

The study addresses the mechanism through which inhibition of LSD1 sensitizes AML cells to retinoic acid–induced differentiation.
Abstract
The histone demethylase LSD1 is deregulated in several tumors, including leukemias, providing the rationale for the clinical use of LSD1 inhibitors. In acute promyelocytic leukemia (APL), pharmacological doses of retinoic acid (RA) induce differentiation of APL cells, triggering degradation of the PML-RAR oncogene. APL cells are resistant to LSD1 inhibition or knockout, but targeting LSD1 sensitizes them to physiological doses of RA without altering of PML-RAR levels, and extends survival of leukemic mice upon RA treatment. The combination of RA with LSD1 inhibition (or knockout) is also effective in other non-APL, acute myeloid leukemia (AML) cells. Nonenzymatic activities of LSD1 are essential to block differentiation, while RA with targeting of LSD1 releases a differentiation gene expression program, not strictly dependent on changes in histone H3K4 methylation. Integration of proteomic/epigenomic/mutational studies showed that LSD1 inhibitors alter the recruitment of LSD1-containing complexes to chromatin, inhibiting the interaction between LSD1 and the transcription factor GFI1.
INTRODUCTION
Histone methylation is dynamically controlled by histone methyltransferases and histone demethylases (KDMs). Among KDMs, lysine-specific demethylase 1 (LSD1; KDM1A) works mainly as a transcriptional co-repressor, which catalyzes the demethylation of mono- and dimethylated histone H3 lysine 4 (1, 2). LSD1 is essential for embryonic development (3) and is required for many physiological processes (4, 5). During hematopoietic differentiation, GFI1 (growth factor independent 1)/GFI1b transcription factors recruit LSD1 to their target genes and maintain lineage-specific gene expression (6–9).
Aberrant histone methylation has been described in cancer (10). LSD1 has been found overexpressed in several tumors and has been shown to be required for maintenance of acute myeloid leukemia (AML) cells (11, 12). Several inhibitors of LSD1, targeting its catalytical activity, have been developed, most of which derive from the monoamine oxidase inhibitor tranylcypromine (10). Treatment of AML cells with LSD1 inhibitors (LSD1i) showed a highly variable pattern of response (13). Sensitivity of AML cells to LSD1 inhibition can be restored by combination with other drugs, such as retinoic acid (RA) (11, 13). LSD1i as single agents or in combination with RA have therefore entered phase 1/2A clinical trials in patients with AML. The molecular mechanisms underlying response of AML cells to LSD1i remain, however, largely unknown. APL (acute promyelocytic leukemia) is an AML subtype caused by the promyelocytic leukemia (PML)–RA receptor (RAR) fusion oncoprotein. PML-RAR works as an altered transcription factor and modifies the epigenetic landscape of APL cells, causing differentiation block at the promyelocytic stage (14). Pharmacological doses of RA lead to degradation of the fusion protein, terminal differentiation of leukemic cells, and, in combination with other agents (chemotherapy or arsenic), definitive cure for most patients with APL, making this form of cancer an attractive model system for the study of tumor cell response to therapy (15, 16).
We showed that APL cells are resistant to pharmacological inhibition of LSD1 and that LSD1 inhibition sensitizes APL cells to physiological concentrations of RA (17). Here, we have investigated the molecular mechanisms underlying the sensitization to RA by LSD1 inhibition in AML cells (APL and non-APL), which are intrinsically resistant to LSD1 inhibition.
RESULTS
LSD1 inhibition sensitizes AML cells to physiological concentrations of RA
We tested the effect of the LSD1i MC_2580 (18, 19), alone or in combination with RA, on a panel of 21 AML cell lines representative of all AML subtypes. LSD1i alone considerably affected the growth (>70% inhibition) of a small subset of cell lines (Kasumi-1, SKNO-1, and EOL-1 cells). Similarly, low doses of RA (0.01 to 0.1 μM) had a minor effect on the great majority of the cell lines, and only a few of them responded markedly to treatment (EOL-1, MonoMac1, and MV4-11 cells). The combination of RA with MC_2580 had a remarkable impact on the viability of almost all cell lines, both resistant and sensitive to LSD1 inhibition alone (Fig. 1A).
Fig. 1. LSD1 inhibition sensitizes AML cells to physiological concentrations of RA.
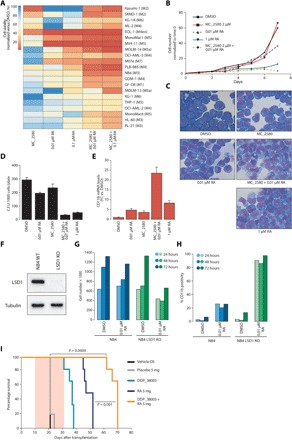
(A) Heat map representing the results of cell proliferation assays performed on 21 AML cell lines treated with MC_2580 (2 μM) and/or RA as indicated. Values are normalized on dimethyl sulfoxide (DMSO) treatment. The AML French-American-British classifications of each cell line are in parentheses. (B) Growth curves of NB4 APL cells treated as indicated. (C) Morphological analysis by May-Grünwald-Giemsa staining of NB4 cells treated for 96 hours in liquid culture as indicated. (D) Colony-forming ability, scored after 7 days, of 1000 NB4 cells plated in methylcellulose medium and treated with MC_2580 (2 μM) and/or 0.01 μM RA and 1 μM RA. Means and SDs of three independent experiments are shown. C.F.U., colony-forming units. (E) Analysis of CD11b mRNA levels in NB4 cells treated for 96 hours in liquid culture as indicated. Values are normalized against GAPDH (glyceraldehyde phosphate dehydrogenase) and referred to DMSO. The graph represents the mean and SD of three independent experiments. FC, fold change. (F) Western blot showing depletion of LSD1 in NB4 cells knocked out for LSD1 (LSD1 KO). WT, wild type. (G) Proliferation assays of NB4 and NB4 LSD1 KO cells after 24, 48, and 72 hours of the indicated treatments (DMSO as control). (H) Expression of CD11b by fluorescence-activated cell sorting (FACS) in NB4 and NB4 LSD1 KO cells after 24, 48, and 72 hours of the indicated treatments (DMSO as control). (I) Kaplan-Meier survival plots for mice transplanted with murine APL cells and treated as indicated (n = 6 for each treatment group). Pink shaded area indicates the duration of RA treatment (21 days, pellet), while LSD1i (DDP_38003) was administrated twice a week orally (OS) for the entire duration of RA treatment (total, six times). P values were obtained using analysis of variance (ANOVA).
Confirming previous findings, APL cells (NB4) were not sensitive to LSD1 inhibition or to physiological doses of RA (0.01 μM, RA low), while pharmacological doses of RA (1 μM, RA high) markedly reduced cell proliferation. The combination of LSD1i with RA low reduced cell proliferation in liquid culture and colony-forming ability in semisolid culture (Fig. 1, B and C). The observed phenotype was due to cell differentiation, as assessed by the induction of the myeloid differentiation marker CD11b and morphological changes associated with neutrophilic differentiation (Fig. 1, D and E).
To assess whether the effect of LSD1i was specific, we depleted LSD1 by CRISPR-Cas9 or by a retroviral mediated knockdown (Fig. 1F and fig. S1). LSD1 depletion did not affect viability of NB4 cells (fig. S1), but it enhanced their sensitivity to RA low, as evidenced by the reduction of cell proliferation and the induction of CD11b (Fig. 1, G and H). LSD1 depletion thus mimics the effects of LSD1 inhibition, confirming the specificity of the LSD1i.
We then measured the effect of LSD1 depletion/inhibition on global levels of histone H3K4 methylation by quantitative mass spectrometry (MS) (fig. S2). We observed, in all cases, an increase in global H3K4me2 and H3K4me3 levels, with the H3K4me3 slight increase likely being a consequence of H3K4me2 accumulation.
Last, we tested the effect of LSD1i and RA combination in vivo. We tested DDP_38003 as LSD1i (20) since MC_2580 cannot be used in vivo. As previously described, all control mice (either treated with placebo or vehicle) died within 3 weeks (median survival, 21 days), and RA treatment induced differentiation and significantly prolonged survival (median survival, 49 days; Fig. 1I). Treatment with the LSD1i alone prolonged survival to a lesser extent than RA (median survival, 37 days), while the combination of RA with LSD1i strongly potentiated the therapeutic effect of RA, confirming the results obtained in vitro (P = 0.001 over RA treatment and P = 0.0009 over placebo; median survival, 70 days; fig. S3).
LSD1 inhibition allows APL cell differentiation bypassing the oncogenic function of PML-RAR
While pharmacological doses of RA (RA high) trigger PML-RAR degradation, physiological doses of RA (RA low) do not (15, 21). PML-RAR protein remained stable upon LSD1 inhibition, whether in the presence or in the absence of RA low (fig. S4A). PML-RAR alters the normal assembly of PML nuclear bodies, inducing a micro-speckled pattern of PML (22). RA high induced degradation of PML-RAR and restoration of the normal pattern of PML nuclear bodies (fig. S4B). Consistent with the results in fig. S4A, MC_2580 did not alter the micro-speckled pattern of PML both in the presence and in the absence of RA low. We then investigated PML-RAR binding to its target genes after the different treatments (fig. S4C). We performed chromatin immunoprecipitation (IP)–quantitative polymerase chain reaction (ChIP-qPCR) on PML-RAR-Regulated Adapter Molecule 1 (PRAM1) and Phosphoinositide 3-kinase delta (PI3KD), two established PML-RAR targets in NB4 cells (23), and observed that LSD1 inhibition and RA low did not alter PML-RAR recruitment on those genes, while RA high displaced PML-RAR from chromatin and/or triggered degradation of the fusion protein.
Comparing their genomic distribution in NB4 cells, we observed that PML-RAR shares most of its binding sites with LSD1 (fig. S4D).We then measured PML-RAR and LSD1 occupancy on genes activated only by the cotreatment and genes activated by RA high: Genes specifically regulated by the cotreatment show a significantly lower PML-RAR occupancy (P < 0.01) compared to genes activated by high concentrations of RA (Fig. 2F).
Fig. 2. LSD1 plays a key role in the control of differentiation of APL cells.
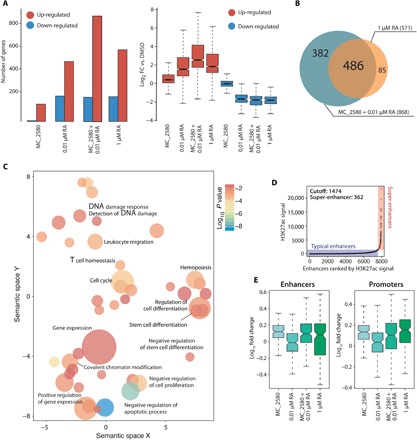
(A) RNA sequencing (RNA-seq) was performed in NB4 cells treated with MC_2580 and/or RA (0.01 and 1 μM) for 24 hours and DMSO as control. Left: The bar plot represents number of genes regulated [up- or down-regulated with respect to control; RPKM (reads per kilobase million) > 0.5; log2(FC) > 1.5] upon the indicated treatments. Right: The box plot shows magnitude of induction by the indicated treatment versus control (DMSO). (B) Venn diagrams indicating the sum of all regulated genes, number of genes regulated by each individual treatment, and number of genes regulated by both treatments in NB4 cells treated with MC_2580 and 0.01 μM RA versus 1 μM RA. (C) Gene Ontology (biological processes) analysis of LSD1 target genes in NB4 cells. Adjusted P values and relative enrichment (color coded) are shown for each class. (D) Scatter plots of super-enhancers in cells treated with MC_2580 + 0.01 μM RA. All stitched regions were ranked by H3K27ac signal. Super-enhancers and typical enhancers were in different colors as indicated. (E) Box plot analysis of H3K4me2 enrichment upon indicated treatment at enhancers (left) and promoters (right) of the 382 genes shown in Fig. 3B. Values are represented as log10FC versus DMSO.
Last, we checked whether PML-RAR is able to recruit LSD1 on specific genomic targets using the U937-derived PR9 cell line, which express PML-RAR under a zinc-inducible promoter (23). LSD1 peaks (97%) in PR9 cells remain stable after PML-RAR induction, showing that LSD1 is not recruited by PML-RAR (fig. S4E).
Together, these results indicate that the combined treatment with LSD1i and RA low allows differentiation and growth arrest of APL without affecting PML-RAR stability and its recruitment on chromatin.
LSD1 regulates differentiation of APL cells
We performed washout experiments to define whether there is a temporal window of LSD1 inhibition sufficient to commit NB4 cells to differentiation in the presence of physiological concentrations of RA. We found that treatment of at least 24 hours in the presence of RA low led to a response comparable to continuous cotreatment (fig. S5). We thus determined the gene expression profile of NB4 cells after 24 hours of treatment with MC_2580 and/or RA by RNA sequencing (RNA-seq). LSD1 inhibition [and LSD1 knockout (KO)] had only a modest effect on gene transcription, both in terms of the number of modulated genes and in the magnitude of their regulation (Fig. 2A and fig. S5). Only a few genes were down-regulated upon LSD1 inhibition (Fig. 2A).
LSD1 inhibition, however, markedly potentiated the effect of physiological concentrations of RA, doubling the number of regulated genes compared to treatment with RA low alone. Although the combination of MC_2580 with RA low regulated almost all of the genes regulated by RA high, it specifically induced a subset of 382 additional genes (Fig. 2B). The large majority of regulated genes falls within gene categories associated with hematopoietic development and differentiation (fig. S5D). These observations suggest that while the combinatorial treatment and RA high induced a similar differentiation program, the combination had a larger impact on transcription than RA high: The combination had a stronger effect on cell proliferation than that observed with RA high alone (Fig. 1B).
H3K4me2 accumulation in regulatory regions after treatment does not correlate with induction of transcription
We assessed the genome-wide distribution of LSD1 in NB4 cells by ChIP sequencing (ChIP-seq). LSD1 binds preferentially regulatory regions (fig. S6) of genes related to hematopoiesis and cell differentiation (Fig. 2C). We next performed ChIP-seq for H3K4me1/me2/me3 and H3K27 acetylation upon the different drug treatments and observed a marked reorganization of the chromatin. In particular, there was an evident rearrangement of active super-enhancers after the combination of LSD1 inhibition and RA low, with 207 newly activated super-enhancers, of which 118 of 207 were uniquely activated in the cotreatment and not by high doses of RA (Fig. 2D). Consistent with the global increase observed by mass spectrometry, we observed a local increase of H3K4me2 at LSD1-bound regulatory regions after LSD1i. We separately analyzed chromatin changes subsequent to the different treatments occurring in the subset of genes specifically induced by the cotreatment (n = 382) and those similarly induced by both RA high and cotreatment (n = 486). Active transcription is positively correlated with the presence of both H3K4me3 and H3K27ac (fig. S6). LSD1 inhibition, alone or in combination with RA low, induced an increase in the levels of H3K4me2. This chromatin change is not directly associated with active transcription, as an increase in its levels is also present after treatment with high doses of RA at the promoters and enhancers of those genes that are not induced by RA high (Fig. 2E). Thus, an increase in H3K4me2 levels is not sufficient to trigger gene expression changes.
LSD1 catalytic activity is dispensable for sensitization to RA-induced differentiation
We then reconstituted LSD1 KO NB4 cells with either wild-type or a catalytically inactive LSD1 mutant (K661A-LSD1) (Fig. 3A). While LSD1 KO cells were hypersensitive to low doses of RA, LSD1 and K661A-LSD1 reconstituted cells were insensitive to RA low, recapitulating the phenotype of NB4 wild-type cells (Fig. 3B). Consistently, after treatment with RA low of both LSD1 and K661A-LSD1 reconstituted cells, we did not observe an increase in CD11b-expressing cells (Fig. 3C). By ChIP-qPCR, we observed that wild-type LSD1, but not the catalytically inactive K661A-LSD1, was able to reduce the levels of H3K4me2 at LSD1 target regions (Fig. 3D). These data demonstrate that LSD1 catalytic function is dispensable for its ability to control differentiation of NB4 cells.
Fig. 3. The catalytic activity of LSD1 is dispensable.
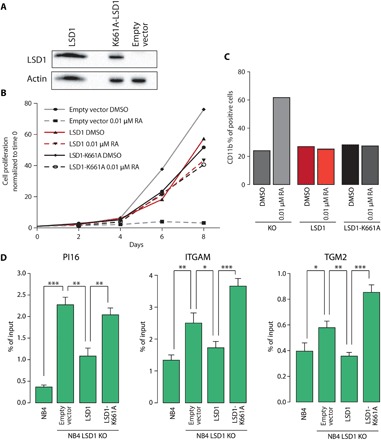
(A) Western blot analysis of LSD1 levels in NB4 LSD1 KO cells transduced with expression vectors encoding for wild-type LSD1 or the catalytically inactive mutant K661A-LSD1. Empty vector was used as control, and actin served as loading control. (B) Growth curves of NB4 LSD1 KO cells transduced with empty vector, LSD1, or K661A-LSD1 and treated with 0.01 μM RA (DMSO as control treatment). (C) Percentage of CD11b-expressing cells assessed by FACS in NB4 LSD1 KO cells transduced with expression vectors encoding for LSD1, K661A-LSD1, or empty vector, treated with 0.01 μM RA (or DMSO as control) for 24 hours. (D) H3K4me2 enrichment at the promoter of PI16 (left), ITGAM (center), and TGM2 (right) in NB4 and NB4 LSD1 KO cells transduced with expression vectors encoding for LSD1, K661A-LSD1, or empty vector, assessed by ChIP-qPCR. The results represent percentage of input chromatin. Error bars indicate SD from triplicate experiments, and P values were determined by a two-tailed unpaired Student’s t test (*P < 0.05, **P < 0.01, and ***P < 0.001).
LSD1 pharmacological inhibition disrupts its interaction with GFI1
The above results demonstrate that nonenzymatic functions of LSD1 are critical for its activities. We hypothesized that LSD1 may have a main scaffolding role, and that LSD1i could somehow interfere with the interaction between LSD1 and other proteins. To verify this hypothesis, we used an approach of Stable Isotope Labelling by Aminoacids in Cell culture (SILAC)–based proteomics in combination with LSD1 IP to study the LSD1 interactome in NB4 cells (Fig. 4A). By using three different biological replicates, we defined 147 LSD1 putative interactors (Fig. 4, B and C). Among them, we enriched the CoREST complex, as expected [histone deacetylase 1 (HDAC1), HDAC2, REST corepressor 1 (RCOR1), and RCOR3]. Western blot analysis of different LSD1 interactors validated the mass spectrometry data (Fig. 4D).
Fig. 4. Pharmacological inhibition of LSD1 disrupts its interaction with GFI1.
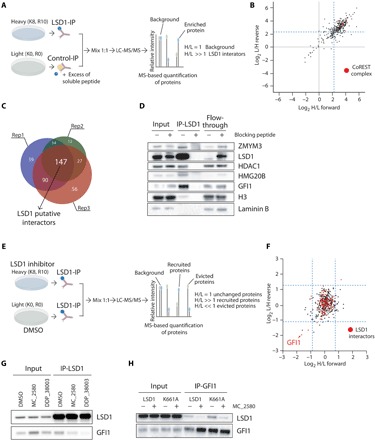
(A) Schematic representation of SILAC mass spectrometry approach to identify LSD1 interactors in NB4 cells. LC-MS/MS, liquid chromatography–tandem mass spectrometry. (B) Scatterplot showing log2 (heavy/light) ratio of forward reaction on the x axis (Rep1) and the log2 (light/heavy) ratio of reverse reaction on the y axis (Rep3). In the top right quadrant are represented LSD1 interactors. The blue dashed lines define the threshold used to define the LSD1 interactors from the background. Proteins belonging to the CoREST complex are shown in red dots. (C) Venn diagrams with numbers of individual and overlapping putative LSD1 interactors identified in the three different SILAC replicates. (D) Western blot analysis of LSD1 and some identified interactors in LSD1 IPs, with or without blocking peptide. Lamin B1 is used as loading control. (E) Schematic representation of SILAC mass spectrometry approach to identify recruited and evicted interactors of LSD1, upon LSD1 pharmacological inhibition with 2 μM MC_2580 for 24 hours. (F) Scatterplot showing log2 (heavy/light) ratio of forward reaction on the x axis and the log2 (light/heavy) ratio of reverse reaction on the y axis. Proteins recruited by LSD1 after inhibition are present in the top right quadrant, while proteins evicted from the interaction with LSD1 after drug treatment are found in the bottom left quadrant. Proteins previously identified as interactors of LSD1 in NB4 are shown as red dots. The blue dashed lines define the threshold used to determine recruited and evicted proteins. (G) Western blot analysis of LSD1 and GFI1 in LSD1 IPs in NB4 cells treated for 24 hours with DMSO, 2 μM MC_2580, or 2 μM DDP_38003. (H) Western blot analysis of LSD1 and GFI1 in GFI1 IPs in NB4 LSD1 KO cells transduced with empty vector, wild-type, or catalytic inactive K661A-LSD1, treated with 2 μM MC_2580 or DMSO.
We then profiled the changes in the LSD1 interactome following treatment with MC_2580 (Fig. 4E). Most of the interactors remained stably associated with LSD1 upon drug treatment. The binding of the known interactor GFI1 to LSD1, however, was strongly affected by the inhibitor (Fig. 4F). IP of endogenous GFI1 with anti-LSD1 antibodies in untreated and treated cells (using two different LSD1i, MC_2580 and DDP_38003) confirmed the results (Fig. 4G). Reciprocal IP using anti-GFI1 antibodies in untreated and treated NB4 cells with the drugs MC_2580 and DDP_38003 validated the previous results (fig. S7A). The catalytic inactive LSD1 mutant (K661A-LSD1) bound to GFI1 in a comparable manner to wild-type LSD1, and the LSD1i reduced the LSD1 mutant/GFI1 interaction (Fig. 4H and fig. S7) as well. These data show that the LSD1i that we used hamper the interaction between LSD1 and GFI1, implying that LSD1i target catalytic and noncatalytic functions of LSD1.
Sensitization of LSD1 inhibition to RA treatment depends on the interaction LSD1-GFI1 in APL and non-APL AML cells
We then analyzed the genome-wide distribution of binding sites for GFI1 and LSD1 by ChIP-seq, which resulted to be largely overlapping (Fig. 5A). Upon treatment with MC_2580, LSD1 was evicted from 732 GFI1-bound regions (Fig. 5B). We analyzed three GFI1 target genes and found that HDAC1 was also displaced (Fig. 5C). GFI1 recruitment to chromatin, in contrast, appears to be less affected (fig. S8).
Fig. 5. The interaction of LSD1 with GFI1 is fundamental for LSD1 activity in APL cells.
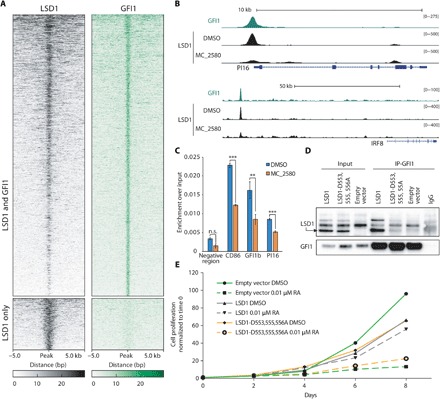
(A) Heat maps showing ChIP-seq signal of LSD1 and GFI1 ranked according to decrescent LSD1 signal. bp, base pairs. (B) Representative examples of overlapping LSD1 and GFI1 binding regions. University of California Santa Cruz Genome Browser profile of LSD1 and GFI1 ChIP-seq on the PI16 gene promoter and interferon regulatory factor 8 (IRF8) putative enhancer is shown. (C) HDAC1 recruitment (measured by ChIP-qPCR) at three genomic regions bound by both LSD1 and GFI1, before and after treatment with MC_2580. n.s., not significant. **P < 0.01 and ***P < 0.001. (D) Western blot analysis of LSD1 and GFI1 in GFI IP assays performed in NB4 LSD1 KO cells reconstituted with wild-type LSD1, LSD1-D553,555,556A mutant, or empty vector (as control). IgG, immunoglobulin G. (E) Growth curves of NB4 LSD1 KO cells transduced with wild-type LSD1, LSD1-D553,555,556A, or empty vector and treated for the indicated time with 0.01 μM RA (or DMSO as control).
To validate our hypothesis that the disruption of LSD1-GFI1 interaction is the causative event for the sensitization to RA by LSD1i, we took advantage of an LSD1 mutant (D553,555,556A) that is not able to bind the SNAIL/GFI1 (SNAG) domain of GFI1, which is required for their interaction (Fig. 5D) (24). Forced expression of D553,555,556A-LSD1 did not rescue NB4 KO cells from their sensitivity to low doses of RA (Fig. 5E), failing to establish a differentiation block and confirming the validity of our hypothesis.
Last, we decided to check whether these findings are restricted to APL or can be observed also in other AML subtypes. We therefore knocked out LSD1 in MOLM13 cells, an acute monocytic leukemia AML cell line resistant to LSD1 inhibition (Fig. 6A). LSD1 depletion did not impair the viability of MOLM13 cells but strongly potentiated the differentiation induced by RA treatment (Fig. 6, B and C). RA treatment of LSD1 KO cells reconstituted with either wild-type LSD1 or the catalytic mutant K661A showed restoration of the phenotype observed for wild-type MOLM13 cells, while reconstitution with the LSD1-D553,555,556A mutant continued to show an enhanced sensitivity to RA (Fig. 6, D to F).
Fig. 6. The interaction of LSD1 with GFI1 is fundamental for LSD1 activity in non-APL AML cells.
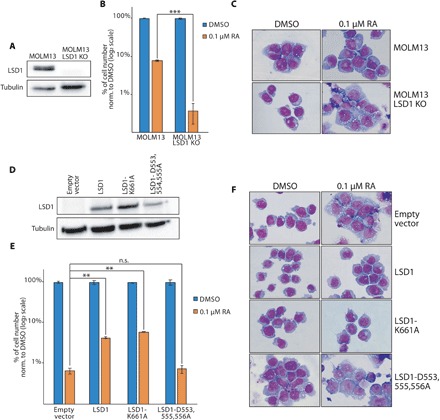
(A) Western blot analysis of LSD1 in MOLM13 wild-type and LSD1 KO cells. Tubulin served as loading control. (B) Treatment of wild-type and LSD1 KO MOLM13 cells with 0.1 μM RA (or DMSO as control). P values were obtained by unpaired Student’s t test (***P < 0.001). (C) Morphological analysis of MOLM13 wild-type and KO cells after 11 days of treatment with 0.1 μM. (D) Western blot analysis of LSD1 in MOLM13 LSD1 KO cells transduced with the indicated vectors. (E) Treatment of MOLM13 LSD1 KO cells transduced with either wild-type LSD1, LSD1-K661A, and LSD1-D553,555,556A vectors and treated with 0.1 μM RA. P values were obtained by unpaired Student’s t test (**P < 0.01). (F) Morphological analysis (May-Grünwald-Giemsa staining) of MOLM13 LSD1 KO cells transduced with the indicated vectors and treated for 11 days with 0.1 μM RA.
Together, these data demonstrate that LSD1-GFI1 interaction is fundamental for the establishment of a differentiation block by LSD1 in AML cells, and that the sensitization to RA triggered by LSD1i relies on the disruption of LSD1-GFI1 interaction.
DISCUSSION
LSD1 has emerged as an interesting target for cancer therapy, and LSD1i have entered clinical trials for treatment of several cancer types, including AML. Only a minority of AML cells are sensitive to LSD1 inhibition as single treatment (25); a strong cooperative action of LSD1i and RA can be, however, measured even in those AML subtypes not responsive to either drug alone (11), justifying a clinical investigation of this approach (ClinicalTrials.gov: NCT02261779, NCT02273102, and NCT02717884). An analysis of the mechanism of action of LSD1i in combination with RA has not been reported so far: Focusing on this phenomenon in cells resistant to LSD1i provides useful insights to optimize the use of LSD1i in a clinical setting.
Here, we show that the combination of RA and LSD1i, or LSD1 genetic KO, leads to complete differentiation of AML cells resistant to LSD1i alone. LSD1 inhibition has almost no impact on global gene expression, but the combination of RA and LSD1i leads to the induction of a large set of genes involved in cellular differentiation and to profound changes in chromatin structure, including activation and decommissioning of super-enhancers. Since these changes occur in the continuous presence of PML-RAR, they must bypass the action of the oncogene, and in fact, we show that this mechanistic model applies also to other AML subtypes, not expressing PML-RAR.
The correlation between transcriptional derepression and LSD1 catalytic activity is an open debate. LSD1 demethylase activity seems to be essential to maintain the undifferentiated state of human embryonic stem cells (26) and to override oncogene-induced senescence (27). In contrast, the catalytic activity of LSD1 is not required to block differentiation of AML cells sensitive to LSD1 inhibition (28).
Here, we analyze the role of LSD1 catalytic activity in AML cells resistant to LSD1 inhibition. We show that reconstitution of LSD1 KO cells with either the wild-type or a catalytically inactive form of the enzyme recovers the differentiation block of leukemic cells, although the inactive LSD1 (unlike the wild-type form) fails to modify chromatin at its targets. We therefore conclude that the catalytic activity of LSD1 is not involved in the establishment of a differentiation block in AML cells resistant to LSD1 inhibition.
We used a proteomic approach to analyze the LSD1 interactome in the absence and in the presence of LSD1 inhibition. We show that while most interactors remain associated with LSD1 upon treatment with MC_2580, the inhibitor leads to the selective dissociation of GFI1 from LSD1. On the basis of these results, we hypothesize that the LSD1 complex is therefore not affected by LSD1 inhibition and that the inhibitor acts on the LSD1-GFI1 direct interaction. In particular, LSD1 interacts with GFI1 through the SNAG domain present at the N terminus of GFI1, which works as a molecular hook to recruit LSD1 (24). Consistently, upon inhibition, LSD1 (and LSD1-associated proteins, such as HDAC1) is evicted from chromatin at GFI1 target genes. The interaction of GFI1 with LSD1 is critical for the differentiation block maintained by LSD1 in the absence of RA. Similarly, the association of LSD1 with GFI1 has been reported to be also critical for maintaining the differentiation block of AML cells (28, 29) and medulloblastoma cells (30) sensitive to LSD1i.
The small-molecule LSD1i used in this study (MC_2580 and DDP_38003) are thus still able to dissociate GFI1 from a catalytically inactive LSD1. This feature can be explained from the three-dimensional structure of the enzyme-inhibitor and enzyme-GFI1 complexes: The inhibitor and the transcription factor binding sites fully overlap. The inhibitor can therefore be expected to create steric hindrance that prevents GFI1 binding (31).
Together, our results point to a model where in AML cells resistant to LSD1 inhibition, LSD1 acts primarily as a scaffolding module to recruit the CoREST complex to transcription factors such as GFI1, which functions as a brake to differentiation stimuli (such as RA). The inhibitors of LSD1 that we used are able to trigger dissociation of the complex from GFI1 and favor differentiation by RA. Although further in vivo studies are needed to fully understand the implications of our observations, these results are of both mechanistical and translational relevance and, since most tumor cells tolerate LSD1 inhibition (25), further support the use of LSD1i in combination therapy.
MATERIALS AND METHODS
Compounds
RA was purchased from Sigma-Aldrich S.r.l. (R2625). MC_2580 and DDP_38003 have been synthesized as previously described (17, 20).
Cell culture
All cell lines were grown according to American Type Culture Collection recommendations. Cultures were maintained in a humidified tissue culture incubator at 37°C in 5% CO2.
SILAC labeling of NB4 cells
For standard SILAC labeling, NB4 cells were grown in “light” and “heavy” SILAC RPMI 1640 (Thermo Fisher Scientific, 89984) supplemented with either l-arginine and l-lysine or their heavy isotope counterparts l-arginine-13C6, 15N4 hydrochloride (Arg10; Sigma-Aldrich, 608033) and l-lysine-13C6, 15N2 hydrochloride (Lys8; Sigma-Aldrich, 608041) (32). All media were supplemented with 10% dialyzed fetal bovine serum (FBS) (26400-044, Gibco, Life Technologies), 1% glutamine, penicillin (100 U/ml), and streptomycin (100 mg/ml).
In vivo studies
For in vivo studies, lineage-negative (lin−) cells were isolated from the bone marrow of 8- to 10-week-old 129SvEv mice, and murine PML-RAR cells were generated using a retroviral transduction and transplantation approach as previously described (33). The animals were checked periodically for clinical signs of disease and for presence of blast cells by May-Grünwald-Giemsa staining of blood smears. All-trans retinoic acid (ATRA) pellets (21-day release, 5 mg) or placebo pellets were implanted subcutaneously by trocar injection 10 days after cell injection. DDP_38003 was dissolved in a vehicle composed of 40% polyethylene glycol, molecular weight 400 in a 5% glucose solution and orally administered at the dose of 17 mg/kg 2 days per week, for 3 weeks. For the combination, mice were dosed with ATRA pellets (21-day release, 5 mg) and DDP_38003 at the dose of 17 mg/kg following the administration schedule of the single treatment. In vivo studies were performed after approval from our fully authorized animal facility and our institutional welfare committee Organismo Preposto al Benessere degli Animali (OPBA) and notification of the experiments to the Ministry of Health (as required by the Italian law; Institutional Animal Care and Use Committee numbers: 759/2015 in accordance with European Union directive 2010/63).
Proliferation, cell viability, methylcellulose plating, and morphological characterization
For proliferation assays, cells were counted in trypan blue (Sigma-Aldrich, St. Louis, MO, USA). CellTiter-Glo luminescent cell viability assays (Promega, Madison, WI, USA) were performed following the manufacturer’s instructions.
Colony-forming assays were performed in methylcellulose medium (MethoCult H4444, Stem Cell Technology, Vancouver, BC, Canada) containing 10% FBS. After 7 to 10 days of culture, colonies were scored.
Cytospin preparations obtained from 200,000 cells per sample were stained for 8 min with May-Grünwald solution, washed three times in deionized water, and then incubated for 40 min with Giemsa solution. After three more washes, samples were air-dried and evaluated.
Flow cytometry
Fluorescence-activated cell sorting (FACS) analyses were performed using a FACSCalibur flow cytometer (BD Biosciences, Oxford, UK). Cell sorting experiments were performed using a FACSAria cell sorter (BD Biosciences, Oxford, UK). The antibody used for flow cytometry is CD11b-PE clone ICRF44 (eBioscience Inc., San Diego, CA).
Gene editing
The CRISPR-Cas9 gene editing vector lentiCRISPR v2 was a gift from F. Zhang (Addgene, plasmid no. 52961). The CRISPR target sequence CGCGGAGGCTCTTTCTTGCG in exon 1 was selected to knock out LSD1 and cloned into lentiCRISPR v2 vector following the manufacturer’s instructions.
Lentiviral vectors, retroviral vectors, and expression constructs
The lentiviral CRISPRv2 vector was cotransfected with vesicular stomatitis virus glycoprotein and dR8.2 plasmids into 293T cells, and the viral supernatants were produced as previously described (33). Cells were spin-infected with viral supernatant and selected with puromycin-containing medium for 3 days. After clonal growth by limiting dilution, sublines were screened by Western blotting against LSD1.
To knock down LSD1, two short hairpin RNA (shRNA) sequences were tested. Interfering sequences were cloned into the LMP vector by Xho–Eco RI double digestion. shRNA transcription in these vectors is RNA polymerase II mediated and is under the control of an long terminal repeat promoter and expresses puromycin resistance cassette (shLSD1#3, TGCTGTTGACAGTGAGCGAAGTGATACTGTGCTTGTCCACTAGTGAAGCCACAGATGTAGTG GACAAGCACAGTATCACTGTGCCTACTGCCTCGGA and shLSD1#5, TGCTGTTGACAGTGAGCGATCTCAGAAGATGAGTATTATTTAGTGAAGCCACAGATGTAAAT AATACTCATCTTCTGAGAGTGCCTACTGCCTCGGA).
LSD1 N-terminal truncated (172-833) wild-type and catalytic mutant (K661A-LSD1) constructs were a gift by E. Battaglioli (University of Milan). Constructs were PCR-amplified from original vectors and cloned into pCR 2.1-TOPO (Invitrogen) using the following primers: LSD1 forward, ATGTCGGGTGTGGAGGGCGCAGCTTTC and LSD1 reverse, TCACATGCTTGGGGACTGCTGTGC.
Products were then subcloned into the Eco RI site of the retroviral PINCO vector for ectopic expression. To make the D553,555,556A triple mutant construct described in (24), the following primers were used in three sequential site-directed mutagenesis reactions using the pCR-TOPO-LSD1WT as the template vector: LSD1_D553A forward, CTTAAGCACTGGGCTCAGGATGATGACTTTGAGTTC; LSD1_D553A reverse, GAACTCAAAGTCATCATCCTGAGCCCAGTGCTTAAG; LSD1_D553,555A forward, CTTAAGCACTGGGCTCAGGCTGATGACTTTGAGTTC; LSD1_D553,555A reverse, GAACTCAAAGTCATCAGCCTGAGCCCAGTGCTTAAG; LSD1_D553,555,556A forward, CTTAAGCACTGGGCTCAGGCTGCTGACTTTGAGTTC; and LSD1_D553,555,556A reverse, GAACTCAAAGTCAGCAGCCTGAGCCCAGTGCTTAAG.
Western blot
Antibodies used for Western blot analyses were as follows: actin (Sigma-Aldrich, no. A4700, clone AC-40), GFI1 (Santa Cruz Biotechnology, sc-376949, clone B9), GSE1 (Proteintech, no. 24947-I-AP), H3 (Abcam, no. 1791), H3K4me1 (Abcam, no. 8895), H3K4me2 (Abcam, no. 32356), H3K4me3 (Active Motif, no. 39159), HDAC1 (Abcam, no. 7028), HMG20B (Proteintech, no. 14582-1-AP), laminin B (Abcam, no. 16048), LSD1 (Cell Signaling Technology, no. 2139 and Abcam, no. 17721), RAR (Santa Cruz Biotechnology, sc-551), tubulin (Santa Cruz Biotechnology, sc-32356), vinculin (Sigma-Aldrich, no. V9131, clone hVIN-1), and ZMYM3 (Abcam, no. 106626).
Immunofluorescence
NB4 cells, treated as indicated for 24 hours, were transferred by cytospin to histological glasses and fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and blocked with 5% normal goat serum. Staining was performed using a mouse anti-human PML antibody (PGM3) to reveal PML nuclear bodies or micro-speckles (34). A Cy3-conjugated goat anti-mouse (Jackson ImmunoResearch Laboratories Inc., West Grove, PA, USA) was used as secondary antibody.
ChIP and ChIP-seq analyses
Detailed protocols for ChIP are given in the Supplementary Materials. For histone modification, 3 × 106 cells per IP and primary antibody (4 μg/ml) were used; for LSD1, GFI1, and PML ChIP, 40 × 106 cells per IP were used (10 μg/ml). The obtained DNA was then quantified by PicoGreen and processed for ChIP-seq library preparation (as described for the Illumina protocol) or used for qPCR. For libraries preparation, 2 ng of immunoprecipitated DNA was used. Antibodies used for ChIP are as follows: H3K4me1 (Abcam, no. 8895), H3K4me2 (Abcam, no. 32356), H3K4me3 (Active Motif, no. 39159), H3K27ac (Abcam, no. 4729), LSD1 (Abcam, no. 17721), GFI1 (Abcam, no. 21061), PML (Santa Cruz Biotechnology, sc-5621), and immunoglobulin G rabbit (Jackson ImmunoResearch, lot no. 134230).
Chromatin immunoprecipitation–quantitative polymerase chain reaction
For the validation of specific regions, ChIP-qPCRs were performed as follows. Immunoprecipitated DNA was diluted in 9.6 μl of H2O per reaction, plus 400 nM primers in a final volume of 20 μl in SYBR Green. Each ChIP experiment was performed at least three times with biological replicates. For ChIP-qPCRs, the following primers were used: negative control forward, AGCTATCTGTCGAGCAGCCAAG; negative control reverse, CATTCCCCTCTGTTAGTGGAAGG; PRAM1 forward, CCACAGAGCCTCCCCTAGA; PRAM1 reverse, TGCAACACCTCCCTGTGA; peptidase inhibitor 16 (PI16) forward, AGCCCTCACAGATGAGGAGA; PI16 reverse: GCCACACTTACCATGTGCAG; Integrin subunit alpha (ITGAM) forward, GGAGGAGAAGTGACATGGCT; ITGAM reverse, AGGCAAAGTGGAGATGGTGA; transgutaminase 2 (TGM2) forward, CAGATACAGACACACGCAGC; TGM2 reverse, TGGGGAGGTGTTCTTGATCC; CD86 forward, ACAGTCATTGCCGAGGAAGG; CD86 reverse, CTCATCCGTGTGTCTGTGCT; GFI1b forward, CAGGGAGGGGAACAGAAGAG; and GFI1b reverse, GAACTGCAAAGCCTCTCTCG.
Active enhancers, active promoters, and super-enhancer identification and analysis
Active enhancers were defined as regions with overlapping peaks of H3K4me1 and H3K27ac that fall into distal genomic regions [defined as −20/−5 kb and +5/+20 kb from transcription start sites (TSS)]. Active promoters were defined as regions with overlapping peaks of H3K4me3 and H3K27ac falling within 5 kb of RefSeq TSS. Coordinates of active enhancers and active promoters overlapping LSD1 peaks were used for histone mark analysis. Read coverage in those regions was computed with BEDTools suite version 2.17.0 (35) and normalized according to sequencing depth of each sample.
MACS peaks of H3K27ac over background were used for super-enhancer identification. Constituent enhancers within 12,500 base pairs were stitched together and ranked by input-subtracted signal of H3K27ac; enhancers located within a window of ±2.5 kb around TSS of RefSeq genes were excluded from the analysis. Super-enhancers were identified as those above the inflection point of the H3K27ac signal versus enhancer rank (36).
RNA extraction and reverse transcription qPCR
Total RNA was extracted from NB4 cells with TRIzol (Invitrogen) and then purified using the RNeasy kit (Qiagen). Reverse transcription was performed with the SuperScript II Kit (Invitrogen), according to the manufacturer’s protocol. qPCRs were performed in triplicates in 20 μl of final reaction volume containing SYBR Green buffer (Applied Biosystems), 20 ng of cDNA retrotranscribed from the RNA, and 0.4 μM of each primer mix. All the qPCR amplifications were performed in the AB-7000 sequence detection system (Applied Biosystems) at 50°C for 2 min and 95°C for 10 min, followed by 40 cycles at 95°C. For reverse transcription qPCRs, the following primers were used: glyceraldehyde phosphate dehydrogenase (GAPDH) forward, GCCTCAAGATCATCAGCAATGC; GAPDH reverse, CCACGATACCAAAGTTGTCATGG; CD11b forward, AACCCCTGGTTCACCTCCT; CD11b reverse, CATGACATAAGGTCAAGGCTGT; GFI1b forward, CAGGGAGGGGAACAGAAGAG; and GFI1b reverse, GAACTGCAAAGCCTCTCTCG.
RNA-seq and data analysis
mRNA-seq libraries were prepared according to the TruSeq low sample protocol (Illumina, San Diego, CA, USA), starting with 1 μg of total RNA per sample. Details are provided in the Supplementary Materials.
Protein coimmunoprecipitation analysis
NB4 nuclear fraction was quantified and diluted in IP buffer [10 mM tris HCl (pH 7.6), 150 mM NaCl, and 0.2% NP-40] supplemented with 1× Roche protease inhibitors and 0.5 mM phenylmethylsulfonyl fluoride (PMSF) to obtain a concentration of 2 mg/ml. Preclearing of the lysate was carried out by 1-hour incubation with protein G magnetic beads. The lysate was requantified and diluted with the IP buffer supplemented with protease inhibitors up to a concentration of 1.3 mg/ml. An aliquot of the input was collected before the addition of the desired antibody. The samples were incubated with the antibody overnight on a rotating wheel at 4°C. The following day, 50 to 100 μl of Dynabeads Protein G preequilibrated in phosphate-buffered saline (PBS) and supplemented with 0.5% bovine serum albumin (BSA) was added to the extracts and incubated for 3 hours on a rotating wheel at 4°C. Beads were then washed three times with IP buffer and once with washing buffer IP [10 mM tris HCl (pH 7.6), 250 mM NaCl, and 0.2% NP-40]. Then, bound material was eluted by incubation with LDS Sample Buffer (NuPAGE), supplemented with 100 mM dithiothreitol (DTT), at 95°C for 5 min. Eluted samples were loaded on SDS–polyacrylamide gel electrophoresis (PAGE) for protein separation and subsequent analysis. Antibodies used for protein coimmunoprecipitation are as follows: LSD1 (LSD1; Abcam, no. 17721) and GFI1 (sc-376949).
LSD1 coimmunoprecipitation for mass spectrometry analysis of protein-protein interactions
The protein content of the nuclear fraction of light and heavy samples was quantified by Bradford assay and diluted to a concentration of 2 mg/ml in IP buffer [10 mM tris HCl (pH 7.6), 150 mM NaCl, and 0.2% NP-40], supplemented with 1× protease inhibitors (Roche) and 0.5 mM PMSF. Preclearing of the lysates was achieved by incubation with protein G magnetic beads for 1 hour on a rotating wheel at 4°C. The lysates were then requantified and diluted with the IP buffer supplemented with protease inhibitors to a concentration of 1.3 mg/ml. A minor fraction of input was collected before adding 10 μg of anti-LSD1 antibody to each sample. All samples were then incubated on a rotating wheel at 4°C overnight. For the acquisition of the basal LSD1 interactome, 120-fold molar excess of LSD1 blocking peptide was incubated together with the antibody as negative control. This peptide competes with the bait and all its coassociated factors for the antibody binding (37). The peptide was added to the light channel in the forward experiment (Rep1 and Rep2) and to the heavy channel in the reverse experiment (Rep3). Instead, for the dynamic LSD1 interactome, the inhibitor MC_2580 was added to the heavy channel in the forward experiment and in the light one in the reverse replicate. The following day, 100 μl of Dynabeads Protein G, preequilibrated in PBS supplemented with 0.5% BSA, was added to each sample and incubated for 3 hours on a rotating wheel at 4°C. Beads were then washed three times with IP buffer and once with the washing buffer IP [10 mM tris HCl (pH 7.6), 250 mM NaCl, and 0.2% NP-40]. In the last washing step, light and heavy samples of each SILAC replicate were mixed, and the coimmunoprecipitated proteins were eluted by incubation at 95°C for 5 min with the LDS Sample Buffer (NuPAGE-Invitrogen) supplemented with 100 mM DTT. Samples were loaded on an SDS-PAGE gradient gel for subsequent protein separation and mass spectrometry analysis.
Antibody used for the preparative LSD1 coimmunoprecipitation was LSD1 (Abcam, no. 17721). The blocking peptide used as mock control was Human KDM1/LSD1 peptide (ab17763).
In-gel digestion of immunoprecipitated proteins
In-gel digestion of gel-separated proteins was carried out with trypsin, as previously described (38). After digestion and extraction from the gel pieces, the digested peptides were desalted and concentrated by reversed-phase chromatography onto C18 Stage Tip microcolumns (39). Peptides were then eluted from the stage tips with buffer B (80% acetonitrile and 0.5% acetic acid), lyophilized, resuspended in 0.1% formic acid (FA), and subjected to liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis.
Liquid chromatography and tandem mass spectrometry
Peptide mixtures were analyzed by online nanoflow LC-MS/MS using an EASY-nLC 1000 (Thermo Fisher Scientific, Odense, Denmark) connected to a quadrupole/Orbitrap mass spectrometer (Q Exactive HF, Thermo Fisher Scientific) through a nanoelectrospray ion source. The nano-LC system was operated in one column setup with EASY-Spray LC Columns (length, 50 cm; inner diameter, 75 μm) packed with C18 reversed-phase resin (2 μm). Solvent A was 0.1% FA in ddH2O, and solvent B was 80% ACN with 0.1% FA. Peptides were injected at a flow rate of 500 nl/min and separated with a gradient of 5 to 30% solvent B over 80 min, followed by a gradient of 30 to 60% for 10 min and 60 to 95% over 3 min at a flow rate of 250 nl/min. The Q-Exactive was set in the data-dependent mode to automatically switch between full-scan MS and MS/MS acquisition. MS experiments consisted of an Orbitrap full scan, followed by the acquisition of the 15 most intense peptide ions (top 15) detected in the survey MS scan. Mass spectrometer conditions for all experiments were as follows: full MS [Automatic Gain Control (AGC), 3 × 106; resolution, 60,000; mass/charge ratio (m/z) range, 375 to 1650; maximum ion time, 20 ms] and MS/MS (AGC, 15,000; maximum ion time, 80 ms; isolation width, 2 m/z with a dynamic exclusion time of 20 s). Singly charged ions and ions for which no charge state could be determined were excluded from the selection. Normalized collision energy was set to 28%. Spray voltage was set to 1.9 kV. No sheath and auxiliary gas flow. Heated capillary temperature was 275°C; S-lens radio frequency level is 50%.
LSD1 interactome data analysis
MaxQuant outputs were manually filtered for the LSD1 interactome analysis according to the following parameters: Proteins were considered in the analysis if identified with more than two peptides of which at least one is unique and ratio count is greater than 1 (RC > 1). Peptides of the antibody used for the IP as well as of epidermal and hair proteins (such as keratin variants and dermcidin), if identified, were removed (i.e., light contaminant). To analyze the putative LSD1 interactors in NB4 cells, the “mixtools” R package was used to define two populations of specific interacting and background proteins based on their SILAC ratios. Mean (μ) and SD (σ) of the interacting protein population were calculated. Proteins with a SILAC ratio higher than μ-σ were considered significant. To identify the modulated proteins after LSD1 pharmacological inhibition, a statistical approach based on the calculation of the median and the SD (σ) of the protein distribution was applied. Proteins with a SILAC ratio higher than the median +2σ were defined as recruited, while proteins with a SILAC ratio lower than the median –2σ were defined as evicted. Once the distributions of each replicate are analyzed, the modulated proteins were filtered on the basis of the results of the basal LSD1 interactome, which sets the list of specific binders. Analysis of the protein distribution of each SILAC replicate and scatter plots were obtained with Perseus software.
Statistical analysis
Data are presented as means ± SD. Statistical comparisons were carried out using two-tailed unpaired Student’s t tests, unless otherwise specified. The number of biological (n) or technical replicates, the type of statistical analyses performed, and statistical significance are reported in the corresponding figures and figure legends.
Supplementary Material
Acknowledgments
Funding: This work was supported by AIRC (grant number no. 19086), CNR Flagship Program EPIGEN, and TRANSCAN DRAMA project JTC 2014-48. Research in T.B.’s group was supported by AIRC (grant number 15741) and CNR Flagship Program EPIGEN. Research in A.Mat.’s laboratory was supported by AIRC (IG-11342) and MIUR (Flagship EPIGEN project and PRIN 20152TE5PK). Research in A. Mai’s group was supported by AIRC (grant no. IG16 19162), MIUR (PRIN 20152TE5PK), and the Italian Ministry of Health (RF-2010-2318330). Author contributions: R.R., L.N., A.H., P.L.R., and R.D.Z. performed experiments. R.R., L.N., E.C., P.L.R., I.B., and L.F. performed data analysis. C.M., M.V., A.Mat., A. Mai, and G.P. provided reagents, technical support, and contributed to experimental plans. S.V. and R.F. synthetized MC_2580. T.B. designed the MS experiments and analyzed the data together with L.N. and contributed to the interpretation of results. R.R. and S.M. conceived the study and wrote the manuscript. S.M. provided project supervision. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD012954. All sequencing data have been deposited into the Gene Expression Omnibus (GEO) repository with the accession number GSE128530. All other data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/15/eaax2746/DC1
REFERENCES AND NOTES
- 1.Shi Y., Lan F., Matson C., Mulligan P., Whetstine J. R., Cole P. A., Casero R. A., Shi Y., Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941–953 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Forneris F., Binda C., Vanoni M. A., Mattevi A., Battaglioli E., Histone demethylation catalysed by LSD1 is a flavin-dependent oxidative process. FEBS Lett. 579, 2203–2207 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Foster C. T., Dovey O. M., Lezina L., Luo J. L., Gant T. W., Barlev N., Bradley A., Cowley S. M., Lysine-specific demethylase 1 regulates the embryonic transcriptome and CoREST stability. Mol. Cell. Biol. 30, 4851–4863 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sprüssel A., Schulte J. H., Weber S., Necke M., Händschke K., Thor T., Pajtler K. W., Schramm A., König K., Diehl L., Mestdagh P., Vandesompele J., Speleman F., Jastrow H., Heukamp L. C., Schüle R., Dührsen U., Buettner R., Eggert A., Göthert J. R., Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia 26, 2039–2051 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Kerenyi M. A., Shao Z., Hsu Y.-J., Guo G., Luc S., O’Brien K., Fujiwara Y., Peng C., Nguyen M., Orkin S. H., Histone demethylase Lsd1 represses hematopoietic stem and progenitor cell signatures during blood cell maturation. eLife 2, e00633 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saleque S., Kim J., Rooke H. M., Orkin S. H., Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol. Cell 27, 562–572 (2007). [DOI] [PubMed] [Google Scholar]
- 7.van der Meer L. T., Jansen J. H., van der Reijden B. A., Gfi1 and Gfi1b: Key regulators of hematopoiesis. Leukemia 24, 1834–1843 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Möröy T., Vassen L., Wilkes B., Khandanpour C., From cytopenia to leukemia: The role of Gfi1 and Gfi1b in blood formation. Blood 126, 2561–2569 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thambyrajah R., Mazan M., Patel R., Moignard V., Stefanska M., Marinopoulou E., Li Y., Lancrin C., Clapes T., Möröy T., Robin C., Miller C., Cowley S., Göttgens B., Kouskoff V., Lacaud G., GFI1 proteins orchestrate the emergence of haematopoietic stem cells through recruitment of LSD1. Nat. Cell Biol. 18, 21–32 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Helin K., Minucci S., The role of chromatin-associated proteins in cancer. Annu. Rev. Cancer Biol. 1, 355–377 (2017). [Google Scholar]
- 11.Schenk T., Chen W. C., Göllner S., Howell L., Jin L., Hebestreit K., Klein H.-U., Popescu A. C., Burnett A., Mills K., Casero R. A. Jr., Marton L., Woster P., Minden M. D., Dugas M., Wang J. C. Y., Dick J. E., Müller-Tidow C., Petrie K., Zelent A., Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 18, 605–611 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris W. J., Huang X., Lynch J. T., Spencer G. J., Hitchin J. R., Li Y., Ciceri F., Blaser J. G., Greystoke B. F., Jordan A. M., Miller C. J., Ogilvie D. J., Somervaille T. C. P., The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell 21, 473–487 (2012). [DOI] [PubMed] [Google Scholar]
- 13.McGrath J. P., Williamson K. E., Balasubramanian S., Odate S., Arora S., Hatton C., Edwards T. M., O’Brien T., Magnuson S., Stokoe D., Daniels D. L., Bryant B. M., Trojer P., Pharmacological inhibition of the histone lysine demethylase KDM1A suppresses the growth of multiple acute myeloid leukemia subtypes. Cancer Res. 76, 1975–1988 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Altucci L., Minucci S., Epigenetic therapies in haematological malignancies: Searching for true targets. Eur. J. Cancer 45, 1137–1145 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Nasr R., Guillemin M.-C., Ferhi O., Soilihi H., Peres L., Berthier C., Rousselot P., Robledo-Sarmiento M., Lallemand-Breitenbach V., Gourmel B., Vitoux D., Pandolfi P. P., Rochette-Egly C., Zhu J., de Thé H., Eradication of acute promyelocytic leukemia-initiating cells through PML-RARA degradation. Nat. Med. 14, 1333–1342 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Ablain J., de The H., Revisiting the differentiation paradigm in acute promyelocytic leukemia. Blood 117, 5795–5802 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Binda C., Valente S., Romanenghi M., Pilotto S., Cirilli R., Karytinos A., Ciossani G., Botrugno O. A., Forneris F., Tardugno M., Edmondson D. E., Minucci S., Mattevi A., Mai A., Biochemical, structural, and biological evaluation of tranylcypromine derivatives as inhibitors of histone demethylases LSD1 and LSD2. J. Am. Chem. Soc. 132, 6827–6833 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Hauser A.-T., Bissinger E.-M., Metzger E., Repenning A., Bauer U.-M., Mai A., Schüle R., Jung M., Screening assays for epigenetic targets using native histones as substrates. J. Biomol. Screen. 17, 18–26 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Valente S., Rodriguez V., Mercurio C., Vianello P., Saponara B., Cirilli R., Ciossani G., Labella D., Marrocco B., Ruoppolo G., Botrugno O. A., Dessanti P., Minucci S., Mattevi A., Varasi M., Mai A., Pure diastereomers of a tranylcypromine-based LSD1 inhibitor: Enzyme selectivity and in-cell studies. ACS Med. Chem. Lett. 6, 173–177 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vianello P., Botrugno O. A., Cappa A., Zuffo R. D., Dessanti P., Mai A., Marrocco B., Mattevi A., Meroni G., Minucci S., Stazi G., Thaler F., Trifiró P., Valente S., Villa M., Varasi M., Mercurio C., Discovery of a novel inhibitor of histone lysine-specific demethylase 1A (KDM1A/LSD1) as orally active antitumor agent. J. Med. Chem. 59, 1501–1517 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Ablain J., Leiva M., Peres L., Fonsart J., Anthony E., de Thé H., Uncoupling RARA transcriptional activation and degradation clarifies the bases for APL response to therapies. J. Exp. Med. 210, 647–653 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Thé H., Chen Z., Acute promyelocytic leukaemia: Novel insights into the mechanisms of cure. Nat. Rev. Cancer 10, 775–783 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Martens J. H. A., Brinkman A. B., Simmer F., Francoijs K.-J., Nebbioso A., Ferrara F., Altucci L., Stunnenberg H. G., PML-RARα/RXR alters the epigenetic landscape in acute promyelocytic leukemia. Cancer Cell 17, 173–185 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Lin Y., Wu Y., Li J., Dong C., Ye X., Chi Y.-I., Evers B. M., Zhou B. P., The SNAG domain of Snail1 functions as a molecular hook for recruiting lysine-specific demethylase 1. EMBO J. 29, 1803–1816 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohammad H. P., Smitheman K. N., Kamat C. D., Soong D., Federowicz K. E., van Aller G. S., Schneck J. L., Carson J. D., Liu Y., Butticello M., Bonnette W. G., Gorman S. A., Degenhardt Y., Bai Y., McCabe M. T., Pappalardi M. B., Kasparec J., Tian X., McNulty K. C., Rouse M., McDevitt P., Ho T., Crouthamel M., Hart T. K., Concha N. O., McHugh C. F., Miller W. H., Dhanak D., Tummino P. J., Carpenter C. L., Johnson N. W., Hann C. L., Kruger R. G., A DNA hypomethylation signature predicts antitumor activity of LSD1 inhibitors in SCLC. Cancer Cell 28, 57–69 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Adamo A., Sesé B., Boue S., Castaño J., Paramonov I., Barrero M. J., Belmonte J. C. I., LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells. Nat. Cell Biol. 13, 652–659 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Yu Y., Schleich K., Yue B., Ji S., Lohneis P., Kemper K., Silvis M. R., Qutob N., van Rooijen E., Werner-Klein M., Li L., Dhawan D., Meierjohann S., Reimann M., Elkahloun A., Treitschke S., Dörken B., Speck C., Mallette F. A., Zon L. I., Holmen S. L., Peeper D. S., Samuels Y., Schmitt C. A., Lee S., Targeting the senescence-overriding cooperative activity of structurally unrelated H3K9 demethylases in melanoma. Cancer Cell 33, 322–336.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maiques-Diaz A., Spencer G. J., Lynch J. T., Ciceri F., Williams E. L., Amaral F. M. R., Wiseman D. H., Harris W. J., Li Y., Sahoo S., Hitchin J. R., Mould D. P., Fairweather E. E., Waszkowycz B., Jordan A. M., Smith D. L., Somervaille T. C. P., Enhancer activation by pharmacologic displacement of LSD1 from GFI1 induces differentiation in acute myeloid leukemia. Cell Rep. 22, 3641–3659 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barth J., Abou-El-Ardat K., Dalic D., Kurrle N., Maier A.-M., Mohr S., Schütte J., Vassen L., Greve G., Schulz-Fincke J., Schmitt M., Tosic M., Metzger E., Bug G., Khandanpour C., Wagner S. A., Lübbert M., Jung M., Serve H., Schüle R., Berg T., LSD1 inhibition by tranylcypromine derivatives interferes with GFI1-mediated repression of PU.1 target genes and induces differentiation in AML. Leukemia 33, 1411–1426 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Lee C., Rudneva V. A., Erkek S., Zapatka M., Chau L. Q., Tacheva-Grigorova S. K., Garancher A., Rusert J. M., Aksoy O., Lea R., Mohammad H. P., Wang J., Weiss W. A., Grimes H. L., Pfister S. M., Northcott P. A., Wechsler-Reya R. J., Lsd1 as a therapeutic target in Gfi1-activated medulloblastoma. Nat. Commun. 10, 332 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baron R., Binda C., Tortorici M., McCammon J. A., Mattevi A., Molecular mimicry and ligand recognition in binding and catalysis by the histone demethylase LSD1-CoREST complex. Structure 19, 212–220 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ong S.-E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M., Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Minucci S., Monestiroli S., Giavara S., Ronzoni S., Marchesi F., Insinga A., Diverio D., Gasparini P., Capillo M., Colombo E., Matteucci C., Contegno F., Lo-Coco F., Scanziani E., Gobbi A., Pelicci P. G., PML-RAR induces promyelocytic leukemias with high efficiency following retroviral gene transfer into purified murine hematopoietic progenitors. Blood 100, 2989–2995 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Di Croce L., Raker V. A., Corsaro M., Fazi F., Fanelli M., Faretta M., Fuks F., Coco F. L., Kouzarides T., Nervi C., Minucci S., Pelicci P. G., Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 295, 1079–1082 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Quinlan A. R., Hall I. M., BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hnisz D., Abraham B. J., Lee T. I., Lau A., Saint-André V., Sigova A. A., Hoke H. A., Young R. A., Super-enhancers in the control of cell identity and disease. Cell 155, 934–947 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soldi M., Bonaldi T., The proteomic investigation of chromatin functional domains reveals novel synergisms among distinct heterochromatin components. Mol. Cell. Proteomics 12, 764–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shevchenko A., Tomas H., Havli J., Olsen J. V., Mann M., In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Rappsilber J., Mann M., Ishihama Y., Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Barozzi I., Termanini A., Minucci S., Natoli G., Fish the ChIPs: A pipeline for automated genomic annotation of ChIP-Seq data. Biol. Direct 6, 51 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Langmead B., Trapnell C., Pop M., Salzberg S. L., Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cesaroni M., Cittaro D., Brozzi A., Pelicci P. G., Luzi L., CARPET: A web-based package for the analysis of ChIP-chip and expression tiling data. Bioinformatics 24, 2918–2920 (2008). [DOI] [PubMed] [Google Scholar]
- 43.Ramírez F., Ryan D. P., Grüning B., Bhardwaj V., Kilpert F., Richter A. S., Heyne S., Dündar F., Manke T., deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trapnell C., Williams B. A., Pertea G., Mortazavi A., Kwan G., van Baren M. J., Salzberg S. L., Wold B. J., Pachter L., Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cox J., Mann M., MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008). [DOI] [PubMed] [Google Scholar]
- 46.Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M., Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Maile T. M., Izrael-Tomasevic A., Cheung T., Guler G. D., Tindell C., Masselot A., Liang J., Zhao F., Trojer P., Classon M., Arnott D., Mass spectrometric quantification of histone post-translational modifications by a hybrid chemical labeling method. Mol. Cell. Proteomics 14, 1148–1158 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/15/eaax2746/DC1