

Soft extracellular matrix boosts the activation of MSCs by TNFα to generate and recruit monocytes in a paracrine manner.
Abstract
Mesenchymal stromal cells (MSCs) modulate immune cells to ameliorate multiple inflammatory pathologies. Biophysical signals that regulate this process are poorly defined. By engineering hydrogels with tunable biophysical parameters relevant to bone marrow where MSCs naturally reside, we show that soft extracellular matrix maximizes the ability of MSCs to produce paracrine factors that have been implicated in monocyte production and chemotaxis upon inflammatory stimulation by tumor necrosis factor–α (TNFα). Soft matrix increases clustering of TNF receptors, thereby enhancing NF-κB activation and downstream gene expression. Actin polymerization and lipid rafts, but not myosin-II contractility, regulate mechanosensitive activation of MSCs by TNFα. We functionally demonstrate that human MSCs primed with TNFα in soft matrix enhance production of human monocytes in marrow of xenografted mice and increase trafficking of monocytes via CCL2. The results suggest the importance of biophysical signaling in tuning inflammatory activation of stromal cells to control the innate immune system.
INTRODUCTION
Mesenchymal stromal cells (MSCs) in bone marrow (BM) consist of different subpopulations with multiple biological functions, including multipotent differentiation (1), maintenance of hematopoietic stem cells (HSCs) (2), and immunomodulation (3). The role of MSCs in immunomodulation has been shown mostly in the context of adoptive transfer where plastic-adherent cells derived from the BM are expanded in vitro and administered to treat diverse inflammatory pathologies (4). Thus, it remains generally unclear how physiologically relevant signals from the extracellular matrix (ECM) in the BM microenvironment regulate these immunomodulatory functions of MSCs. Since MSCs show preclinical efficacy in some disease models without long-term engraftment (5), it has been hypothesized that one key mode of action by MSCs is through paracrine secretion of soluble factors. MSCs express a range of receptors to recognize inflammatory signals, including tumor necrosis factor–α (TNFα), interferon-γ (IFN-γ), and lipopolysaccharide (LPS) (4). Upon inflammatory stimulation, MSCs produce chemokines to recruit immune cells, followed by factors that polarize or differentiate immune cells (6). This mode of action is highlighted in regulation of monocytes and their lineages by MSCs (4). For instance, nestin+ MSCs in BM secrete CCL2 in response to inflammation in vivo, causing monocytes to emigrate into circulation (7). Adoptively transferred MSCs secrete a number of factors to polarize macrophages such as prostaglandin E2 (PGE2) and TNFα-stimulated gene 6 (TSG-6), which, in turn, attenuate sepsis (8) and regenerate damaged tissues (9), respectively. Therefore, understanding how signals from the ECM regulate MSCs to direct monocyte functions will help inform both fundamental insights and previously unidentified strategies to control inflammation and program tissue regeneration (10).
Genetic labeling and ablation studies show that some MSC subpopulations are localized like pericytes in the vasculature of the BM (11) and contribute to both adipogenic and osteogenic differentiation in vivo (1). Other MSC populations are localized more exclusively at the rigid endosteal surface and contribute to bone maintenance and regeneration (12). However, vascular and endosteal regions often overlap, and hence, some MSCs contact both regions simultaneously (2). Recent studies show that the BM exhibits a diversity of biophysical cues contributed by the ECM. First, the BM shows a range of stiffness (Young’s modulus, E) from 2 (“soft”) to 100 (“stiff”) kPa (13). In addition, the central regions of the BM may act more like viscoelastic solids than the outer regions of the BM due to fluid transport and exchange through large central vessels (14). Advances in biomaterial design have enabled investigators to independently control these biophysical parameters, leading to insights that MSCs are highly sensitive to stiffness (15) and stress relaxation (16) of matrices, all of which subsequently influence multilineage differentiation. Previous studies show that matrix stiffness regulates secretions of proangiogenic factors (17) and factors relevant to hematopoietic recovery (18), while substrate dimensionality or topography regulates secretions of factors involved in immunomodulation (19). MSCs primed on soft substrates increase survival after radiation-induced hematopoietic failure in vivo (20), while a bone-derived scaffold maintains functionality of hematopoietic progenitors when cultured together with MSCs (21). However, it remains generally unclear how mechanical cues of the ECM coordinate with biochemical cues during inflammation to program MSC secretomes and, subsequently, to achieve specific functional outcomes. In addition, the relevance of matrix biophysical properties to immunomodulatory functions of MSCs in vivo remains unclear.
By leveraging alginate-based engineered hydrogels, we tested the hypothesis that physiologically relevant matrix stiffness affects the ability of MSCs to modulate monocyte functions under inflammation by paracrine signaling. We show that soft matrix maximizes TNFα-mediated induction of both chemokines and cytokines in MSCs involved in recruitment and differentiation of monocytes, respectively. Through experiments and modeling, we elaborate that soft matrix primarily increases clustering of TNF receptors (TNFRs) and binding of TNFα to the cell surface, thereby enhancing activation of nuclear factor κB (NF-κB) to up-regulate downstream genes. Mechanistic studies show that actin polymerization and lipid rafts are involved in mechanosensitive activation of MSCs with TNFα, but not myosin-II contractility. Consistent with these results, we show that human MSCs primed with TNFα in soft matrix enhance in vivo production of human monocytes in marrow of xenografted mice and trafficking of monocytes via CCL2.
RESULTS
Matrix stiffness regulates TNFα-induced gene expression in MSCs
Since BM exhibits diverse mechanical properties (Fig. 1A) (13), we used an alginate-based hydrogel system with a conjugated minimal integrin adhesion ligand containing Arg-Gly-Asp (RGD) (“alginate-RGD”). MSCs were encapsulated in the ionically cross-linked alginate-RGD hydrogel so that they could be exposed to a three-dimensional (3D) environment with viscoelastic properties (16) typical of marrow (“3D gels”) (14). By varying ionic cross-linking density, the initial elastic modulus of 3D gels (here, we use the term “matrix stiffness”) was tuned as follows: “soft matrix,” E = ~2 kPa, and “stiff matrix,” E = ~35 kPa (fig. S1A). We first tested whether matrix stiffness regulates constitutive protein secretion by using primary human BM MSCs transduced with Gaussia luciferase (~20 kDa). Matrix stiffness does not affect diffusion of Gaussia luciferase proteins alone (fig. S1B) and does not alter the ability of genetically engineered MSCs to constitutively produce Gaussia luciferase over time (fig. S1C). We then determined whether matrix stiffness affects expression of monocyte regulatory genes in MSCs upon inflammatory stimulation in vitro. TNFα was chosen as the stimulatory cytokine because myeloid cells are known to produce it shortly after inflammation, and it activates MSCs to affect monocyte differentiation and trafficking (22). Recombinant TNFα (~50%) diffuses rapidly (t1/2 = ~1.2 hours) from the media into 3D gels regardless of their stiffness (fig. S1D). MSCs encapsulated in soft matrix show a higher fold increase in mRNA expression of monocyte regulatory genes, including chemokines [CCL2 and CCL7; monocyte trafficking (7)], interleukins [IL6 and IL8; monocyte differentiation (23)], and an anti-inflammatory factor [TSG6 (9)] (Fig. 1B), in response to TNFα. Genes that are known to regulate myeloid lineage differentiation (CSF1, 2) and hematopoiesis (SCF, CXCL12, and IL11) also show a similar trend, but with a lower fold increase in gene expression by TNFα (fig. S1E). In contrast, genes involved in angiogenesis (VEGF and ANGPT1) and macrophage polarization (IL10) are up-regulated by TNFα to the same extent regardless of matrix stiffness (fig. S1E). Since CCL2 and IL6 show the highest up-regulation by TNFα with sensitivity to matrix stiffness, subsequent studies have focused on these two genes.
Fig. 1. 3D matrix stiffness regulates expression of TNFα-inducible genes implicated in monocyte functions.
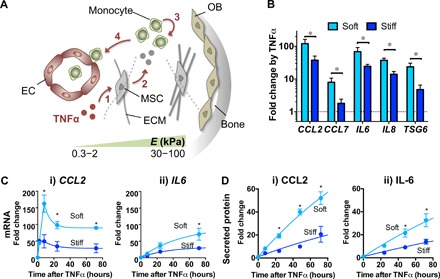
(A) Schematic showing that matrix stiffness in the bone marrow (BM) microenvironment can potentially influence MSC activation by TNFα to modulate monocyte functions in marrow. The extracellular matrix (ECM) in the central marrow and vascular [endothelial cell (EC)] regions is softer (Young’s modulus E = 0.3 to 2 kPa), while that near the bone surface [osteoblast (OB)] is stiffer (E = 30 to 100 kPa) (13). Upon inflammation, TNFα could activate MSCs (1) to produce secreted factors (2) that can influence monocyte production (3) and trafficking (4) before systemic distribution. (B) Effects of 3D matrix stiffness on TNFα-mediated up-regulation of gene expression in MSCs. MSCs were encapsulated in soft or stiff alginate-RGD 3D hydrogels, incubated for 1 day, and treated with TNFα (100 ng/ml) for 3 days. Transcript levels of the indicated genes were measured by qPCR and normalized against glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Fold change by TNFα for each group was then calculated from the untreated control (in log scale). (C) Effects of 3D matrix stiffness on mRNA expression kinetics in response to TNFα. (i) CCL2: Data from 8 to 72 hours were fitted to standard exponential decay curves. t1/2 and plateau for soft: 5.2 hours, 90-fold; stiff: 8.9 hours, 28-fold. (ii) IL6: Data were fitted to standard one-phase association curves. t1/2 and plateau for soft: 36.5 hours, 95-fold; stiff: 23.2 hours, 33-fold. (D) Effects of 3D matrix stiffness on protein secretion kinetics in response to TNFα. Data were fitted to standard one-phase association curves. (i) CCL2: t1/2 = 126 hours for both soft and stiff; plateau for soft = 156-fold and for stiff = 61-fold. (ii) IL-6: t1/2 = 204 hours for both soft and stiff; plateau for soft = 143-fold and for stiff = 58-fold. Fold change refers to increase over untreated condition. For (B) to (D), paired t test, *P < 0.05 soft versus stiff at each time point (n = 3 donors). Error bars, ±SEM.
Consistent with the results from 3D gels, MSCs plated on the 2D soft gel show higher up-regulation of CCL2 and IL6 in response to TNFα than MSCs on the 2D stiff gel or plastic culture (E ~ GPa) (fig. S1F). MSCs in soft matrix show ~150-fold increase in CCL2 mRNA after 8 hours of TNFα stimulation, followed by stabilization at ~90-fold at 24 to 72 hours (Fig. 1C, i). MSCs in soft matrix show ~3-fold higher CCL2 mRNA than those in stiff matrix between 8 and 72 hours. Unlike CCL2, TNFα gradually increases IL6 mRNA (Fig. 1C, ii). MSCs in soft matrix show ~3-fold increase in the maximal level of IL6 mRNA compared with those in stiff matrix. Consistent with the mRNA kinetics, CCL2 protein reaches its half-maximal value faster than IL-6 protein in response to TNFα (Fig. 1D). For both proteins, MSCs in soft matrix show ~2.5-fold higher expression than those in stiff matrix. We chose to use a single MSC donor in the subsequent studies because different MSC donors all exhibited higher expression of CCL2 protein upon TNFα stimulation when encapsulated in soft rather than stiff matrix (fig. S1G). The percentage of viable cells remains >70% in both soft and stiff matrices after culturing for 3 days (fig. S1H). TNFα does not affect the viability of MSCs encapsulated in either soft or stiff matrix (fig. S1H). The effects of matrix stiffness appear to be more selective for TNFα signaling since it does not alter downstream activation of signal transducers and activators of transcription 1 (STAT1) in MSCs by another inflammatory cytokine IFN-γ (fig. S1I). Therefore, matrix stiffness is a key biophysical parameter that regulates TNFα-induced gene expression.
Matrix stiffness regulates TNFα-induced NF-κB activation via TNFR clustering
Since soft matrix enhances TNFα-mediated up-regulation of both CCL2 and IL-6 in a persistent manner (Fig. 1, C and D), we hypothesized that soft matrix enhances activation of NF-κB, a well-known transcription factor of the TNFα signaling pathway (24). ML-210B, an inhibitor of IκB kinase β (IKKβ), suppresses TNFα-induced expression of CCL2 and IL-6 at median inhibitory concentration (IC50) 0.5 to 1 μM in both soft and stiff matrices (fig. S2A), confirming that NF-κB activation is required for up-regulation of downstream targets by TNFα. As expected (24), the experimental data show that NF-κB becomes maximally activated as early as 10 min after stimulation with TNFα, followed by decrease in 60 min. Consistent with our hypothesis, MSCs in soft matrix show ~1.6-fold higher levels of activated NF-κB in all of the tested time points at t ≥ 10 min (Fig. 2A and fig. S2B).
Fig. 2. Soft matrix increases NF-κB activation by facilitating TNFR1 clustering in response to TNFα.
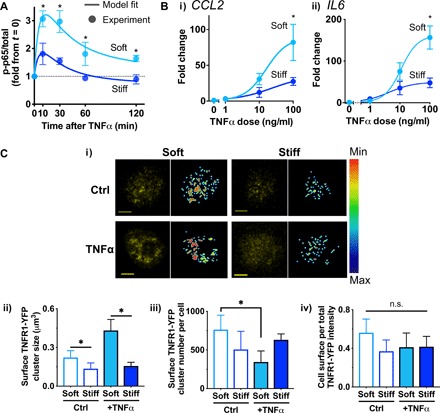
(A) NF-κB activation kinetics of MSCs in soft or stiff 3D gels in response to TNFα (100 ng/ml) evaluated by p65 phosphorylation at Ser536 (p-p65). The experimental results from soft and stiff alginate hydrogels are fitted to the I1-FFL–based model derived analytically (R2 > 0.8; Supplementary Materials). (B) Dose-response curves of TNFα to up-regulate mRNA expression in soft and stiff alginate-RGD hydrogels after the 3-day continuous treatment. (i) CCL2: IC50 and maximum fold increase for soft = 17 μM, 88-fold, and for stiff = 20 μM, 35-fold. (ii) IL6: IC50 and maximum fold increase for soft = 9 μM, 160-fold, and for stiff = 7 μM, 50-fold. *P < 0.05, paired t test. For (A) and (B), paired t test, *P < 0.05 soft versus stiff at each time point or dose (n = 3 experiments). Error bars, ±SEM. (C) Cell surface distribution and clustering of TNFR1-YFP in MSCs encapsulated in soft or stiff 3D gels. (i) Representative confocal images for each group showing maximum projection of TNFR1-YFP (left, yellow) and cell surface TNFR1-YFP after filtering (right, scale bar, min: 0, max: 255; fig. S2H). (ii) Cluster size of TNFR1-YFP on the cell surface. (iii) Cluster number of TNFR1-YFP per cell. (iv) Cell surface per total TNFR1-YFP intensity. P < 0.05 one-way Brown-Forsythe and Welch ANOVA for (ii) and (iii) with Dunnett T3 multiple comparisons test, *P < 0.05 (n ≥ 15 cells from two experiments). Error bars, ±SD. n.s., not significant.
We developed a parsimonious mathematical model using a minimal number of functions and variables to gain further insights into how matrix stiffness may regulate NF-κB activation by TNFα. Because NF-κB activation occurs in a pulse-like manner (24) (Fig. 2A), we devised a set of ordinary differential equations describing an incoherent type-1 feed-forward loop (I1-FFL) (25), a simple network motif where activation of TNFRs not only drives NF-κB activation through the IKK followed by IκB degradation (24) but also recruits putative factors that inhibit NF-κB activation (fig. S2C, i). From this model, the governing equation describing the NF-κB activation kinetics under I1-FFL can be derived analytically (fig. S2C, ii, and Supplementary Materials). Fitting the data to the equation (Fig. 2A) suggests that matrix stiffness primarily changes the maximal level of activated NF-κB but not the other parameters of the I1-FFL model (fig. S2D and Supplementary Materials).
Matrix stiffness may affect the maximal level of activated NF-κB by tuning either the constitutive activation rate of NF-κB or dose response of TNFα binding to its receptors (Supplementary Materials). The basal level of activated NF-κB in the absence of TNFα remains the same when MSCs are encapsulated in either soft or stiff matrix (fig. S2E), suggesting that the constitutive level of activated NF-κB is less likely influenced by matrix stiffness. Therefore, we tested whether matrix stiffness regulates the TNFα dose response under continuous treatment for 3 days. While soft matrix increases the maximum response of CCL2 and IL6 mRNA expression, matrix stiffness does not affect the dose required for the half-maximum response (i.e., TNFα potency) (Fig. 2B). Since matrix stiffness does not alter total TNF receptor 1 (TNFR1) expression (fig. S2F), the data suggest that matrix stiffness may regulate surface receptor clustering or density, but not intrinsic receptor affinity or conformation. To further investigate this idea, we performed confocal imaging analysis of hydrogel-encapsulated live MSCs that overexpress TNFR1 fused with yellow fluorescent protein (YFP). We labeled gel-encapsulated live MSCs with a cell-tracking dye to label the cytoplasmic compartment so that TNFR1-YFP in the cell surface and the cytoplasmic compartments could be distinguished by image processing (fig. S3A and Materials and Methods). As predicted, the cluster size of cell surface TNFR1-YFP is generally larger when MSCs are encapsulated in soft than stiff matrix and cultured for 1 day even in the absence of TNFα (Fig. 2C, i and ii). In response to TNFα, MSCs in soft matrix show ~2-fold increase in the cluster size of surface TNFR1-YFP (Fig. 2C, ii) and ~2-fold decrease in the number of surface TNFR1-YFP clusters per cell (Fig. 2C, iii). Since the level of surface TNFR1-YFP relative to that of the total TNFR1-YFP remains unchanged by TNFα (Fig. 2C, iv), the results suggest that TNFR1-YFP undergoes consolidation by lateral clustering in response to TNFα when MSCs are in soft matrix. In contrast, TNFα does not influence TNFR1-YFP clustering when MSCs are in stiff matrix (Fig. 2C). The same phenotypes are also observed with HeLa cells (fig. S3B), which are known to express α5β1 integrins (26) and undergo TNFR1 clustering (27), suggesting that the effect of matrix stiffness on TNFR1 clustering in response to TNFα may be generalizable to other adherent cells. Thus, soft matrix is an enabling environment for MSCs to respond to TNFα by TNFR clustering, thereby facilitating activation of downstream NF-κB and subsequent gene expression.
Matrix stiffness regulates TNFα binding to cell surface via actin polymerization and lipid rafts
Since actin polymerization and contractility are important for the ability of cells to sense matrix stiffness (15), we tested whether these processes are required for mechanosensitive activation of MSCs by TNFα. Consistent with increased TNFα-induced TNFR1 clustering in soft matrix (Fig. 2C), more TNFα binds to MSCs in soft matrix than in stiff matrix (Fig. 3A). While inhibition of actin polymerization by latrunculin A suppresses TNFα ligand binding when the hydrogel is soft, inhibition of myosin-II contractility by blebbistatin does not affect TNFα ligand binding (Fig. 3A). Consistent with these results, short-term treatment of latrunculin A is sufficient to disrupt up-regulation of CCL2 mRNA by TNFα selectively in the soft hydrogel (Fig. 3B). However, blebbistatin does not alter CCL2 mRNA up-regulation by TNFα (Fig. 3C). Thus, the results suggest that actin polymerization, but not myosin-II contractility, mediates enhanced responsiveness of MSCs to TNFα in soft matrix.
Fig. 3. Actin polymerization and lipid rafts mediate responsiveness of MSCs to TNFα in soft matrix.
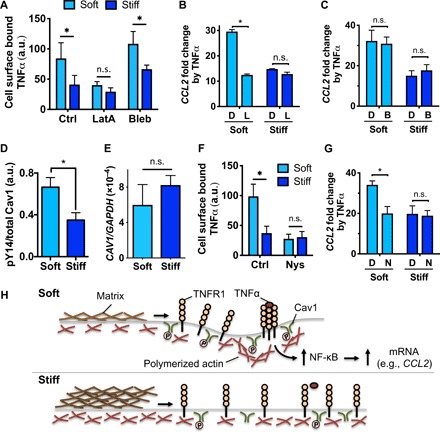
(A) TNFα binding to MSCs in soft or stiff 3D gels in the presence of DMSO (Ctrl), latrunculin A (LatA, 0.25 μM), or blebbistatin (Bleb, 10 μM) for 3 hours. P < 0.05 two-way ANOVA with Sidak’s multiple comparison test, *P < 0.05 (n = 3 experiments). (B) Effects of inhibiting actin polymerization on TNFα-induced CCL2 mRNA expression. MSCs in soft or stiff 3D gels were preincubated with DMSO (“D”) or latrunculin A (“L”) for 1 hour, treated with or without TNFα in the presence of the drugs for 2 hours, followed by washout and incubating in media for 1 day prior to qPCR. (C) Effects of inhibiting myosin-II contractility on TNFα-induced CCL2 protein expression. Gel-encapsulated MSCs were treated with TNFα in the presence of DMSO (“D”) or blebbistatin (“B”) in the same manner as (B). (D) Phosphorylation level of caveolin-1 (Cav1) at Tyr14 in MSCs after 1 day of encapsulation in soft or stiff 3D gels. (E) Gene expression of CAV1 relative to GAPDH measured by qPCR after culturing MSCs in soft or stiff 3D gels for 1 day. (F) TNFα binding to MSCs in soft or stiff 3D gels in the presence of DMSO (Ctrl) or nystatin (Nys, 50 μM) for 2 hours, followed by washout and incubating in media for 1 day prior to qPCR. (G) Effects of inhibiting lipid rafts on TNFα-induced CCL2 mRNA expression in the presence of DMSO (“D”) or nystatin (“N”) in the same manner as (B). For (B) to (G), *P < 0.05, paired t test; n = 3 experiments; and error bars, ±SEM. (H) Model of actin polymerization and lipid rafts in mediating matrix stiffness–dependent binding of TNFα to the cell surface. In soft matrix, lipid rafts mediate redistribution of actin polymerization to facilitate clustering of TNFR1 in response to TNFα. In contrast, stiff matrix impedes this process, thereby impairing TNFR1 clustering and TNFα binding. a.u., arbitrary unit.
To understand what other mechanisms may be involved in regulating mechanosensitive TNFα activation, we explored roles of lipid rafts, which are known to facilitate receptor clustering (28) and to recruit TNFR1 for NF-κB activation by TNFα (29). Lipid rafts are sensitive to biophysical stimuli as they rapidly disappear when intracellular tension is high (30). Consistent with this notion, MSCs in the soft matrix exhibit a higher phospho-Y14 level of caveolin-1 than the stiff matrix in the absence of TNFα, indicating higher lipid raft activation in soft matrix (Fig. 3D). The expression level of caveolin-1 remains similar after MSCs are cultured in soft and stiff matrices for 1 day (Fig. 3E). Disrupting lipid rafts with nystatin reduces TNFα binding to MSCs in soft, but not stiff, matrix (Fig. 3F). Consistently, treating MSCs with nystatin decreases TNFα-mediated expression of CCL2 in MSCs encapsulated in the soft matrix (Fig. 3G).
To further understand how actin polymerization and lipid rafts mediate TNFR1 clustering as a function of matrix stiffness, MSCs overexpressing both TNFR1-YFP and LifeAct–red fluorescent protein (RFP) that binds to F-actin (31) were studied. F-actin is greater in MSCs in stiff matrix than MSCs in soft matrix (fig. S4A). Most TNFR1-YFP cluster on the cell surface at regions of high F-actin (fig. S4B). Dual expression of TNFR1-YFP and LifeAct-red reveals a similar effect of matrix stiffness on TNFR1-YFP clustering upon TNFα stimulation as in Fig. 2C (fig. S4C). Consistent with the results in Fig. 3F, nystatin inhibits TNFα-induced clustering of TNFR1-YFP on the cell surface in soft matrix (fig. S4C). F-actin colocalized with TNFR1-YFP near the cell surface is increased in soft matrix, but not stiff matrix, in response to TNFα; this effect is abolished by nystatin (fig. S4D). Concomitantly, F-actin is decreased near the cell surface in regions where TNFR1-YFP clusters are not present, when MSCs in soft matrix are treated with TNFα—nystatin also reverses this effect (fig. S4E). Thus, soft matrix enables lipid rafts to enhance TNFR1 clustering upon TNFα stimulation by mediating redistribution of actin polymerization (Fig. 3H).
Matrix stiffness regulates MSC paracrine modulation of monocyte production and chemotaxis
Since matrix stiffness mediates TNFα-induced production of cytokines implicated in monocyte differentiation and polarization (Fig. 1B), we tested whether matrix stiffness influences the ability of MSCs to affect hematopoiesis in vitro by conducting coculture experiments. After priming human MSCs in different culture environments with or without TNFα, they were retrieved and cocultured with cord blood CD34+ hematopoietic and stem progenitor cells (HSPCs) in the viscous alginate-RGD fluid without cross-linking, which mimics the known viscosity value of blood or marrow (32) (fig. S5A, i). Most MSCs remain viable after a 4-day culture in the alginate-RGD fluid (fig. S5A, ii). Multicolor flow cytometry was used to quantify different hematopoietic subpopulations as described (33, 34). While MSCs do not affect early HSPCs, multipotent progenitors, and common myeloid progenitors, they generally increase granulocyte/monocyte progenitors and differentiated CD14+ monocytes—in particular, MSCs primed in soft matrix with TNFα shows the highest number of differentiated monocytes compared with MSCs primed in stiff matrix or on 2D plastic culture (fig. S5B). To confirm these results in vivo, we established human hematopoiesis prior to MSC delivery by injecting cord blood CD34+ cells in the nonobese diabetic (NOD)/severe combined immunodeficient (SCID)/IL-2γ−/− (NSG) mice. In this model, human myeloid lineage becomes detectable after 14 days (35). After 10 days of transplanting cord blood CD34+ cells, we encapsulated human MSCs in soft or stiff 3D alginate-RGD hydrogels, cultured for 1 day, and treated with or without TNFα for 3 days. Alginate lyase was then used to retrieve MSCs from gels, followed by injection of MSCs into the right tibial BM after 14 days of cord blood CD34+ transplantation (Fig. 4A). Phosphate-buffered saline (PBS) was delivered into the left tibial BM as a control. Four days after delivering MSCs, the total percentage of human CD45+ cells in marrow is 20 to 30% on average with no significant difference between the tibias or across the experimental groups (fig. S6A). Previous studies show that three monocyte subsets exist in humans based on expression of CD14 and CD16 during differentiation (36). Consistent with these studies, we show that the majority of the monocytes in marrow across the tested groups are classical (CD14+CD16−), while ≤5% of CD14+ cells (or ≤1% of total human CD45+ cells) are intermediate (CD14+CD16+). Nonclassical (CD14−CD16+) monocytes are not detectable in the NSG mice, as shown previously (37). Delivery of TNFα-primed MSCs from either soft or stiff matrix leads to ~3-fold higher percentage of classical monocytes in the cell-injected tibia compared with that of unprimed MSCs or PBS control (Fig. 4B, i). The PBS-injected tibia also shows increased classical monocytes upon delivery of TNFα-primed MSCs from soft, but not stiff, matrix to the other tibia (Fig. 4B, i). In addition, TNFα-primed MSCs from the soft matrix increase percentage of intermediate monocytes in the cell-injected tibia to the known physiological level (38) (1 to 2% of CD14+ cells, or 0.2 to 0.4% of total human CD45+ cells) (Fig. 4B, ii). Intermediate monocyte levels are also increased in the PBS-injected left tibia when TNFα-primed MSCs from the soft matrix are delivered to the right tibia (Fig. 4B, ii). Live imaging via IVIS shows that MSCs remain localized within the cell-injected tibia after delivery for at least 4 days (Fig. 4C). Since the effect of MSCs on monocyte production in a distal BM compartment is not seen when they are stimulated with TNFα in 3D stiff matrix, it is less likely that the effect will be seen with MSCs primed on 2D matrices or plastic culture (fig. S1F) due to an order of magnitude lower level of up-regulation in monocyte regulatory factors as compared with MSCs in 3D gels. Together, the results show that ex vivo priming of MSCs with TNFα in soft matrix, but not stiff matrix, enhances monocyte production in both local and distal BM compartments in vivo.
Fig. 4. Soft matrix promotes the ability of MSCs to produce and recruit monocytes upon TNFα stimulation.
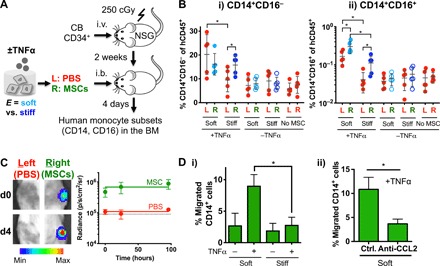
(A) Experimental scheme. Sublethally irradiated (250 cGy) NSG mice were injected intravenously (i.v.) with 20,000 human cord blood HSPCs per 20 g of mouse. After 2 weeks, MSCs that were cultured in soft or stiff gels for 1 day followed by ±TNFα treatment (100 ng/ml) for three additional days were retrieved and delivered to the right (R) tibia by an intrabone (i.b.) route, while the left (L) tibia was delivered with PBS. After 4 days, both tibias were analyzed for human monocyte subsets. (B) Percentage of (i) classical (CD14+CD16−) and (ii) intermediate (CD14+CD16+) human monocytes in tibias. P < 0.05, two-way ANOVA with Sidak’s multiple comparison test, *P < 0.05. n = 5 recipients from two independent experiments. (C) MSCs remain localized in one tibia after intrabone delivery to NSG mice. Firefly luciferase–transduced MSCs were delivered in the right tibia, while PBS was injected in the left tibia, followed by IVIS imaging over 4 days. Left: Representative images at days 0 and 4 after MSC delivery. Scale bar, radiance (p/sec/cm2/sr, min: 1 M, max: 15 M). Right: Quantification of radiance signals over 4 days. The signal from the left tibia remains at the background level (~90,000, dotted line), while that from the right tibia remains ~10-fold higher over the 4 days. (D) TNFα-treated MSCs in soft matrix increase migration of human peripheral blood CD14+ monocytes via secretion of CCL2. MSCs in soft or stiff alginate hydrogel were treated with TNFα (100 ng/ml) for 1 day prior to washout and collection of the media for 1 day for the Transwell migration assay. (i) The extent of CD14+ migration under chemotactic gradient through 3-μm pores for 3 hours. P < 0.05 from one-way ANOVA with Tukey’s post hoc test, *P < 0.05 (n = 3 experiments). (ii) CCL2 is required for increased chemotaxis of CD14+ cells by the media from TNFα-treated MSCs in soft matrix. The media were treated with a neutralizing antibody against CCL2 (anti-CCL2) or isotype control (ctrl) for 3 hours prior to the Transwell assay. *P < 0.05, paired t test (n = 3 experiments). All error bars for both (i) and (ii), ±SEM.
Once monocytes are generated, they need to be recruited to blood vessels so that they can enter circulation. Since CCL2 secreted from MSCs plays a critical role in this process (7), we tested whether matrix stiffness regulates the ability of MSCs to recruit monocytes and other blood lineages. CD14+ monocytes (~2.5%) from human peripheral blood mononuclear cells migrate through 3-μm pores when the conditioned media from human MSCs are applied—priming human MSCs in the soft matrix with TNFα prior to collecting the media enhances CD14+ cell migration to ~10%, while doing so in the stiff matrix or on 2D plastic culture shows no effect (Fig. 4D, i, and fig. S6B). The neutralizing antibody against CCL2 is sufficient to eliminate the phenotype (Fig. 4D, ii). Migration of lymphocyte lineages is generally low (<2.5%) toward the conditioned media from MSCs on 2D plastic culture, but priming with TNFα increases migration of CD4+ and CD8+ T cells and CD56+ natural killer cells to ~15%, while that of CD19+ B cells remains ~2.5% (fig. S6B). In contrast, 5 to 25% of lymphocytes are migrated toward the conditioned media from MSCs cultured in soft or stiff matrix even in the absence of TNFα, while TNFα does not further enhance migration (fig. S6C). These results suggest that, in addition to enhancing production of monocytes, soft matrix increases the ability of MSCs to selectively recruit them upon TNFα activation.
DISCUSSION
Stromal cells play important roles in regulating immune cells across different tissues. Previous studies have mostly focused on how a variety of biochemical cues enable stromal cells to modulate immune cells (4, 6). Here, we show that physiological relevant mechanical cues regulate the sensitivity of BM MSCs to an inflammatory cytokine TNFα and their subsequent ability to modulate monocytes. Presence of TNFα has been closely linked to controlling monocyte turnover and functions. Myeloid rather than lymphoid cells were shown to be mainly responsible for in vivo production of TNFα during inflammation (39), which in turn regulates in vivo survival and functions of monocytes (40). CCL2-producing stromal cells in vivo are strategically localized near the vasculature in the BM to recruit and egress monocytes during inflammation (7). Consistent with this notion, our results show that matrices mimicking stiffness of the soft vascular niche lead to higher production of monocyte regulatory factors in MSCs upon TNFα stimulation than those mimicking the more rigid endosteal niche (Fig. 1). Thus, the results suggest the important role of biophysical cues in the BM in modulating TNFα-mediated production of monocyte regulatory factors in MSCs.
The activation kinetics of NF-κB in response to TNFα has been extensively studied with quantitative modeling (24). An incoherent feed-forward loop from competitive binding between NF-κB and an inhibitory transcription factor to promoter regions allows cells to tune TNFα-induced transcription in response to fold change rather than absolute abundance of activated NF-κB (41). However, our analysis suggests a possibility that an incoherent feed-forward loop may also play an upstream role in the NF-κB pathway when TNFα activates and clusters TNFRs (fig. S2C). The exact identity of the putative inhibitory components in the loop remains to be elucidated, but previous studies suggest potential candidates, including wild-type p53-induced phosphatase 1 (WIP1) (42) and protein phosphatase 4 regulatory subunit 1 (PP4R1) (43). While soft matrix persistently increases NF-κB activation by ~1.6-fold compared with stiff matrix after 10 min of TNFα stimulation (fig. S2B), this effect appears to be amplified downstream as shown by more than twofold higher up-regulation in target genes (Fig. 1) and paracrine monocyte production and migration (Fig. 4). Future studies will help elucidate how the effect of matrix stiffness on TNFα activation can result in amplified downstream effects.
Our studies point to potential mechanisms by which matrix stiffness regulates TNFα ligand binding to the cell surface. A previous study shows that EDTA decreases binding of biotinylated LPS to monocytes after 60 min of incubation (44). Here, EDTA was used to rapidly digest alginate gels so that encapsulated MSCs can be retrieved in ~5 min prior to analyzing TNFα ligand binding on the cell surface (Materials and Methods). While this short time frame of EDTA treatment may less likely affect TNFα binding to TNFR, fluorescently tagged TNFα may be used in future studies to directly visualize TNFα binding to MSCs in gels. Consistent with this idea, confocal imaging of live MSCs in intact gels shows that soft matrix increases surface TNFR1 clustering (Fig. 2C). Previous studies show that cells cultured on soft matrices internalize integrin β1 receptors through lipid rafts (45) and integrin β3 receptors through clathrin (46), both in a myosin-II–dependent manner. Cells on less contractile 2D geometry were recently shown to increase TNFα-induced activation of NF-κB by decreasing myosin-II–mediated contractility (47). However, our results suggest a distinct regulatory mode, where matrix stiffness modulates cell sensitivity to an inflammatory signal in a myosin-II independent but actin polymerization– and caveolae–dependent manner (Fig. 3). Actin polymerization and caveolae may synergize with each other to facilitate TNFR clustering and TNFα binding in soft matrix, since polymerized actin is known to stabilize the neck part of caveolae to concentrate surface receptors (28). As the ECM becomes stiffer, actin polymerization may instead be used to increase cortical tension (15, 48), which is shown to decrease caveolae formation (30). Elucidating mechanisms behind how actin polymers and lipid rafts interact as a function of matrix stiffness will be important to further understand roles of matrix stiffness in regulating surface receptor dynamics and its implications in MSC functions.
In addition to the NF-κB pathway, matrix stiffness can potentially influence other TNFα-activated downstream signaling pathways, including mitogen-activated protein kinases (e.g., p38 and JNK) and caspases (49). In particular, JNK and caspases-8 are required to induce apoptosis in response to TNFα in immune cells, while NF-κB activation is known to inhibit cell death (50). However, our results show that TNFα does not affect the viability of MSCs (fig. S1H). TNFR1 can activate both survival and apoptotic pathways depending on interactions with distinct adaptor proteins, such as TNFR-associated death domain protein (TRADD) and Fas-associated death domain (FADD) (49). Thus, future studies may help identify adaptor proteins that bind to TNFR1 as a function of matrix stiffness and how this process can selectively affect downstream signaling.
While TNFα directly acts on HSCs to regulate hematopoiesis under both baseline and inflammatory conditions (51), our results suggest that stromal cells together with matrix stiffness may also contribute to monocyte generation from hematopoietic progenitors in response to TNFα. The NSG model used in this study shows a transient increase in monocyte subpopulations between 2 and 3 weeks after transplantation of human hematopoietic progenitors (37). Since injection of human TNFα-primed MSCs from soft matrix leads to the physiological level of human intermediate monocytes (Fig. 4B, ii), this model can potentially be used to study clinical relevance of this population, which was previously implicated in liver regeneration (38). While the NSG model is appropriate to study the short-term effect of MSCs on human monocyte production, other emerging models, such as mice engineered to express human cytokines (37), can be used in future studies to study more long-term effects. In addition, genetic knockout strategies to delete TNFRs specifically in MSCs in situ could help validate their contribution to controlling monocyte turnover.
The approach presented here can broadly serve as a platform to prime MSCs with a combination of biophysical and biochemical cues and deliver them orthotopically to the BM to control immune cell turnover in vivo. Different routes of administration have been explored to test therapeutic efficacy of MSCs, including intravascular routes, which have been pursued by ~40% of the clinical trials (52). The intravascular routes are commonly used to deliver mononuclear blood cells as in hematopoietic transplantation, since they can readily deform and squeeze through confined spaces, including capillaries (53). In contrast, MSCs are larger and more rigid than blood cells, hindering their trafficking through small pores (54). Consistent with these observations, MSCs delivered intravascularly are known to be localized primarily to the lungs due to microembolism and to be cleared within 24 hours (5) without successful engraftment in marrow (55). Since monocyte lineages play a role in clearance, it is possible that phagocytosis of MSCs that fail to engraft after intravascular injection can potentially affect the phenotype of monocytes or macrophages as suggested (56). In contrast, our results inform a fundamentally distinct strategy where delivering MSCs via the intrabone route not only maintains their in vivo residence in local marrow but also encourages production of monocytes from human hematopoietic progenitors in vivo. Generalizability of this strategy will depend on in vivo demonstration with multiple MSC donors in the future. In addition, it will be important to successfully retrieve cells from gels while maintaining viability and functionality after retrieval. While alginate lyase used in this study is known to be orthogonal to mammalian cells because they do not express alginate (57), introducing more natural mechanisms in hydrogel design such as protease degradable sequences (58) can be useful to retrieve cells without a need to use an external degradation mechanism. MSCs in soft hydrogel maintain higher CCL2 expression for 1 day even after transiently stimulated with TNFα for 2 hours (Fig. 3), suggesting a possibility that MSCs can potentially maintain their secretomes even after ex vivo priming is complete. However, strategies to locally specify insoluble biophysical cues around cells in vivo, such as microfluidic thin gel coating (59), can help present MSCs with predefined biochemical and matrix biophysical cues in a continuous manner in vivo. Since monocytes generated from the BM contribute to the pool of tissue macrophages in various disease conditions including tissue damage and fibrosis (10), the ability to systematically control monocyte turnover, as exemplified by our approach, may help inform therapeutic strategies for these diseases.
MATERIALS AND METHODS
MSC isolation and expansion
MSCs were derived by plastic adherence (“passage 0”) of mononucleated cells from human BM aspirate donors (Lonza). After plastic adherence for 3 days, nonadherent cells were washed out, and adherent cells were cultured at 37°C in 5% CO2 in the MSC medium: α-minimal essential medium (αMEM) supplemented with 20% fetal bovine serum (Atlanta Biologicals), 1% penicillin/streptomycin (P/S; Thermo Fisher Scientific), and 1% GlutaMAX (Thermo Fisher Scientific). After reaching 70 to 80% confluence at around 10 to 14 days, cells were split and expanded in the MSC medium. MSCs became >80% confluent within a week after ~500,000 cells were plated on a 175-cm2 flask. No difference was observed among tested donors in terms of the rate of proliferation. Passage numbers up to “4” or total culture day from initial plastic adherence up to 43 were used for experiments. After this passage number, MSCs undergo slower cell proliferation and were not used for experiments.
Alginate-based hydrogels for cell culture
Alginate from brown algae was purchased from a commercial vendor (FMC BioPolymer). Sodium alginate with a high molecular weight (LF200) and a low molecular weight (LF10/60) was dialyzed against deionized water for 3 days (molecular weight cutoff of 3500 Da), treated with activated charcoal, sterile filtered, and lyophilized. Before encapsulation, lyophilized alginate was reconstituted in FluoroBrite Dulbecco’s modified Eagle’s medium (DMEM) at 3 to 4% (w/v) as a stock solution. For LF200 alginate, an RGD peptide (GGGGRGDSP, Peptides International) was conjugated using carbodiimide chemistry at 20 RGD peptides per alginate chain [or ~750 μM RGD in 1% (w/v) alginate]. The coupling efficiency was previously characterized (48). For gelation, 1% (w/v) LF10/60 alginate was combined with 1% (w/v) LF200-RGD alginate [total 2% (w/v)], so that all hydrogels contain the same amount of RGD ligand, and rapidly mixed with DMEM containing the appropriate concentration of calcium sulfate. The mixed solution was deposited between two glass plates with 1-mm thickness. After 1.5 hours, hydrogels were punched into discs. For 2D hydrogels, discs were placed in ultralow attachment plates (Corning), so that cells could be seeded selectively on gels but not on plastic surface. For 3D hydrogels, cells were mixed with alginate prior to gelation. Cells on or in gels were incubated in FluoroBrite DMEM (Thermo Fisher Scientific) supplemented with 1% P/S and 1% GlutaMAX.
Mechanical characterization
To determine Young’s modulus (E) of hydrogels, a gel disc of 5 mm by 1 mm was first placed onto a polydimethylsiloxane mold on a glass slide and immersed in a drop of DMEM. The slide was then placed in an MFP-3D system (Asylum Research) to perform atomic force microscopy analysis with silicon nitride cantilevers (Bruker Model MLCT). A spring constant of the cantilever was determined from thermal fluctuations at room temperature (RT) (20 to 40 mN/m). The cantilever was brought toward the hydrogel surface at 1 μm/s and indented on the surface until it reached the trigger voltage (0.5 V), followed by retraction. Force-indentation curves were fitted using the Hertzian model with a pyramid indenter.
Establishing MSCs that express exogenous genes
To introduce firefly or Gaussia luciferase in MSCs, premade lentiviral particles containing mCherry–internal ribosomal entry site (IRES)–Firefly luciferase or cyan fluorescent protein–IRES-Gaussia luciferase were purchased from the Mass General Hospital Vector Core. MSCs were incubated with viral particles for 2 days. Fluorescent MSCs were then isolated by fluorescence-activated cell sorting and expanded for further analysis.
Quantification of viable and dead cells
Hydrogels with encapsulated cells were digested with alginate lyase (3.4 mg/ml; Sigma-Aldrich) in Hanks’ balanced salt solution (HBSS) for 30 min at 37°C. The digested solutions were directly added to HBSS that contains calcein AM (1:2000; Biotium), ethidium bromide (1:2000; Thermo Fisher Scientific), and a predefined number of allophycocyanin (APC) beads (BD). After incubation at RT for 10 min, the samples were analyzed by flow cytometry.
Gene expression analysis
At a desired time point, culture media were removed, and gel discs containing 3D encapsulated cells were washed with HBSS twice. Gel digestion and cell lysis were done simultaneously using 50 mM EDTA in ribonuclease (RNase)–free water for less than 1 min. The samples were then directly added to TRIzol (Thermo Fisher Scientific). Chloroform (200 μl) was added per 1 ml of TRIzol to perform phase separation, followed by centrifugation for 15 min at 12,500 rpm, 4°C. The top layer was collected to a new tube, and RNA was precipitated with 250 μl of isopropanol and 250 μl of solution of 0.8 M sodium citrate and 1.2 M sodium chloride for at least 15 min at 4°C. Samples were then centrifuged at 12,500 rpm for 15 min at 4°C. The supernatant was removed, and the precipitated RNA was washed with 75% EtOH, followed by centrifugation for 5 min at 7500 rpm, 4°C. EtOH was then removed, and the purified RNA was resuspended in 15 μl of RNase-free water. RNA concentration was quantified by the NanoDrop spectrophotometer. Complementary DNA (cDNA) was reverse transcribed by SuperScript-III reverse transcriptase (Thermo Fisher Scientific). Quantitative polymerase chain reaction (qPCR) was performed in the ViiA7 qPCR system with Power SYBR Green master mix (Applied Biosystems). For each qPCR experiment, samples were analyzed in triplicates with 50 ng of cDNA per well. Relative gene expression was computed by the delta-delta Ct method by comparing Ct values to reference gene (GAPDH). See table S1 for the list of primers for qPCR.
Measuring levels of secreted proteins
To determine constitutive secretion of Gaussia luciferase from engineered MSCs, collected media were diluted ~100-fold and added to a white, opaque 96-well plate. The sample (10 μl) was mixed with 100 μl of coelenterazine h substrate (10 μg/ml; Biotium) in PBS immediately prior to measurement by using a luminometer that can programmatically inject the substrate (BioTek). Photon counts were acquired for 10 s. To determine levels of secreted human CCL2 and IL-6 in culture media, a sandwich enzyme-linked immunosorbent assay was used as described by the manufacturer’s (Peprotech) protocol. Briefly, samples were diluted ~10-fold and incubated overnight in a 96-well plate (Nunc MaxiSorp, Thermo Fisher Scientific) coated with primary capture antibody. Samples were then incubated with primary detection antibody for 2 hours, followed by secondary antibody conjugated to horseradish peroxidase (HRP) for 30 min. Samples were washed out between each step with PBS with 0.05% Tween 20. At the final step, the HRP substrate, 3,3′,5,5′-tetramethylbenzidine (TMB), was added to detect HRP activity. After developing for 10 to 15 min, the equal volume of 1 M HCl was added to neutralize the reaction. A plate reader (PHERAstar) was used to read absorbance at 450 nm. The media incubated without cells were used to determine background signals. The total protein amount of each sample was normalized to total viable cell number.
Characterization of NF-κB activation kinetics
After culturing cells in gels for 1 day, recombinant TNFα (100 ng/ml; Peprotech) was added. At indicated time points, cells were rapidly retrieved from alginate hydrogels by adding 30 mM EDTA in DMEM with 1% bovine serum albumin (BSA) for 5 min on ice. During this process, dead cells were labeled with the LIVE/DEAD fixable violet dead cell stain (1:1000; Thermo Fisher Scientific). Cells were then washed out with DMEM with 1% BSA and fixed with 4% paraformaldehyde in HBSS at RT for 10 min. After washing two times with PBS/0.1% BSA, cells were stained with either phospho–NF-κB p65 (Ser536) (93H1) or total NF-κB p65 (D14E12) antibody (both from Cell Signaling Technology) at 1:100 dilution in the staining buffer (HBSS/0.1% saponin/0.1% BSA) at RT for 2 hours. They were then washed out once with the staining buffer and incubated with a secondary antibody (donkey Alexa 647 anti-rabbit, Thermo Fisher Scientific) for 30 min at RT, followed by washing out and resuspension in HBSS. Flow cytometry analysis was done using LSRFortessa (BD). The sample incubated with the isotype control [rabbit immunoglobulin G (IgG), DA1E, Cell Signaling Technology] was used as a negative control. Signals from live cell fractions were used for analysis.
Quantification of protein localization in gel-encapsulated live cells
To visualize TNFR1 or F-actin, cells were first transfected with TNFR1-YFP or mTagRFP-T-LifeAct-7 (“LifeAct-RFP”). A plasmid that contains TNFR1-YFP was a gift from J. A. Schmid (Addgene plasmid #111209). mTagRFP-T-Lifeact-7 was a gift from M. Davidson (Addgene plasmid # 54586). Each plasmid (2 μg) and 500,000 cells were mixed in 100 μl of nucleofection buffer from the human MSC nucleofector kit (VPE-1001, Lonza) and electroporated using a high-viability program in Amaxa (Lonza). Transfected cells were then cultured overnight. After encapsulating cells in soft or stiff alginate-RGD gels, they were cultured for an additional day. Cells in 3D gels were then stained with 1 μM CellTrace Far Red (C34572, Thermo Fisher Scientific) for 1 hour in 37°C to label the cytoplasm. Prior to imaging, 1 μM ethidium bromide was added to exclude dead cells. 3D scanning of encapsulated cells expressing TNFR1-YFP and/or LifeAct-RFP was done on a Zeiss laser scanning microscope (LSM 710) with 40× objective. To quantify clustering of TNFR1 in response to TNFα, TNFα (100 ng/ml) was added to cells in 3D gels, and imaging was done after 20 min. BioImageXD, an open-source software, was used as described (60) to determine the outer boundary of the CellTrace Far Red signals by thresholding and Gaussian filtering so that TNFR1-YFP molecules localized on the boundary (“cell surface”) versus those inside the cytoplasm could be distinguished. The built-in algorithms were then used after automatic thresholding (level 0.5) to quantify signal intensity, cluster size, and cluster number of TNFR1-YFP. To quantify both F-actin and TNFR1 near the cell surface, gel-encapsulated cells were preincubated with either dimethyl sulfoxide (DMSO) or nystatin (50 μM) for 2 hours, followed by TNFα stimulation for 20 min. Cells that express both TNFR1-YFP and LifeAct-RFP were then imaged, followed by analysis using Imaris 7.7 (Bitplane). Thresholding values were kept constant across all samples. Gaussian filtering was used to smooth the CellTrace signal, and the outer boundary of the reconstructed voxel was defined as the cell surface. A TNFR1 cluster was defined as any distinct TNFR1-YFP signal on the cell surface with voxel volume >0.1 μm3. Analysis was done on every single TNFR1-YFP cluster for each cell. For each TNFR1-YFP cluster on the cell surface, a region of interest (2 μm by 1 μm by 1 μm) was drawn, and the corresponding LifeAct-RFP volume within the region was analyzed. As a control, LifeAct-RFP signals were also quantified near the cell surface regions where TNFR1-YFP clusters were not present.
Evaluation of TNFα binding to cells
After culturing gel-encapsulated cells for 1 day, DMSO or chemical inhibitors (latrunculin A, blebbistatin, or nystatin, purchased from Cayman Chemical) were added and incubated for 2 hours at 37°C. Cells were then rapidly retrieved on ice as described in characterization of NF-κB activation kinetics, but they were not fixed. Retrieved cells were incubated with biotinylated TNFα (R&D Systems) at saturating concentration (1 μg/ml) for 1 hour, followed by avidin–fluorescein isothiocyanate (FITC) for 30 min on ice. Flow cytometry analysis was then performed. The sample incubated with avidin-FITC alone was used as a negative control.
Coculture studies to evaluate effects of MSCs on hematopoiesis in vitro
Human cord blood CD34+ cells from mixed donors were purchased from ZenBio (SER-CD34-F). Cord blood cells were first precultured in the StemSpan-II medium (#09605, STEMCELL Technology) supplemented with stem cell factor (SCF; 50 ng/ml), thrombopoietin (Tpo; 50 ng/ml), and 1% P/S for 8 days. In parallel, human MSCs were either plated on 2D plastic culture or encapsulated in 3D soft or stiff alginate-RGD gel for 1 day, followed by stimulation with or without TNFα (100 ng/ml) for 3 days. Precultured cord blood cells (5000) and 1677 primed MSCs (3:1 ratio) were then retrieved and cocultured in the 0.5% LF200 alginate-RGD fluid dissolved in the StemSpan-II medium supplemented with SCF (20 ng/ml), Tpo (20 ng/ml), and 1% P/S on the ultralow-binding 96-well plate (Sigma-Aldrich) for 4 days. Cells were then analyzed for human hematopoietic stem and progenitor cell subpopulations and monocyte lineages by flow cytometry after staining with the following antibodies based on previous studies (33, 34): APC/Cy7 anti-human CD34 (581), phycoerythrin (PE)/Cy7 anti-human CD38 (HIT8a), APC anti-human CD90 (5E10), PE CD45RA (HI100), PE/Dazzle CD135 (BV10A4H2), FITC anti-human CD14 (HCD14), and Pacific Blue anti-human CD16 (3G8), all purchased from BioLegend.
Xenograft studies to evaluate effects of human MSCs on human monocyte production in vivo
All animal work was performed in compliance with the National Institutes of Health and institutional guidelines approved by the ethical committee from the University of Illinois at Chicago. Human cord blood CD34+ cells (20,000) were injected retroorbitally into sublethally irradiated 8- to 12-week-old NOD/SCID/IL-2γ−/− (NSG) mice. After 2 weeks, all of the hair surrounding the knee joint was removed. MSCs from gels were retrieved by digesting them with alginate lyase in DMEM for 20 min at 37°C. Ex vivo conditioned human MSCs (10,000) were resuspended in 10 μl of PBS and delivered into the marrow cavity of the right tibia through the patellar tendon using an insulin syringe (28-gauge needle). PBS with no cell was delivered to the left tibia. After 4 days, mice were euthanized, the marrow from each tibia was flushed out, and cells were analyzed for human monocyte lineages by flow cytometry after staining with 7-AAD for cell viability and the following antibodies: APC anti-mouse CD45 (30-F11), PE-Cy7 anti-human CD45 (HI30), FITC anti-human CD14 (HCD14), and Pacific Blue anti-human CD16 (3G8), all purchased from BioLegend.
Tracking biodistribution of transplanted cells in vivo
After delivering firefly-expressing MSCs to NSG mice, their in vivo residence kinetics was evaluated. At different time points, 1.5 mg of d-luciferin in 200 μl of PBS was injected retroorbitally per 20 g of mouse followed by luminescence imaging with the IVIS Spectrum (PerkinElmer) within 10 min of injection. Average radiance (photons/s/cm2/sr) from each time point was measured from each tibia.
Transwell migration assay
Transwell inserts with polycarbonate membrane with 3-μm pore size (Corning) were coated with fibronectin (10 μg/ml) in HBSS overnight at 4°C, followed by blocking with 1% BSA in HBSS for 1 hour at RT. The conditioned media were collected from gel-encapsulated human MSCs that are cultured in the presence or absence of TNFα (100 ng/ml) for 1 day, followed by washout and cultured in the absence of TNFα for another day. The same cell number per gel (~50,000 per 8-mm disc) was used to collect the conditioned media. Fresh human peripheral mononuclear cells (~100,000) were added to the top wells in serum-free FluoroBrite DMEM, while the conditioned media were added to the bottom wells. In some experiments, the conditioned media were incubated with the neutralizing antibody against human CCL2 (MAB679, R&D Systems) or the mouse IgG2B isotype control (MAB004, R&D Systems) for 3 hours at 4°C prior to the migration assay. The cells were allowed to migrate for 3 hours at 37°C, 5% CO2. The cells collected from the bottom wells were subject to flow cytometry analysis for different lineages using the following anti-human antibodies: FITC CD14 (HCD14), APC CD4 (RPA-T4), APC-cy7 CD8 (SK1), PE CD19 (HIB19), and PE-cy7 CD56 (HCD56), all purchased from BioLegend. The cell number per sample was counted by adding a predefined number of polystyrene beads (Spherotech). The fraction of migrated cells was calculated by dividing with the total number of cells for each lineage added to each well.
Statistical analyses
All statistical analyses were performed using GraphPad Prism 6. Unless otherwise noted, all statistical comparisons were made from at least three independent experiments by one-way analysis of variance (ANOVA), followed by Tukey’s post hoc testing, and were considered significant if P < 0.05.
Supplementary Material
Acknowledgments
We gratefully acknowledge the Research Open Access Publishing (ROAAP) Fund of the University of Illinois at Chicago for financial support towards the open access publishing fee for this article. The atomic force microscopy work was performed in part at the Center for Nanoscale Systems (CNS), a member of the National Nanotechnology Coordinated Infrastructure Network (NNCI), which is supported by the National Science Foundation under NSF award no. 1541959. CNS is part of Harvard University. Funding: This work was supported by NIH grant R01HL141255 (J.-W.S.), R00HL125884 (J.-W.S.), and R01EB023287 (D.J.M.). Author contributions: S.W.W., S.L., M.H.C., D.J.M., and J.-W.S. designed the research. S.W.W., S.L., M.H.C., and J.-W.S. performed the research. S.W.W., S.L., D.J.M., and J.-W.S. wrote the paper. Competing interests: All authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/15/eaaw0158/DC1
References and notes
- 1.Yue R., Zhou B. O., Shimada I. S., Zhao Z., Morrison S. J., Leptin receptor promotes adipogenesis and reduces osteogenesis by regulating mesenchymal stromal cells in adult bone marrow. Cell Stem Cell 18, 782–796 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Méndez-Ferrer S., Michurina V., Ferraro F., Mazloom A. R., Macarthur B. D., Lira S. A., Scadden D. T., Ma’ayan A., Enikolopov G. N., Frenette P. S., Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466, 829–834 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartholomew A., Sturgeon C., Siatskas M., Ferrer K., McIntosh K., Patil S., Hardy W., Devine S., Ucker D., Deans R., Moseley A., Hoffman R., Mesenchymal stem cells suppress lymphocyte proliferation in vitro and prolong skin graft survival in vivo. Exp. Hematol. 30, 42–48 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Le Blanc K., Mougiakakos D., Multipotent mesenchymal stromal cells and the innate immune system. Nat. Rev. Immunol. 12, 383–396 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Parekkadan B., Milwid J. M., Mesenchymal stem cells as therapeutics. Annu. Rev. Biomed. Eng. 12, 87–117 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y., Chen X., Cao W., Shi Y., Plasticity of mesenchymal stem cells in immunomodulation: Pathological and therapeutic implications. Nat. Immunol. 15, 1009–1016 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Shi C., Jia T., Mendez-Ferrer S., Hohl T. M., Serbina N. V., Lipuma L., Leiner I., Li M. O., Frenette P. S., Pamer E. G., Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll-like receptor ligands. Immunity 34, 590–601 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Németh K., Leelahavanichkul A., Yuen P. S., Mayer B., Parmelee A., Doi K., Robey P. G., Leelahavanichkul K., Koller B. H., Brown J. M., Hu X., Jelinek I., Star R. A., Mezey E., Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat. Med. 15, 42–49 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee R. H., Pulin A. A., Seo M. J., Kota D. J., Ylostalo J., Larson B. L., Semprun-Prieto L., Delafontaine P., Prockop D. J., Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell 5, 54–63 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mosser D. M., Edwards J. P., Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ding L., Saunders T. L., Enikolopov G., Morrison S. J., Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481, 457–462 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park D., Spencer J. A., Koh B. I., Kobayashi T., Fujisaki J., Clemens T. L., Lin C. P., Kronenberg H. M., Scadden D. T., Endogenous bone marrow MSCs are dynamic, fate-restricted participants in bone maintenance and regeneration. Cell Stem Cell 10, 259–272 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ivanovska I. L., Swift J., Spinler K., Dingal D., Cho S., Discher D. E., Cross-linked matrix rigidity and soluble retinoids synergize in nuclear lamina regulation of stem cell differentiation. Mol. Biol. Cell 28, 2010–2022 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhong Z., Akkus O., Effects of age and shear rate on the rheological properties of human yellow bone marrow. Biorheology 48, 89–97 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Engler A. J., Sen S., Sweeney H. L., Discher D. E., Matrix elasticity directs stem cell lineage specification. Cell 126, 677–689 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Chaudhuri O., Gu L., Klumpers D., Darnell M., Bencherif S. A., Weaver J. C., Huebsch N., Lee H. P., Lippens E., Duda G. N., Mooney D. J., Hydrogels with tunable stress relaxation regulate stem cell fate and activity. Nat. Mater. 15, 326–334 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abdeen A. A., Weiss J. B., Lee J., Kilian K. A., Matrix composition and mechanics direct proangiogenic signaling from mesenchymal stem cells. Tissue Eng. Part A 20, 2737–2745 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu F. D., Pishesha N., Poon Z., Kaushik T., Van Vliet K. J., Material viscoelastic properties modulate the mesenchymal stem cell secretome for applications in hematopoietic recovery. ACS Biomater Sci. Eng. 3, 3292–3306 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Valles G., Bensiamar F., Crespo L., Arruebo M., Vilaboa N., Saldaña L., Topographical cues regulate the crosstalk between MSCs and macrophages. Biomaterials 37, 124–133 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu F. D., Tam K., Pishesha N., Poon Z., Van Vliet K. J., Improving hematopoietic recovery through modeling and modulation of the mesenchymal stromal cell secretome. Stem Cell Res. Ther. 9, 268 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang X., Zhu B., Wang X., Xiao R., Wang C., Three-dimensional co-culture of mesenchymal stromal cells and differentiated osteoblasts on human bio-derived bone scaffolds supports active multi-lineage hematopoiesis in vitro: Functional implication of the biomimetic HSC niche. Int. J. Mol. Med. 38, 1141–1151 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ren G., Zhao X., Wang Y., Zhang X., Chen X., Xu C., Yuan Z. R., Roberts A. I., Zhang L., Zheng B., Wen T., Han Y., Rabson A. B., Tischfield J. A., Shao C., Shi Y., CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFα. Cell Stem Cell 11, 812–824 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakamura K., Kouro T., Kincade P. W., Malykhin A., Maeda K., Coggeshall K. M., Src homology 2-containing 5-inositol phosphatase (SHIP) suppresses an early stage of lymphoid cell development through elevated interleukin-6 production by myeloid cells in bone marrow. J. Exp. Med. 199, 243–254 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoffmann A., Levchenko A., Scott M. L., Baltimore D., The IkappaB-NF-kappaB signaling module: Temporal control and selective gene activation. Science 298, 1241–1245 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Mangan S., Itzkovitz S., Zaslaver A., Alon U., The incoherent feed-forward loop accelerates the response-time of the gal system of Escherichia coli. J. Mol. Biol. 356, 1073–1081 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Bax D. V., Messent A. J., Tart J., van Hoang M., Kott J., Maciewicz R. A., Humphries M. J., Integrin alpha5beta1 and ADAM-17 interact in vitro and co-localize in migrating HeLa cells. J. Biol. Chem. 279, 22377–22386 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Morton P. E., Perrin C., Levitt J., Matthews D. R., Marsh R. J., Pike R., McMillan D., Maloney A., Poland S., Ameer-Beg S., Parsons M., TNFR1 membrane reorganization promotes distinct modes of TNFα signaling. Sci. Signal. 12, eaaw2418 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Foti M., Porcheron G., Fournier M., Maeder C., Carpentier J. L., The neck of caveolae is a distinct plasma membrane subdomain that concentrates insulin receptors in 3T3-L1 adipocytes. Proc. Natl. Acad. Sci. U.S.A. 104, 1242–1247 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Legler D. F., Micheau O., Doucey M. A., Tschopp J., Bron C., Recruitment of TNF receptor 1 to lipid rafts is essential for TNFα-Mediated NF-κB activation. Immunity 18, 655–664 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Sinha B., Köster D., Ruez R., Gonnord P., Bastiani M., Abankwa D., Stan R. V., Butler-Browne G., Vedie B., Johannes L., Morone N., Parton R. G., Raposo G., Sens P., Lamaze C., Nassoy P., Cells respond to mechanical stress by rapid disassembly of caveolae. Cell 144, 402–413 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riedl J., Crevenna A. H., Kessenbrock K., Yu J. H., Neukirchen D., Bista M., Bradke F., Jenne D., Holak T. A., Werb Z., Sixt M., Wedlich-Soldner R., Lifeact: A versatile marker to visualize F-actin. Nat. Methods 5, 605–607 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shin J. W., Mooney D. J., Extracellular matrix stiffness causes systematic variations in proliferation and chemosensitivity in myeloid leukemias. Proc. Natl. Acad. Sci. U.S.A. 113, 12126–12131 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manz M. G., Miyamoto T., Akashi K., Weissman I. L., Prospective isolation of human clonogenic common myeloid progenitors. Proc. Natl. Acad. Sci. U.S.A. 99, 11872–11877 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Majeti R., Park C. Y., Weissman I. L., Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell 1, 635–645 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glimm H., Eisterer W., Lee K., Cashman J., Holyoake T. L., Nicolini F., Shultz L. D., von Kalle C., Eaves C. J., Previously undetected human hematopoietic cell populations with short-term repopulating activity selectively engraft NOD/SCID-β2 microglobulin-null mice. J. Clin. Invest. 107, 199–206 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patel A. A., Zhang Y., Fullerton J. N., Boelen L., Rongvaux A., Maini A. A., Bigley V., Flavell R. A., Gilroy D. W., Asquith B., Macallan D., Yona S., The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J. Exp. Med. 214, 1913–1923 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rongvaux A., Willinger T., Martinek J., Strowig T., Gearty S. V., Teichmann L. L., Saito Y., Marches F., Halene S., Palucka A. K., Manz M. G., Flavell R. A., Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. 32, 364–372 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schauer D., Starlinger P., Zajc P., Alidzanovic L., Maier T., Buchberger E., Pop L., Gruenberger B., Gruenberger T., Brostjan C., Monocytes with angiogenic potential are selectively induced by liver resection and accumulate near the site of liver regeneration. BMC Immunol. 15, 50 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grivennikov S. I., Tumanov A. V., Liepinsh D. J., Kruglov A. A., Marakusha B. I., Shakhov A. N., Murakami T., Drutskaya L. N., Förster I., Clausen B. E., Tessarollo L., Ryffel B., Kuprash D. V., Nedospasov S. A., Distinct and nonredundant in vivo functions of TNF produced by t cells and macrophages/neutrophils: Protective and deleterious effects. Immunity 22, 93–104 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Wolf Y., Shemer A., Polonsky M., Gross M., Mildner A., Yona S., David E., Kim K. W., Goldmann T., Amit I., Heikenwalder M., Nedospasov S., Prinz M., Friedman N., Jung S., Autonomous TNF is critical for in vivo monocyte survival in steady state and inflammation. J. Exp. Med. 214, 905–917 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee R. E., Walker S. R., Savery K., Frank D. A., Gaudet S., Fold change of nuclear NF-κB determines TNF-induced transcription in single cells. Mol. Cell 53, 867–879 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chew J., Biswas S., Shreeram S., Humaidi M., Wong E. T., Dhillion M. K., Teo H., Hazra A., Fang C. C., López-Collazo E., Bulavin D. V., Tergaonkar V., WIP1 phosphatase is a negative regulator of NF-kappaB signalling. Nat. Cell Biol. 11, 659–666 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Brechmann M., Mock T., Nickles D., Kiessling M., Weit N., Breuer R., Müller W., Wabnitz G., Frey F., Nicolay J. P., Booken N., Samstag Y., Klemke C. D., Herling M., Boutros M., Krammer P. H., Arnold R., A PP4 holoenzyme balances physiological and oncogenic nuclear factor-kappa B signaling in T lymphocytes. Immunity 37, 697–708 (2012). [DOI] [PubMed] [Google Scholar]
- 44.Brunialti M. K., Kallas E. G., Freudenberg M., Galanos C., Salomao R., Influence of EDTA and heparin on lipopolysaccharide binding and cell activation, evaluated at single-cell level in whole blood. Cytometry 50, 14–18 (2002). [PubMed] [Google Scholar]
- 45.Du J., Chen X., Liang X., Zhang G., Xu J., He L., Zhan Q., Feng X. Q., Chien S., Yang C., Integrin activation and internalization on soft ECM as a mechanism of induction of stem cell differentiation by ECM elasticity. Proc. Natl. Acad. Sci. U.S.A. 108, 9466–9471 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu C. H., Rafiq N. B. M., Cao F., Zhou Y., Krishnasamy A., Biswas K. H., Ravasio A., Chen Z., Wang Y. H., Kawauchi K., Jones G. E., Sheetz M. P., Integrin-beta3 clusters recruit clathrin-mediated endocytic machinery in the absence of traction force. Nat. Commun. 6, 8672 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mitra A., Venkatachalapathy S., Ratna P., Wang Y., Jokhun D. S., Shivashankar G. V., Cell geometry dictates TNFα-induced genome response. Proc. Natl. Acad. Sci. U.S.A. 114, E3882–E3891 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huebsch N., Arany P. R., Mao A. S., Shvartsman D., Ali O. A., Bencherif S. A., Rivera-Feliciano J., Mooney D. J., Harnessing traction-mediated manipulation of the cell/matrix interface to control stem-cell fate. Nat. Mater. 9, 518–526 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sabio G., Davis R. J., TNF and MAP kinase signalling pathways. Semin. Immunol. 26, 237–245 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deng Y., Ren X., Yang L., Lin Y., Wu X., A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell 115, 61–70 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Pronk C. J., Veiby O. P., Bryder D., Jacobsen S. E., Tumor necrosis factor restricts hematopoietic stem cell activity in mice: Involvement of two distinct receptors. J. Exp. Med. 208, 1563–1570 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bailey A. M., Mendicino M., Au P., An FDA perspective on preclinical development of cell-based regenerative medicine products. Nat. Biotechnol. 32, 721–723 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Shin J. W., Spinler K. R., Swift J., Chasis J. A., Mohandas N., Discher D. E., Lamins regulate cell trafficking and lineage maturation of adult human hematopoietic cells. Proc. Natl. Acad. Sci. U.S.A. 110, 18892–18897 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harada T., Swift J., Irianto J., Shin J. W., Spinler K. R., Athirasala A., Diegmiller R., Dingal P. C. D. P., Ivanovska I. L., Discher D. E., Nuclear lamin stiffness is a barrier to 3D migration, but softness can limit survival. J. Cell Biol. 204, 669–682 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Muguruma Y., Yahata T., Miyatake H., Sato T., Uno T., Itoh J., Kato S., Ito M., Hotta T., Ando K., Reconstitution of the functional human hematopoietic microenvironment derived from human mesenchymal stem cells in the murine bone marrow compartment. Blood 107, 1878–1887 (2006). [DOI] [PubMed] [Google Scholar]
- 56.de Witte S. F. H., Luk F., Sierra Parraga J. M., Gargesha M., Merino A., Korevaar S. S., Shankar A. S., O’Flynn L., Elliman S. J., Roy D., Betjes M. G. H., Newsome P. N., Baan C. C., Hoogduijn M. J., Immunomodulation by therapeutic Mesenchymal Stromal Cells (MSC) is triggered through phagocytosis of MSC By monocytic cells. Stem Cells 36, 602–615 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Wong T. Y., Preston L. A., Schiller N. L., Alginate lyase: Review of major sources and enzyme characteristics, structure-function analysis, biological roles, and applications. Annu. Rev. Microbiol. 54, 289–340 (2000). [DOI] [PubMed] [Google Scholar]
- 58.Lutolf M. P., Lauer-Fields J. L., Schmoekel H. G., Metters A. T., Weber F. E., Fields G. B., Hubbell J. A., Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: Engineering cell-invasion characteristics. Proc. Natl. Acad. Sci. U.S.A. 100, 5413–5418 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mao A. S., Shin J. W., Utech S., Wang H., Uzun O., Li W., Cooper M., Hu Y., Zhang L., Weitz D. A., Mooney D. J., Deterministic encapsulation of single cells in thin tunable microgels for niche modelling and therapeutic delivery. Nat. Mater. 16, 236–243 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kankaanpää P., Paavolainen L., Tiitta S., Karjalainen M., Päivärinne J., Nieminen J., Marjomäki V., Heino J., White D. J., BioImageXD: An open, general-purpose and high-throughput image-processing platform. Nat. Methods 9, 683–689 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/15/eaaw0158/DC1