immunoglobulin G (IgG)–horseradish peroxidase (HRP) (1:400; ZB-2301, ZSGB-BIO, Beijing, China), and anti-rat IgG-HRP (G2514; Santa Cruz Biotechnology, CA, USA). Negative controls were carried out by substituting a normal IgG for the primary antibody, including rat IgG isotype control (10700, Invitrogen, USA), rabbit IgG isotype control (10500C, Invitrogen, USA), and mouse IgG isotype control (NI03; Millipore, USA).
Western blot analysis
DPC spheres, DPCs P2, and monolayer cells were collected, and total proteins were extracted in lysis buffer (catalog no. KGP702-100, KeyGen Biotech, China) and quantified using the BCA (bicinchoninic acid) Protein Assay Kit (catalog no. KGP902, KeyGen Biotech, China) according to the manufacturer’s instructions. Then, equal amounts (20 μg) of protein lysates from each sample were separated by SDS–polyacrylamide gel electrophoresis. The proteins were then transferred onto polyvinylidene difluoride membranes. The membranes were blocked with 5% skim milk in TBST (Tris-Buffered Saline and Tween 20 medium) and then incubated with primary antibodies. Anti–glyceraldehyde-3-phosphate dehydrogenase (catalog no. 200306-7E4, RRID: AB_2722713, Zen Bioscience, China) was used as the internal control. Samples were then incubated with HRP-conjugated secondary antibodies and visualized with Clarify Western ECL Substrate (catalog no. 1705061, Bio-Rad, USA) according to the manufacturer’s protocol. Images were captured with Image Quant LAS 4000 Mini (GE Healthcare Life Sciences) and quantified by scanning densitometry (ImageQuant TL, GE Healthcare Life Sciences). Experiments were independently performed for each sample, and at least three technical replicates were performed for each of the treated samples and controls.
Transcriptome analysis by RNA sequencing
RNA purity was measured using NanoDrop, and RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Agilent Bioanalyer 2100 system (Agilent Technologies, CA, USA). mRNA and RNA sequencing were conducted to investigate the global expression profile of monolayer cells, DPC spheres, and DPCs P2 (n = 3), and 150–base pair paired-end reads were generated. The adaptors and low-quality reads from raw reads of each sample were trimmed to obtain clean reads. The clean reads were mapped against the mouse genome (Mus musculus, GRCm38) using HISAT2 v2.1.0 (22). The expression level of each gene was quantified guided by reference annotation (M. musculus, GRCm38.91) using featureCounts v1.6.0 (39). The differentially expressed genes were analyzed using R and DESeq2 package (39) by the cutoff of log fold change > 2 and P adjusted value of <0.01. Functional annotation and enrichment analysis of the significantly differentially expressed genes were performed with R and clusterProfiler package (40). CD markers were collected from the Cell Surface Protein Atlas database (http://wlab.ethz.ch/cspa/) and Human and Mouse CD Marker Handbook (BD Biosciences). The complete sequencing dataset has been submitted to the Gene Expression Omnibus database (GSE138372).
RT-PCR and quantitative real-time PCR
Total RNAs were extracted using the TRIzol method (catalog no. 15596026, Thermo Fisher Scientific, USA). After extraction, 1 μg of total RNAs was used for first-strand complementary DNA using SuperScript II (catalog no. 18064014, Thermo Fisher Scientific, USA). RT-PCR was performed to visualize the expression of pluripotent-related genes in spheres with DreamTaq Green PCR Master Mix (catalog no. K1082, Thermo Fisher Scientific, USA) and Mastercycler nexus gradient PCR cycler (Eppendorf, Germany). ES (embryonic stem) cells served as a positive control, and H2O served as a negative control. qPCR was performed using SYBR Premix Ex Taq (catalog no. 172-5121, Bio-Rad, USA). The primer sequences used are listed in table S1.
Flow cytometry
Cells were washed twice with ice-cold PBS. Approximately 5 × 105 cells were incubated with primary antibody (rat anti-CD24a, ab64064, Abcam, USA) for 30 min on ice. Cells were then washed two to three times with ice-cold PBS and incubated with the secondary antibody (donkey anti-rat IgG–fluorescein isothiocyanate, catalog no. A21208, Thermo Fisher Scientific, USA) for 30 min. Rat normal IgG (catalog no. 10700, Thermo Fisher Scientific, USA) was used as the negative control. Cells were further washed three times with ice-cold PBS, resuspended thoroughly, and filtered through a 70-μm nylon mesh. FACS was performed using Becton-Dickinson Accuri C6 (BD Biosciences, USA) for analysis and BD Aria III (BD Biosciences, USA) for sorting. The data were analyzed by FlowJo (version 7.6.1).
Lentiviral RNA interference
At least two shRNAs (table S2) per target gene were designed according to The RNAi Consortium (TRC) library and then cloned into pLKO.1-puro lentiviral backbone (plasmid no. 8453, Addgene, USA). To generate lentivirus, 293FT cells were seeded into six-well plates at a density of 7.5 × 105 per well. The next day, the packaging plasmid (rev, gag, and vsv-g) and lentiviral vectors (shRNAs or nontargeting and copGFP) were transfected into 293FT cells in a molar ratio of 5:1 with liposomes. Viral supernatant was harvested at 48 hours after transfection, centrifuged at 4000 rpm for 5 min to remove cell debris, and further concentrated 20-fold in basal medium with Lenti-X (catalog no. 632180, Takara, Japan). Primary DPCs were transduced with the concentrated virus and polybrene to enhance viral infection (final concentration, 4 μg/ml) and centrifuge at 750g for 45 min. Viral medium was changed to fresh culture medium the next day, and the transduced cells were drug selected by puromycin (3.5 μg/ml). To evaluate the knockdown efficiency, shRNA-infected cells were harvested at day 3 for RNA isolation and reverse transcription. shRNA knockdown efficiency was measured by qPCR with primers listed in table S2.
Statistical analysis
Data are presented as the means ± SD from at least three independent experiments. Statistical comparison was performed using Student’s t test. Statistical significance was analyzed using the SPSS 22.0 software package (IBM Corporation, Armonk, NY). P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We would like to extend our gratitude to all members of Li Lab for the support and useful discussions. Funding: This study is supported by grants from National Key Research and Development Program of China (no. 2017YFA0104801), Strategic Priority Research Program of the Chinese Academy of Sciences (XDA16010505), National Natural Science Foundation of China (no. 31401262), “One Thousand Talents” program from the Chinese Central Government and Sichuan Province, and the Fundamental Research Funds for the Central Universities (SCU2019D013). Author contributions: Z.L., G.C., and H.C. conceived the study. H.C. performed most of the experiments, collected and analyzed the data, and drafted the manuscript. H.F. performed the RNA sequencing data analysis and produced corresponding transcriptome figures. X.W. performed lentiviral RNA interference experiments and proved the function of Sp7. Y.D. and S.Z. participated in the animal experiments. Y.L. and H.H. helped with FACS validation. T.W. and Y.Y. helped with paraffin tissue section. Z.L. and G.C. designed the experiments and oversaw the collection of results and data interpretation. Z.L. revised the manuscript. Z.L. and W.T. provided experimental platform and financial supports. All authors have seen and approved the final version of the paper. Competing interests: Z.L. and H.C. are inventors on a patent application related to this work filed by Sichuan University (no. 201910674796.1, filed 25 July 2019). The other authors declare that they have no competing interests. Data and materials availability: Tissue sections, cell lines, and plasmids related to the paper can be provided by Z.L. from Sichuan University pending scientific review and a completed material transfer agreement. Requests should be submitted to Zhonghan.Li@scu.edu.cn. All data needed to evaluate the conclusions in the paper are present in the paper and the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/15/eaay1514/DC1
REFERENCES AND NOTES
- 1.Simon S. R. J., Berdal A., Cooper P. R., Lumley P. J., Tomson P. L., Smith A. J., Dentin-pulp complex regeneration: From lab to clinic. Adv. Dent. Res. 23, 340–345 (2011). [DOI] [PubMed] [Google Scholar]
- 2.European Society of Endodontology , Quality guidelines for endodontic treatment: consensus report of the European Society of Endodontology. Int. Endod. J. 39, 921–930 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Sonoyama W., Liu Y., Yamaza T., Tuan R. S., Wang S., Shi S., Huang G. T.-J., Characterization of the apical papilla and its residing stem cells from human immature permanent teeth: A pilot study. J. Endod. 34, 166–171 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miura M., Gronthos S., Zhao M., Lu B., Fisher L. W., Robey P. G., Shi S., SHED: Stem cells from human exfoliated deciduous teeth. Proc. Natl. Acad. Sci. 100, 5807–5812 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morsczeck C., Gotz W., Schierholz J., Zeilhofer F., Kuhn U., Mohl C., Sippel C., Hoffmann K. H., Isolation of precursor cells (PCs) from human dental follicle of wisdom teeth. Matrix Biol. 24, 155–165 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Seo B. M., Miura M., Gronthos S., Bartold P. M., Batouli S., Brahim J., Young M., Robey P. G., Wang C. Y., Shi S., Investigation of multipotent postnatal stem cells from human periodontal ligament. Lancet 364, 149–155 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Gronthos S., Mankani M., Brahim J., Robey P. G., Shi S., Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 97, 13625–13630 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tziafas D., Kodonas K., Differentiation potential of dental papilla, dental pulp, and apical papilla progenitor cells. J. Endod. 36, 781–789 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Liu J., Yu F., Sun Y., Jiang B., Zhang W., Yang J., Xu G. T., Liang A., Liu S., Concise reviews: Characteristics and potential applications of human dental tissue-derived mesenchymal stem cells. Stem Cells 33, 627–638 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Huang G. T., Yamaza T., Shea L. D., Djouad F., Kuhn N. Z., Tuan R. S., Shi S., Stem/progenitor cell-mediated de novo regeneration of dental pulp with newly deposited continuous layer of dentin in an in vivo model. Tissue Eng. Part A 16, 605–615 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xuan K., Li B., Guo H., Sun W., Kou X., He X., Zhang Y., Sun J., Liu A., Liao L., Liu S., Liu W., Hu C., Shi S., Jin Y., Deciduous autologous tooth stem cells regenerate dental pulp after implantation into injured teeth. Sci. Transl. Med. 10, eaaf3227 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Takahashi A., Nagata M., Gupta A., Matsushita Y., Yamaguchi T., Mizuhashi K., Maki K., Ruellas A. C., Cevidanes L. S., Kronenberg H. M., Ono N., Ono W., Autocrine regulation of mesenchymal progenitor cell fates orchestrates tooth eruption. Proc. Natl. Acad. Sci. U.S.A. 116, 575–580 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bianco P., Robey P. G., Simmons P. J., Mesenchymal stem cells: Revisiting history, concepts, and assays. Cell Stem Cell 2, 313–319 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inman J. L., Robertson C., Mott J. D., Bissell M. J., Mammary gland development: Cell fate specification, stem cells and the microenvironment. Development 142, 1028–1042 (2015). [DOI] [PubMed] [Google Scholar]
- 15.van der Flier L. G., Clevers H., Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol. 71, 241–260 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Nunes M. C., Roy N. S., Keyoung H. M., Goodman R. R., McKhann G. II, Jiang L., Kang J., Nedergaard M., Goldman S. A., Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat. Med. 9, 439–447 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Bredenoord A. L., Clevers H., Knoblich J. A., Human tissues in a dish: The research and ethical implications of organoid technology. Science 355, eaaf9414 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Debnath S., Yallowitz A. R., McCormick J., Lalani S., Zhang T., Xu R., Li N., Liu Y., Yang Y. S., Eiseman M., Shim J. H., Hameed M., Healey J.-H., Bostrom M. P., Landau D. A., Greenblatt M. B., Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature 562, 133–139 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bae J.-M., Clarke J. C., Rashid H., Adhami M. D., Cullough K. M., Scott J. S., Chen H., Sinha K. M., de Crombrugghe B., Javed A., Specificity Protein 7 Is Required for Proliferation and Differentiation of Ameloblasts and Odontoblasts. J. Bone Miner. Res. 33, 1126–1140 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuroda Y., Kitada M., Wakao S., Nishikawa K., Tanimura Y., Makinoshima H., Goda M., Akashi H., Inutsuka A., Niwa A., Shigemoto T., Nabeshima Y., Nakahata T., Nabeshima Y., Fujiyoshi Y., Dezawa M., Unique multipotent cells in adult human mesenchymal cell populations. Proc. Natl. Acad. Sci. U.S.A. 107, 8639–8643 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nombela-Arrieta C., Ritz J., Silberstein L. E., The elusive nature and function of mesenchymal stem cells. Nat. Rev. Mol. Cell Biol. 12, 126–131 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iseki M., Kushida Y., Wakao S., Akimoto T., Mizuma M., Motoi F., Asada R., Shimizu S., Unno M., Chazenbalk G., Dezawa M., Muse cells, nontumorigenic pluripotent-like stem cells, have liver regeneration capacity through specific homing and cell replacement in a mouse model of liver fibrosis. Cell Transplant. 26, 821–840 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kinoshita K., Kuno S., Ishimine H., Aoi N., Mineda K., Kato H., Doi K., Kanayama K., Feng J., Mashiko T., Kurisaki A., Yoshimura K., Therapeutic potential of adipose-derived SSEA-3-positive muse cells for treating diabetic skin ulcers. Stem Cells Transl. Med. 4, 146–155 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uchida N., Kushida Y., Kitada M., Wakao S., Kumagai N., Kuroda Y., Kondo Y., Hirohara Y., Kure S., Chazenbalk G., Dezawa M., Beneficial Effects of Systemically Administered Human Muse Cells in Adriamycin Nephropathy. J. Am. Soc. Nephrol. 28, 2946–2960 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carbon S., Ireland A., Mungall C. J., Shu S., Marshall B., Lewis S.; AmiGO Hub; Web Presence Working Group , AmiGO: Online access to ontology and annotation data. Bioinformatics 25, 288–289 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Snippert H. J., Haegebarth A., Kasper M., Jaks V., van Es J. H., Barker N., van de Wetering M., van den Born M., Begthel H., Vries R. G., Stange D. E., Toftgard R., Clevers H., Lgr6 marks stem cells in the hair follicle that generate all cell lineages of the skin. Science 327, 1385–1389 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Huang P. Y., Kandyba E., Jabouille A., Sjolund J., Kumar A., Halliwill K., McCreery M., DelRosario R., Kang H. C., Wong C. E., Seibler J., Beuger V., Pellegrino M., Sciambi A., Eastburn D. J., Balmain A., Lgr6 is a stem cell marker in mouse skin squamous cell carcinoma. Nat. Genet. 49, 1624–1632 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang W., Sui X., Zhang D., Liu M., Ding M., Shi Y., Deng H., CD24: A novel surface marker for PDX1-positive pancreatic progenitors derived from human embryonic stem cells. Stem Cells 29, 609–617 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Shakiba N., White C. A., Lipsitz Y. Y., Yachie-Kinoshita A., Tonge P. D., Hussein S. M., Puri M. C., Elbaz J., Morrissey-Scoot J., Li M., Munoz J., Benevento M., Rogers I. M., Hanna J. H., Heck A. J., Wollscheid B., Nagy A., Zandstra P. W., CD24 tracks divergent pluripotent states in mouse and human cells. Nat. Commun. 6, 7329 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuang R., Zhang Z., Jin X., Hu J., Shi S., Ni L., Ma P. X., Nanofibrous spongy microspheres for the delivery of hypoxia-primed human dental pulp stem cells to regenerate vascularized dental pulp. Acta Biomater. 33, 225–234 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cordeiro M. M., Dong Z., Kaneko T., Zhang Z., Miyazawa M., Shi S., Smith A. J., Nör J. E., Dental pulp tissue engineering with stem cells from exfoliated deciduous teeth. J. Endod. 34, 962–969 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Guo W., He Y., Zhang X., Lu W., Wang C., Yu H., Liu Y., Li Y., Zhou Y., Zhou J., Zhang M., Deng Z., Jin Y., The use of dentin matrix scaffold and dental follicle cells for dentin regeneration. Biomaterials 30, 6708–6723 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Nakashima M., Iohara K., Recent progress in translation from bench to a pilot clinical study on total pulp regeneration. J. Endod. 43, S82–S86 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Pevsner-Fischer M., Levin S., Zipori D., The origins of mesenchymal stromal cell heterogeneity. Stem Cell Rev. 7, 560–568 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Fang X., Zheng P., Tang J., Liu Y., CD24: From A to Z. Cell. Mol. Immunol. 7, 100–103 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee T. K., Castilho A., Cheung V. C., Tang K. H., Ma S., Ng I. O., CD24+ liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell 9, 50–63 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Sonoyama W., Liu Y., Fang D., Yamaza T., Seo B. M., Zhang C., Liu H., Gronthos S., Wang C.-Y., Wang S., Shi S., Mesenchymal stem cell-mediated functional tooth regeneration in swine. PLOS ONE 1, e79 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Y., Wu F., Zhang L., Wu X., Li D., Xin J., Xie J., Kong F., Wang W., Wu Q., Zhang D., Wang R., Gao S., Li W., Transcriptional defects and reprogramming barriers in somatic cell nuclear reprogramming as revealed by single-embryo RNA sequencing. BMC Genomics 19, 734 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dissanayaka W. L., Zhu L., Hargreaves K. M., Jin L., Zhang C., Scaffold-free prevascularized microtissue spheroids for pulp regeneration. J. Dent. Res. 93, 1296–1303 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fennema E., Rivron N., Rouwkema J., van Blitterswijk C., de Boer J., Spheroid culture as a tool for creating 3D complex tissues. Trends Biotechnol. 31, 108–115 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/15/eaay1514/DC1